

Abstract
Changes in protein ubiquitination have been linked to cancer. Deubiquitinating enzymes (DUBs) counteract E3 ligase activities and have emerged as promising targets for cancer treatment. Ubiquitin-specific peptidase 5 (USP5) is a member of the DUBs family and has been implicated in promoting tumorigenesis in numerous cancers. However, the clinical significance and biological function of USP5 in osteosarcoma (OS) remains unclear. Here, we found elevated USP5 expression in OS tissues compared with normal bone tissues. Furthermore, we observed significant associations of elevated USP5 levels with increased mortality and more malignant phenotypes in OS patients. Moreover, our results revealed that USP5 could facilitate metastasis and cell progression in OS by activating the hedgehog (Hh) signaling pathway using cultured cells and animal tumor models. Mechanistically, USP5 appeared to stabilize and deubiquitinate Gli1, a key mediator of the Hh signaling pathway. Additionally, the oncogenic effect of USP5 in OS was dependent on Gli1 stability. Our findings support the model where USP5 contributes to OS pathogenesis by activating the Hh/Gli1 signaling pathway, making USP5 a potential diagnostic and therapeutic target for OS.
Keywords: Osteosarcoma, USP5, hedgehog signaling pathway, Gli1, ubiquitination
Introduction
Osteosarcoma (OS) is a type of bone cancer that often develops in bone-forming cells, and a common primary solid malignant tumor with high risks of metastatic progression and recurrence [1]. Although combining surgery with chemotherapy has greatly improved the outcomes of OS patients, the five-year overall survival rate of patients with metastatic or recurrent OS remains unfavorable [2,3]. Consequently, investigating and understanding the molecular mechanisms of OS tumorigenesis is crucial to the development of novel and effective strategies for the diagnosis and treatment of OS patients.
The highly conserved hedgehog (Hh) signaling pathway is vital to tissue development and homeostasis [4]. The canonical Hh pathway is activated upon ligand binding to the 12-pass transmembrane receptors (Patched1 and Patched2), which leads to the release of the 7-pass transmembrane protein Smoothened receptor and the nuclear translocation of the Gli transcription factors (Gli1, 2, and 3) that ultimately activate the transcription of downstream target genes [5]. Dysregulation of the activation of the Hh pathway, either through mutations of key players or ligand overexpression, constitutes a major contributor to cancer initiation and progression [6,7]. For instance, RUNX3 has been shown to suppress the metastasis and stemness of colorectal cancer cells by inhibiting the Hh signaling pathway [8]. Similarly, Li et al. demonstrated that MT1M could promote gastric cancer progression and stemness by modulating the Hh pathway protein Gli1 [9].
As a member of the DUBs family, USP5 can modulate multiple cellular events and act as a pathophysiologic agent in diseases such as cancer [10-12]. Previous studies have demonstrated the diagnostic value of USP5, linking USP5 with poor clinical outcomes in several cancers including ovarian [13], pancreatic [14], and liver cancers [15]. USP5 binds directly to β-catenin and catalyzes its deubiquitination, thereby promoting cancer stem cell-like properties in lung cancer [16]. Despite these observations, few studies to date have demonstrated a direct role of USP5 as a carcinogenic factor.
In the present study, we found that high levels of USP5 were strongly correlated with disease progression and poor outcomes in OS patients. Additionally, our data indicate that USP5 could promote the growth and metastasis of OS cells by activating the Hh/Gli1 signaling pathway. Taken together, our results point to USP5 as a promising target for developing novel strategies for the diagnosis and treatment of OS.
Materials and methods
Patients and tissue specimens
Normal bone and OS tissue samples were collected from patients at the Second Affiliated Hospital of Nanchang University with written consent. Samples were immediately stored in liquid nitrogen until further tests. Study protocols were approved by the Ethics and Research Committee of the Second Affiliated Hospital of Nanchang University. Patient clinical characteristics are listed in Table 1.
Table 1.
Correlation between USP5 and clinicopathologic characteristics of 82 OS patients
| Parameters | n | USP5 expression | P value | |
|---|---|---|---|---|
|
| ||||
| Low (n=28) | High (n=54) | |||
| Age (years) | P=0.753 | |||
| ≤18 | 66 | 22 | 44 | |
| >18 | 16 | 6 | 10 | |
| Sex | P=0.641 | |||
| Female | 41 | 13 | 28 | |
| Male | 41 | 15 | 26 | |
| Tumor size (cm) | P<0.0001 | |||
| <3 | 32 | 23 | 9 | |
| ≥3 | 50 | 5 | 45 | |
| Location | P=0.118 | |||
| Upper limb bone | 56 | 16 | 40 | |
| Lower limb bone | 26 | 12 | 14 | |
| Clinical stage | P=0.0002 | |||
| I/II | 33 | 19 | 14 | |
| III/IV | 49 | 9 | 40 | |
| Pathological differentiation | P=0.344 | |||
| Well/Moderately | 38 | 15 | 23 | |
| Poor | 44 | 13 | 31 | |
| Recurrence | P=0.086 | |||
| Absence | 39 | 17 | 22 | |
| Presence | 43 | 11 | 32 | |
Cell lines and cell culture
The normal human osteoblasts (hFOB 1.19) and OS cell lines (U2OS, Saos-2, 143B, and MG-63) were procured from ATCC (Rockville, MD, USA). Cell lines were maintained in DMEM supplemented with 10% fetal bovine serum (FBS; Thermo fisher scientific, Waltham, MA, USA) at 37°C and 5% CO2.
Over-expression constructs, and lentivirus shRNA infection
USP5 or Gli1 overexpression plasmids were generated by subcloning full-length cDNAs of human USP5 or Gli1 into the pRNAT-U6.1/Neo vector. OS cells were transfected with these constructs using Lipofectamine 3000. Because the pRNAT-U6.1/Neo vector confers G418 resistance, cells were selected in G418 to enrich for positively transfected clones.
For stable knockdown of USP5, the shRNAs targeting USP5 (shUSP5), 5’-CCGGGACCACACGATTTGCCTCATTCTCGAGAATGAGGCAAATCGTGTGGTCTTTTTG-3’ (shUSP5-1) and 5’-CCGGCCTGTCTGTAAGGAGACTTTGCTCGAGCAAAGTCTCCTTACAGACAGGTTTTTG-3’ (shUSP5-2) and a negative control (synthetized by GenePharma), were cloned into pLKO.1 puro vector (Addgene, Cambridge, USA). OS cells were infected with high-titer lentiviral stocks expressing shUSP5. And puromycin was used for stable cell selection.
Real-time quantitative polymerase chain reaction (qRT-PCR)
The total RNA extraction utilized a standard TRIzol-based protocol. PrimeScript RT Reagent Kit (Thermo fisher scientific, Waltham, MA, USA) was used to reverse transcribe the RNA according to the manufacturer’s instructions. The qRT-PCR employed SYBR Premix Ex Taq (TaKaRa, Dalian, China), and data was analyzed by the ABI PRISM 7900 HT sequence detection system. The primer details are as follows: USP5 (sense: 5’-CGAGTCTACTTGCACCTCCG-3’, antisense: 5’-CGCCTTCAACACCAATAGCC-3’), Gli1 (sense: 5’-GACAGAGGCCCACTCTTTTC-3’, antisense: 5’-TCCGACAGAGGTGAGATGGA-3’), and GAPDH (sense: 5’-GAAAGCCTGCCGGTGACTAA-3’, antisense: 5’-AGGAAAAGCATCACCCGGAG-3’).
Western blot assay and IHC staining
Western blot assays were performed using the following antibodies: anti-USP5 (1:1000, Santa Cruz), anti-Gli1 (1:1000, Abcam), anti-Tubulin (1:1000, Santa Cruz), and anti-Gli2 (1:1000, Abcam). Tubulin served as a loading control. Signal detection was analyzed by enhanced chemiluminescence (Pierce, USA).
For IHC staining, tissue samples were fixed, embedded, sectioned, and deparaffinized. Subsequently, sections were blocked in serum-free protein blocking buffer (DAKO, CA, USA) for 30 minutes and stained with anti-USP5 (1:200, Santa Cruz). All samples were examined by three different researchers to independently verify pathological information using the German semiquantitative scoring method. Each specimen was scored for the intensity of nucleic, cytoplasmic, and membrane staining (no staining =0, weak staining =1, moderate staining =2, strong staining =3) and for the extent of stained cells (0%=0, 1%-24%=1, 25%-49%=2, 50%-74%=3, 75%-100%=4). Final IHC scores were the product of intensity scores multiplied by extent scores.
Colony formation and CCK8 assay
The cell viability analysis employed Cell Counting Kit 8 (CCK8). OS cells with 1×103 cells/well density were seeded in 6-well plates and cultured for two weeks. Cells were then washed thrice with phosphate-buffered saline, fixed in methanol, and stained with 1% crystal violet. Colonies that stained purple were counted and quantified.
Cell migration and invasion assay
OS cells were examined for their metastatic capacity in a Transwell Boyden chamber (pore size of 8 mm, BD Biosciences). The polycarbonate membrane in the upper layer of the chamber was pre-coated with substrate gel for the cell invasion assay.
Tumorigenicity assay and bioluminescence imaging
OS cells stably expressing the firefly luciferase gene (generated through viral transduction) were used for injection into animals and luciferase expression was monitored in vivo by bioluminescence imaging to assess tumor development. For in vivo tumorigenicity assays, nude mice (male BALB/c-nu/nu, aged six to eight weeks) were injected subcutaneously with OS cells (1×106, 100 ml PBS) in the abdomen. Subsequently, the mice were anesthetized using isoflurane and examined by Lumina Series III IVIS (In Vivo Imaging System; PerkinElmer, MA, USA) to detect luciferase signals in vivo signals. The experimental protocols using animal models were approved by the Ethics Committee for Animal Experiments of the Second Affiliated Hospital of Nanchang University.
In vivo metastasis assay
OS cells (5×105) were re-suspended in DMEM (50 μL) and injected into the tail vein of BALB/c nude mice. The mice were euthanized after 12 weeks followed by the lung excision for H&E staining and pathological examination. Animal tests were approved by the Ethics Committee for Animal Experiments of the Second Affiliated Hospital of Nanchang University and were conducted as per the “Guide for the Care and Use of Laboratory Animals”.
Co-IP and in vivo ubiquitination assay
For Co-IP analysis, cell lysates were incubated with primary antibodies overnight at 4°C and subsequently with protein A/G-Sepharose beads (Santa Cruz, USA). Co-precipitated proteins were examined by immunoblotting using appropriate antibodies. For in vivo ubiquitination assays, cells were co-transfected with appropriate plasmids and then treated with the proteasome inhibitor MG132 (50 μg/mL) for 12 hours. Cell lysates were then collected for immunoprecipitation with anti-Gli1 antibodies. The ubiquitination level of Gli1 was determined with an anti-Ubiquitin (linkage-specific K48) antibody (1:1000, abcam, ab140601).
Statistical analysis
Data were analyzed using GraphPad Prism 6 (GraphPad Software, USA) and represented as mean ± standard error. Statistical significance was determined by the two-tailed distribution analysis and Student’s t-test. Survival curves were calculated via the Kaplan-Meier approach, and the significance was determined through a log-rank test. Values with P<0.05 were considered statistically significant.
Results
High USP5 expression is linked to poor prognosis in OS patients
To investigate the role of USP5 in OS development, we first determined USP5 expression in normal bone vs. primary OS tissues. qRT-PCR data demonstrated that USP5 mRNA levels were elevated in primary OS compared to normal bone tissues (Figure 1A and 1B). Likewise, USP5 protein levels were also markedly upregulated in most cases (Figure 1C and 1D). Consistent with these data, IHC staining also revealed upregulated USP5 expression in primary OS (Figure 1E and 1F).
Figure 1.

Overexpression of USP5 is correlated with poor outcomes in OS patients. (A and B) USP5 mRNA expression in OS and normal tissues was determined by qRT-PCR analysis. ***P<0.001. (C and D) Identification and quantification of USP5 protein levels in OS and normal tissues by western blotting assay. Tubulin served as a loading control. **P<0.01. (E and F) Representative image (E) and quantification (F) of IHC staining of USP5 in OS and normal tissues. Scale bar, 50 μm. **P<0.01. (G and H) Overall and progression-free survival Kaplan-Meier curves of 82 OS patients plotted according to the expression level of USP5.
To test whether USP5 could serve as a prognostic factor in OS, the relationship between USP5 and the clinicopathological characteristics of OS patients was probed. Results demonstrated that USP5 expression levels could indeed be linked to tumor size (P<0.0001) and clinical stage (P=0.0002; Table 1). Moreover, data from 82 OS patients found a negative correlation between high levels of USP5 and survival (five-year and disease-free) (Figure 1G and 1H). Collectively, these findings indicate USP5 overexpression in OS tissues, suggesting that ISP5 may be a promising prognostic marker for OS patients.
USP5 facilitates OS cell development in vitro and tumorigenesis in vivo
To explore the biological function of USP5 in OS, we next examined its expression in four OS cell lines (U2OS, Saos-2, 143B, and MG-63) and normal human osteoblasts (hFOB 1.19) (Figure 2A and 2B). We then transfected Saos-2 and U2OS cells with lentiviral vectors encoding USP5-specific shRNAs (Figure 2C-E). CCK-8 assays indicated that USP5 silencing substantially inhibited cell proliferation (Figure 2F and 2G). Moreover, USP5 knockdown also significantly suppressed the growth of OS cells in colony formation assays (Figure 2H and 2I). Importantly, tumorigenicity assays in nude mice demonstrated that USP5-knockdown led to significantly decreased tumor size and growth (Figure 2J-L). Additionally, Ki67 staining assay found a decline in cell proliferation as well (Figure 2M). Collectively, our results suggest that USP5 is a player in OS tumorigenicity regulation in vivo and in vitro.
Figure 2.

Effects of USP5 on OS cell growth. (A and B) Protein and mRNA levels of USP5 were detected in four OS cell lines and the normal human osteoblast line hFOB 1.19. Tubulin served as a loading control. ***P<0.001, **P<0.01. (C-E) Western blot, and qRT-PCR analyses were used to monitor the expression level of USP5 in U2OS and Saos-2 cells stably transfected with the USP5 knockdown vector. Tubulin served as a loading control. **P<0.01. (F and G) CCK-8 assays were performed in USP5-knockdown OS cells. **P<0.01. (H and I) Representative images (left) and quantification (right) of colony formation assays using OS cells transfected with the shUSP5 vector. **P<0.01. (J-L) USP5-knockdown OS cells were injected into the flanks of each mouse (J). The tumors were weighed at experimental endpoints (K), and tumor size was monitored at the indicated time points (L). Scale bar, 50 μm. n=6, *P<0.05, **P<0.01. (M) Representative images of Ki67 staining in tumour tissues isolated from the USP5-knockdown animal group. Scale bar, 50 μm.
USP5 facilitates the ability of OS cells to metastasize in vivo and in vitro
To further confirm the role of USP5 in OS tumor progression, we assessed the effect of USP5 knockdown on the metastasis of OS cells via Transwell invasion and migration assays. As illustrated in Figure 3A-F, USP5-knockdown in Saos-2 and U2OS cell lines significantly reduced the ability of these cells to invade and migrate. We further investigated the impact of altered USP5 expression on OS metastasis in vivo. After six weeks, the average number of metastatic lesions in lung tissue sections decreased in the USP5-knockdown group compared to the control group (Figure 3G and 3H). Collectively, these results suggest that USP5 can facilitate OS cell metastasis and invasion in vivo and in vitro, supporting its oncogene role in OS.
Figure 3.
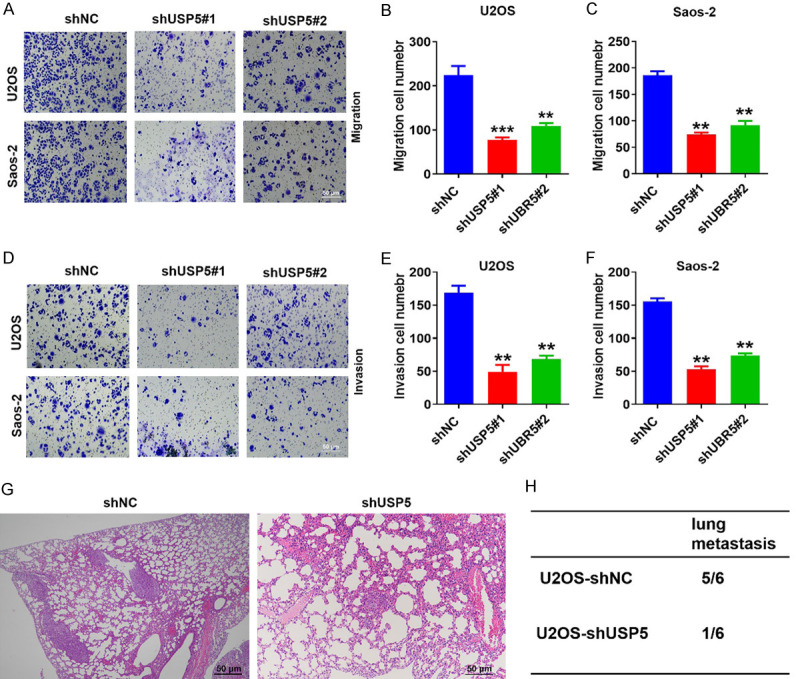
USP5 accelerates the migration and invasion of OS cells in vitro and in vivo. (A-F) Transwell migration and invasion assays using U2OS and Saos2 cells transfected with the USP5 knockdown vector. The image was captured at 400× magnification. Scale bar, 50 μm. **P<0.01, ***P<0.001. (G and H) USP5-knockdown OS cells were injected into the tail vein of nude mice, and the incidence of lung metastasis was measured after six to eight weeks. Representative images (G) and quantification (H) of H&E staining of lung metastatic nodules are shown here. Scale bar, 50 μm. N=6/group.
The Hh pathway functions downstream of USP5 and facilitates its role in OS cells
To uncover the mechanisms of USP5-mediated OS tumorigenesis, we explored the GSEA and TCGA databases. Based on our analysis, the genomes of Hallmark_Hh pathway_Targets were highly enriched in OS tissues with higher expression levels of USP5 (Figure 4A), indicating that the Hh pathway was involved in USP5-mediated function. Additionally, USP5 knockdown inhibited the mRNA expression of target genes downstream of the Hh signaling pathway (Figure 4B and 4C). Interestingly, USP5 depletion had no obvious effects on the protein level of Gli2, whereas Gli1 protein levels were significantly decreased (Figure 4D and 4E). Moreover, we investigated Gli1 expression in OS tissues exhibiting high expression levels of USP5 by Western blotting assays (Figure 4F). Statistical analysis revealed positive correlation between Gli1 and USP5 expression in OS tissues (Figure 4G and 4H). Moreover, IHC analysis found reduced Gli1 staining with USP5 knockdown in mice (Figure 4I). Collectively, the above findings suggest that the Hh/Gli1 signaling pathway may be crucial for the oncogenic role of USP5 in OS.
Figure 4.

USP5 activates Hh/Gli1 signaling in OS cells. (A) GSEA comparing the gene sets of Hh pathway targets in USP5-high OS patients. Data were obtained from the TCGA database. NES means normalized enrichment score. (B and C) qRT-PCR assays showing the expression levels of USP5, PTCH1, VEGFC, Cyclin D1, and Snail in USP5-knockdown OS cells. **P<0.01. (D and E) Western blot analyses were used to detect the expression levels of USP5, Gli1, and Gli2 in OS cells stably transfected with the USP5 knockdown vector. Tubulin served as a loading control. (F and G) Identification (F) and quantification (G) of USP5 and Gli1 protein levels in OS and normal tissues by the western blot assay. Tubulin served as a loading control. **P<0.01. (H) Scatter plots show a positive correlation between USP5 and Gli1 at the protein level in OS tissues. (I) Representative images of Gli1 staining in tumour tissues isolated from the USP5 silencing animal group. Scale bar, 50 μm.
USP5 can directly interact with and deubiquitinate Gli1
Our results thus far demonstrated that silencing USP5 did not affect Gli1 mRNA levels in OS cells, suggesting a possible role for USP5 in modulating Gli1 levels post-transcriptionally (Supplementary Figure 1). In addition, treatment with the autophagy inhibitor chloroquine (CQ) did not impact Gli1 upregulation in USP5-overeexpression OS cells (Supplementary Figure 2), indicating that USP5 likely positively regulates Gli1 expression in a lysosomal degradation pathway-independent manner. As expected, USP5 overexpression enhanced Gli1 expression in cycloheximide (CHX) treated OS cells (Figure 5A-D). Furthermore, in the presence of the proteasome inhibitor MG132, Gli1 protein levels remained comparable in USP5-overexpressing vs. knockdown cells (Figure 5E and 5F). Moreover, in co-IP assays using antibodies against endogenous Gli1 and USP5, we observed co-precipitation of Gli1 and USP5 in OS cells (Figure 5G and 5H). As shown in Figure 5I, GST pull-down further confirmed that USP5 could bind to Gli1 under cell-free conditions. Additionally, co-localization of USP5 and Gli1 was clearly evident in OS cells (Figure 5J). Finally, ectopic expression of USP5 reduced the K48-linked ubiquitination level of Gli1, whereas the opposite was observed with USP5 silencing in OS cells (Figure 5K and 5L). In addition, overexpression of USP5WT but not the catalytic-inactive mutant USP5C335A significantly upregulated the protein level of Gli1 in OS cells (Supplementary Figure 3). Our findings combined suggest that USP5 can act as a deubiquitinating enzyme to stabilize Gli1 in OS.
Figure 5.

USP5 interacts with Gli1 and stabilizes Gli1 expression via deubiquitination. (A-D) Representative (A and C) and quantitative (B and D) results of Gli1 protein levels in USP5-knockdown OS cells. At the indicated time points, CHX-treated cells were analyzed by Western blotting. **P<0.01. (E and F) USP25-overexpressing (E) or USP5-knockdown (F) OS cells were treated with MG132 (10 μM) for 6 h. Cells were then collected and analyzed by Western blotting with the indicated antibodies. (G and H) The correlation between USP5 and Gli1 was determined in U2OS cells by co-IP assays. (I) GST pull-down assays were performed to detect interactions between USP5 and Gli1 in HEK293 cells. (J) Co-localization studies of OS cells using an anti-USP5 antibody (1:100, green) and an anti-Gli1 antibody (1:100, red), followed by DAPI nuclear counterstaining (blue). The merged images of USP5 (green) and Gli1 (red) with DAPI (blue) are also shown. Scale bar, 50 μm. (K and L) Lysates from U2OS cells transduced with Flag-USP5 (K) or shUSP5 (L) were immunoprecipitated with an anti-Ub antibody and immunoblotted with the anti-Gli1 antibody.
USP5-mediated tumorigenic effects on OS cells depend on Gli1 stability
We next performed rescue experiments to further validate the tumor-promoting funciton of USP5 in OS. As shown in Figure 6A, USP5-knockdown cells exhibited elevated Gli1 expression in the presence of a Gli1 overexpression vector. This rescued the viability and metastatic ability of these USP5-knockdown OS cells (Figure 6B-E). Subsequently, the Hh signaling pathway ectopically expressing USP5 was inhibited with the Gli1 inhibitor GANT61 (Figure 6F), which reversed the elevated viability and metastatic ability of OS cells (Figure 6G-J), confirming the importance of Gli1 to USP5-mediated OS carcinogenesis.
Figure 6.

The oncogenic effect of USP5 is dependent on Gli1 activation. (A) After overexpressing Gli1 in USP5-knockdown U2OS cells, the protein levels of USP5 and Gli1 were examined. Tubulin served as a loading control. (B-E) Restoring Gli1 expression impaired the anti-tumor effect from USP5 knockdown, as determined by CCK8 (B), colony formation (C), and Transwell assays in U2OS cells. **P<0.01. (F) The protein levels of USP5 and Gli1 were detected in USP5-overexpression U2OS cells after treatment with or without the Gli1 inhibitor GANT61 (30 μM). Tubulin served as a loading control. (G-J) Quantification for CCK8 (G), colony formation (H) and Transwell assays (I and J) of USP5-overexpression U2OS cells transfected with or without GANT61. *P<0.05, **P<0.01. (K) A proposed model of USP5 function in promoting Hh/Gli1 pathway signaling in OS.
Discussion
Understanding OS pathogenesis is crucial for devising advanced and effective treatment. An overactive Hh/Gli1 signaling pathway is frequently seen in many tumors; therefore, uncovering the mechanism of its activation in OS may facilitate the discovery of new therapeutic strategies for OS patients. Our results showed that USP5 could activate the Hh/Gli1 signaling pathway to promote carcinogenesis, making a possible prognostic marker and promising target for treating patients with OS.
Aberrant regulation of ubiquitination is associated with various cancers [17]. Ubiquitination involves a series of enzymes, including E3s, E2s, and E1s, and an array of DUBs [18]. DUBs are attractive therapeutic targets because of their ability to inhibit the actions of E3 ligases [19,20]. USP5 is an oncogene implicated in the pathogenesis of several types of cancer [8,12]. However, less is known about the molecular mechanism or function of USP5 in OS. To the best of our knowledge, our study is the first to investigate the role of USP5 in OS. Higher levels of USP5 expression were observed in tumor samples from OS patients compared with normal bone tissues. And a significant association was observed between elevated USP5 expression and decreased patient survival, distant metastases, clinical stage, and tumor size. Furthermore, results from cultured cells and animal tumor models revealed that USP5 could activate the Hh/Gli1 signaling pathway, thereby promoting OS proliferation and metastasis. Collectively, these findings suggest the possibility of using USP5 as a new marker of severe OS.
Gli1, a member of the GLI family, is known to be up-regulated in malignant gliomas, and its abnormal activation a critical process in the Hh signaling pathway [7,21]. In normal cells, the Gli1 protein activity is tightly modulated, spatiotemporally and quantitatively. Its dysregulation has been found to contribute to malignant features in several cancers [22]. Recent studies have shown the importance of post-translational modifications, particularly ubiquitination, to the regulation of Gli1 [23,24]. Although several E3-ubiquitin ligases may ubiquitinate and thereby leading to the degradation of Gli1, little is known about the regulation of Gli1 through the deubiquitination pathway [25,26]. Our results demonstrate that USP5 is a specific Gli1 deubiquitinase in OS cells. USP5 has been suggested to control the ubiquitination of other substrates in a variety of malignancies [27,28]. For instance, USP5 promotes progression of non-small cell lung cancer by stabilizing PD-L1 [29]. In the present study, we found that USP5 interacted with Gli1 directly and inhibited its poly-ubiquitination, thereby stabilizing Gli1 expression in OS cells. Importantly, rescue experiments further confirmed that the oncogenic effect of USP5 in OS was dependent on Gli1 stability. Thus, these findings support Gli1 as a crucial target of USP5 in OS. Although the broader role of USP5 in regulating other targets in OS remains to be determined, the identification of USP5 as a Gli1 deubiquitinase should spur more research on USP5 as a therapeutic target in OS.
In summary, our work has revealed that USP5 activates the Hh signaling pathway, a key carcinogenesis regulator in many malignancies. This is accomplished through Gli1’s ubiquitination-regulated processing, which is induced by functional cooperation of USP5 and Gli1 (Figure 6K). Considering the pivotal role of the Hh signaling pathway in tumor development, targeting molecular players to down-regulate the Hh signaling should be evaluated clinically for anticancer treatment. Our findings should provide insight into the mechanisms underlying the relationship between metastatic and advanced OS and highlight USP5 as a tumorigenesis promotor and Gli1 as an oncogenic factor.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (81960486 and 82160480), Natural Science Foundation of Jiangxi (2019BAB205030) and the Project of Health and Family Planning Commission of Jiangxi Provincial (No. 2014A028).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Eaton BR, Schwarz R, Vatner R, Yeh B, Claude L, Indelicato DJ, Laack N. Osteosarcoma. Pediatr Blood Cancer. 2021;68(Suppl 2):e28352. doi: 10.1002/pbc.28355. [DOI] [PubMed] [Google Scholar]
- 2.Yang C, Tian Y, Zhao F, Chen Z, Su P, Li Y, Qian A. Bone microenvironment and osteosarcoma metastasis. Int J Mol Sci. 2020;21:6985. doi: 10.3390/ijms21196985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beird HC, Bielack SS, Flanagan AM, Gill J, Heymann D, Janeway KA, Livingston JA, Roberts RD, Strauss SJ, Gorlick R. Osteosarcoma. Nat Rev Dis Primers. 2022;8:77. doi: 10.1038/s41572-022-00409-y. [DOI] [PubMed] [Google Scholar]
- 4.Ingham PW. Hedgehog signaling. Curr Top Dev Biol. 2022;149:1–58. doi: 10.1016/bs.ctdb.2022.04.003. [DOI] [PubMed] [Google Scholar]
- 5.Sigafoos AN, Paradise BD, Fernandez-Zapico ME. Hedgehog/GLI signaling pathway: transduction, regulation, and implications for disease. Cancers (Basel) 2021;13:3410. doi: 10.3390/cancers13143410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang J. Hedgehog signaling mechanism and role in cancer. Semin Cancer Biol. 2022;85:107–122. doi: 10.1016/j.semcancer.2021.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doheny D, Manore SG, Wong GL, Lo HW. Hedgehog signaling and truncated GLI1 in cancer. Cells. 2020;9:2114. doi: 10.3390/cells9092114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim BR, Na YJ, Kim JL, Jeong YA, Park SH, Jo MJ, Jeong S, Kang S, Oh SC, Lee DH. RUNX3 suppresses metastasis and stemness by inhibiting Hedgehog signaling in colorectal cancer. Cell Death Differ. 2020;27:676–694. doi: 10.1038/s41418-019-0379-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li K, Sun S, Lu Y, Liang W, Xu X, Zhang H, Chang Z, Wang C, Gao Y, Chen L. MT1M regulates gastric cancer progression and stemness by modulating the Hedgehog pathway protein GLI1. Biochem Biophys Res Commun. 2023;670:63–72. doi: 10.1016/j.bbrc.2023.05.121. [DOI] [PubMed] [Google Scholar]
- 10.Cai B, Zhao J, Zhang Y, Liu Y, Ma C, Yi F, Zheng Y, Zhang L, Chen T, Liu H, Liu B, Gao C. USP5 attenuates NLRP3 inflammasome activation by promoting autophagic degradation of NLRP3. Autophagy. 2022;18:990–1004. doi: 10.1080/15548627.2021.1965426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun HL, Zhu AC, Gao Y, Terajima H, Fei Q, Liu S, Zhang L, Zhang Z, Harada BT, He YY, Bissonnette MB, Hung MC, He C. Stabilization of ERK-phosphorylated METTL3 by USP5 increases m(6)A methylation. Mol Cell. 2020;80:633–647. e637. doi: 10.1016/j.molcel.2020.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Wang Y, Luo Y, Liu Y, Yi Y, Li J, Pan Y, Li W, You W, Hu Q, Zhao Z, Zhang Y, Cao Y, Zhang L, Yuan J, Xiao ZJ. USP5-Beclin 1 axis overrides p53-dependent senescence and drives Kras-induced tumorigenicity. Nat Commun. 2022;13:7799. doi: 10.1038/s41467-022-35557-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du Y, Lin J, Zhang R, Yang W, Quan H, Zang L, Han Y, Li B, Sun H, Wu J. Ubiquitin specific peptidase 5 promotes ovarian cancer cell proliferation through deubiquitinating HDAC2. Aging (Albany NY) 2019;11:9778–9793. doi: 10.18632/aging.102425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li XY, Wu HY, Mao XF, Jiang LX, Wang YX. USP5 promotes tumorigenesis and progression of pancreatic cancer by stabilizing FoxM1 protein. Biochem Biophys Res Commun. 2017;492:48–54. doi: 10.1016/j.bbrc.2017.08.040. [DOI] [PubMed] [Google Scholar]
- 15.Meng J, Ai X, Lei Y, Zhong W, Qian B, Qiao K, Wang X, Zhou B, Wang H, Huai L, Zhang X, Han J, Xue Y, Liang Y, Zhou H, Chen S, Sun T, Yang C. USP5 promotes epithelial-mesenchymal transition by stabilizing SLUG in hepatocellular carcinoma. Theranostics. 2019;9:573–587. doi: 10.7150/thno.27654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue S, Wu W, Wang Z, Lu G, Sun J, Jin X, Xie L, Wang X, Tan C, Wang Z, Wang W, Ding X. USP5 promotes metastasis in non-small cell lung cancer by inducing epithelial-mesenchymal transition via Wnt/β-Catenin pathway. Front Pharmacol. 2020;11:668. doi: 10.3389/fphar.2020.00668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mansour MA. Ubiquitination: friend and foe in cancer. Int J Biochem Cell Biol. 2018;101:80–93. doi: 10.1016/j.biocel.2018.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Park HB, Baek KH. E3 ligases and deubiquitinating enzymes regulating the MAPK signaling pathway in cancers. Biochim Biophys Acta Rev Cancer. 2022;1877:188736. doi: 10.1016/j.bbcan.2022.188736. [DOI] [PubMed] [Google Scholar]
- 19.Harrigan JA, Jacq X, Martin NM, Jackson SP. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat Rev Drug Discov. 2018;17:57–78. doi: 10.1038/nrd.2017.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Niu K, Fang H, Chen Z, Zhu Y, Tan Q, Wei D, Li Y, Balajee AS, Zhao Y. USP33 deubiquitinates PRKN/parkin and antagonizes its role in mitophagy. Autophagy. 2020;16:724–734. doi: 10.1080/15548627.2019.1656957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carpenter RL, Lo HW. Hedgehog pathway and GLI1 isoforms in human cancer. Discov Med. 2012;13:105–113. [PMC free article] [PubMed] [Google Scholar]
- 22.Avery JT, Zhang R, Boohaker RJ. GLI1: a therapeutic target for cancer. Front Oncol. 2021;11:673154. doi: 10.3389/fonc.2021.673154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gulino A, Di Marcotullio L, Canettieri G, De Smaele E, Screpanti I. Hedgehog/Gli control by ubiquitination/acetylation interplay. Vitam Horm. 2012;88:211–227. doi: 10.1016/B978-0-12-394622-5.00009-2. [DOI] [PubMed] [Google Scholar]
- 24.Liu H, Yan S, Ding J, Yu TT, Cheng SY. DeSUMOylation of Gli1 by SENP1 attenuates sonic hedgehog signaling. Mol Cell Biol. 2017;37:e00579–16. doi: 10.1128/MCB.00579-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Q, Shi Q, Chen Y, Yue T, Li S, Wang B, Jiang J. Multiple Ser/Thr-rich degrons mediate the degradation of Ci/Gli by the Cul3-HIB/SPOP E3 ubiquitin ligase. Proc Natl Acad Sci U S A. 2009;106:21191–21196. doi: 10.1073/pnas.0912008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mazzà D, Infante P, Colicchia V, Greco A, Alfonsi R, Siler M, Antonucci L, Po A, De Smaele E, Ferretti E, Capalbo C, Bellavia D, Canettieri G, Giannini G, Screpanti I, Gulino A, Di Marcotullio L. PCAF ubiquitin ligase activity inhibits Hedgehog/Gli1 signaling in p53-dependent response to genotoxic stress. Cell Death Differ. 2013;20:1688–1697. doi: 10.1038/cdd.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang W, Liu X, Zhang Y, Deng M, Li G, Chen G, Yu L, Jin L, Liu T, Wang Y, Chen Y. USP5 promotes breast cancer cell proliferation and metastasis by stabilizing HIF2α. J Cell Physiol. 2022;237:2211–2219. doi: 10.1002/jcp.30686. [DOI] [PubMed] [Google Scholar]
- 28.Li G, Yang T, Chen Y, Bao J, Wu D, Hu X, Feng C, Xu L, Li M, Li G, Jin M, Xu Y, Zhang R, Qian G, Pan J. USP5 sustains the proliferation of glioblastoma through stabilization of cyclinD1. Front Pharmacol. 2021;12:720307. doi: 10.3389/fphar.2021.720307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan J, Qiao Y, Chen C, Zang H, Zhang X, Qi F, Chang C, Yang F, Sun M, Lin S, Tang Q, Li L, Wang M, Wu M, Liu Y, Lai C, Chen J, Chen G. USP5 facilitates non-small cell lung cancer progression through stabilization of PD-L1. Cell Death Dis. 2021;12:1051. doi: 10.1038/s41419-021-04356-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.