

Abstract
5-Methylcytosine (m5C) is an RNA modification prevalent on tRNAs, where it can protect tRNAs from endonucleolytic cleavage to maintain protein synthesis. The NSUN family (NSUN1–7 in humans) of RNA methyltransferases is responsible for installing the methyl group onto the C5 position of cytosine in RNA. NSUNs are implicated in a wide range of (patho)physiological processes, but selective and cell-active inhibitors of these enzymes are lacking. Here, we use cysteine-directed activity-based protein profiling (ABPP) to discover azetidine acrylamides that act as stereoselective covalent inhibitors of human NSUN2. Despite targeting a conserved catalytic cysteine in the NSUN family, the NSUN2 inhibitors show negligible cross-reactivity with other human NSUNs and exhibit good proteome-wide selectivity. We verify that the azetidine acrylamides inhibit the catalytic activity of recombinant NSUN2, but not NSUN6, and demonstrate that these compounds stereoselectively disrupt NSUN2-tRNA interactions in cancer cells, leading to a global reduction in tRNA m5C content. Our findings thus highlight the potential to create isotype-selective and cell-active inhibitors of NSUN2 with covalent chemistry targeting a conserved catalytic cysteine.
Keywords: activity-based protein profiling, NSUN2, covalent inhibitor, 5-methylcytosine, RNA methylation
Graphical Abstract

Activity-based protein profiling identifies azetidine acrylamides that covalently inhibit NSUN2 through targeting a conserved catalytic cysteine. The azetidine acrylamides show high isotype selectivity and perturb NSUN2-tRNA interactions and the m5C content of RNA in cancer cells.
Introduction
RNAs are subject to >150 different post-transcriptional modifications that collectively regulate RNA stability and translation, as well as many downstream facets of cell biology[1]. A major form of RNA modification is RNA methylation, which involves dedicated sets of deposition (writer) and reversal (eraser) enzymes, as well as binding proteins (readers)[2]. 5-methylcytosine (m5C), which was historically studied as a DNA modification, has also been found through analytical methods such as LC-MS/MS[3] and RNA bisulfite sequencing[4] to be a prevalent modification on tRNAs, where it serves to regulate protein translational integrity in the cell[5]. m5C modification can also be found on other RNAs (snRNAs, mRNAs) and is catalyzed by enzymes belonging to the NOL1/NOP2/SUN domain (NSUN) family and the DNA methyltransferase homolog DNMT2[6].
The NSUN family contains seven members that share ~30–35% sequence identity and a catalytic mechanism involving the transfer of a methyl group from the co-substrate S-adenosylmethionine (SAM) to the C5 position of cytosine[7], which requires two conserved, active-site cysteine residues (C271 and C321 in human NSUN2). NSUN2 is one of the more extensively studied members of the NSUN family and has been found to regulate diverse cellular processes[6, 8] that include gene expression[9] and cell proliferation[10] and migration[11]. NSUN2 introduces specific m5C modifications into tRNAs, a metabolic function that is non-redundant with other NSUNs[12], and can also act on mRNAs and noncoding RNAs[6, 12–13]. Despite the central functions that NSUNs play in RNA modification and cell physiology, inhibitors of these enzymes have not yet, to our knowledge, been described.
Here, we report the chemical proteomic discovery and functional characterization of azetidine acrylamides as covalent inhibitors of NSUN2 that stereoselectively react with the catalytic C271, while showing negligible cross-reactivity with other NSUNs in human cells.
Results and Discussion
In the course of evaluating the proteome-wide reactivity of a set of stereochemically defined azetidine acrylamides (referred to hereafter as ‘stereoprobes’; Fig. 1A) by cysteine-directed activity-based protein profiling (ABPP)[14], we discovered that one of the stereoprobes (MY-1B) stereoselectively liganded C271 of NSUN2 (Fig. 1B). In these experiments, cysteines were considered to be covalently liganded if they showed substantial (> 66.7%) and stereoselective (> 2.5-fold between enantiomers) reductions in reactivity with an iodoacetamide-desthiobiotin (IA-DTB) probe in proteomic lysates from cells treated with the stereoprobes. Near-complete engagement of NSUN2_C271 (> 90%) was observed in the human prostate cancer cell line 22Rv1 treated with 5 or 20 µM of MY-1B (3 h), while the enantiomer MY-1A only showed partial engagement (~50%) at 20 µM, and the diastereomers MY-3A and MY-3B were inactive at 20 µM (Fig. 1B, C). Other cysteines in NSUN2 showed minimal changes in IA-DTB reactivity, with the exception of the second catalytic cysteine C321, which also showed a partial (~75%) stereoselective loss of reactivity in MY-1B-treated cells (Fig. 1D). The cysteine-directed ABPP experiments more broadly quantified > 15,000 cysteines in 22Rv1 cells, including catalytic cysteines in NOP2 (C463), NSUN3 (C214, C265), NSUN4 (C258), NSUN5 (C308, C359), and none of these other NSUN cysteines were engaged by MY-1B (Fig. 1D, Supplementary Fig. 1A, and Supplementary Dataset 1). Finally, we corroborated the stereoselective and isotype-restricted liganding of NSUN2 by MY-1B using a complementary protein-directed ABPP method[17] that quantified the enrichment of proteins engaged by alkyne analogs of MY-1A/1B/3A/3B (MY-11A/11B/12A/12B) and the blockade of this enrichment by pre-treatment with MY-1A/1B/3A/3B. Protein-directed ABPP revealed the stereoselective enrichment of NSUN2 by MY-11B and the blockade of this enrichment by MY-1B (Fig. 1E, F). Other NSUNs (NSUN3–6) were quantified in this protein-directed ABPP experiment, but did not show stereoselective enrichment or competition by MY-11B/MY-1B (Fig. 1E, F and Supplementary Dataset 1). Across the greater proteome, MY-1B showed good overall selectivity, with stereoselective reactivity being limited to a handful of additional cysteines (10 cysteines at 5 µM) (Fig. 1G, H and Supplementary Dataset 1). We sought to verify the stereoselective engagement of NSUN2_C271 by MY-1B by recombinantly expressing a Flag epitope-tagged NSUN2 protein in HEK293T cells by transient transfection and treating lysates from these cells and mock-transfected cells with alkynylated stereoprobes MY-11A/11B/12A/12B followed by gel-ABPP. This experiment revealed robust, stereoselective labeling of recombinant NSUN2 by MY-11B (Fig. 2A), and this labeling was blocked by pretreatment with MY-1B, but not MY-1A (Fig. 2B).
Figure 1.
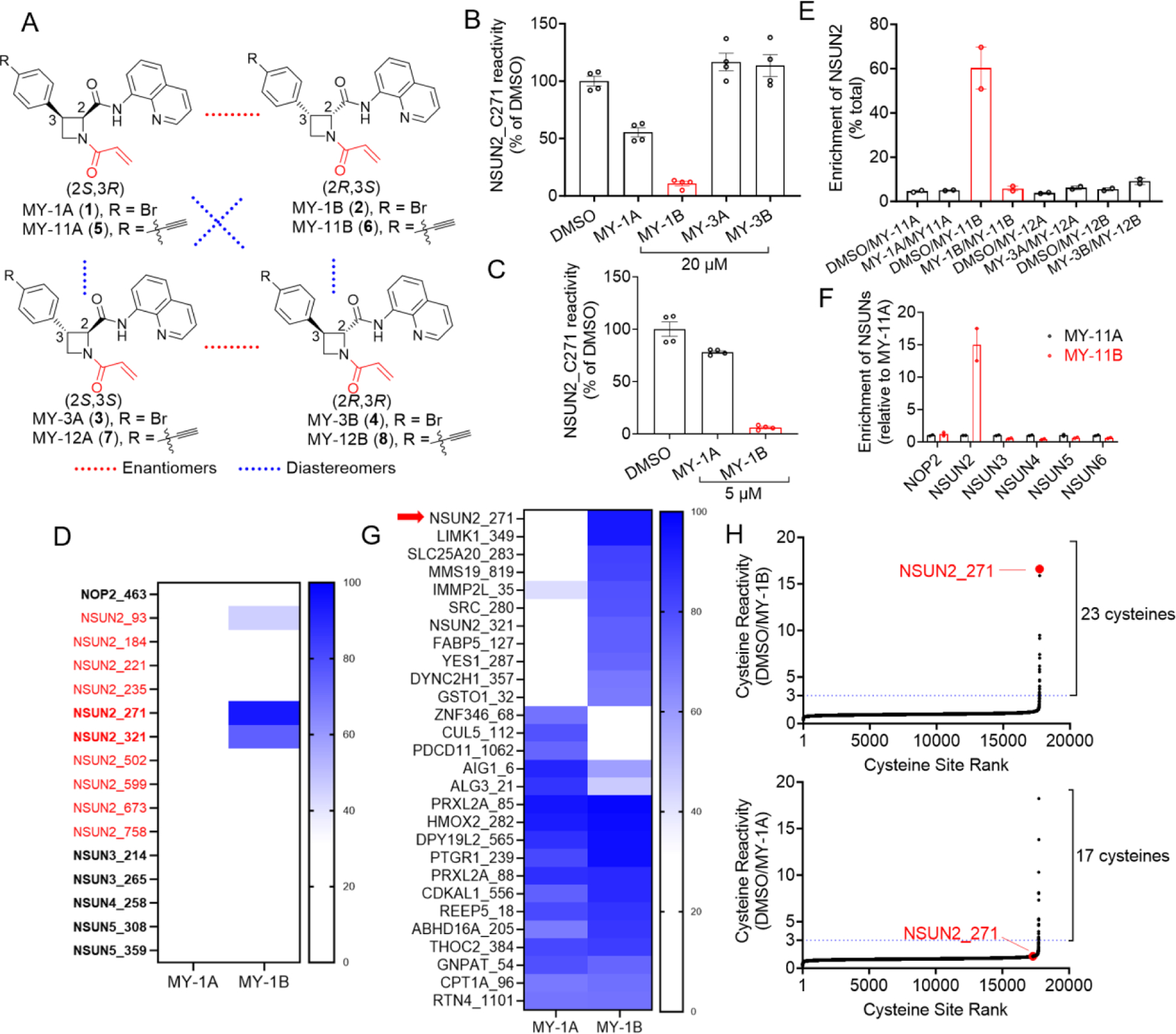
ABPP of human cancer cells identifies azetidine acrylamides that stereo- and site-selectively engage C271 of NSUN2. (A) Structures of a set of azetidine acrylamide stereoprobes MY-1A (1), MY-1B (2), MY-3A (3), and MY-3B (4) and respective alkynylated analogs MY-11A (5), MY-11B (6), MY-12A (7), and MY-12B (8). (B, C) Cysteine-directed ABPP data for NSUN2_C271 from 22Rv1 cells treated with the indicated azetidine acrylamides (20 µM, 3 h for (B), 5 µM, 3 h for (C)) relative to control 22Rv1 cells treated with DMSO. For (B-C), data were taken from ref. 14B and represent average values ± SEM from two independent experiments each performed with two technical replicates. (D) Heatmap showing engagement data for MY-1A and MY-1B (5 µM, 3 h) with the indicated cysteines in NSUN2 (red) and catalytic cysteines of other NSUNs (bold type) as determined by cysteine-directed ABPP experiments described in (C). Data represent average values from two independent experiments each performed with two technical replicates. (E) Protein-directed ABPP data for NSUN2 from Ramos cells treated with DMSO or the indicated azetidine acrylamides (MY-1A/1B/3A/3B) (20 µM, 2 h) followed by corresponding alkyne analogs (MY-11A/11B/12A/12B) (5 µM, 1 h). Enrichment values for each condition indicate the fraction of signal intensity for that condition relative to the total signal intensities for all conditions. (F) Protein-directed ABPP data for indicated NSUN proteins from DMSO/MY-11A or DMSO/MY-11B experimental groups in (E). Data for each NSUN in the DMSO/MY-11A group were set to a value of 1 and DMSO/MY-11B data were normalized to these values. For (E) and (F), data represent average values ± SEM for two replicate experiments. (G) Heatmap showing data for cysteines exhibiting > 67% engagement with either MY-1A or MY-1B (5 µM, 3 h) as determined by cysteine-directed ABPP of stereoprobe-treated 22RV1 cells (% changes in cysteine reactivity are reported relative to DMSO-treated cells). Data represents average values from two independent experiments each performed with two technical replicates. (H) Waterfall plots of cysteine-directed ABPP data showing IA-DTB reactivity ratios (DMSO/MY-1A or DMSO/MY-1B) for all quantified cysteines in the proteome of 22RV1 cells treated with MY-1A or MY-1B (5 µM, 3 h). The blue dashed line denotes a three-fold decrease in cysteine reactivity (67% engagement). NSUN2_271 is highlighted in red. Data represents average values from two independent experiments each performed with two technical replicates.
Figure 2.

Azetidine acrylamides stereoselectively engage recombinant NSUN2. (A) Gel-ABPP showing the reactivity of alkynylated azetidine acrylamide stereoprobes MY-11A/11B/12A/12B (1 h) with lysate of WT-NSUN2-expressing HEK293T cells. Lysates from mock (empty vector)-transfected cells treated with MY-11B are shown for comparison. Stereoprobe-labeled proteins were visualized by copper-catalyzed azide-alkyne cycloaddition (CuAAC)[15] to an azide-rhodamine tag followed by SDS-PAGE and in-gel fluorescence scanning[16]. Left, representative gel-ABPP result. Right, quantification of data presented as mean values ± SEM from three independent experiments. Statistical significance was calculated with unpaired two-tailed Student’s t-tests. ****P < 0.0001. (B) Gel-ABPP showing the concentration-dependent effects of MY-1A and MY-1B on MY-11B reactivity with recombinant WT-NSUN2 in HEK293T cells. Cells were treated in situ with MY-1A/B for 3 h, followed by MY-11B (10 µM, 1 h) and analyzed as described in (A). Left, representative gel-ABPP result. Right, quantification of data presented as mean values ± SEM from three independent experiments. Uncropped images can be found in the Supporting Information.
We next aimed to clarify the cysteine residue in NSUN2 that was covalently engaged by azetidine acrylamide stereoprobes using site-directed mutagenesis. As noted above, both catalytic cysteines (C271 and C321) showed stereoselective reductions in IA-DTB reactivity in cells treated with MY-1B. Mechanistically, C321 first adds to the C6 position of a cytosine in RNA, promoting C5-mediated methyl transfer from SAM and forming an enzyme-RNA intermediate that requires C271 for release[6] (Fig. 3A). We found that a C321-NSUN2 mutant maintained reactivity with MY-11B and competitive blockade of this interaction by MY-1B, while a C271A-NSUN2 mutant, as well as a C271A/C321A-NSUN2 double mutant, failed to react with MY-11B (Fig. 3B). These results support a model where azetidine acrylamides directly react with C271, and this covalent adduct sterically hinders IA-DTB reactivity with C321, which is located in close proximity to C271 based on an AlphaFold[18] structural model of NSUN2 (Supplementary Fig. 1B, C). We also used the AlphaFold NSUN2 structure to generate a docking model that provided a rationale for the stereoselective reactivity with MY-1B and highlighted several active-site residues that are different between NSUN2 and NSUN6 (the most closely related NSUN family member to NSUN2) (Supplementary Fig. 1B, C). We additionally screened a C267A-NSUN2 mutant and found that it maintained reactivity with MY-11B and MY-1B (Fig. 3B). This additional control was performed because the MY-1B-sensitive NSUN2 tryptic peptide quantified in cysteine-directed ABPP experiments contains both C267 and C271. Finally, and consistent with previous studies[19], the C271A-NSUN2 mutant was found to be trapped as an enzyme-RNA adduct that displayed an upward shift in Western blots (Fig. 3B). The addition of nuclease partially resolved this migration change, and MY-1B did not alter the RNA-adducted proteoforms of the C271A-NSUN2 mutant (Supplementary Fig. 2). These data, taken together, indicate that MY-1B and MY-11B stereoselectively and site-specifically engage C271 of NSUN2. In further support of this conclusion, our protein-directed ABPP experiments revealed that all quantified tryptic peptides in NSUN2 (including the tryptic peptide containing C321) showed consistent stereoselective enrichment in MY-11B-treated cells with the exception of the tryptic peptide containing C271, which was not stereoselectively enriched (consistent with this peptide being covalently modified by MY-11B) (Supplementary Fig. 3).
Figure 3.

Azetidine acrylamides engage C271 of NSUN2. (A) Schematic showing methyltransferase mechanism of NSUN2 where C321 forms an adduct with cytidine in RNA that requires subsequent release mediated by C271. (B) Gel-ABPP showing MY-11B (in vitro, 10 µM, 1 h) reactivity with recombinant WT-NSUN2 and NSUN2 mutants, with or without the addition of nuclease or pre-treatment with MY-1B (in vitro, 10 µM, 1 h). Samples were analyzed as described in Fig. 2A. Top, representative gel-ABPP results and Western blots corresponding to the ABPP gel. Lower graph, quantification of data presented as mean values ± SEM from two independent experiments. Uncropped images can be found in the Supporting Information.
We performed an initial exploration of the structure-activity relationship (SAR) for (2R, 3S)-azetidine acrylamide-NSUN2 interactions by gel-ABPP, which revealed that replacement of the acrylamide with alternative electrophiles such as an α-chloroacetamide or butynamide resulted in loss of engagement with NSUN2 (Supplementary Fig. 4). The bromine substituent on the azetidine core could be replaced by an OCH3 group (DO-26B (9)) with minimal loss in apparent potency, but larger substitutions decreased NSUN2 engagement (Supplementary Fig. 4). Most of the tested replacements for the quinoline also impaired NSUN2 engagement to varying degrees, although an isopropyl substitution (WX-04–196 (15)) maintained discernible interactions (Supplementary Fig. 4). We quantified IC50 values for MY-1B, DO-26B, and WX-04–196 of 1.3 ± 0.1, 3.5 ± 0.1, and 8.5 ± 1.3 µM, respectively, by gel-ABPP (Fig. 4A, B and Supplementary Fig. 4). We additionally confirmed that DO-26B displayed stereoselective engagement of NSUN2 (Fig. 4A).
Figure 4.

Potency and initial SAR for azetidine acrylamide reactivity with NSUN2. (A) Gel-ABPP showing the concentration-dependent effects of MY-1B and DO-26B on MY-11B reactivity with NSUN2-WT. Also shown are the effects of MY-1A and DO-26A tested at a single concentration (20 µM). Lysate of WT-NSUN2-expressing HEK293T cells (or mock-transfected cells) were treated with azetidine compounds (1 h), followed by MY-11B (10 µM, 1 h) and analyzed by gel-ABPP as described in Fig. 2A. (B) Quantification of data presented as mean values ± SEM from two independent experiments with calculated IC50 values and 95% confidence limits. Uncropped images can be found in the Supporting Information.
We found that the reactivity of MY-11B with NSUN2 was blocked in a concentration-dependent manner by the product of NSUN2 catalysis S-adenosylhomocysteine (SAH) (Fig 5A, B), suggesting that azetidine acrylamides interact with the co-substrate binding pocket of the enzyme. We then established an in vitro RNA methylation assay[13b] to measure the inhibitory activity of azetidine acrylamides with NSUN2. Lysates from HEK293T cells recombinantly expressing Flag-WT-NSUN2 were treated with active (MY-1B and DO-26B) or inactive (MY-1A and DO-26A) azetidine acrylamides (20 µM, 1 h), or DMSO, after which NSUN2 was enriched by anti-Flag agarose, and methylation activity measured on-bead using a synthetic tRNA substrate (Gly-CCC)[20]. Mock and C271A-NSUN2 mutant-expressing HEK293T lysates were similarly evaluated. The reactions were then digested to single nucleosides by treatment with a nucleoside digestion mix and the ratio of 5-methylcytidine (m5Cy) to cytidine (Cy) determined by LC-MS/MS[3, 21]. These assays revealed much greater m5Cy formation in WT-NSUN2-expressing lysate compared to mock or C271A-NSUN2 expressing lysates, and MY-1B and DO-26B, but not the enantiomers MY-1A and DO-26A, inhibited the m5Cy production of WT-NSUN2 (Fig. 5C and Supplementary Fig. 5). The estimated IC50 value for MY-1B in this RNA methylation assay (2.4 ± 0.9 µM) generally matched the IC50 value measured by ABPP (1.3 ± 0.3 µM; Fig. 4B). We did not observe complete blockade of NSUN2 activity at the highest concentrations of MY-1B or DO-26B, which instead plateaued at ~80% inhibition (Fig. 5C). We are unsure why this might be the case, but it is possible that a small portion of recombinant NSUN2 is insensitive to the azetidine acrylamides. Similar RNA methylation assays performed with recombinant NSUN6 using a Cys-GCA tRNA substrate[22] showed that neither MY-1A or MY-1B (20 µM, 1 h) inhibited this NSUN (Fig. 5D). Measuring the effect of MY-1B on the methyltransferase activity of recombinant NSUN2 across different preincubation times and concentrations[23] rendered a kinact/KI value of 67 ± 15 M−1s−1 (Supplementary Fig. 6).
Figure 5.

Azetidine acrylamides inhibit NSUN2 methyltransferase activity. (A) Gel-ABPP showing the concentration-dependent effects of SAH on MY-11B reactivity with recombinant WT-NSUN2 in HEK293T cell lysates. Samples were analyzed as described in Fig. 2A. (B) Quantification of gel-ABPP data presented as mean values ± SEM from two independent experiments. (C) Effects of azetidine acrylamides on the methyltransferase activity of NSUN2. Lysate of Flag-WT-NSUN2-expressing HEK293T cells were treated with MY-1A or DO-26A/B (20 µM, 1 h), or MY-1B (various concentrations, 1 h) and then NSUN2 was enriched using anti-Flag agarose and on-bead methyltransferase assays performed with a synthetic tRNA substrate (Gly-CCC) and analyzed by LC-MS/MS. Left, m5Cy/Cy ratios calculated for the indicated samples. Right, concentration-dependent effects of MY-1B on m5Cy/Cy ratio. Data presented as mean values ± SEM from three independent experiments. Statistical significance was calculated with unpaired two-tailed Student’s t-tests. ****P < 0.0001 (D) The effect of azetidine acrylamides on the methyltransferase activity of recombinant NSUN6, where assays were performed as described in panel (C), but with a Cys-GCA tRNA substrate. Quantification of data presented as mean values ± SEM from three independent experiments. Uncropped images can be found in the Supporting Information.
We finally evaluated the impact of NSUN2 inhibitors on RNA substrate interactions in human cells using the Aza-IP method[24], where a metabolically incorporated cytidine analog 5-azacytidine (5-AzaC) forms an irreversible covalent bond with catalytic C321 of NSUN2 (Fig. 7A), enabling sequencing of bound RNAs following affinity isolation of NSUN2. THP-1 cells were pulse-labeled with 5-AzaC (10 µM) and NSUN2 inhibitors (MY-1B, DO-26B) or inactive enantiomers (MY-1A, DO-26A) or DMSO for 2 h, after which the distribution of annotated 5-AzaC peaks in each treatment group was determined by RNA sequencing. The profiles revealed that, under control conditions (DMSO), NSUN2 mainly bound to tRNA (~70% of the sequencing reads), and that treatment with either MY-1B or DO-26B, but not MY-1A or DO-26A, strongly diminished tRNA interactions (Fig. 7B). We examined two high-confidence tRNA targets of NSUN2[24–25], which confirmed blockade of NSUN2 binding in MY-1B- and DO-26B-treated cells, but not in MY-1A and DO-26A-treated cells (Supplementary Fig. 7).
We also evaluated the bulk m5C content of extracted tRNA from THP-1 cells by bisulfite sequencing at various time points after treatment with azetidine acrylamides. For these studies, we compared the effects of DO-26A an DO-26B, as we found these compounds to have somewhat lower anti-proliferative effects than MY-1A and MY-1B in THP-1 cells (Supplementary Fig. 8 and Supplementary Table 1), which facilitated analyses of m5C content across a two-day period. which revealed a stereoselective decrease in m5C at 48 h for DO-26B-treated cells compared to DO-26A-treated cells (Supplementary Fig. 9A). We used DO-26B for these experiments because cells appeared to tolerate this compound better than MY-1B over the course of a two-day treatment. The extended time required to observe a change in m5C content is consistent with the long half-lives of tRNAs in human cells (> 30 h)[26]. A similar stereoselective decrease in m5C content was observed for two high-confidence NSUN2 target tRNAs (tRNA_TRG-GCC3:46 and tRNA_TRV-CAC3:48) in DO-26B-treated cells (Supplementary Fig. 9B). We finally confirmed by protein-directed ABPP that NSUN2 remained at least partially engaged by DO-26B over the 48 h treatment period in THP-1 cells (Supplementary Fig. 9C).
Conclusion
We report herein the chemical proteomic discovery and initial functional characterization of stereoselective covalent inhibitors of the RNA methyltransferase NSUN2. These compounds modify one of the two catalytic cysteines in NSUN2, but surprisingly do not cross-react with other human NSUNs. Future studies assessing the effects of mutating unique active site residues found in NSUN2 (Supplementary Fig. 1B) may shed further light on the isotype-restricted reactivity of azetidine acrylamides with this protein. Our data indicate that MY-1B and DO-26B can be used to study the specific functions of NSUN2 in human cells, at least over a short treatment time where substantial NSUN2 inhibition is maintained (~24–48 h for DO-26B). We should, however, qualify that further improvements in the potency and selectivity of covalent NSUN2 inhibitors may be needed for some cellular experiments. For instance, we observed IC50 values for effects on cell growth that ranged from ~10–30 µM in human cancer cell lines treated with the azetidine acrylamides for 48 h (Supplementary Fig. 8 and Supplementary Table 1). These cell growth effects did not show evidence of stereoselectivity, indicating they are unrelated to NSUN2 inhibition and possibly instead reflective of general electrophile stress. Enhancing the potency of covalent NSUN2 inhibitors by 5–10-fold should allow for a broader array of cell biological experiments at concentrations that do not cause electrophile stress. With the current set of compounds, we emphasize the importance of restricting interpretation of NSUN2-related biological effects to those that show stereoselectivity. Also, because C271 is a catalytic residue, we were unable to generate an inhibitor-resistant form of NSUN2 by mutating this cysteine, which can serve as another valuable control for biological experiments[27]. Future studies may identify other mutations in the NSUN2 active site that can block azetidine acrylamide binding without affecting basal NSUN2 activity. We also call attention to complementary chemical biology approaches to study NSUN2 such as the use of non-natural amino acid incorporation to study the functional effects of mutating C271[28]. Finally, our findings provide an additional compelling example of the utility of stereochemically defined libraries of electrophilic small molecules (“stereoprobes“) in combination with ABPP for the expedited discovery of first-in-class functional ligands for human proteins[14b, 17, 27c, 29]. We anticipate that continued exploration of structurally diverse stereoprobes will offer a valuable path for expanding the ligandability of the human proteome.
Supplementary Material
Fig 6.

Azetidine acrylamides stereoselectively suppress NSUN2-tRNA interactions in human cells. (A) Fraction of mapped reads of NSUN2-bound RNA from Aza-IP experiments performed in THP-1 cells treated with active (MY-1B, DO-26B) or inactive control (MY-1A, DO-26A) azetidine acrylamides (10 µM, 2 h) or DMSO. Total reads for experimental group were: 39,159,526 (Input #1), 36,529,622 (Input #2), 43,180,843 (DMSO #1), 49,513,712 (DMSO #2), 57,287,020 (MY-1A #1), 44,913,838 (MY-1A #2), 36,000,565 (MY-1B #1), 45,057,427 (MY-1B #2), 42,262,985 (DO-26A #1), 50,975,977 (DO-26A #2), 37,076,827 (DO-26B #1), and 42,223,295 (DO-26B #2). (B) Relative fractions of tRNA bound to NSUN2 in cells treated with indicated compounds. #1 and 2 refer to independent replicate experiments. Input refers to RNA sample before immunoprecipitation.
Acknowledgements
We thank the Cravatt and He lab for helpful discussions. This work was supported by the NIH (CA231991, HG008935). C. He is a Howard Hughes Medical Institute Investigator.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Supporting Information
The authors have cited additional references within the Supporting Information[30].
Competing interests
Dr. Cravatt is a founder and advisor to Vividion Therapeutics. C.H. is a scientific founder, a member of the scientific advisory board and equity holder of Aferna Green, Inc. and AccuaDX Inc., and a scientific co-founder and equity holder of Accent Therapeutics, Inc. Drs. Cravatt, Melillo, and Schreiber are founders and/or scientific advisors to Magnet Therapeutics.
References
- [1].Frye M, Harada BT, Behm M, He C, Science 2018, 361, 1346–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Shi H, Wei J, He C, Mol. Cell 2019, 74, 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Thüring K, Schmid K, Keller P, Helm M, Methods 2016, 107, 48–56. [DOI] [PubMed] [Google Scholar]
- [4].Schaefer M, Pollex T, Hanna K, Lyko F, Nucleic Acids Res. 2009, 37, e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Motorin Y, Lyko F, Helm M, Nucleic Acids Res. 2010, 38, 1415–1430; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Guo G, Pan K, Fang S, Ye L, Tong X, Wang Z, Xue X, Zhang H, Mol. Ther. Nucleic. Acids 2021, 26, 575–593; [DOI] [PMC free article] [PubMed] [Google Scholar]; c)Squires JE, Preiss T, Epigenomics 2010, 2, 709–715; [DOI] [PubMed] [Google Scholar]; d)Trixl L, Lusser A, Wiley Interdiscip. Rev. RNA 2019, 10, e1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bohnsack KE, Höbartner C, Bohnsack MT, Genes 2019, 10, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Walsh CT, ACS Chem. Biol. 2022, 17, 2686–2703. [DOI] [PubMed] [Google Scholar]
- [8].a) Chellamuthu A, Gray SG, Cells 2020, 9, 1758; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Tong X, Xiang Y, Hu Y, Hu Y, Li H, Wang H, Zhao K-N, Xue X, Zhu S, Front. Oncol. 2022, 12, 788801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sun Z, Xue S, Xu H, Hu X, Chen S, Yang Z, Yang Y, Ouyang J, Cui H, Epigenomics 2019, 11, 439–453. [DOI] [PubMed] [Google Scholar]
- [10].Tang H, Fan X, Xing J, Liu Z, Jiang B, Dou Y, Gorospe M, Wang W, Aging (Albany N.Y.) 2015, 7, 1143–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Yang L, Ma Y, Han W, Li W, Cui L, Zhao X, Tian Y, Zhou Z, Wang W, Wang H, J. Biol. Chem. 2015, 290, 26627–26637; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Xu X, Zhang Y, Zhang J, Zhang X, J. Biol. Chem. 2020, 295, 18134–18147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tuorto F, Liebers R, Musch T, Schaefer M, Hofmann S, Kellner S, Frye M, Helm M, Stoecklin G, Lyko F, Nat. Struct. Mol. Biol. 2012, 19, 900–905. [DOI] [PubMed] [Google Scholar]
- [13].a) Müller M, Samel-Pommerencke A, Legrand C, Tuorto F, Lyko F, Ehrenhofer-Murray AE, RNA Biol. 2019, 16, 249–256; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Shinoda S, Kitagawa S, Nakagawa S, Wei F-Y, Tomizawa K, Araki K, Araki M, Suzuki T, Suzuki T, Nucleic Acids Res. 2019, 47, 8734–8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Tao Y, Remillard D, Vinogradova EV, Yokoyama M, Banchenko S, Schwefel D, Melillo B, Schreiber SL, Zhang X, Cravatt BF, J. Am. Chem. Soc. 2022, 144, 18688–18699; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Lazear MR, Remsberg JR, Jaeger MG, Rothamel K, Her HL, DeMeester KE, Njomen E, Hogg SJ, Rahman J, Whitby LR, Won SJ, Schafroth MA, Ogasawara D, Yokoyama M, Lindsey GL, Li H, Germain J, Barbas S, Vaughan J, Hanigan TW, Vartabedian VF, Reinhardt CJ, Dix MM, Koo SJ, Heo I, Teijaro JR, Simon GM, Ghosh B, Abdel-Wahab O, Ahn K, Saghatelian A, Melillo B, Schreiber SL, Yeo GW, Cravatt BF, Mol. Cell 2023, 83, 1725–1742 e1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rostovtsev VV, Green LG, Fokin VV, Sharpless KB, Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- [16].a) Speers AE, Cravatt BF, ChemBioChem 2004, 5, 41–47; [DOI] [PubMed] [Google Scholar]; b)Speers AE, Adam GC, Cravatt BF, J. Am. Chem. Soc. 2003, 125, 4686–4687. [DOI] [PubMed] [Google Scholar]
- [17].Cravatt B, Njomen E, Hayward R, DeMeester K, Ogasawara D, Dix M, Nguyen T, Ashby P, Simon G, Schreiber S, Melillo B, ChemRxiv. 2023, 10.26434/chemrxiv-22023-s26446n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D, Nature 2021, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Hussain S, Sajini AA, Blanco S, Dietmann S, Lombard P, Sugimoto Y, Paramor M, Gleeson JG, Odom DT, Ule J, Frye M, Cell Rep. 2013, 4, 255–261; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Hussain S, Aleksic J, Blanco S, Dietmann S, Frye M, Genome Biol. 2013, 14, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Auxilien S, Guérineau V, Szweykowska-Kulińska Z, Golinelli-Pimpaneau B, RNA Biol. 2012, 9, 1331–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Thüring K, Schmid K, Keller P, Helm M, in RNA Methylation, Springer, 2017, pp. 3–18; [Google Scholar]; b)Kellner S, Ochel A, Thüring K, Spenkuch F, Neumann J, Sharma S, Entian K-D, Schneider D, Helm M, Nucleic Acids Res. 2014, 42, e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Haag S, Warda AS, Kretschmer J, Günnigmann MA, Höbartner C, Bohnsack MT, RNA 2015, 21, 1532–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].a) Mons E, Roet S, Kim RQ, Mulder MPC, Curr. Protoc. 2022, 2, e419; [DOI] [PubMed] [Google Scholar]; b)Hartung IV, Rudolph J, Mader MM, Mulder MPC, Workman P, J. Med. Chem. 2023, 66, 9297–9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Khoddami V, Cairns BR, Nat. Biotechnol. 2013, 31, 458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, Suter CM, Preiss T, Nucleic Acids Res. 2012, 40, 5023–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Abelson H, Johnson L, Penman S, Green H, Cell 1974, 1, 161–165. [Google Scholar]
- [27].a) Kathman SG, Koo SJ, Lindsey GL, Her HL, Blue SM, Li H, Jaensch S, Remsberg JR, Ahn K, Yeo GW, Ghosh B, Cravatt BF, Nat. Chem. Biol. 2023, 19, 825–836; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Kavanagh ME, Horning BD, Khattri R, Roy N, Lu JP, Whitby LR, Ye E, Brannon JC, Parker A, Chick JM, Eissler CL, Wong AJ, Rodriguez JL, Rodiles S, Masuda K, Teijaro JR, Simon GM, Patricelli MP, Cravatt BF, Nat. Chem. Biol. 2022, 18, 1388–1398; [DOI] [PMC free article] [PubMed] [Google Scholar]; c)Feldman HC, Merlini E, Guijas C, DeMeester KE, Njomen E, Kozina EM, Yokoyama M, Vinogradova E, Reardon HT, Melillo B, Schreiber SL, Loreto A, Blankman JL, Cravatt BF, Proc. Natl. Acad. Sci. U.S.A. 2022, 119, e2208457119; [DOI] [PMC free article] [PubMed] [Google Scholar]; d)Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS, Chem. Biol. 2013, 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhao J, Hu H, Wang S, Wang L, Wang R, Genes 2021, 12, 1488.34680884 [Google Scholar]
- [29].a) Vinogradova EV, Zhang X, Remillard D, Lazar DC, Suciu RM, Wang Y, Bianco G, Yamashita Y, Crowley VM, Schafroth MA, Yokoyama M, Konrad DB, Lum KM, Simon GM, Kemper EK, Lazear MR, Yin S, Blewett MM, Dix MM, Nguyen N, Shokhirev MN, Chin EN, Lairson LL, Melillo B, Schreiber SL, Forli S, Teijaro JR, Cravatt BF, Cell 2020, 182, 1009–1026.e1029; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Chen Y, Craven GB, Kamber RA, Cuesta A, Zhersh S, Moroz YS, Bassik MC, Taunton J, Nat. Chem. 2023, in press. [DOI] [PubMed] [Google Scholar]
- [30].a) Khoddami V, Cairns BR, Nat. Protoc. 2014, 9, 337–361; [DOI] [PubMed] [Google Scholar]; b)Maetani M, Zoller J, Melillo B, Verho O, Kato N, Pu J, Comer E, Schreiber SL, J. Am. Chem. Soc. 2017, 139, 11300–11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.