

Abstract
INTRODUCTION:
Human accelerated regions (HARs) are evolutionarily conserved sequences that acquired an unexpectedly high number of nucleotide substitutions in the human genome since divergence from our common ancestor with chimpanzees. Prior work has established that many HARs are gene regulatory enhancers that function during embryonic development, particularly in neurodevelopment, and that most HARs show signatures of positive selection. However, the events that caused the sudden change in selective pressures on HARs remain a mystery.
RATIONALE:
Because HARs acquired many substitutions in our ancestors after millions of years of extreme constraint across diverse mammals, we reasoned that their conserved roles in regulating development of the brain and other organs must have changed during human evolution. One mechanism that could drive such a functional shift is enhancer hijacking, whereby the target gene repertoire of a noncoding sequence is changed through alterations in three-dimensional genome folding. The regulatory information encoded in a hijacked enhancer would likely need to change to avoid deleterious expression of the altered target gene while also possibly supporting modified expression patterns. Structural variants—large genomic insertions, deletions, and rearrangements—are the greatest sources of sequence differences between the human and chimpanzee genomes, and they have the potential to affect how a region of the genome folds and localizes in the nucleus. We therefore hypothesized that some HARs were generated through enhancer hijacking triggered by nearby human-specific structural variants (hsSVs).
RESULTS:
We leveraged an alignment of hundreds of mammalian genomes plus a Nextflow pipeline that we wrote for automating the detection of lineage-specific accelerated regions to identify 312 high-confidence HARs (zooHARs). Through massively parallel reporter assays and machine learning integration of hundreds of epigenomic datasets, we showed that many zooHARs function as neurodevelopmental enhancers and that their human substitutions alter transcription factor binding sites, consistent with previous studies. We further mapped zooHARs to specific cell types and tissues using single-cell open chromatin and gene expression data, and we found that they represent a more diverse set of neurodevelopmental processes than a parallel set of chimpanzee accelerated regions.
To test the enhancer hijacking hypothesis, we first examined the three-dimensional neighborhoods of zooHARs using publicly available chromatin capture (Hi-C) data, finding a significant enrichment of zooHARs in domains with hsSVs. This motivated us to use deep learning to predict how hsSVs changed genome folding in the human versus the chimpanzee genomes. We found that 30% of zooHARs occur within 500 kb of an hsSV that substantially alters local chromatin interactions, and we confirmed this association in Hi-C data that we generated in human and chimpanzee neural progenitor cells. Finally, we showed that chromatin domains containing zooHARs and hsSVs are enriched for genes differentially expressed in human versus chimpanzee neurodevelopment.
CONCLUSION:
The origin of many HARs may be explained by human-specific structural variants that altered three-dimensional genome folding, causing evolutionarily conserved enhancers to adapt to different target genes and regulatory domains.
Graphical Abstract
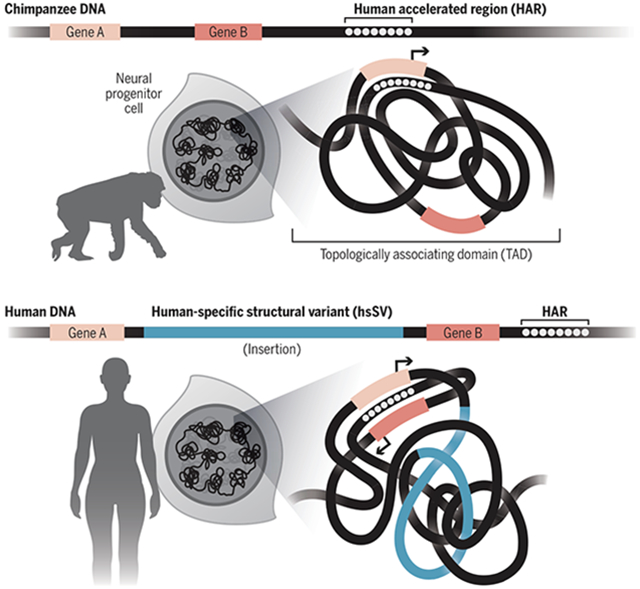
Example of HAR enhancer hijacking. The HAR is nearby and regulates gene A, but not gene B, as the chimpanzee genome folds. An insertion in the human genome brings the HAR closer to gene B, causing expression of gene B. The HAR adapts to being in gene B’s regulatory domain through substitutions to previously conserved nucleotides.
Human accelerated regions (HARs) are conserved genomic loci that evolved at an accelerated rate in the human lineage and may underlie human-specific traits. We generated HARs and chimpanzee accelerated regions with an automated pipeline and an alignment of 241 mammalian genomes. Combining deep learning with chromatin capture experiments in human and chimpanzee neural progenitor cells, we discovered a significant enrichment of HARs in topologically associating domains containing human-specific genomic variants that change three-dimensional (3D) genome organization. Differential gene expression between humans and chimpanzees at these loci suggests rewiring of regulatory interactions between HARs and neurodevelopmental genes. Thus, comparative genomics together with models of 3D genome folding revealed enhancer hijacking as an explanation for the rapid evolution of HARs.
Human accelerated regions (HARs) are genomic loci that were conserved over millions of years of vertebrate evolution but evolved quickly in the human lineage and thus are of great interest based on their potential to underlie human-specific traits (1-8). Many HARs are predicted to function as gene enhancers, particularly for genes implicated in neural development (9). Furthermore, most HARs appear to have evolved under positive selection due to having more human substitutions than expected given the local neutral rate (10)—an indication that the sequence changes were beneficial to ancient humans. However, the mechanisms facilitating their shift in selective pressure after millions of years of constraint remains to be determined.
Structural variation is a substantial driver of genome evolution. The majority of genomic differences between humans and our closest extant relatives, chimpanzees and bonobos, derive from structural variation, largely in the noncoding genome (11). Changes to genome organization mediated by structural variants can rewire gene regulatory networks through enhancer hijacking—also called enhancer adoption—through which genes gain or lose regulatory signals, affecting spatiotemporal gene expression (12-14). Enhancer hijacking has been identified as a contributing factor to cancer and other human diseases (13, 15-17), and previous work has proposed that it may be a driver of species evolution (7, 18, 19). For example, the locus containing the cluster of Hox genes is encompassed in a single topologically associating domain (TAD) in the bilaterian ancestor, but vertebrates have two separate TADs; this difference may have driven evolutionary innovations in developmental body patterning specific to vertebrates (18, 20, 21).
Motivated by these findings, we hypothesized that some HAR enhancers were hijacked as a result of human-specific structural variants (hsSVs) altering their three-dimensional (3D) contacts. This could have changed the HAR’s target gene repertoire and subjected it to different selective pressures in humans, thus driving its human-specific accelerated evolution. Testing this complex hypothesis is now possible because of the confluence of recent datasets and technologies. First, the Zoonomia Consortium generated an alignment of 241 mammalian genomes (22), which provided the opportunity to detect lineage-specific evolutionary patterns at an unprecedented scale. Second, recent work comparing multiple great ape genomes has identified a high-quality set of 17,789 hsSVs (23). Third, publicly available epigenomic, transcriptomic, and chromatin interaction datasets for many cell types and tissues enable machine learning predictions of how lineage-specific sequence changes affect genome function (24). Finally, we had access to primary tissue from the human midgestation telencephalon to validate our predictions. In this study, we combine these experimental and computational resources to demonstrate that HARs and hsSVs occur in the same TAD significantly more often than expected and that these TADs are enriched for genes that are differentially expressed between humans and chimpanzees. These results implicate enhancer hijacking as a genetic mechanism to explain the lineage-specific accelerated evolution of many HARs, potentially underlying human-specific neurodevelopmental phenotypes.
Human and chimpanzee accelerated regions share features consistent with function as neurodevelopmental enhancers
To test HAR loci for enhancer hijacking, we first sought to generate an updated set of HARs from the Zoonomia alignment (zooHARs) alongside a consistently inferred set of chimpanzee accelerated regions (zooCHARs). The identification of species-specific accelerated regions in alignments containing many species with large genomes requires substantial computational resources. The necessary methods are implemented in the Phylogenetic Analysis with Space/Time models (PHAST) software package (25), but users need to combine multiple methods and runtime parameters to manipulate multiple sequence alignments, fit phylogenetic models, identify conserved elements, and perform statistical tests for acceleration. These requirements are limiting how many researchers can conduct these analyses. To assist with implementation on high-performance computing and automate previously developed scripts for detecting accelerated regions (1, 25-27), we developed a Nextflow pipeline that is portable to different parallel computing environments (28). This required optimizing modeling parameters in the PHAST software package for large, multiple-sequence alignments (25). The resulting open-source software tool, called AcceleratedRegionsNF (29), enables automated, reproducible, and streamlined identification of accelerated regions in any species or lineage on any computing platform (Fig. 1A) (29).
Fig. 1. hsSVs are enriched in zooHAR chromatin domains and predicted to change the 3D genome.

(A) Pipeline to identify lineage-specific accelerated regions. Blue circles indicate initial input data, purple hexagons are intermediate results, and the green square is the final output. (B) Odds ratio of chromatin contact domains in GM12878 cells (33) containing hsSVs and zooHARs (green line) compared with a null distribution (shaded blue region) of odds ratios for chromatin contact domains containing conserved (phastCons) elements and hsSVs from 1000 random draws of phastCons equaling the number of zooHARs. (C) Akita prediction of a locus [hg38.chr4:26614489-27531993; hsSV, human-specific insertion hsSV1 from (23, 30)] with a human-specific insertion (Original), with the human-specific insertion deleted in silico (hsSV deleted), and a subtraction matrix (Original - hsSV deleted) comparing the chromatin contact matrices with and without the human-specific insertion. White boxes indicate regions that change in the original compared with the hsSV deleted sequences. Log(observed/expected) contact values are shown in the heatmaps.
Using AcceleratedRegionsNF (29), we leveraged the Zoonomia alignment of 241 mammal genomes (22) to identify 312 zooHARs (table S1). The zooHARs demonstrate similar features to previous sets of HARs, including being mainly noncoding and being located near genes involved in developmental and neurological processes (fig. S1A and fig. S2; see additional discussion in the supplementary text) (6, 9, 30). The majority of zooHARs (86%) also have signatures of positive selection, here defined as having a substitution rate that significantly exceeds a local estimate of neutral rate and not showing a substitution pattern consistent with GC-biased gene conversion (fig. S1B). We assessed evidence for selection, GC-biased gene conversion (faster than neutral substitution rate with a strong bias toward A/T to G/C changes), and loss of constraint (approximately neutral substitution rate in the human lineage versus conservation in other mammals) using a previously published model (10). Supporting roles in neurodevelopment, approximately one-third of zooHARs are transcribed in the developing human neocortex (fig. S1C).
To compare accelerated evolution in the human and chimpanzee genomes side by side, we next used the Zoonomia alignment (22) and AcceleratedRegionsNF (29) to identify 141 zooCHARs. The median distance between zooHARs and zooCHARs is significantly less than expected (1.05 Mb; bootstrap P value = 0.02, both in hg38), as observed in previous sets of primate accelerated regions (31). We then annotated the zooCHARs (in hg38) with the same datasets as zooHARs and observed that these two sets of species-specific accelerated regions have similar genomic and epigenomic features (fig. S1, D and E; fig. S3; and table S2). These annotations are strongly indicative of zooCHARs being regulatory elements in the developing brain and other tissues, similar to zooHARs, despite a human bias in the available annotation datasets. Genes near both zooHARs and zooCHARs are significantly enriched for roles in transcriptional regulation (hypergeometric tests; figs. S2 and S3). Orthologous regions to zooCHARs are also transcribed in the developing human neocortex (fig. S1F). These findings suggest that distinct sets of evolutionarily conserved enhancers regulating transcription factors and other neurodevelopmental genes evolved under positive selection in both the human and chimpanzee genomes.
Despite these notable similarities, we also observed some differences between zooHARs and zooCHARs. The annotations of genes nearby zooHARs suggest connections to a broader diversity of developmental processes compared with zooCHARs (figs. S2 and S3), which may indicate that enhancer evolution affected more aspects of neurobiology and development in humans compared with chimpanzees. Another difference is the smaller number of zooCHARs. A similar number of conserved elements were used in the zooHAR versus zooCHAR analyses (225,317 and 225,287, respectively), but a smaller percentage of conserved elements qualified as zooCHARs (0.06% compared with 0.1% for zooHARs). Although it is tempting to speculate that the higher number of zooHARs is because of more adaptive evolution in the human versus chimpanzee lineage, it may instead be attributable to the lower quality of the chimpanzee reference genome and the strict quality control filtering we performed when running AcceleratedRegionsNF (29). Prior work has found that the number of accelerated regions identified in different primates is related to how deeply the genomes were sequenced (31). Future improvements to genome assemblies for nonhuman primates will enable reliable estimates of the relative levels of genomic acceleration across species. Together, these analyses demonstrate that zooHARs identified from an alignment of 241 mammals have features consistent with previous studies proposing functionality as gene regulatory elements, particularly in neurodevelopment, and possibly with broader downstream consequences than can be linked to zooCHARs.
HARs are enriched in 3D TADs with hsSVs
Genomic loci near duplicated genes have been shown to evolve rapidly (32), which suggests that there is synergy between structural variation and nucleotide-level genome evolution. To explore this, we sought to determine whether zooHARs and hsSVs tended to colocate in the context of the 3D genome. Using a high-quality set of TADs from lymphoblastoid cells (33), we found that zooHARs are strongly enriched in TADs with hsSVs relative to the set of phastCons conserved elements from which zooHARs are identified (odds ratio = 3.0, bootstrap P < 0.001; Fig. 1B). This enrichment is robust to repeating the analysis with TADs from other cell types, including primary midgestation telencephalon, and a different TAD-calling method (fig. S4). To determine whether the enrichment is simply driven by localization of hsSVs near zooHARs in the linear genome sequence, we replaced the TADs with random, size-matched windows and found that zooHARs were not significantly enriched in this context relative to phastCons elements (fig. S4). Thus, we conclude that zooHARs are specifically enriched in TADs with hsSVs, which suggests that 3D genome organization and structural variation may be linked to the accelerated evolution of HARs.
hsSVs are predicted to have changed the 3D chromatin environment of zooHARs
Structural variation is the main contributor to genome-wide genetic divergence between the human and chimpanzee genomes (11), and it has the potential to generate large changes in 3D genome organization through the disruption of insulating boundaries or other structural motifs (34). Based on our observation that zooHARs are enriched in TADs with hsSVs (Fig. 1B), we sought to determine whether hsSVs may have generated changes in 3D genome folding in loci with zooHARs. Using Akita, a neural network–based deep learning model trained on six cell types to predict 3D genome contact matrices from DNA sequence (35), we assessed the effect of hsSVs (table S3). For each variant, we predicted the chromatin contact matrices for the DNA sequence with and without the variant and computed the mean squared distance between the two matrices (Fig. 1C and table S3). Many hsSVs are predicted to change 3D genome organization near zooHARs, and 30% of zooHARs occur within 500 kb of a hsSV with a disruption score in the top decile of all disruption scores for hsSVs. These results suggest that human-specific 3D genome structures are encoded in DNA sequence and are modified through hsSVs.
High-resolution Hi-C data from humans and chimpanzees validates 3D genome reorganization near zooHARs and zooCHARs
To validate the predicted changes to 3D genome organization mediated by hsSVs near zooHARs, we generated chromatin capture (Hi-C) data from neural progenitor cells (NPCs) differentiated from two human and two chimpanzee induced pluripotent stem cell (iPSC) lines at matched developmental time points. Together, these experiments generated more than 3.4 billion individually mapped chromatin contacts (table S4). All lines were from male individuals, and two technical replicates were generated per sample. Stratum-adjusted correlation coefficients (36) demonstrated high concordance of data between replicates and individuals from the same species (fig. S5), so we merged data from all replicates and samples of each species for downstream analyses. The cis/trans interaction ratio and distance-dependent interaction frequency decay indicate that the data are high quality (table S4 and fig. S6).
Conservation of 3D genome structures, such as A and B compartments and TAD boundaries, has been demonstrated in various species. However, our understanding of the extent of this conservation is still developing, with gene regulatory interactions inside TADs appearing to be somewhat dynamic across cell types and species (33, 37-42). Analyzing our NPC Hi-C data, we found 10% of chromatin loops and 8% of TAD boundaries to be human specific (table S5). This is slightly less than the 14% identified in a recent study comparing human and macaque chromatin organization (40), likely because chimpanzees are more closely related to humans than are macaques. Thus, the majority of chromatin loops, also called dots or peaks (43), are conserved or partially conserved between the human and chimpanzee NPCs (table S5 and fig. S7) (44, 45). These results support the idea of conservation of large-scale chromatin structures between human and chimpanzee, although differences are detectable in specific loci.
We next confirmed the enrichment of zooHARs in TADs containing hsSVs in our Hi-C data from human NPCs (fig. S4E and table S5). This enrichment was also observed between zooCHARs and chimpanzee-specific structural variants (23) in TADs from the chimpanzee data (odds ratio = 4.8, bootstrap P = 0.04), indicating that colocation of lineage-specific structural variants and accelerated regions is not a human-specific phenomenon. As structural variants and Hi-C data are generated for more species, it will be possible to use the tools from this study to quantify this notable association across diverse Eukaryotes. Finally, we used our NPC Hi-C data (table S5) to associate zooHARs and zooCHARs with genes and found significant enrichment for transcriptional regulators of developmental processes, confirming and extending our gene ontology (GO) results based on nearby genes (table S6).
Hijacked zooHARs associated with differentially expressed genes
Based on the idea that zooHARs are regulatory elements that control gene expression, we sought to determine whether genes that are differentially expressed between humans and chimpanzees are linked to zooHARs in the 3D genome. We compiled a compendium of matched human and chimpanzee RNA sequencing (RNA-seq) datasets and converted these into lists of genes that are differentially expressed between the two species in various tissues and cell types. Intersecting these with our NPC TAD calls (table S5), we observed that TADs containing zooHARs and hsSVs are enriched for genes differentially expressed between humans and chimpanzees in NPCs (chi-squared P = 0.018; table S7) (46) and cerebral organoids (chi-squared P = 0.003; table S7) (47). By contrast, genes differentially expressed between human and chimpanzee adult brain tissue (48), iPSCs, iPSC-derived cardiomyocytes, and heart tissue (49) are not enriched in TADs containing zooHARs and hsSVs (table S7) (23, 46-49). These results support our enhancer hijacking hypothesis while suggesting that the effects of enhancer hijacking may be developmental stage and cell type specific.
The loci encompassing zooHAR.126 and zooHAR.15 are two clear examples of how hsSVs can alter 3D regulatory interactions between HAR enhancers and neurodevelopmental genes. Each locus has a strong Akita prediction of altered genome folding in the presence of a hsSV, which is highly similar to the differences observed in NPC Hi-C data (Fig. 2, A and B) (35). The average disruption, which measures differences between the human and chimpanzee Hi-C data, is greatest at specific genomic elements within the 1-Mb region (Fig. 2, C and D), including at species-specific loops and the promoters of genes differentially expressed between humans and chimpanzees (Fig. 2, E and F, and fig. S8). For example, the Tourette’s syndrome gene NECTIN3 (50) is in the same TAD with a hsSV and zooHAR.126, and it is down-regulated in human versus chimpanzee NPCs (fig. S8) (46). Similarly, the developmental gene MAF, implicated in Ayme-Gripp syndrome, is differentially expressed between humans and chimpanzees in inhibitory neurons, NPCs, iPSCs, and iPSC-derived cardiomyocyte progenitors (46, 47, 49), and it is in a TAD encompassing a hsSV and zooHAR.15, which overlaps previously identified 2xHAR.21 (51). To determine with higher confidence that the observed changes in 3D structure at these loci were human derived, we assessed the orthologous loci in previously published rhesus macaque fetal brain cortex plate (40). For both loci, the human-specific changes to 3D genome organization described here were not observed in the rhesus macaque data (40), which suggests that they are human derived as a result of the hsSVs, as predicted by Akita (fig. S9) (35). Together, these results establish that the 3D genome changes in these loci are human specific, associated with gene expression changes, and likely caused by the hsSVs.
Fig. 2. hsSVs change the 3D genome around zooHARs and zooCHARs.

White boxes highlight differences between the species. Log(observed/expected) values are shown in the heatmaps. (A and B) Subtraction matrices for the in silico predicted change due to the human-specific insertion (left) and observed chromatin contact maps in human compared with chimpanzee NPC Hi-C (right) for the loci containing zooHAR.126 [hg38.chr4:26614489-27531993; hsSV1 from (23, 30)] and zooHAR.15 [hg38.chr16:79237694-80155198; hsSV2 from (23, 30)], respectively. (C and D) Human (top) and chimpanzee (bottom) log (observed/expected) Hi-C contact frequencies in each locus, with the disruption score (10-kb resolution) in between. (E and F) zooHAR locations denoted by vertical lines adjacent to their names. Conserved (blue), chimpanzee-specific (green), and human-specific (orange) loops are shown [5-kb resolution, loops called with Mustache (44)].
Many zooHARs are neurodevelopmental enhancers with cell type–specific activity
To define the cell types and tissues that may be affected by hijacked HARs, we expanded on previous work demonstrating enhancer-associated epigenomic signatures of HARs in specific cell types and tissues (51) and predicting HAR enhancer activity (9, 50). We annotated a 1500–base pair (bp) genomic window centered at the midpoint of each zooHAR by overlap with recently generated datasets of open chromatin [61 assay for transposase-accessible chromatin with sequencing (ATAC-seq), 40 deoxyribonuclease 1 hypersensitive sites sequencing (DNase-seq)], chromatin-bound proteins [204 chromatin immunoprecipitation sequencing (ChIP-seq) experiments for histone modifications and transcription factors], and 3D chromatin interactions [4 proximity ligation–assisted ChIP-seq (PLAC-seq), 4 promoter-capture Hi-C] (52-59). This window size was chosen to match the typical size of in vivo validated enhancers (60). Collectively, these annotations cover 44 human cell types, including multiple brain regions from specific developmental time points. To explore the gene regulatory pathways of zooHARs, we further annotated them with previously published transcription factor footprints (55).
First, we used these annotations to explore the cell types in which zooHARs may function as gene regulatory elements. Even against a stringent background set of phastCons elements, which themselves tend to be enriched for gene regulatory marks related to development (9), zooHARs are enriched for annotations indicative of neurodevelopmental regulatory activity, including ATAC-seq peaks and promoter-capture Hi-C interactions in multiple neuronal cell types (centered odds ratio range, 2.20 to 55.9; bootstrap P < 0.05; fig. S10). As one example, zooHAR.126 overlaps numerous regulatory epigenomic marks and footprints for seven transcription factors (Fig. 3A). Over all zooHAR footprints, enriched transcription factors included inhibitory neuron specifier DLX1 (61), master brain regulator and telencephalon marker FOXG1, and cortical and striatal projection neuron marker MEIS2 (62, 63) (Fig. 3B and table S8). Thus, zooHARs do have epigenetic signatures consistent with developmental enhancer activity, particularly in the embryonic brain, consistent with prior HAR studies.
Fig. 3. zooHARs in human brain development.

(A) Transcription factor footprints (55) and epigenomic marks (59) overlapping zooHAR.126. NSC, neural stem cell. (B) Subset of enriched transcription factor footprints in zooHARs relative to phastCons elements (Fisher’s exact P ≤ 0.05). Full set is available in table S8. (C) Cell types in which zooHARs are predicted to regulate gene expression based on CellWalker analysis of data from the developing human telencephalon. (D) Cell type assignments for zooCHARs based on CellWalker analysis of data from the developing human telencephalon. Unlike with HARs, no CHARs map to late-stage excitatory neurons. Cell types are abbreviated as follows: excitatory neurons (ENs) derived from primary visual cortex (V1) or prefrontal cortex (PFC), newborn excitatory neurons (nENs), inhibitory cortical interneurons (IN-CTXs) originating in the medial/caudal ganglionic eminence (MGE/CGE), newborn interneurons (nINs), intermediate progenitor cells (IPCs), and truncated/ventral/outer radial glia (tRG/vRG/oRG). More cell type information is available at hhttps://cells.ucsc.edu/?ds=cortex-dev (58, 66).
Next, we used these epigenetic annotations to build a new machine learning model for predicting neurodevelopmental enhancers (materials and methods) (30). The epigenetic datasets were used as features, and the in vivo validated VISTA enhancers (64) served as examples of neurodevelopmental enhancers for training the model. After validating the model on held-out VISTA enhancers, we used it to predict that 197/312 zooHARs (63.1%) function as neurodevelopmental enhancers based on their epigenetic profiles (table S1). This increases the proportion of HARs with predicted regulatory activity in the brain relative to predictions from previous work (9, 24).
To further specify cell types in the human brain, where zooHARs likely function as regulatory elements, we applied the CellWalker method to map them to cell types using single-cell ATAC-seq with RNA-seq from the developing human telencephalon surveyed at midgestation (58, 65-67). We found the highest number of zooHARs assigned to newborn interneurons, radial glia, excitatory neurons from the prefrontal cortex, and medial ganglionic eminence intermediate progenitors (Fig. 3C and table S9). Repeating this analysis for zooCHARs, cell types were largely similar to those assigned to zooHARs, but many fewer zooCHARs mapped to excitatory neurons from the prefrontal cortex (Fig. 3D and table S9). This difference may provide clues toward the mechanisms underlying species-specific neurodevelopmental traits, such as increased plasticity and protracted maturation in the human brain. However, these results must be interpreted with the caveat that cell type assignments were made from human data because parallel chimpanzee data are not available. Finally, we repeated the CellWalker analysis using single-cell ATAC-seq and RNA-seq from the human adult brain (68, 69) and heart (70). Very few accelerated regions mapped to adult heart cell types. In the adult brain, fewer zooCHARs were assigned cell types compared with zooHARs, with the largest species difference being in excitatory neurons, mirroring our finding in the midgestation brain (fig. S11 and table S9).
Massively parallel validation of zooHARs in human primary cortical cells
To validate these predictions, we performed a massively parallel reporter assay (MPRA) to test the enhancer activity of all 312 zooHARs in five replicates of human primary cells from midgestation (gestational week 18) telencephalon (71). After stringent quality control, we obtained RNA/DNA ratios of 276 zooHARs and found that 139 (50.1%) drove reporter gene expression to a level indicative of enhancer activity as determined by the median activity of a set of externally validated positive controls in the MPRA experiment (materials and methods and table S8) (30, 71). Thus, many zooHARs are capable of driving gene expression in the human telencephalon at midgestation. On the basis of our machine learning predictions and epigenetic profiling of zooHARs, we expect that additional zooHARs are active enhancers in other brain regions and developmental stages.
Next, we compared MPRA activity with the results of our machine learning predictions for the same zooHARs (table S1). Of the 175 zooHARs predicted to function as neurodevelopmental enhancers and passing MPRA quality control, 88 (50.3%) drove reporter gene expression to a level indicative of enhancer activity (30, 71). This high-confidence set of human accelerated enhancers active in human neurodevelopment includes zooHAR.133, zooHAR.138, and zooHAR.156, all of which are in TADs with developmental genes (EFNA5, EN1, and PBX3, respectively) that have differential contacts in our human versus chimpanzee NPC Hi-C data. Prior studies precisely reconstructing human-specific mutations at the endogenous locus in the mouse have validated zooHAR.1 (also known as HACNS1, HAR2, 2xHAR.3) as an enhancer of GBX2 and zooHAR.138 (also known as 2xHAR.20, HAR19, HAR80) as an enhancer of EN1. Other zooHARs with enhancer-like epigenetic signatures but lower MPRA activity may function in different developmental stages or in cell types poorly represented in our telencephalon samples, or their activity may be underestimated by MPRA because of our use of 270-bp sequences and random integration sites. Despite these limitations, our MPRA data strongly support the conclusion that many zooHARs function as enhancers in cell types of the developing brain.
Altogether, this work demonstrates that hsSVs cluster in TADs with HARs that likely function as regulatory elements in neurodevelopment, and these hsSVs can change 3D regulatory interactions of HARs. Our findings demonstrate that HARs, which have multiple lines of evidence suggesting enhancer activity in neurodevelopment, cluster in TADs with hsSVs that may drive differential 3D interactions of HARs specifically in humans.
Discussion
Lineage-specific accelerated regions represent sequence-based evolutionary innovations in the genome that may underlie traits that define each species. The Nextflow pipeline introduced in this work enables reproducible identification of accelerated regions in any species in very large alignments, as demonstrated with the Zoonomia dataset of 241 mammals (22).
By integrating dozens of public and newly developed datasets, a machine learning model of enhancer activity, a network-based cell type labeling method, and MPRA experiments performed on primary cells from the human midgestation telencephalon, we refined our understanding of which HARs may function as regulatory elements, at which developmental stages, and in what cell types. Viewing accelerated regions through the lens of 3D genome organization revealed an enrichment of zooHARs and zooCHARs in TADs containing species-specific structural variants. Generation of the high-resolution cross-species Hi-C in matched NPCs from humans and chimpanzees enabled the further discovery that hsSVs predicted by a deep learning model to change 3D genome organization nearby HARs and CHARs correspond to true differences between human and chimpanzee NPCs. Because HARs are active enhancers in diverse cell types and the majority of them contact putative target genes in a cell type–specific manner (72), future investigations of more cell types may uncover further perturbations.
There are interesting questions to be asked about the sequence of genomic events in loci with hsSVs and HARs. One possibility is that, in some cases, the hsSV altered the 3D chromatin contacts of a conserved regulatory element that then underwent rapid adaptation through point mutations in the same species to adjust to its altered target genes. With available data, however, we cannot rule out the possibility that the accelerated region changed before the structural variant. We also cannot confidently infer that the structural variant and 3D genome changes caused accelerated sequence evolution of the regulatory element. It is important to note that most TADs containing hsSVs with high disruption scores do not contain zooHARs, and approximately one-third contain phastCons elements that are not human accelerated. Nonetheless, our integrative data analysis points to enhancer hijacking as a potential genetic mechanism to explain HARs and other lineage-accelerated, conserved noncoding regions. Further experimentation will be needed to ascertain the validity of this hypothesis. However, it is clear that the evolution of genome sequence and 3D organization do not occur in isolation.
Materials and methods summary
To identify zooHARs, we ran AcceleratedRegionsNF (29) on the genome-wide, multiple-sequence alignments of 241 mammals from the Zoonomia Consortium (22), specifying the branch from the chimpanzee-human ancestor to modern humans as the lineage to test for acceleration and using a false discovery rate threshold of 5%. The phastCons conserved elements from which zooHARs were identified served as a background distribution for enrichment tests. zooCHARs were discovered and characterized in a similar manner. AcceleratedRegionsNF is available as an open-source, Nextflow pipeline that automates the computation of accelerated regions on large, multiple-sequence alignments through code that is easily ported to any computing environment (28, 29).
The effects of hsSVs on 3D genome folding were predicted using the Akita model (35). Genome sequences with and without each hsSV were provided to Akita, and the mean squared error (disruption score) between the resulting two contact matrices was computed.
NPCs were differentiated from two human (WTC11 and HS1) and two chimpanzee (C3649 and Pt2a) iPSC lines. Hi-C was performed using the Arima Genomics Hi-C kit according to the manufacturer’s instructions, libraries were sequenced with paired-end, 150-bp reads using two lanes of an Illumina NovaSeq6000 S2.
A 1500-bp window centered on each zooHAR was annotated with publicly available epigenetic and gene expression data plus chromatin loops, TADs, and compartments called in our NPC Hi-C data. These annotations were used for enrichment tests and as features in a machine learning model trained to distinguish neurodevelopmental enhancers from enhancers active in other tissues plus nonenhancers downloaded from the VISTA Enhancer Browser (64). We estimated the neurodevelopmental cell types in which zooHARs are active using CellWalker (66). Each zooHAR was assessed for evidence for positive selection versus GC-biased gene conversion or loss of constraint using a previously published model based on population genetic dynamics (10).
To test human zooHAR sequences for enhancer activity, lentivirus-based MPRAs were performed in cultured primary cells that were dissociated from human telencephalon tissue harvested at midgestation (73). Additional methodological details are available in the supplementary materials (30).
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Pittman, G. Fudenberg, A. Lind, E. McArthur, R. Ziffra, T. Capra, and S. Lyalina for helpful discussions, sharing code, and suggestions toward the results shown in this work. We thank G. Maki and T. Tolpa for assistance with figures and visualization.
Funding:
This study received support from a Discovery Fellowship (K.C.K.), National Institute of Mental Health grants R01MH109907 and U01MH116438 (N.A., K.S.P., and K.C.K.), National Institute of Mental Health grant DP2MH122400-01 (A.P. and T.F.), National Institute of Human Genome Research grant R01HG008742 (E.K.), Gladstone Institutes (K.S.P.), the Schmidt Futures Foundation (A.P. and T.F.), the Shurl and Kay Curci Foundation (A.P. and T.F.), and a Swedish Research Council Distinguished Professor Award (K.L.-T.).
Zoonomia Consortium
Gregory Andrews1, Joel C. Armstrong2, Matteo Bianchi3, Bruce W. Birren4, Kevin R. Bredemeyer5, Ana M. Breit6, Matthew J. Christmas3, Hiram Clawson2, Joana Damas7, Federica Di Palma8,9, Mark Diekhans2, Michael X. Dong3, Eduardo Eizirik10, Kaili Fan1, Cornelia Fanter11, Nicole M. Foley5, Karin Forsberg-Nilsson12,13, Carlos J. Garcia14, John Gatesy15, Steven Gazal16, Diane P. Genereux4, Linda Goodman17, Jenna Grimshaw14, Michaela K. Halsey14, Andrew J. Harris5, Glenn Hickey18, Michael Hiller19,20,21, Allyson G. Hindle11, Robert M. Hubley22, Graham M. Hughes23, Jeremy Johnson4, David Juan24, Irene M. Kaplow25,26, Elinor K. Karlsson1,4,27, Kathleen C. Keough17,28,29, Bogdan Kirilenko19,20,21, Klaus-Peter Koepfli30,31,32, Jennifer M. Korstian14, Amanda Kowalczyk25,26, Sergey V. Kozyrev3, Alyssa J. Lawler4,26,33, Colleen Lawless23, Thomas Lehmann34, Danielle L. Levesque6, Harris A. Lewin7,35,36, Xue Li1,4,37, Abigail Lind28,29, Kerstin Lindblad-Toh3,4, Ava Mackay-Smith38, Voichita D. Marinescu3, Tomas Marques-Bonet39,40,41,42, Victor C. Mason43, Jennifer R. S. Meadows3, Wynn K. Meyer44, Jill E. Moore1, Lucas R. Moreira1,4, Diana D. Moreno-Santillan14, Kathleen M. Morrill1,4,37, Gerard Muntané24, William J. Murphy5, Arcadi Navarro39,41,45,46, Martin Nweeia47,48,49,50 Sylvia Ortmann51, Austin Osmanski14, Benedict Paten2, Nicole S. Paulat14, Andreas R. Pfenning25,26, BaDoi N. Phan25,26,52, Katherine S. Pollard28,39,53, Henry E. Pratt1, David A. Ray14, Steven K. Reilly38, Jeb R. Rosen22, Irina Ruf54, Louise Ryan23, Oliver A. Ryder55,56, Pardis C. Sabeti4,57,58, Daniel E. Schäffer25, Aitor Serres24, Beth Shapiro59,60, Arian F. A. Smit22, Mark Springer61, Chaitanya Srinivasan25, Cynthia Steiner55, Jessica M. Storer22, Kevin A. M. Sullivan14, Patrick F. Sullivan62,63, Elisabeth Sundström3, Megan A. Supple59, Ross Swofford4, Joy-El Talbot64, Emma Teeling23, Jason Turner-Maier4, Alejandro Valenzuela24, Franziska Wagner65, Ola Wallerman3, Chao Wang3, Juehan Wang16, Zhiping Weng1, Aryn P. Wilder55, Morgan E. Wirthlin25,26,66, James R. Xue4,57, Xiaomeng Zhang4,25,26
1Program in Bioinformatics and Integrative Biology, UMass Chan Medical School, Worcester, MA 01605, USA. 2Genomics Institute, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 3Department of Medical Biochemistry and Microbiology, Science for Life Laboratory, Uppsala University, Uppsala 751 32, Sweden. 4Broad Institute of MIT and Harvard, Cambridge, MA 02139, USA. 5Veterinary Integrative Biosciences, Texas A&M University, College Station, TX 77843, USA. 6School of Biology and Ecology, University of Maine, Orono, ME 04469, USA. 7The Genome Center, University of California Davis, Davis, CA 95616, USA. 8Genome British Columbia, Vancouver, BC, Canada. 9School of Biological Sciences, University of East Anglia, Norwich, UK. 10School of Health and Life Sciences, Pontifical Catholic University of Rio Grande do Sul, Porto Alegre 90619-900, Brazil. 11School of Life Sciences, University of Nevada Las Vegas, Las Vegas, NV 89154, USA. 12Biodiscovery Institute, University of Nottingham, Nottingham, UK. 13Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala 751 85, Sweden. 14Department of Biological Sciences, Texas Tech University, Lubbock, TX 79409, USA. 15Division of Vertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA. 16Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA. 17Fauna Bio Incorporated, Emeryville, CA 94608, USA. 18Baskin School of Engineering, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 19Faculty of Biosciences, Goethe-University, 60438 Frankfurt, Germany. 20LOEWE Centre for Translational Biodiversity Genomics, 60325 Frankfurt, Germany. 21Senckenberg Research Institute, 60325 Frankfurt, Germany. 22Institute for Systems Biology, Seattle, WA 98109, USA. 23School of Biology and Environmental Science, University College Dublin, Belfield, Dublin 4, Ireland. 24Department of Experimental and Health Sciences, Institute of Evolutionary Biology (UPF-CSIC), Universitat Pompeu Fabra, Barcelona 08003, Spain. 25Department of Computational Biology, School of Computer Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 26Neuroscience Institute, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 27Program in Molecular Medicine, UMass Chan Medical School, Worcester, MA 01605, USA. 28Department of Epidemiology & Biostatistics, University of California San Francisco, San Francisco, CA 94158, USA. 29Gladstone Institutes, San Francisco, CA 94158, USA. 30Center for Species Survival, Smithsonian’s National Zoo and Conservation Biology Institute, Washington, DC 20008, USA. 31Computer Technologies Laboratory, ITMO University, St. Petersburg 197101, Russia. 32Smithsonian-Mason School of Conservation, George Mason University, Front Royal, VA 22630, USA. 33Department of Biological Sciences, Mellon College of Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 34Senckenberg Research Institute and Natural History Museum Frankfurt, 60325 Frankfurt am Main, Germany. 35Department of Evolution and Ecology, University of California Davis, Davis, CA 95616, USA. 36John Muir Institute for the Environment, University of California Davis, Davis, CA 95616, USA. 37Morningside Graduate School of Biomedical Sciences, UMass Chan Medical School, Worcester, MA 01605, USA. 38Department of Genetics, Yale School of Medicine, New Haven, CT 06510, USA. 39Catalan Institution of Research and Advanced Studies (ICREA), Barcelona 08010, Spain. 40CNAG-CRG, Centre for Genomic Regulation, Barcelona Institute of Science and Technology (BIST), Barcelona 08036, Spain. 41Department of Medicine and Life Sciences, Institute of Evolutionary Biology (UPF-CSIC), Universitat Pompeu Fabra, Barcelona 08003, Spain. 42Institut Català de Paleontologia Miquel Crusafont, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Barcelona, Spain. 43Institute of Cell Biology, University of Bern, 3012 Bern, Switzerland. 44Department of Biological Sciences, Lehigh University, Bethlehem, PA 18015, USA. 45BarcelonaBeta Brain Research Center, Pasqual Maragall Foundation, Barcelona 08005, Spain. 46CRG, Centre for Genomic Regulation, Barcelona Institute of Science and Technology (BIST), Barcelona 08003, Spain. 47Department of Comprehensive Care, School of Dental Medicine, Case Western Reserve University, Cleveland, OH 44106, USA. 48Department of Vertebrate Zoology, Canadian Museum of Nature, Ottawa, ON K2P 2R1, Canada. 49Department of Vertebrate Zoology, Smithsonian Institution, Washington, DC 20002, USA. 50Narwhal Genome Initiative, Department of Restorative Dentistry and Biomaterials Sciences, Harvard School of Dental Medicine, Boston, MA 02115, USA. 51Department of Evolutionary Ecology, Leibniz Institute for Zoo and Wildlife Research, 10315 Berlin, Germany. 52Medical Scientist Training Program, University of Pittsburgh School of Medicine, Pittsburgh, PA 15261, USA. 53Chan Zuckerberg Biohub, San Francisco, CA 94158, USA. 54Division of Messel Research and Mammalogy, Senckenberg Research Institute and Natural History Museum Frankfurt, 60325 Frankfurt am Main, Germany. 55Conservation Genetics, San Diego Zoo Wildlife Alliance, Escondido, CA 92027, USA. 56Department of Evolution, Behavior and Ecology, School of Biological Sciences, University of California San Diego, La Jolla, CA 92039, USA. 57Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA 02138, USA. 58Howard Hughes Medical Institute, Harvard University, Cambridge, MA 02138, USA. 59Department of Ecology and Evolutionary Biology, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 60Howard Hughes Medical Institute, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 61Department of Evolution, Ecology and Organismal Biology, University of California Riverside, Riverside, CA 92521, USA. 62Department of Genetics, University of North Carolina Medical School, Chapel Hill, NC 27599, USA. 63Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden. 64Iris Data Solutions, LLC, Orono, ME 04473, USA. 65Museum of Zoology, Senckenberg Natural History Collections Dresden, 01109 Dresden, Germany. 66Allen Institute for Brain Science, Seattle, WA 98109, USA.
Footnotes
Data and materials availability: The Zoonomia data are available at https://zoonomiaproject.org/the-project/. The Nextflow pipeline to identify lineage-specific accelerated regions is available at https://github.com/keoughkath/AcceleratedRegionsNF (29) The Hi-C data are available at GSE183137. The MPRA data are available at Dryad (73). All other data are available in the main text or the supplementary materials.
Competing interests: K.C.K. is currently an employee of Fauna Bio. The other authors declare no competing interests.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Pollard KS et al. , Forces shaping the fastest evolving regions in the human genome. PLOS Genet. 2, e168 (2006). doi: 10.1371/journal.pgen.0020168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pollard KS et al. , An RNA gene expressed during cortical development evolved rapidly in humans. Nature 443, 167–172 (2006). doi: 10.1038/nature05113 [DOI] [PubMed] [Google Scholar]
- 3.Prabhakar S, Noonan JP, Pääbo S, Rubin EM, Accelerated evolution of conserved noncoding sequences in humans. Science 314, 786 (2006). doi: 10.1126/science.1130738 [DOI] [PubMed] [Google Scholar]
- 4.Bird CP et al. , Fast-evolving noncoding sequences in the human genome. Genome Biol. 8, R118 (2007). doi: 10.1186/gb-2007-8-6-r118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bush EC, Lahn BT, A genome-wide screen for noncoding elements important in primate evolution. BMC Evol. Biol 8, 17 (2008). doi: 10.1186/1471-2148-8-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hubisz MJ, Pollard KS, Exploring the genesis and functions of Human Accelerated Regions sheds light on their role in human evolution. Curr. Opin. Genet. Dev 29, 15–21 (2014). doi: 10.1016/j.gde.2014.07.005 [DOI] [PubMed] [Google Scholar]
- 7.Franchini LF, Pollard KS, Human evolution: The non-coding revolution. BMC Biol. 15, 89 (2017). doi: 10.1186/s12915-017-0428-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reilly SK, Noonan JP, Evolution of gene regulation in humans. Annu. Rev. Genomics Hum. Genet 17, 45–67 (2016). doi: 10.1146/annurev-genom-090314-045935 [DOI] [PubMed] [Google Scholar]
- 9.Capra JA, Erwin GD, McKinsey G, Rubenstein JLR, Pollard KS, Many human accelerated regions are developmental enhancers. Phil. Trans. R. Soc. B 368, 20130025 (2013). doi: 10.1098/rstb.2013.0025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kostka D, Hubisz MJ, Siepel A, Pollard KS, The role of GC-biased gene conversion in shaping the fastest evolving regions of the human genome. Mol. Biol. Evol 29, 1047–1057 (2012). doi: 10.1093/molbev/msr279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chimpanzee Sequencing and Analysis Consortium, Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87 (2005). doi: 10.1038/nature04072 [DOI] [PubMed] [Google Scholar]
- 12.Hnisz D et al. , Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351, 1454–1458 (2016). doi: 10.1126/science.aad9024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Affer M et al. , Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia 28, 1725–1735 (2014). doi: 10.1038/leu.2014.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zimmerman MW et al. , MYC drives a subset of high-risk pediatric neuroblastomas and is activated through mechanisms including enhancer hijacking and focal enhancer amplification. Cancer Discov. 8, 320–335 (2018). doi: 10.1158/2159-8290.CD-17-0993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lupiáñez DG, Spielmann M, Mundlos S, Breaking TADs: How alterations of chromatin domains result in disease. Trends Genet. 32, 225–237 (2016). doi: 10.1016/j.tig.2016.01.003 [DOI] [PubMed] [Google Scholar]
- 16.Ibn-Salem J et al. , Deletions of chromosomal regulatory boundaries are associated with congenital disease. Genome Biol. 15, 423 (2014). doi: 10.1186/s13059-014-0423-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franke M et al. , Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538, 265–269 (2016). doi: 10.1038/nature19800 [DOI] [PubMed] [Google Scholar]
- 18.Acemel RD et al. , A single three-dimensional chromatin compartment in amphioxus indicates a stepwise evolution of vertebrate Hox bimodal regulation. Nat. Genet 48, 336–341 (2016). doi: 10.1038/ng.3497 [DOI] [PubMed] [Google Scholar]
- 19.Maeso I, Acemel RD, Gómez-Skarmeta JL, Cis-regulatory landscapes in development and evolution. Curr. Opin. Genet. Dev 43, 17–22 (2017). doi: 10.1016/j.gde.2016.10.004 [DOI] [PubMed] [Google Scholar]
- 20.Acemel RD, Maeso I, Gómez-Skarmeta JL, Topologically associated domains: A successful scaffold for the evolution of gene regulation in animals. WIREs Dev. Biol 6, e265 (2017). doi: 10.1002/wdev.265 [DOI] [PubMed] [Google Scholar]
- 21.Lonfat N, Duboule D, Structure, function and evolution of topologically associating domains (TADs) at HOX loci. FEBS Lett. 589, 2869–2876 (2015). doi: 10.1016/j.febslet.2015.04.024 [DOI] [PubMed] [Google Scholar]
- 22.Christmas MJ et al. , Evolutionary constraint and innovation across hundreds of placental mammals. Science 380, eabn3943 (2023). doi: 10.1123/science.abn3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kronenberg ZN et al. , High-resolution comparative analysis of great ape genomes. Science 360, eaar6343 (2018). doi: 10.1126/science.aar6343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whalen S et al. , Machine-Learning Dissection of Human Accelerated Regions in Primate Neurodevelopment (Cell Press, 2022); https://ssrn.com/abstract=4149954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hubisz MJ, Pollard KS, Siepel A, PHAST and RPHAST: Phylogenetic analysis with space/time models. Brief. Bioinform 12, 41–51 (2011). doi: 10.1093/bib/bbq072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siepel A, Pollard KS, Haussler D, in Research in Computational Molecular Biology, Apostolico A, Guerra C, Istrail S, Pevzner PA, Waterman M, Eds., vol. 3909 of Lecture Notes in Computer Science (Springer, 2006), pp. 190–205. [Google Scholar]
- 27.Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A, Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121 (2010). doi: 10.1101/gr.097857.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Tommaso P et al. , Nextflow enables reproducible computational workflows. Nat. Biotechnol 35, 316–319 (2017). doi: 10.1038/nbt.3820 [DOI] [PubMed] [Google Scholar]
- 29.Keough K, keoughkath/AcceleratedRegionsNF: Release for Zenodo, version 1.0, Zenodo; (2022); 10.5281/zenodo.7478724. [DOI] [Google Scholar]
- 30.See the supplementary materials.
- 31.Kostka D, Holloway AK, Pollard KS, Developmental loci harbor clusters of accelerated regions that evolved independently in ape lineages. Mol. Biol. Evol 35, 2034–2045 (2018). doi: 10.1093/molbev/msy109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kostka D, Hahn MW, Pollard KS, Noncoding sequences near duplicated genes evolve rapidly. Genome Biol. Evol 2, 518–533 (2010). doi: 10.1093/gbe/evq037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rao SSP et al. , A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). doi: 10.1016/j.cell.2014.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spielmann M, Lupiáñez DG, Mundlos S, Structural variation in the 3D genome. Nat. Rev. Genet 19, 453–467 (2018). doi: 10.1038/s41576-018-0007-0 [DOI] [PubMed] [Google Scholar]
- 35.Fudenberg G, Kelley DR, Pollard KS, Predicting 3D genome folding from DNA sequence with Akita. Nat. Methods 17, 1111–1117 (2020). doi: 10.1038/s41592-020-0958-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang T et al. , HiCRep: Assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome Res. 27, 1939–1949 (2017). doi: 10.1101/gr.220640.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dixon JR et al. , Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012). doi: 10.1038/nature11082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vietri Rudan M et al. , Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 10, 1297–1309 (2015). doi: 10.1016/j.celrep.2015.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eres IE, Luo K, Hsiao CJ, Blake LE, Gilad Y, Reorganization of 3D genome structure may contribute to gene regulatory evolution in primates. PLOS Genet. 15, e1008278 (2019). doi: 10.1371/journal.pgen.1008278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo X et al. , 3D Genome of macaque fetal brain reveals evolutionary innovations during primate corticogenesis. Cell 184, 723–740.e21 (2021). doi: 10.1016/j.cell.2021.01.001 [DOI] [PubMed] [Google Scholar]
- 41.Eres IE, Gilad Y, A TAD skeptic: Is 3D genome topology conserved? Trends Genet. 37, 216–223 (2021). doi: 10.1016/j.tig.2020.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoencamp C et al. , 3D genomics across the tree of life reveals condensin II as a determinant of architecture type. Science 372, 984–989 (2021). doi: 10.1126/science.abe2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mirny LA, Imakaev M, Abdennur N, Two major mechanisms of chromosome organization. Curr. Opin. Cell Biol 58,142–152 (2019). doi: 10.1016/j.ceb.2019.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roayaei Ardakany A, Gezer HT, Lonardi S, Ay F, Mustache: Multi-scale detection of chromatin loops from Hi-C and Micro-C maps using scale-space representation. Genome Biol. 21, 256 (2020). doi: 10.1186/s13059-020-02167-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diehl AG, Ouyang N, Boyle AP, Transposable elements contribute to cell and species-specific chromatin looping and gene regulation in mammalian genomes. Nat. Commun 11, 1796 (2020). doi: 10.1038/s41467-020-15520-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marchetto MC et al. , Species-specific maturation profiles of human, chimpanzee and bonobo neural cells. eLife 8, e37527 (2019). doi: 10.7554/eLife.37527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pollen AA et al. , Establishing cerebral organoids as models of human-specific brain evolution. Cell 176, 743–756.e17 (2019). doi: 10.1016/j.cell.2019.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kanton S et al. , Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 574, 418–422 (2019). doi: 10.1038/s41586-019-1654-9 [DOI] [PubMed] [Google Scholar]
- 49.Pavlovic BJ, Blake LE, Roux J, Chavarria C, Gilad Y, A comparative assessment of human and chimpanzee iPSC-derived cardiomyocytes with primary heart tissues. Sci. Rep 8, 15312 (2018). doi: 10.1038/s41598-018-33478-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Doan RN et al. , Mutations in human accelerated regions disrupt cognition and social behavior. Cell 167, 341–354.e12 (2016). doi: 10.1016/j.cell.2016.08.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lindblad-Toh K et al. , A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482 (2011). doi: 10.1038/nature10530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Castelijns B et al. , Hominin-specific regulatory elements selectively emerged in oligodendrocytes and are disrupted in autism patients. Nat. Commun 11, 301 (2020). doi: 10.1038/s41467-019-14269-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Markenscoff-Papadimitriou E et al. , A chromatin accessibility atlas of the developing human telencephalon. Cell 182, 754–769.e18 (2020). doi: 10.1016/j.cell.2020.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Forrest MP et al. , Open chromatin profiling in hiPSC-derived neurons prioritizes functional noncoding psychiatric risk variants and highlights neurodevelopmental loci. Cell Stem Cell 21, 305–318.e8 (2017). doi: 10.1016/j.stem.2017.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Funk CC et al. , Atlas of transcription factor binding sites from ENCODE DNase hypersensitivity data across 27 tissue types. Cell Rep. 32, 108029 (2020). doi: 10.1016/j.celrep.2020.108029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song M et al. , Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nat. Genet 51, 1252–1262 (2019). doi: 10.1038/s41588-019-0472-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Song M et al. , Cell-type-specific 3D epigenomes in the developing human cortex. Nature 587, 644–649 (2020). doi: 10.1038/s41586-020-2825-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ziffra RS et al. , Single-cell epigenomics reveals mechanisms of human cortical development. Nature 598, 205–213 (2021). doi: 10.1038/s41586-021-03209-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kundaje A et al. , Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). doi: 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pennacchio LA et al. , In vivo enhancer analysis of human conserved non-coding sequences. Nature 444, 499–502 (2006). doi: 10.1038/nature05295 [DOI] [PubMed] [Google Scholar]
- 61.Petryniak MA, Potter GB, Rowitch DH, Rubenstein JLR, Dlx1 and Dlx2 control neuronal versus oligodendroglial cell fate acquisition in the developing forebrain. Neuron 55, 417–433 (2007). doi: 10.1016/j.neuron.2007.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmitz MT et al. , The development and evolution of inhibitory neurons in primate cerebrum. Nature 603, 871–877 (2022). doi: 10.1038/s41586-022-04510-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shibata M et al. , Regulation of Prefrontal Patterning, Connectivity and Synaptogenesis by Retinoic Acid. bioRxiv 2019.12.31.891036 [Preprint] (2019). 10.1101/2019.12.31.891036. [DOI] [Google Scholar]
- 64.Visel A, Minovitsky S, Dubchak I, Pennacchio LA, VISTA Enhancer Browser—A database of tissue-specific human enhancers. Nucleic Acids Res. 35, D88–D92 (2007). doi: 10.1093/nar/gkl822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Przytycki PF, Pollard KS, CellWalkR: An R package for integrating and visualizing single-cell and bulk data to resolve regulatory elements. Bioinformatics 38, 2621–2623 (2022). doi: 10.1093/bioinformatics/btac150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Przytycki PF, Pollard KS, CellWalker integrates single-cell and bulk data to resolve regulatory elements across cell types in complex tissues. Genome Biol. 22, 61 (2021). doi: 10.1186/s13059-021-02279-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nowakowski TJ et al. , Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323 (2017). doi: 10.1126/science.aap8809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hodge RD et al. , Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019). doi: 10.1038/s41586-019-1506-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tasic B et al. , Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78 (2018). doi: 10.1038/s41586-018-0654-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hocker JD et al. , Cardiac cell type-specific gene regulatory programs and disease risk association. Sci. Adv 7, eabf1444 (2021). doi: 10.1126/sciadv.abf1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Deng C et al. , Massively parallel characterization of psychiatric disorder-associated and cell-type-specific regulatory elements in the developing human cortex. bioRxiv 2023.02.15.528663 [Preprint] (2023). 10.1101/2023.02.15.528663. [DOI] [Google Scholar]
- 72.Won H, Huang J, Opland CK, Hartl CL, Geschwind DH, Human evolved regulatory elements modulate genes involved in cortical expansion and neurodevelopmental disease susceptibility. Nat. Commun 10, 2396 (2019). doi: 10.1038/s41467-019-10248-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Keough K, Supporting data for: Three-dimensional genome re-wiring in loci with Human Accelerated Regions, dataset, Dryad; (2023); 10.7272/Q6057D5N. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.