

Abstract
Previous studies have revealed tight metabolic complementarity between bivalves and their endosymbiotic chemosynthetic bacteria, but little is known about their interactions with ectosymbionts. Our analysis of the ectosymbiosis between a deep-sea scallop (Catillopecten margaritatus) and a gammaproteobacterium showed that bivalves could be highly interdependent with their ectosymbionts as well. Our microscopic observation revealed abundant sulfur-oxidizing bacteria (SOB) on the surfaces of the gill epithelial cells. Microbial 16S rRNA gene amplicon sequencing of the gill tissues showed the dominance of the SOB. An analysis of the SOB genome showed that it is substantially smaller than its free-living relatives and has lost cellular components required for free-living. Genomic and transcriptomic analyses showed that this ectosymbiont relies on rhodanese-like proteins and SOX multienzyme complex for energy generation, mainly on the Calvin–Benson–Bassham (CBB) cycle and peripherally on a phosphoenolpyruvate carboxylase for carbon assimilation. Besides, the symbiont encodes an incomplete tricarboxylic acid (TCA) cycle. Observation of the scallop’s digestive gland and its nitrogen metabolism pathways indicates it does not fully rely on the ectosymbiont for nutrition. Analysis of the host’s gene expression provided evidence that it could offer intermediates for the ectosymbiont to complete its TCA cycle and some amino acid synthesis pathways using exosomes, and its phagosomes, endosomes, and lysosomes might be involved in harvesting nutrients from the symbionts. Overall, our study prompts us to rethink the intimacy between the hosts and ectosymbionts in Bivalvia and the evolution of chemosymbiosis in general.
Keywords: chemosynthesis, cold seep, ectosymbiosis, glass scallop, thiotrophy
Introduction
Chemosymbioses between lithoautotrophic bacteria and animals have been widely recognized as a driving force for the ecological adaptation and evolution of invertebrates ranging from sponges, sea anemones, flatworms, nematodes, arthropods, annelids, and molluscs [1-3]. Still, the factors that enable the initiation, maintenance, and development of chemosymbioses remain poorly understood [1].
Chemosymbiosis is most phylogenetically widespread in the molluscan class Bivalvia, with nine families spanning from the early branching protobranchs to the recent venerids known to host chemosynthetic bacteria [4-6]. The chemosymbionts of bivalves are diverse, with some being methane-oxidizing bacteria (MOB) and others sulfur-oxidizing bacteria (SOB) or both MOB and SOB [7]. They also exhibit different levels of intimacy with their hosts from loosely attached to the gill surfaces (e.g. thyasirids) to enclose within epithelial gill cells (e.g. vesicomyids) [3]. Bivalves adopt different modes of symbiont transmission—from environmental transmission to strictly maternal transmission through the eggs [8] and consequently, the symbionts display varying degrees of bottleneck-driven genome reduction [5, 7, 9, 10].
The prevalence and diversity of chemosymbioses in Bivalvia make them prominent models to investigate some of the key questions within the field of symbiosis research: (i) whether endosymbiosis gradually evolves from ectosymbiosis [11, 12] and (ii) whether vertical symbiont transmission evolves from horizontal symbiont transmission [9, 11]. Answering these questions will be facilitated by uncovering the full extent of the chemosymbiotic diversity in bivalves and resolving their evolutionary histories. Despite a growing body of research, we do not know whether symbiosis has evolved multiple times within the group or from a single common ancestor [13]. To this end, it is imperative to screen for chemosynthesis in the 113 currently recognized families of Bivalvia, especially in the majority (104 families) where no such symbiotic relationship has yet been reported [14].
Here, we report chemosymbiosis in the order Pectinida, a large group of Bivalvia commonly known as scallops and are widely present from shallow water to the deep sea, including chemosynthetic habitats [15-19]. The lack of morphological specializations in the gill of a vent-dwelling scallop has been suggested to indicate an absence of chemosymbiosis [16]. Yet, the discoveries of simple homorhabdic gills bearing chemosymbionts in thysarids [20] and a heterotrophic alphaproteobacterial endosymbiont in a shallow-water scallop [21] supported further investigation of chemosymbiosis in vent and seep scallops. Catillopecten margaritatus—a deep-sea glass scallop that inhabits the Haima seep in the South China Sea— is commonly found on the empty shells of the vesicomyid Archivesica marissinica and around the tubeworm Sclerolinum annulatum aggregation [22-24]. Our previous analyses of the stable isotopes of C. margaritatus [23] revealed δ13C and δ34S values typical of the SOB symbiont-bearing bivalves that are known to rely solely or mainly on SOB for nutrition [12, 23, 25]. In this study, we aimed to characterize the chemosymbionts of C. margaritatus. First, we used a combination of 16S rRNA gene amplicon sequencing and microscopic analyses to identify and locate the SOB. Then, we sequenced the genome of the SOB and compared it with those of other bivalve chemosymbionts. Lastly, we conducted de novo meta-transcriptomics to reconstruct the holobiont metabolism. Our results indicate that the gill of C. margaritatus hosts a single epibiont phylotype related to those associated with bathymodioline mussels but divergent to those of other co-occurring invertebrates. This symbiont is primarily reliant on thiosulfate for sulfide oxidation and lacks hydrogenotrophic capabilities. Besides, we found that despite the extracellular localization of the symbiont and the host's use of external food sources, its genome is relatively small compared with its free-living relatives and the level of host-symbiont metabolic complementarity was high. Together, these results suggest an obligate association between the host and the bacteria. Given that previous omics studies of symbiosis in Bivalvia have focused on endosymbionts with expected tight host-symbiont metabolic integration [5, 8, 10], our results are significant because they not only reveal a new evolutionary path from asymbiosis to symbiosis in scallops, but also open a gate for comparative studies of bivalve ectosymbionts that are widespread in Thyasiridae and small Bathymodiolinae [20, 26], and considered as the early stages of bivalve-chemosynthetic bacteria symbioses [4].
Materials and methods
Sample collection
Three C. margaritatus individuals were collected from the Haima seep (16°54.04’N, 110°28.47′E) during a dive by the remotely operated vehicle Pioneer on board research vessel (R/V) Xiangyanghong 01 at 1433–1441 m water depth in September 2022. The samples were dissected onboard the R/V to separate the gill and gonad tissues, stored at −80°C for DNA and RNA sequencing, fixed by 4% paraformaldehyde (PFA) for hematoxylin–eosin (HE) staining and fluorescence in situ hybridization (FISH), and 2.5% glutaraldehyde for transmission electron microscopy (TEM), respectively.
HE staining, FISH, and TEM observation
The HE staining and FISH were conducted to visualize the morphology and symbiont distribution in gill and gonad tissues from two specimens. The PFA-fixed gill and gonad samples were dehydrated with ethanol and soaked in xylene, then embedded in Paraplast (Sigma, USA). Paraffin block was cut into 6 μm sections using an RM 2126 microtome (Leica, Germany). Paraplast was removed using xylene and rehydrated and the sections were stained with hematoxylin (Abcam, UK) and observed under a differential interference microscope (Olympus BX51, Japan). For FISH, the rehydrated sections were treated with 0.1% tween 20 in phosphate-buffered saline (PBST) to increase permeability, and hybridized in formamide hybridization buffer (0.9 M NaCl, 0.02 M Tris–HCl, 0.01% sodium dodecyl sulfate (SDS), 35% deionized formamide, 0.5 μM of probe BangT-642 [27] labeled by Cy5 dye targeting thiotrophic Gammaproteobacteria and 0.5 μM of negative control probe IMedM-138 [27] labeled by Cy3 dye targeting methanotrophic Gammaprobacteria) for 1 h at 46°C. The sections were rinsed with washing buffer (0.1 M NaCl, 0.02 M Tris–HCl, 5 mM ethylenediaminetetraacetic acid (EDTA), 0.01% SDS) for 15 min at 48°C, and stained using 4',6-diamidino-2-phenylindole (DAPI, Sigma, USA) for 3 min at room temperature. Then the sections were embedded in Antifade Mounting Medium (Beyotime, China) under a cover slip. The sections were observed under an LSM 710 NLO laser scanning confocal microscope (ZEISS, Germany).
For TEM observation, the gill sample preserved in glutaraldehyde was washed in PBS, transferred to 1% osmic acid for further fixation at 4°C for 2 h, rinsed in PBS, then dehydrated in a gradient of ethanol solutions and embedded in epoxy resin (EPON 812). After polymerizing at 37°C, 45°C, and 65°C for 24 h, respectively, the resin blocks were cut into 70 nm ultrathin sections with an EM UC7 ultramicrotome (Leica, Germany), and then stained by uranyl acetate then by lead nitrate and observed with a JEM-1200EX transmission electron microscope (JEOL, Japan).
DNA extraction, 16S rRNA gene amplicon analysis, and phylogenetic reconstruction
Bacterial 16S rRNA gene amplicon sequencing for the gill samples were conducted to determine the bacterial composition. Genomic DNA was extracted from three individuals using the cetyltrimethylammonium bromide (CTAB) method [28]. The V3–V4 region of the bacterial 16S rRNA gene was amplified with the primers 341F and 806R targeting bacteria [29, 30], and the libraries were generated using a TruSeq DNA PCR-Free Sample Preparation Kit (Illumina, USA). Sequencing was conducted on a NovaSeq6000 platform (Illumina, USA) under the PE250 mode in Novogene (Tianjin, China), generating 84 443, 92 176, and 93 095 raw reads, respectively (Table S1).
The 16S rRNA gene amplicon analysis was performed using the QIIME2 v2023.9 pipeline [31] to generate an amplicon sequence variants (ASV) table. The adapters were removed using the cutadapt trim-paired command. Paired-end sequence reads were merged using the vsearch’s merge_pairs function and filtered using the quality-filter q-score command. The Deblur workflow was employed for sequence quality control, utilizing a 16S rRNA gene reference as a positive filter. Reads were classified by taxon using the Greengene2 2022.10 (https://forum.qiime2.org/t/introducing-greengenes2-2022-10/25291) and visualized using the taxa barplot command to generate a barplot of bacterial abundances.
Phylogenetic analysis for the 16S rRNA gene sequences of the dominant and unique SOBs of the scallop and the symbiotic bacteria of related hosts (Table S2-S3) was performed using PhyloSuite v1.2.2 [32] to determine its phylogenic position. MAFFT v7.520 [33] was applied under the “auto” option to align gene fragments. Gblocks v0.91b [34] was applied to remove ambiguously aligned fragments in batches. The Bayesian Inference (BI) analysis was performed using MrBayes v3.2.6 [35] implemented in PhyloSuite for 10 million generations, with the initial 25% of the sampled data discarded as burn-in, and the best-fit substitution model GTR + I + Γ + F determined by ModelFinder implemented in PhyloSuite based on the Bayesian information criterion (BIC). The maximum likelihood (ML) analysis was performed using IQ-TREE v2.1.2 [36] implemented in PhyloSuite under the best-fit substitution model TIM3 + I + Γ4 + F selected by ModelFinder and ran for 1 000 ultrafast bootstraps.
DNA library construction and metagenomic sequencing
Total DNA samples of the gill (G3) containing the host and symbiont DNA, and the gonad tissues (SG1 and SG2, Table S1) were used for the construction of whole-genome shotgun libraries with an insert size of 350 bp, using NEBNext Ultra DNA Library Prep Kit for Illumina (NEB, USA). The libraries were sequenced on a NovaSeq 6000 sequencer (Illumina, USA) in Novogene (Tianjin, China). A Nanopore library of G3 was constructed using a Ligation Sequencing Kit 1D (PM) following the manufacturer's protocol. The library was sequenced on an Oxford Nanopore PromethION platform in Novogene (Tianjin, China), with 1.0 μg of the prepared library loaded onto a FLO-PRO002 flow cell (ID: FLO-PRO002).
Genomic assembly, mapping, annotation, and phylogeny
The adaptors and low-quality Illumina reads were removed using Trimmomatic v0.39 [37] (settings: LEADING:15 TRAILING:15 SLIDINGWINDOW:4:20 MINLEN:40). The Nanopore reads were base-called using Guppy v6.3.9 under the high-accuracy mode, and the reads were corrected and trimmed using Canu v2.1.1 [38] under the default settings, and then assembled using Canu with the genome size and maxInputCoverage set to 1.4 Mb (estimated based on the assembly of Illumina reads) and 1200, respectively. A single, circular bacterial genome was obtained, and two sequential rounds of error correction against the trimmed reads were applied using Minimap2 v2.27 [39] and Racon v1.4.13 [40] under the default settings. Then, the polished genome and the Illumina reads were aligned using BWA v0.7.17 [41], parsed using Samtools v1.12 [42], and base-call polishing using Pilon v1.23 [43] with a mindepth of 1. The genome quality was estimated using CheckM2 v1.0.1 [44], and the genome was annotated using Prokka v1.13.4 [45]. Then Pfam, COG, GO, and KEGG annotations were conducted using eggNOG-MAPPER v5.0 [46] against the eggNOG HMMs database. Besides, KEGG Mapper [47] was used to reconstruct the metabolic pathways. The average nucleotide identity (ANI) values among the selected genomes were calculated using JspeciesWS [48] to determine their phylogenetic distances. To further demonstrate the absence of symbiont in the gonad, metagenomic sequencing reads of two gonad tissue samples were also assembled and binned like above, and mapped to the symbiont genome using Bowtie2 v2.3.3.1 [49].
Phylogenomic analyses were performed as in a pipeline [50], including orthologous groups (OGs) identification using Orthofinder v2.2.7 [51], sequence alignment using MAFFT v7.520 [33], trimming using Gblocks v0.91b [34], and removal of paralogues using Phylopyprunner (https://gitlab.com/fethalen/phylopypruner). Protein sequences from the C. margaritatus symbiont genome, 24 available symbiotic or free-living SOB genomes, and two outgroups were used (Table S3). Two matrices (50% and 80% orthologue occupancy) were prepared. Single-copy OGs were sorted using GenesortR [52], and 600 OGs were selected to build the ML phylogenetic tree using IQ-Tree 2 v2.1.2 [36] under the MFP option for model selection and then run for 1 000 ultrafast bootstraps [53]. To compare the genomic structures of the C. margaritatus symbiont and its close relatives, Mauve v2.4.0 [54] was applied with a match seed weight of 15 and a minimum LCB score of 30 000.
RNA extraction, library construction, and metatranscriptomic sequencing
Metatranscriptomic sequencing was applied to quantify the symbiont gene expression levels and recover the host transcriptome. The total RNA of three gill samples (G1-G3) was extracted using the Trizol reagent (TAKARA, Japan). The metatranscriptomic sequencing was performed in Novogene (Tianjin, China). Briefly, ribosomal RNA was removed using NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, USA), and RNA molecules were fragmented into 250–300 bp and reverse-transcribed into cDNA. The libraries were sequenced on a NovaSeq 6000 sequencer (Illumina, USA) to produce approximately 63.2 million 150-bp paired-end reads per sample (Table S1).
De novo metatranscriptomic assembly and analyses
The adaptors and low-quality reads of Illumina sequencing were removed using Trimmomatic v0.39 [37] (settings: LEADING:15 TRAILING:15 SLIDINGWINDOW:4:20 MINLEN:40). The clean reads were used for de novo meta-assembly using Trinity v2.8.5 [55] under the default settings. De novo assembly generated 972 872 transcripts. The protein-coding genes (PCGs) were predicted using TransDecoder v5.5.0 [55] under the default settings. The highly redundant, bacterial, and potential contamination transcripts were removed. Briefly, Cd-Hit-Est v4.7 [56] was applied to remove redundant contigs using 95% as the sequence similarity cutoff. The PCGs were BLASTx searched against the NR database with an E-value threshold of 1e-10 using DIAMOND v0.9.24 [57], and the bacterial and other potential contaminant hits were removed to generate the host transcripts. After the filtering, 41 024 unigenes were retained. Gene annotation was conducted as in section 2.5, and a total of 25 473 (62.1%, the final host transcriptome) were annotated against at least one public database. A BUSCO v5.4.2 [58] analysis with the Metazoan_odb10 database showed that the transcriptome contained 87.5% complete (including 1.5% duplicated) and 5.6% fragmented metazoan BUSCOs. To quantify the gene expression, the clean reads were mapped to the host transcriptome and symbiont genes under the default settings, respectively, and expressed as transcripts per million (TPM) using Salmon v0.14.1 [59]. Besides, the Pearson correlation was used to evaluate the consistency in gene expression among the three transcriptome samples, which showed a high consistency between G2 and G3 (r = 0.94), but very low consistency between G1 and G2 (r = 0.02) and G1 and G3 (r = 0.10). The RNA data of sample G1 was not used for further analysis due to its low quality (Table S1). The final gene expression levels were determined using the average TPM values of G2 and G3. The host and symbiont genes with TPM values larger than 100 and 300, respectively, were defined as highly expressed genes (HEGs) [6]. The KEGG enrichments of these HEGs were conducted using TBtools-II v2.008 under the default settings [60].
Results and discussion
Symbionts are thiotrophic and located outside the scallop’s gill cells
Our HE staining analysis of the gill sections revealed abundant basophilic particles (deep purple colour indicating DNA) on the surface and between the microvilli of the gill epithelial cells (Fig. 1B-D). Our TEM analysis confirmed aggregations of bacteria on the surface and between the microvilli of the gill epithelial cells (Fig. 1E-H). The observed spherical bacteria were not methanotrophs as they did not contain intracellular concentric stacks [61]. Besides, we found endosomes containing bacterial cells in some sections (Fig. 1G and H), indicating that the host may harvest the symbionts by endocytosis. Our FISH analysis, using a fluorescent probe specific for thioautotrophic Gammaproteobacteria and a negative control targeting methanotrophic Gammaproteobacteria [27], confirmed that they belong to sulfur-oxidizing Gammaproteobacteria and further supported their extracellular distribution on the host’s gill epithelial cells (Fig. 2). Overall, these observations indicate that the symbionts of C. margaritatus are extracellular sulfur-oxidizing Gammaproteobacteria associated with its gill epibacteriocytes. We also conducted FISH analysis of the gonad but did not detect any SOB signal (Fig. S1), which suggests that the symbionts may not be vertically transmitted via germ cells. This observation is consistent with the dominance of horizontal symbiont transmission mode in chemosynthetic ectosymbionts [62]. Among bathymodioline mussels, both extracellular and intracellular symbioses were identified, with Gigantidas species hosting intracellular MOB [10], Bathymodiolus species hosting intracellular SOB and/or MOB [63], and small bathymodiolines like Adipicola, Idas, and Nypamodiolus harboring ectosymbiotic SOB [26, 64]. Previous studies hypothesized that these small bathymodiolines were the intermediate forms between their asymbiotic shallow-water ancestors and the bigger deep-sea vent- and seep-species adopting intracellular symbiosis [12, 65]. Therefore, our discovery of ectosymbiosis in C. margaritatus provides a model to test the hypothesis of symbiont acquisition during the shallow-water to deep-sea transition in bivalves.
Figure 1.
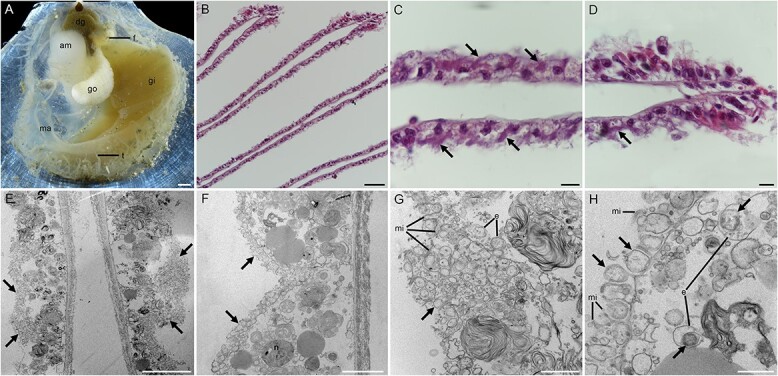
Anatomy features of Catillopecten margaritatus and photomicrographs showing the extracellular localization of the SOB. A: Anatomy features of C. margaritatus showing the gill tissue, left valve. B-D: Hematoxylin–eosin (HE) stain of gill sections showing a gross view of the gill filaments. E-H: Transmission electron microscopy (TEM) of gill filament showing the extracellular distribution of the SOBs. The bacteria are in the extracellular spaces filled by microvilli, indicated by arrows. Scale bar: A: 1 mm; B: 50 μm; C-D: 10 μm; E: 10 μm; F: 5 μm; G: 2 μm; H: 1 μm. Am, adductor muscle; b: Bacteria; dg, digestive gland; e: Endocytosis; f, foot; go, gonad; gi, gill; ma, mantle; mi: Microvillus; t, tentacle.
Figure 2.
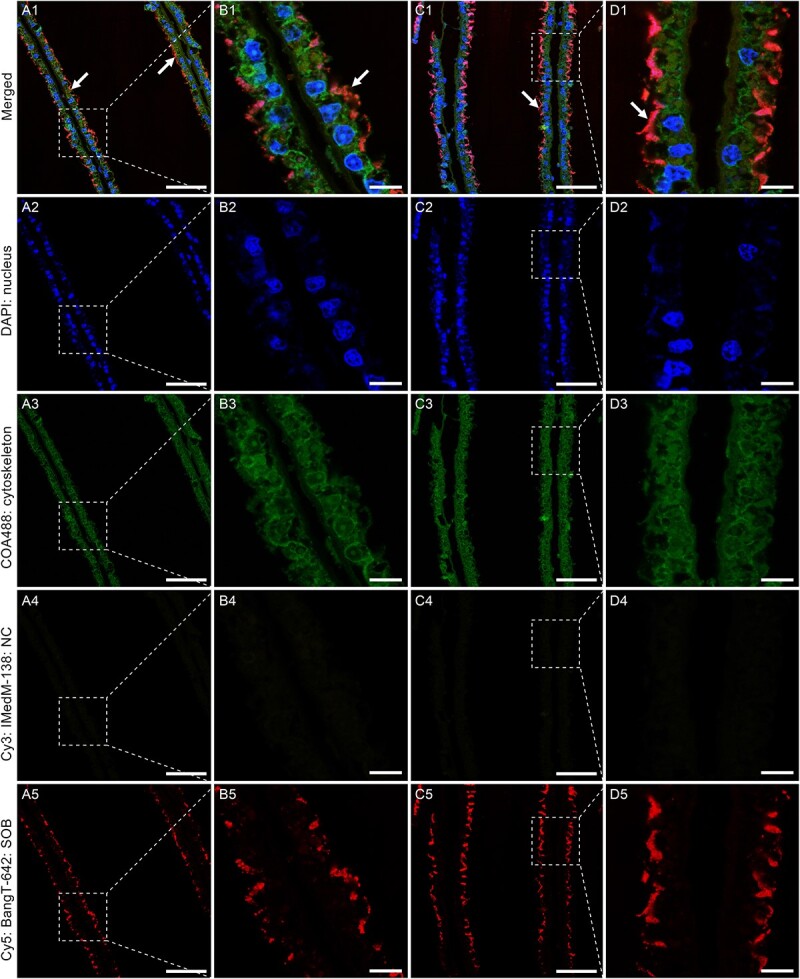
Fluorescence in situ hybridization (FISH) showing the extracellular localization of the SOB. The DAPI channels show the locations of the nuclei and the COA488 channels represent the gill cytoskeleton. The Cy3 channels show the negative control (NC) using the IMedM-138 probe targeting methanotrophic Gammaproteobacteria. The Cy5 channels indicate the bacteria based on the BangT-642 probe targeting thiotrophic Gammaproteobacteria (SOB). The bacteria are indicated by arrows, and the weak bacterial signals inside the gill cells might be the ectosymbionts endocytosed by the host, as indicated by the TEM micrographs (Fig. 1G and H). Scale bar: A & C: 50 μm; B & D: 10 μm.
Scallop gill harbors one dominant thiotrophic symbiont strain
A microbial 16S rRNA gene amplicon analysis revealed the dominance of a thiotrophic gammaproteobacterium belonging to the genus Thiodubiliella in the three samples, accounting for 72.5%, 62.8%, and 74.6% of the total reads, respectively (Fig. 3A and B, Table S4). Of the less abundant bacteria, we identified Desulfobacterota, Campylobacterota, and methane-oxidizing Proteobacteria with the ASV number of 18, 5, and 1, respectively (Table S4). Some of these chemosynthetic bacteria were also identified from the animal and sediment samples collected from the Haima seep [10, 66, 67], and most of them were present in only one of the three samples with low abundance (<1%) (Table S4), indicating they are environmental contaminants. Our phylogenetic analyses showed that the dominant thiotrophic gammaproteobacterium was nested in a clade containing mainly gill symbiotic SOB of bathymodiolines (Fig. S2).
Figure 3.
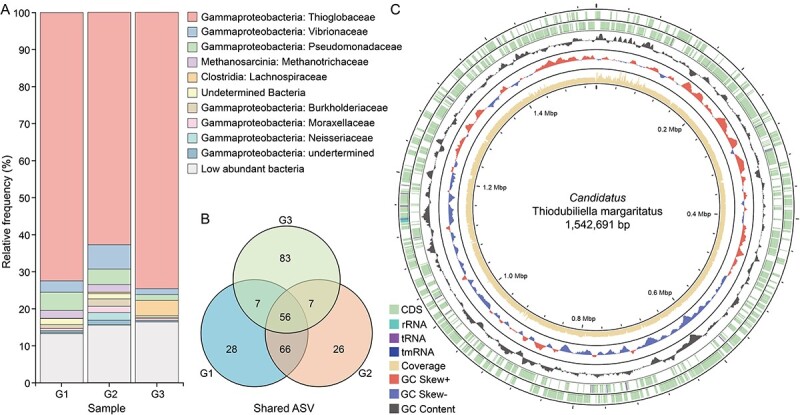
Gill bacterial community composition of Catillopecten margaritatus and genomic overview of the ectosymbiont Candidatus Thiodubiliella margaritatus. A: 16S rRNA gene amplicon sequencing results showing the relative frequency of the SOB symbiont and other bacteria in three individuals at the family level. The most abundant amplicon sequence variant (ASV) is a sulfur-oxidizing bacteria from the family Thioglobaceae (Table S4). B: Venne diagram showing the shared ASVs among the three samples. C: Circus plot of the ectosymbiont genome with its features. From the outer to inner circle: CDS, contigs, GC content, GC skew, genomic size.
Glass scallop’s ectosymbiont is absent in the gonad and phylogenetically close to bathymodioline symbionts
De novo assembly using both Nanopore and Illumina reads recovered a complete and circular gammaproteobacterial genome in a single contig measuring 1.54 Mb (Fig. 3C; Table S5). The genome is substantially smaller than those of the free-living SOB Thiomicrospira crunogena [68] and the environmentally acquired intracellular bathymodioline SOB [10], but only slightly smaller than that of the B. septemdierum and Conchocele bisecta symbionts which were considered in an intermediate state between extra- and intracellular symbiosis [69, 70]. CheckM2 analysis showed that it had 99.99% completeness and 0% contamination, indicating its high quality compared to other published SOB genomes (Table S3). Mapping the Illumina reads to the symbiont genome showed that it represented 43.3% of the reads from the gill sample, indicating that the symbiont is abundant on the gill surface, consistent with microscopic observation (Figs 1 and 2). Gene prediction showed that the symbiont genome contained 1 577 protein-coding genes (PCGs), 3 rRNAs, 36 tRNAs, and 1 tmRNA. The PCGs were well annotated (1 527 PCGs, 96.8%). The COG annotation indicated that the functional composition of the scallop symbiont genome was similar to that of bathymodioline SOB (Table S6).
De novo assembly and binning using Illumina reads of gonad tissues (Table S1) did not generate any bacterial genome. The mapping of Illumina reads to the ectosymbiont genome produced above showed that only a few ectosymbiont reads could be identified in the gonad samples (218 and 684 in 34.81 and 38.34 million reads, respectively). The proportion of ectosymbiont reads in the gonad was extremely small (about 0.001% on average) compared to the gill (43.3%), indicating these gonad-associated reads were contaminants and the ectosymbiont is not vertically transmitted via germ cells.
Consistent with the 16S rRNA gene amplicon results, phylogenetic analyses of 27 SOB genomes (Table S3) showed that this scallop symbiont belongs to the genus Thiodubiliella of the family Thioglobaceae (Fig. 4A), being sister to the SOB symbionts of Bathymodiolus and most closely related to those of B. septemdierum (Fig. 4A). Nevertheless, the symbiont genome of C. margaritatus and the two closest symbiont genomes of B. septemdierum had ANI values of ~80% only (Fig. 4B) – much lower than the recommended threshold of 95% for conspecific bacteria [71], indicating that these symbionts belong to different species. Therefore, we proposed Candidatus Thiodubiliella margaritatus as the name for the SOB symbiont of C. margaritatus. Besides bathymodiolines, symbiotic Thiodubiliella is also known to associate extracellularly with the vent thyasirid clam C. bisecta [69]. These symbionts are closely related to those of B. azoricus and B. puteoserpentis (Fig. 4), underscoring the genus’s ability to form a broad range of associations with bivalves. Catillopecten margaritatus usually lives on the empty shells of vesicomid A. marissinica or the sediment around aggregations of the tubeworm S. annulatum [24]. Although they co-occur, the SOB symbionts of these three species showed huge phylogenetic divergences [5, 22, 24], which may reflect their distinct evolutionary histories and subtle physiological differences allowing them to exploit sulfur resources in the heterogeneous habitats.
Figure 4.
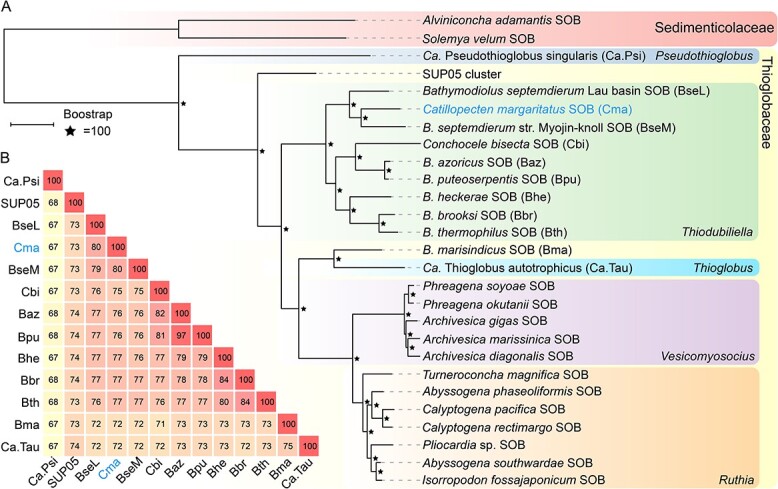
Phylogenetic relationships and genetic distances between the scallop ectosymbiont and other symbiotic and free-living sulfur-oxidizing bacteria (SOB). A: Maximum-likelihood tree constructed using 600 single-copy orthologs, with the endosymbionts of Solemya velum and Alvinconcha adamantis as the outgroups (Table S3). The scale bar (0.1) indicates the mean number of amino acid substitutions per site. B: The ANI values between the ectosymbiont of Catillopecten margaritatus and its relatives. The symbiont names are abbreviated based on their host’s names indicated in A.
Scallop and bathymodioline symbiont genomes are structurally divergent yet contain conserved central metabolism blocks
Whole genome alignment revealed different genomic structures among Ca. T. margaritatus and two available complete bathymodioline symbiont genomes, with multiple insertions, translocations, and inversions among them (Fig. 5; Fig. S3; Table S7). This contrasts with vesicomyid symbiont genomes which are highly conserved in genomic structure, except for a single block of 20 genes/pseudogenes missing in one clade and present in the other [5, 9, 72]. Nevertheless, the Ca. T. margaritatus genome contained eight conserved blocks (B1–8), including sulfur oxidation pathways – the SOX multienzyme complex and reverse dissimilatory sulfite reduction (rDSR) pathway (B2 & B7). The high conservation of these genomic blocks among the SOB symbiont genomes of the three bivalve species indicates selection may have favored the preservation of their organization due to their conserved roles in energy generation. Besides, large and small subunits of RuBisCO form I (rbcLS)—the key genes of the Calvin-Benson-Bassham (CBB) cycle—and several nitrite reductases were also conserved in block B4 between Ca. T. margaritatus and B. thermophilus SOB, whereas this block was inversely translocated in B. septemdierum. Several other genes and pathways of critical functions, including genetic expression (B1–2), ammonium transporter (B3), and some nutrient biosynthesis pathways (B3, B5–6, & B8), were also identified in the conserved blocks (Fig. 5; Table S7).
Figure 5.
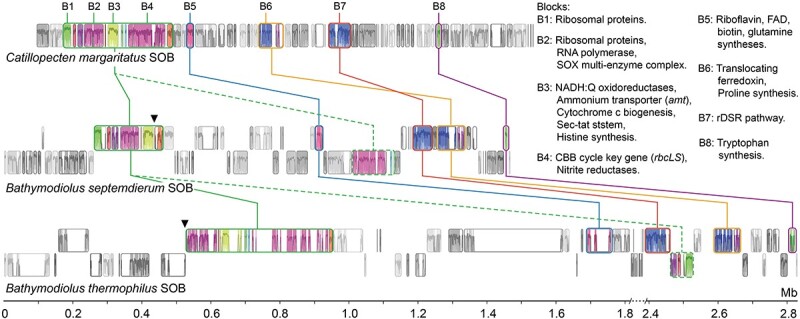
Conserved gene blocks among the symbionts of Catillopecten margaritatus and its two closely related bathymodioline symbiont genomes. The diagram is based on whole genome alignment, with the largest conserved blocks as an anchor. The crucial genes and pathways located in the conserved blocks were indicated at the upper right. The inverse or transposed blocks are labeled using dashed lines, and their original locations are indicated by triangles.
Chemosynthetic capabilities of the scallop ectosymbiont
Chemosynthesis is crucial for many deep-sea vent and seep holobionts. Although the genome of Ca. T. margaritatus is substantially smaller than the bathymodioline SOBs, they all encode the core genes and pathways for carbon fixation and energy production (Fig. 6A, Table S8–9).
Figure 6.

Predicted metabolic map and gene expression levels of the ectosymbiont associated with Catillopecten margaritatus. A: Putative metabolic map of Ca. T. margaritatus. The pathways and enzymes are indicated by solid arrows and bold font, and the missing pathways and enzymes are indicated by dashed arrows. B: Transcriptional levels of the central metabolic pathways, expressed as log2-transferred average transcripts per million (log2(TPM)) values (Table S9).
Sulfur metabolism is primarily reliant on thiosulfate oxidation
All transcriptomic analyses were based on two replicates (G2 and G3, Table S1). The KEGG enrichment of the highly expressed genes showed that the scallop’s ectosymbiont was actively engaged in sulfur metabolism and carbon fixation (Fig. 7; Tables S10–S11). Significantly, sulfur oxidation was the most highly expressed metabolic process (Fig. 6B), with two rhodanese-like proteins suggested to catalyze thiosulfate to sulfite [7, 73] ranked the top second and third in transcription in the scallop’s ectosymbiont (Fig. 6; Table S10). The sulfite produced by rhodanese-like proteins can be further oxidized by the adenylylsulfate reductase (AprAB) (Fig. 6A). Besides, the genes of L-cysteine S-thiosulfotransferase (soxAX) and sulfur-oxidizing protein (soxYZ) were also highly expressed, ranked after rhodanese-like proteins, and the SOX multienzyme complex was more active than the rDSR pathway (Fig. 6B; Table S10). Both the SOX multienzyme complex and rDSR pathway are widely used for sulfur oxidation by the SOBs of deep-sea bathymodioline mussels [74-76], vesicomyid clams [5, 77], and siboglinid tubeworms [78, 79]. The relative transcriptional levels of these two systems differ among these symbionts, which might be related to their different habitats and material sources. Those SOBs associated with animals capable of obtaining hydrogen sulfide from the sediment tend to show high transcriptional of the rDSR pathway, such as those of the clams A. marissinica [5] and Solemya velum [77], and the tubeworms Riftia pachyptila [78], Paraescarpia echinospica [79], and S. annulatum [24]. By contrast, the SOX multienzyme complex, utilizing thiosulfate from ambient water, is more transcriptionally active than rDSR pathway in bathymodioline symbionts [7]. Therefore, the ectosymbiont of C. margaritatus living on the seafloor or the shells of A. marisinica has the potential to use thiosulfate from seawater instead of sulfide from sediment, indicating its physiological adaptation to the availability of sulfur in the environment.
Figure 7.
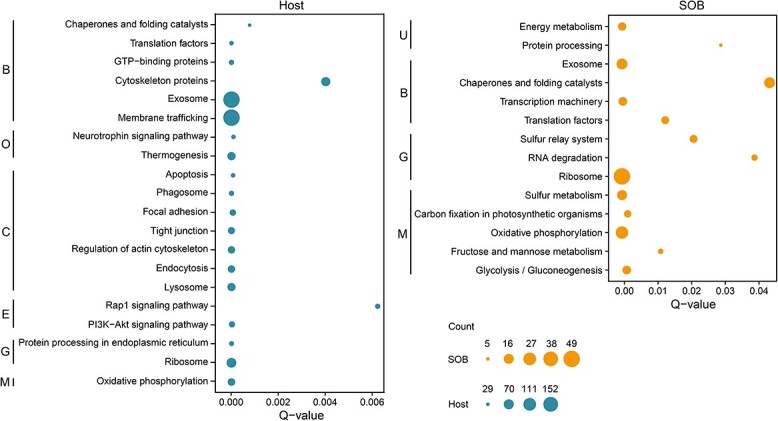
Functional enrichment of highly expressed genes (HEGs) in the gill of host and ectosymbiont. Only pathways with a Q-value <0.05 were considered significant. Only the top 20 abundant pathways of the host were presented (Table S11). The count refers to the number of genes in each category. B: Brite hierarchies; C: Cellular processes; E: Environmental information processing; G: Genetic information processing; M: Metabolism; O: Organismal systems; U: Unclassified.
Carbon is assimilated via the CBB cycle supplied by a phosphoenolpyruvate carboxylase
The CBB cycle is used by most autotrophs for carbon fixation. In Ca. T. margaritatus, the CBB cycle was transcriptionally active, as shown by the high expressional levels of rbcLS, glyceraldehyde 3-phosphate dehydrogenase (gadph), and fructose bisphosphate aldolase (fba) (Figs 6B and 7; Table S10). Similar to the endosymbiotic SOB of other bivalves [5, 6, 8], fpb is absent in the genome of Ca. T. margaritatus. Nevertheless, the CBB cycle is functional and active in the symbiont for fixing carbon dioxide and provision of intermediates and biomass for the holobiont. In addition, the reductive tricarboxylic acid (rTCA) cycle—another carbon assimilation pathway in prokaryotes—is incomplete in Ca. T. margaritatus, with most of the genes missing. However, phosphoenolpyruvate carboxylase (ppc)—the key gene of the rTCA cycle—is identified in the genome of Ca. T. margaritatus (Table S9), which encodes a carboxylase that catalyzes the carboxylation of phosphoenolpyruvate to oxaloacetate and fixes carbon dioxide simultaneously [80]. This gene was lowly expressed with an average TPM value of 56, compared to the high level of the key genes rbcL (TPM = 11 642) and rbcS (TPM = 6 765) in the CBB cycle. Therefore, ppc may play a subsidiary role in inorganic carbon assimilation for the ectosymbiont.
Holobiont may require filter-feeding to meet its nitrogen requirement
The thiotrophic symbionts of vesicomyids, bathymodiolines, and siboglinids encode complete dissimilatory nitrate reduction pathways to provide electron accepters and ammonia [6, 24, 74-76, 79]. Our previous study found that the δ15N value of C. margaritatus (9.9‰) was substantially higher than those of clams, mussels, and tubeworms reliant on SOB for nutrients only [23]. Because our observation of the scallop showed that it had a full digestive gland (Fig. 1A), it is likely mixotrophic, relying partially on filter-feeding to meet its nutritional requirements. Filter-feeding has also been suggested to be responsible for a part of the nutrition of the deep-sea bathymodioline mussels [81] and anemones [82] hosting SOB symbionts. We found that Ca. T. margaritatus genome encodes an incomplete dissimilatory nitrate reduction pathway for nitrite reduction producing ammonium, and an ammonium transporter to intake ammonium from ambient water (Fig. 6A). Most of the genes involved in nitrogen metabolism, including the key genes nitrate/nitrite transporter (nrt) and nitrite reductase (NADH) large and small subunits (nirBD), were lowly expressed (Fig. 6B). By contrast, ammonium transporter (amt) and glutamine synthetase (glnA) were transcriptionally active, with TPM values of 2 605 and 7 770, respectively (Fig. 6B), indicating that ectosymbiont is more dependent on ammonium from the water column for glutamine biosynthesis than on nitrite reduction. In addition, two nitroreductases with TPM values of 392 and 189 were identified, indicating that this scallop SOB may have the ability to degrade toxic nitro-containing compounds like trinitrotoluene [83] from the environment to obtain additional nitrogen. Therefore, C. margaritatus may be mixotrophic, relying on both chemosynthesis and filter-feeding to obtain nitrogen materials. Further investigation is desired to pinpoint the nitrogen sources and nitrogen assimilation pathways in the scallop holobiont.
Ectosymbiont lacks the hydrogenotrophic capability
The fluids from vents typically have higher hydrogen concentrations than those from seeps [84, 85], therefore it has been hypothesized that this energy source is a driver for the differences in chemosynthetic bacteria between the two habitat types, with certain bacteria from hydrothermal vents possessing hydrogenase for energy production. This may explain why the SOB symbionts of some chemosynthetic invertebrates (i.e. mussels B. septemdierum, B. puteoserpentis, and B. thermophilus) inhabiting vents encode hydrogenases [74, 86, 87], whereas seep-dwelling clam Thyasira sp. and tubeworm S. annulatum lack these genes [6, 24]. In line with this hypothesis, we found that the genome of Ca. T. margaritatus lacks hydrogenases. Nevertheless, a [NiFe] hydrogenase was found in the SOB symbionts of the seep tubeworm P. echinospica [79]. Therefore, more chemosymbionts should be screened for the presence/absence of hydrogenases to better understand their roles in the evolution of chemosynthesis.
Ectosymbiosis is probably obligate and the host and symbiont are metabolically interdependent
Obligate symbiotic bacteria are typically small and lack genes and pathways essential for free living, unlike facultative symbionts [88, 89]. For instance, the obligate endosymbionts of the vesicomids are only ~1 Mb in genome size and have lost many genes involved in cellular envelope, motility, and heavy metal resistance [5, 9]. In the Ca. T. margaritatus genome, key genes for the flagellum, pilus, and chemotaxis systems are also missing (Fig. 6A; Tables S8–S9), indicating that the ectosymbiont lacks motility and environmental sensing. Although the ectosymbiotic Ca. T. margaritatus is not transmitted vertically via germ cells, the missing of these genes does not allow this symbiont to maintain an extended free-living period in the ambient water, as indicated for the ectosymbiont symbionts of thyasirids where these components essential for free-living are also absent [6, 69]. Therefore, we suggest that Ca. T. margaritatus is likely an obligate symbiont transmitted horizontally. This is possible as obligate symbionts have been shown capable of surviving outside the host for a short period [90], and horizontal transmission of obligated symbionts have been reported in several groups of marine invertebrates [91, 92]. For instance, whole-genome analyses of S. velum revealed signatures of frequent horizontal transmission of its obligate symbionts [93]. In vent tubeworms R. pachyptila, Oasisia alvinae, and Tevnia jerichonana, obligate endosymbionts colonize the developing tube of the settled larvae and horizontally enter the host through the skin [94]. Besides, the horizontally transmitted thiotrophic symbiont of B. azoricus is suggested to be an obligate symbiont [7]. Therefore, it is possible that the ectosymbionts of C. margaritatus disperse and colonize the scallop larvae in the water column within a short period of their release from the host. That the scallops usually live in aggregations could provide such opportunities for close contact [22].
Previous studies of chemosynthesis have revealed tight metabolic interdependence between the hosts and endosymbionts [5, 8, 79]. Here, we showed that the metabolic complementarity between a deep-sea scallop and its ectosymbiotic SOB could be more intimate than previously thought. In the scallop’s ectosymbiont, the TCA cycle is incomplete without malate dehydrogenase (mdh), succinate dehydrogenase/fumarate reductase (sdh), and 2-oxoglutarate dehydrogenase (ogdh). Previous studies have found the absence of these genes in the intracellular symbiont of the mussel B. azoricus [7] and the ectosymbiont of the clam Thyasira sp. [6], and thus they could not synthesize oxaloacetate, fumarate, and succinate. In B. azoricus, these intermediates could be supplied from the host cytosol via intaking by C4-dicarboxylate transporter (TRAP transporter) and citrate transporter [7]. We found these transporters in the Ca. T. margaritatus genome and they were highly expressed (480, 202, 3 300, and 568 TPM for DctQ, DctM, DctP subunits of the TRAP transporter, and citrate transporter respectively, Table S9), which indicates that these missing intermediates of the symbiont’s TCA cycle must be supplied exogenously, except oxaloacetate may be compensated by ppc. Although the extracellular location of Ca. T. margaritatus prevents it from accessing the intermediates in the host’s cytosol directly, our enrichment of HEGs in C. margaritatus gill tissues showed that the genes involved in exosome and membrane trafficking were highly expressed, being the top two enriched pathways (Fig. 7). Exosomes are extracellular vesicles carrying bioactive molecules like DNA, RNA, and functional proteins, which play significant roles in intercellular communication and immune response [95]. In human-associated bacteria, exosomes can take up many different TCA cycle intermediates including succinate and fumarate from the host [96]. Thus, we propose that exosomes and membrane trafficking genes are involved in the transport of succinate and fumarate from the host’s gill cytosol to the microvillus space, which are then taken up by the ectosymbiont to complete its TCA cycle via transporters. Nevertheless, further evidence is needed to support this hypothesis.
Deep-sea molluscs hosting endosymbionts usually lose the capability to synthesize some amino acids and cofactors [6, 10, 79]. We examined the scallop’s transcriptome and the ectosymbiont genome to investigate their nutrient biosynthetic capabilities (Table S12). Our analysis of the host’s transcriptome showed that the biosynthetic pathways of 12 amino acids (asparagine, aspartate, chorismate, histidine, leucine, lysine, ornithine, phenylalanine, proline, and tryptophan) and eight cofactors (FAD, folate, lipoic acid, pantothenate, protoheme, riboflavin, pyridoxine phosphate, and ubiquinone) were incomplete (Fig. S4; Table S12). These results were only based on the gill transcriptomic data with moderate completeness (87.5% of complete metazoan BUSCOs), and it is desirable to analyze the host’s genome to reveal its metabolic potentials. Nevertheless, the ectosymbiont genome encodes the genes for biosynthesis of all these nutrients (except for ubiquinone) (Fig. 7). The symbionts could be captured by the host via endocytosis or phagocytosis, as indicated by our microscopic observation (Fig. 1F and G), and enrichment and high expression of many genes related to the phagosome, endocytosis, and lysosome in the host gill (Fig. 7). By contrast, the Ca. T. margaritatus genome lacks some genes essential for the biosynthesis of cysteine, methionine, and threonine (Fig. S4; Table S12), indicating that the symbiont might obtain them from the host. This might be carried out by the host's exosomes, as indicated by the expression of the related genes (Fig. 7). However, exactly how these nutrients are transported from the host to symbionts requires further study.
Conclusion
We provided microscopic and molecular evidence for scallop-associated thiotrophic bacterial ectosymbiont. This ectosymbiont is phylogenomically related to the thiotrophic symbionts of bathymodiolines, but their genomic structures are substantially different. Because its TCA cycle, several amino acid biosynthetic pathways, secretion system, and motility system are incomplete, it is likely an obligate symbiont, dependent on the host to provide some of the missing nutrients. The ectosymbiont’s rhodanese-like proteins and SOX multienzyme complex were highly expressed in energy generation, consistent with sulfur oxidization as its main source of energy. Its CBB cycle was also highly expressed, indicating this is the primary pathway for inorganic carbon assimilation. Nevertheless, we also found evidence that its incomplete rTCA cycle plays a subsidiary role in inorganic carbon assimilation. The host might obtain energy and nutrients from the symbionts by harvesting them via phagosomes, endosomes, and lysosomes, whereas providing specific amino acids and some metabolic intermediates to the symbiont. The discovery of this obligate ectosymbiosis with a tight host-bacteria metabolic complementarity not only enables comparative genomic studies to test various hypotheses on the evolution history of symbiosis in Bivalvia but also prompts more multi-omics research on the roles of ectosymbionts in host nutrition.
Supplementary Material
Acknowledgements
We thank the First Institute of Oceanography, MNR (Qingdao) for organizing the cruise and the captain and crew of R/V Xiangyanghong 01 for assistance in sampling.
Contributor Information
Yi-Tao Lin, Department of Biology, Hong Kong Baptist University, Hong Kong SAR, 999077, China.
Jack Chi-Ho Ip, Science Unit, Lingnan University, Hong Kong SAR, 999077, China.
Xing He, Institute of Evolution & Marine Biodiversity, Ocean University of China, Qingdao 266003, China.
Zhao-Ming Gao, Deep-sea Science Division, Institute of Deep-sea Science and Engineering, Chinese Academy of Sciences, Sanya 572000, China.
Maeva Perez, Department of Biology, Hong Kong Baptist University, Hong Kong SAR, 999077, China.
Ting Xu, Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou), Guangzhou 511458, China; Department of Ocean Science, The Hong Kong University of Science and Technology, Hong Kong SAR, 999077, China.
Jin Sun, Institute of Evolution & Marine Biodiversity, Ocean University of China, Qingdao 266003, China.
Pei-Yuan Qian, Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou), Guangzhou 511458, China; Department of Ocean Science, The Hong Kong University of Science and Technology, Hong Kong SAR, 999077, China.
Jian-Wen Qiu, Department of Biology, Hong Kong Baptist University, Hong Kong SAR, 999077, China.
Author contributions
J-WQ conceived and designed this project. Y-TL, Z-MG, and TX collected the samples. Y-TL conducted experiments. Y-TL, JCHI, and XH performed data analyses. Z-MG, MP, JS, and P-YQ provided critical comments. Y-TL drafted the manuscript. All authors contributed to the article and approved the submitted version.
Conflicts of interest
All authors declare no conflict of interest.
Funding
This study was supported by Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (SMSEGL24SC01), the Fundamental Research Funds for the Central Universities (202172 002 and 202241 002), the National Key R&D Program of China (2022YFC2805505), the Collaborative Research Fund (C2013-22GF), and the General Research Fund (16101822, 12101021, 12102623) of Hong Kong SAR.
Data availability
The data sets presented in this study can be found in online repositories. All amplicon, Illumina, Nanopore, RNA sequencing data, symbiont genome, and its annotation were deposited in the National Centre for Biotechnology Information (NCBI) under the BioProject PRJNA1029732. We have released all the data submitted to NCBI and Figshare. The functional annotations and expressional levels were deposited in Figshare under DOI: 10.6084/m9.figshare.24406429.
References
- 1. Dubilier N, Bergin C, Lott C. Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat Rev Microbiol 2008;6:725–40. 10.1038/nrmicro1992 [DOI] [PubMed] [Google Scholar]
- 2. Breusing C, Schultz DT, Sudek Set al. High-contiguity genome assembly of the chemosynthetic gammaproteobacterial endosymbiont of the cold seep tubeworm Lamellibrachia barhami. Mol Ecol Resour 2020;20:1432–44. 10.1111/1755-0998.13220 [DOI] [Google Scholar]
- 3. Sogin EM, Kleiner M, Borowski Cet al. Life in the dark: phylogenetic and physiological diversity of chemosynthetic symbioses. Ann Rev Microbiol 2021;75:695–718. 10.1146/annurev-micro-051021-123130 [DOI] [PubMed] [Google Scholar]
- 4. Roeselers G, Newton ILG. On the evolutionary ecology of symbioses between chemosynthetic bacteria and bivalves. Appl Microbiol Biotechnol 2012;94:1–10. 10.1007/s00253-011-3819-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ip JCH, Xu T, Sun Jet al. Host–endosymbiont genome integration in a deep-sea chemosymbiotic clam. Mol Biol Evol 2021;38:502–18. 10.1093/molbev/msaa241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li Y, He X, Lin Yet al. Reduced chemosymbiont genome in the methane seep thyasirid and the cooperated metabolisms in the holobiont under anaerobic sediment. Mol Ecol Resour 2023;23:1853–67. 10.1111/1755-0998.13846 [DOI] [PubMed] [Google Scholar]
- 7. Ponnudurai R, Kleiner M, Sayavedra Let al. Metabolic and physiological interdependencies in the Bathymodiolus azoricus symbiosis. ISME J 2017;11:463–77. 10.1038/ismej.2016.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Russell SL, McCartney E, Cavanaugh CM. Transmission strategies in a chemosynthetic symbiosis: detection and quantification of symbionts in host tissues and their environment. Proc R Soc B 2018;285:20182157. 10.1098/rspb.2018.2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Perez M, Breusing C, Angers Bet al. Divergent paths in the evolutionary history of maternally transmitted clam symbionts. Proc R Soc B 2022;289:20212137. 10.1098/rspb.2021.2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lin YT, Xu T, Ip JCHet al. Interactions among deep-sea mussels and their epibiotic and endosymbiotic chemoautotrophic bacteria: insights from multi-omics analysis. Zool Res 2023;44:106–25. 10.24272/j.issn.2095-8137.2022.279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith DC. From extracellular to intracellular: the establishment of a symbiosis. Proc Biol Sci 1979;204:115–30. 10.1098/rspb.1979.0017 [DOI] [PubMed] [Google Scholar]
- 12. Fujiwara Y, Kawato M, Noda Cet al. Extracellular and mixotrophic symbiosis in the whale-fall mussel Adipicola pacifica: a trend in evolution from extra-to intracellular symbiosis. PLoS One 2010;5:e11808. 10.1371/journal.pone.0011808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Distel DL. Evolution of chemoautotrophic endosymbioses in bivalves. Bioscience 1998;48:277–86. 10.2307/1313354 [DOI] [Google Scholar]
- 14. Combosch DJ, Collins TM, Glover EAet al. A family-level tree of life for bivalves based on a sanger-sequencing approach. Mol Phylogenet Evol 2017;107:191–208. 10.1016/j.ympev.2016.11.003 [DOI] [PubMed] [Google Scholar]
- 15. Schein-Fatton E. Decouverte Sur la ride du Pacifique oriental a 13 N d’un Pectinidae (Bivalvia, Pteromorphia) d’affinites paleozoiques. Comptes rendus de l’Académie des sciences Série 3, Sciences de la vie 1985;301:491–6. [Google Scholar]
- 16. Beninger PG, Dufour SC, Decottignies Pet al. Particle processing mechanisms in the archaic, peri-hydrothermal vent bivalve Bathypecten vulcani, inferred from cilia and mucocyte distributions on the gill. Mar Ecol Prog Ser 2003;246:183–95. 10.3354/meps246183 [DOI] [Google Scholar]
- 17. Kelly SRA, Blanc E, Price SPet al. Early cretaceous giant bivalves from seep-related limestone mounds, Wollaston Forland, Northeast Greenland. Geol Soc Spec Publ 2000;177:227–46. 10.1144/GSL.SP.2000.177.01.13 [DOI] [Google Scholar]
- 18. Dijkstra HH, Marshall BA. The recent Pectinoidea of the New Zealand region (Mollusca: Bivalvia: Propeamussiidae, Pectinidae and Spondylidae). Molluscan Res 2008;28:1–88. 10.11646/mr.28.1.1 [DOI] [Google Scholar]
- 19. Kiel S, Hybertsen F, Hyzny Met al. Mollusks and a crustacean from early Oligocene methane-seep deposits in the Talara Basin, northern Peru. Acta Palaeontol Pol 2020;65:109–38. 10.4202/app.00631.2019 [DOI] [Google Scholar]
- 20. Dufour SC. Gill anatomy and the evolution of symbiosis in the bivalve family Thyasiridae. Biol Bull 2005;208:200–12. 10.2307/3593152 [DOI] [PubMed] [Google Scholar]
- 21. Shu Y, Wang Y, Wei Zet al. A bacterial symbiont in the gill of the marine scallop Argopecten irradians irradians metabolizes dimethylsulfoniopropionate. mLife 2023;2:178–89. 10.1002/mlf2.12072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin YT, Li YX, Sun Yet al. A new species of the genus Catillopecten (Bivalvia: Pectinoidea: Propeamussiidae): morphology, mitochondrial genome, and phylogenetic relationship. Front Mar Sci 2023;10:1168991. 10.3389/fmars.2023.1168991 [DOI] [Google Scholar]
- 23. He X, Xu T, Chen Cet al. Same (sea) bed different dreams: biological community structure of the Haima seep reveals distinct biogeographic affinities. Innov Geosci 2023;1:100019. 10.59717/j.xinn-geo.2023.100019 [DOI] [Google Scholar]
- 24. Gao ZM, Xu T, Chen HGet al. Early genome erosion and internal phage-symbiont-host interaction in the endosymbionts of a cold-seep tubeworm. iScience 2023;26:107033. 10.1016/j.isci.2023.107033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ritt B, Duperron S, Lorion Jet al. Sarrazin J integrative study of a new cold-seep mussel (Mollusca: Bivalvia) associated with chemosynthetic symbionts in the Marmara Sea. Deep Sea Res Part I Oceanogr Res Pap 2012;67:121–32. 10.1016/j.dsr.2012.05.009 [DOI] [Google Scholar]
- 26. Duperron S, Laurent MC, Gaill Fet al. Sulphur-oxidizing extracellular bacteria in the gills of Mytilidae associated with wood falls. FEMS Microbiol Ecol 2008;63:338–49. 10.1111/j.1574-6941.2008.00438.x [DOI] [PubMed] [Google Scholar]
- 27. Duperron S, Nadalig T, Caprais JCet al. Dual symbiosis in a Bathymodiolus sp. mussel from a methane seep on the Gabon continental margin (Southeast Atlantic): 16S rRNA phylogeny and distribution of the symbionts in gills. Appl Environ Microbiol 2005;71:1694–700. 10.1128/AEM.71.4.1694-1700.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stewart CN, Via LE. A rapid CTAB DNA isolation technique useful for RAPD fingerprinting and other PCR applications. BioTechniques 1993;14:748–50. [PubMed] [Google Scholar]
- 29. Muyzer G, De Waal EC, Uitterlinden AG. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 1993;59:695–700. 10.1128/aem.59.3.695-700.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Caporaso JG, Lauber CL, Walters WAet al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA 2011;108:4516–22. 10.1073/pnas.1000080107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bolyen E, Rideout JR, Dillon MRet al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 2019;37:852–7. 10.1038/s41587-019-0209-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang D, Gao F, Jakovlić Iet al. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour 2020;20:348–55. 10.1111/1755-0998.13096 [DOI] [PubMed] [Google Scholar]
- 33. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Ecol Resour 2013;30:772–80. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 2007;56:564–77. 10.1080/10635150701472164 [DOI] [PubMed] [Google Scholar]
- 35. Ronquist F, Teslenko M, Van Der Mark Pet al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 2012;61:539–42. 10.1093/sysbio/sys029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nguyen LT, Schmidt HA, von Haeseler Aet al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 2015;32:268–74. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–20. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Koren S, Walenz BP, Berlin Ket al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 2017;27:722–36. 10.1101/gr.215087.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 2018;34:3094–100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vaser R, Sović I, Nagarajan Net al. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 2017;27:737–46. 10.1101/gr.214270.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009;25:1754–60. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li H, Handsaker B, Wysoker Aet al. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–9. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Walker BJ, Abeel T, Shea Tet al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 2014;9:e112963. 10.1371/journal.pone.0112963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chklovski A, Parks DH, Woodcroft BJet al. CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat Methods 2023;20:1203–12. 10.1038/s41592-023-01940-w [DOI] [PubMed] [Google Scholar]
- 45. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 2014;30:2068–9. 10.1093/bioinformatics/btu153 [DOI] [PubMed] [Google Scholar]
- 46. Huerta-Cepas J, Szklarczyk D, Heller Det al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 2019;47:D309–14. 10.1093/nar/gky1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moriya Y, Itoh M, Okuda Set al. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 2007;35:W182–5. 10.1093/nar/gkm321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Richter M, Rosselló-Móra R, Oliver Glöckner Fet al. JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016;32:929–31. 10.1093/bioinformatics/btv681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods 2012;9:357–9. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sun Y, Sun J, Yang Yet al. Genomic signatures supporting the symbiosis and formation of chitinous tube in the deep-sea tubeworm Paraescarpia echinospica. Mol Biol Evol 2021;38:4116–34. 10.1093/molbev/msab203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 2019;20:238. 10.1186/s13059-019-1832-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mongiardino KN. Phylogenomic subsampling and the search for phylogenetically reliable loci. Mol Biol Evol 2021;38:4025–38. 10.1093/molbev/msab151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang C, Rabiee M, Sayyari Eet al. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform 2018;19:153–30. 10.1186/s12859-018-2129-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Darling AC, Mau B, Blattner FRet al. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 2004;14:1394–403. 10.1101/gr.2289704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Haas BJ, Papanicolaou A, Yassour Met al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat Protoc 2013;8:1494–512. 10.1038/nprot.2013.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fu L, Niu B, Zhu Zet al. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 2012;28:3150–2. 10.1093/bioinformatics/bts565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods 2015;12:59–60. 10.1038/nmeth.3176 [DOI] [PubMed] [Google Scholar]
- 58. Manni M, Berkeley MR, Seppey Met al. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol 2021;38:4647–54. 10.1093/molbev/msab199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Patro R, Duggal G, Love MIet al. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 2017;14:417–9. 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen C, Wu Y, Li Jet al. TBtools-II: a "one for all, all for one" bioinformatics platform for biological big-data mining. Mol Plant 2023;16:1733–42. 10.1016/j.molp.2023.09.010 [DOI] [PubMed] [Google Scholar]
- 61. Cavanaugh CM, Levering PR, Maki JSet al. Symbiosis of methylotrophic bacteria and deep-sea mussels. Nature 1987;325:346–8. 10.1038/325346a0 [DOI] [Google Scholar]
- 62. Bright M, Bulgheresi S. A complex journey: transmission of microbial symbionts. Nat Rev Microbiol 2010;8:218–30. 10.1038/nrmicro2262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Franke MA. Changing Perspectives-from Ecology to Cellular Biology in the Bathymodioline Symbiosis Doctoral dissertation,. Universität Bremen, Germany, 2021 [Google Scholar]
- 64. Génio L, Rodrigues CF, Guedes IFet al. Mammal carcasses attract a swarm of mussels in the deep Atlantic: insights into colonization and biogeography of a chemosymbiotic species. Mar Ecol 2015;36:71–81. 10.1111/maec.12217 [DOI] [Google Scholar]
- 65. Distel DL, Baco AR, Chuang Eet al. Do mussels take wooden steps to deep-sea vents? Nature 2000;403:725–6. 10.1038/35001667 [DOI] [PubMed] [Google Scholar]
- 66. Niu M, Fan X, Zhuang Get al. Methane-metabolizing microbial communities in sediments of the Haima cold seep area, northwest slope of the South China Sea. FEMS Microbiol Ecol 2017;93:fix101. 10.1093/femsec/fix101 [DOI] [PubMed] [Google Scholar]
- 67. Dong X, Zhang C, Peng Yet al. Phylogenetically and catabolically diverse diazotrophs reside in deep-sea cold seep sediments. Nat Commun 2022;13:4885. 10.1038/s41467-022-32503-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Newton ILG, Woyke T, Auchtung TAet al. The Calyptogena magnifica chemoautotrophic symbiont genome. Science 2007;315:998–1000. 10.1126/science.1138438 [DOI] [PubMed] [Google Scholar]
- 69. Guo Y, Meng L, Wang Met al. Hologenome analysis reveals independent evolution to chemosymbiosis by deep-sea bivalves. BMC Biol 2023;21:51. 10.1186/s12915-023-01551-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ikuta T, Amari Y, Tame Aet al. Inside or out? Clonal thiotrophic symbiont populations occupy deep-sea mussel bacteriocytes with pathways connecting to the external environment. ISME Commun 2021;1:38. 10.1038/s43705-021-00043-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA 2009;106:19126–31. 10.1073/pnas.0906412106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Newton IL, Girguis PR, Cavanaugh CM. Comparative genomics of vesicomyid clam (Bivalvia: Mollusca) chemosynthetic symbionts. BMC Genomics 2008;9:585. 10.1186/1471-2164-9-585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Harada M, Yoshida T, Kuwahara Het al. Expression of genes for sulfur oxidation in the intracellular chemoautotrophic symbiont of the deep-sea bivalve Calyptogena okutanii. Extremophiles 2009;13:895–903. 10.1007/s00792-009-0277-8 [DOI] [PubMed] [Google Scholar]
- 74. Ikuta T, Takaki Y, Nagai Yet al. Heterogeneous composition of key metabolic gene clusters in a vent mussel symbiont population. ISME J 2016;10:990–1001. 10.1038/ismej.2015.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ponnudurai R, Heiden SE, Sayavedra Let al. Comparative proteomics of related symbiotic mussel species reveals high variability of host–symbiont interactions. ISME J 2020;14:649–56. 10.1038/s41396-019-0517-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Romero Picazo D, Werner A, Dagan Tet al. Pangenome evolution in environmentally transmitted symbionts of deep-sea mussels is governed by vertical inheritance. Genome Biol Evol 2022;14:evac098. 10.1093/gbe/evac098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Stewart FJ, Dmytrenko O, DeLong EFet al. Metatranscriptomic analysis of sulfur oxidation genes in the endosymbiont of Solemya velum. Front Microbiol 2011;2:134. 10.3389/fmicb.2011.00134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gardebrecht A, Markert S, Sievert SMet al. Physiological homogeneity among the endosymbionts of Riftia pachyptila and Tevnia jerichonana revealed by proteogenomics. ISME J 2012;6:766–76. 10.1038/ismej.2011.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yang Y, Sun J, Sun Yet al. Genomic, transcriptomic, and proteomic insights into the symbiosis of deep-sea tubeworm holobionts. ISME J 2020;14:135–50. 10.1038/s41396-019-0520-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Burgsdorf I, Sizikov S, Squatrito Vet al. Lineage-specific energy and carbon metabolism of sponge symbionts and contributions to the host carbon pool. ISME J 2022;16:1163–75. 10.1038/s41396-021-01165-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang X, Guan H, Qiu JWet al. Macro-ecology of cold seeps in the South China Sea. Geosystems Geoenviron 2022;1:100081. 10.1016/j.geogeo.2022.100081 [DOI] [Google Scholar]
- 82. Goffredi SK, Motooka C, Fike DAet al. Mixotrophic chemosynthesis in a deep-sea anemone from hydrothermal vents in the Pescadero Basin. Gulf of California BMC Biol 2021;19:8. 10.1186/s12915-020-00921-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Roldán MD, Pérez-Reinado E, Castillo Fet al. Reduction of polynitroaromatic compounds: the bacterial nitroreductases. FEMS Microbiol Rev 2008;32:474–500. 10.1111/j.1574-6976.2008.00107.x [DOI] [PubMed] [Google Scholar]
- 84. Charlou JL, Donval JP, Fouquet Yet al. Geochemistry of high H2 and CH4 vent fluids issuing from ultramafic rocks at the rainbow hydrothermal field (36 14′ N, MAR). Chem Geol 2002;191:345–59. 10.1016/S0009-2541(02)00134-1 [DOI] [Google Scholar]
- 85. Nakamura K, Takai K. Theoretical constraints of physical and chemical properties of hydrothermal fluids on variations in chemolithotrophic microbial communities in seafloor hydrothermal systems. Prog Earth Planet Sci 2014;1:5. 10.1186/2197-4284-1-5 [DOI] [Google Scholar]
- 86. Petersen JM, Zielinski FU, Pape Tet al. Hydrogen is an energy source for hydrothermal vent symbioses. Nature 2011;476:176–80. 10.1038/nature10325 [DOI] [PubMed] [Google Scholar]
- 87. Patra AK, Perez M, Jang SJet al. A regulatory hydrogenase gene cluster observed in the thioautotrophic symbiont of Bathymodiolus mussel in the East Pacific rise. Sci Rep 2022;12:22232. 10.1038/s41598-022-26669-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Jäckle O, Seah BKB, Tietjen Met al. Chemosynthetic symbiont with a drastically reduced genome serves as primary energy storage in the marine flatworm Paracatenula. Proc Natl Acad Sci USA 2019;116:8505–14. 10.1073/pnas.1818995116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Osman EO, Weinnig AM. Microbiomes and obligate symbiosis of deep-sea animals. Annu Rev Anim Biosci 2022;10:151–76. 10.1146/annurev-animal-081621-112021 [DOI] [PubMed] [Google Scholar]
- 90. Rasgon JL, Gamston CE, Ren X. Survival of Wolbachia pipientis in cell-free medium. Appl Environ Microbiol 2006;72:6934–7. 10.1128/AEM.01673-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Breusing C, Genetti M, Russell SLet al. Horizontal transmission enables flexible associations with locally adapted symbiont strains in deep-sea hydrothermal vent symbioses. Proc Natl Acad Sci USA 2022;119:e2115608119. 10.1073/pnas.2115608119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Russell SL. Transmission mode is associated with environment type and taxa across bacteria-eukaryote symbioses: a systematic review and meta-analysis. FEMS Microbiol Lett 2019;366:fnz013. 10.1093/femsle/fnz013 [DOI] [PubMed] [Google Scholar]
- 93. Russell SL, Corbett-Detig RB, Cavanaugh CM. Mixed transmission modes and dynamic genome evolution in an obligate animal–bacterial symbiosis. ISME J 2017;11:1359–71. 10.1038/ismej.2017.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Nussbaumer A, Fisher C, Bright M. Horizontal endosymbiont transmission in hydrothermal vent tubeworms. Nature 2006;441:345–8. 10.1038/nature04793 [DOI] [PubMed] [Google Scholar]
- 95. Gurung S, Perocheau D, Touramanidou Let al. The exosome journey: from biogenesis to uptake and intracellular signalling. Cell Commun Signal 2021;19:47. 10.1186/s12964-021-00730-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ebner P, Götz F. Bacterial excretion of cytoplasmic proteins (ECP): occurrence, mechanism, and function. Trends Microbiol 2019;27:176–87. 10.1016/j.tim.2018.10.006 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets presented in this study can be found in online repositories. All amplicon, Illumina, Nanopore, RNA sequencing data, symbiont genome, and its annotation were deposited in the National Centre for Biotechnology Information (NCBI) under the BioProject PRJNA1029732. We have released all the data submitted to NCBI and Figshare. The functional annotations and expressional levels were deposited in Figshare under DOI: 10.6084/m9.figshare.24406429.