

Abstract
Phenol-soluble modulins (PSMs) are amphipathic, alpha-helical peptides that are secreted by staphylococci in high amounts in a quorum-sensing-controlled fashion. Studies performed predominantly in Staphylococcus aureus showed that PSMs structure biofilms, which results in reduced biofilm mass, while it has also been reported that S. aureus PSMs stabilize biofilms due to amyloid formation. We here analyzed the roles of PSMs in in-vitro and in-vivo biofilms of Staphylococcus epidermidis, the leading cause of indwelling device-associated biofilm infection. We produced isogenic deletion mutants for every S. epidermidis psm locus and a sequential deletion mutant in which production of all PSMs was abolished. In-vitro analysis substantiated the role of all PSMs in biofilm structuring. PSM-dependent biofilm expansion was not observed, in accordance with our finding that no S. epidermidis PSM produced amyloids. In a mouse model of indwelling device-associated infection, the total psm deletion mutant had a significant defect in dissemination. Notably, the total psm mutant produced a significantly more substantial biofilm on the implanted catheter than the wild-type strain. Our study, which for the first time directly quantified the impact of PSMs on biofilm expansion on an implanted device, shows that the in-vivo biofilm infection phenotype in S. epidermidis is in accordance with the PSM biofilm structuring and detachment model, which has important implications for the potential therapeutic application of quorum-sensing blockers.
Keywords: amyloids, device-associated infection, cyclophosphamide, neutrophils, quorum sensing
Introduction
Infection of indwelling medical devices is one of the leading types of nosocomial infections. These infections are often caused by opportunistic pathogens in patients that are immunocompromised due to underlying medical conditions or medical interventions such as surgery. Often, the infected device needs to be replaced, which leads to significantly increased costs for public health systems and a considerable burden for the patient. The main reason for the need to replace rather than treat the infected device with antibiotics stems from the fact that these infections commonly proceed with the development of biofilms, which are surface-attached bacterial aggregates that are inherently resistant to antibiotics [1–3].
Staphylococci are major nosocomial pathogens. Staphylococcus epidermidis in particular, an otherwise innocuous and even beneficial normal component of the skin microbiota, is the most frequent pathogen involved with device-associated infections [4, 5]. This situation has been explained by the facts that these bacteria are (i) abundant common inhabitants of human epithelia [6], and (ii) have exceptional ability to form biofilms [5, 7]. Biofilm-forming capacity has been linked to the variety and abundance of the molecules S. epidermidis secretes to attach to human matrix proteins and form a biofilm matrix [7]. Probably the most important components of the S. epidermidis biofilm matrix are the poly-N-acetylglucosamine polymeric exopolysaccharide PIA/PNAG [8] and the fibril-forming accumulation-associated protein Aap [9], as has been shown by both in-vitro and in-vivo research [10–12]. Some matrix proteins, such as Aap or the small basic protein (Sbp), form amyloid-like fibril scaffolds [13–15]. However, no molecule appears to be absolutely required for S. epidermidis biofilm formation. For example, isolates without the PIA/PNAG-encoding ica genes have been isolated from device-associated infections and ica-negative strains form in-vitro biofilms, although biofilms without PIA/PNAG are weaker [10, 16].
The formation of a mature biofilm, however, does not solely include biofilm matrix formation, but also requires structuring [17]. Biofilm-structuring forces lead to the formation of channels, which are deemed important for nutrient delivery throughout the biofilm, and thus to keep deeper biofilm layers alive. They also lead to detachment, which is a prerequisite for the systemic dissemination of a biofilm infection. In staphylococci, biofilm structuring has been linked to two molecule families: 1) enzymes that degrade biofilm matrix molecules, above all secreted proteases [18, 19], while enzymes that degrade PIA/PNAG are not known in staphylococci, and 2) the surfactant phenol-soluble modulin (PSM) peptides [20–23].
PSMs are peptides with pronounced amphipathy and α-helicity, properties that give them surfactant character [24, 25]. They are categorized into α- and β-type PSMs according to their length. The former are ~ 20 −25 and the latter ~ 45 amino acid in length. Every staphylococcal species produces a species-specific repertoire of PSMs, some but not all of which of share sequence similarities with homologous PSMs in other species. In-vitro research has shown that all PSMs of S. aureus, as well as the S. epidermidis PSMβ peptides, which are strongly produced in that species, structure biofilms. The same PSMs are so far also the only bacterial factors for which roles in the in-vivo dissemination of biofilm-associated infection could be directly demonstrated [20, 21].
However, there are several important outstanding questions as for the role of PSMs in biofilms. First, the difficulties associated with producing isogenic deletion mutants in S. epidermidis has so far not allowed investigating the roles of PSMs other than the PSMβ peptides in S. epidermidis. Second, the effect that the absence of PSMs has on biofilm expansion, which is very pronounced in vitro, has so far not been analyzed in vivo on infected devices. Third, there is a model describing the role of PSMs in biofilm formation that to a certain extent contradicts that described above and which is based on the formation of amyloids by S. aureus PSMs in in-vitro setups [26]. While the role of PSM amyloids in S. aureus biofilm formation is controversial [27], whether S. epidermidis PSM form amyloids with a potential impact on biofilms has not yet been investigated. With some S. epidermidis PSMs showing higher similarity to amyloid-forming S. aureus PSMs (for example, S. epidermidis PSMδ to S. aureus PSMα3) than some of those to each other, amyloid formation by S. epidermidis PSMs appears possible and warrants investigation.
Therefore, we here put considerable efforts into the construction of isogenic mutants of the psm genes in S. epidermidis. Notably, we also constructed a total psm mutant, in which production of all PSMs of S. epidermidis is abolished. We tested the contribution of single and total mutants to in-vitro biofilm formation and also analyzed the contribution of PSMs to in-vivo biofilm formation and dissemination using the total PSM deletion strain and a mouse device-associated infection model. Our results demonstrate a significant contribution of all S. epidermidis PSMs to biofilm structuring processes and for the first time directly show that PSMs impact biofilm formation on an indwelling medical device in vivo. Furthermore, according to our results, S. epidermidis PSMs do not form amyloids and the PSM-dependent biofilm phenotypes S. epidermidis displays in-vitro and in-vivo are at odds with the predictions of the PSM amyloid model that was established in S. aureus.
Results
Production of psm deletion strains
S. epidermidis generally produces six PSMs: PSMα, PSMδ, PSMε, PSMβ1, PSMβ2, and the δ-toxin (sometimes called PSMγ) [25, 28–30]. The psmβ operon contains three genes, coding for the PSMβ1 and PSMβ2 peptides, while the psmβ3 gene appears not to be expressed, as we never found PSMβ3 production in any of many analyzed S. epidermidis culture filtrates. Sometimes, the psmβ1 gene is duplicated, which is, however, not the case in the standard strain 1457 [31] that we and others have used for many previous studies and which we chose as the background strain to construct psm deletions. Further, S. epidermidis contains a homologue of the S. aureus psmα operon, but in which in contrast to S. aureus, only two peptides, PSMα and PSMδ, are encoded. PSMε is another α-type PSM that is encoded in a gene not connected with other psm genes. The δ-toxin is encoded within RNAIII, the intracellular effector of the accessory gene regulator (Agr) quorum-sensing system [32]. Agr strictly controls expression of all PSMs [33] (Fig. 1a,b).
Fig. 1. S. epidermidis PSMs and construction of isogenic psm deletion mutants.
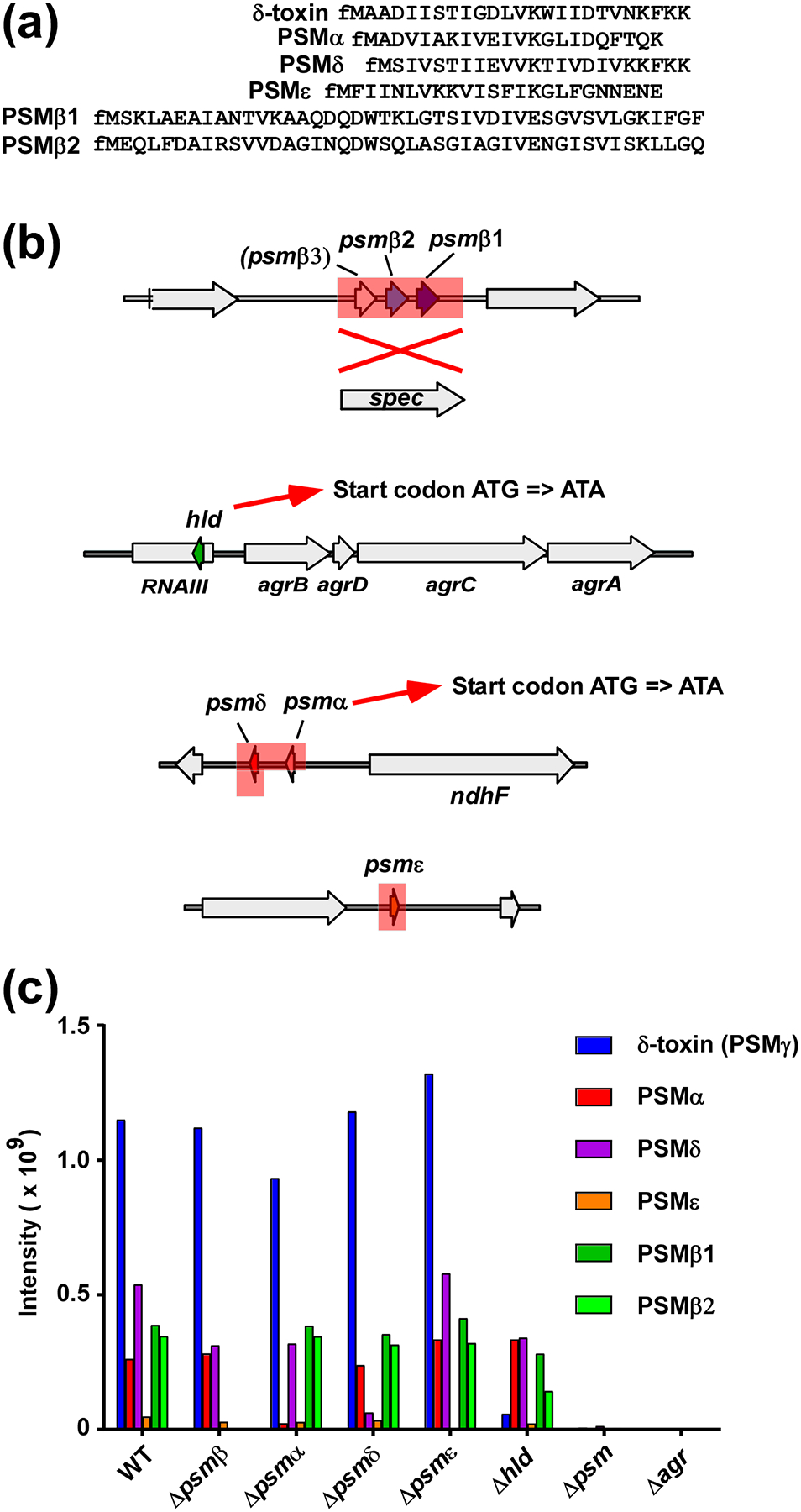
(a) Amino acid sequences of S. epidermidis PSMs. Note all PSMs have an N-terminal formylated methionine (fM) due to export by a dedicated ABC transporter without a signal peptide [55]. (b) S. epidermidis psm deletion mutants. The psmβ deletion mutant was constructed by replacement of the entire psmβ operon with a spectinomycin resistance cassette. Note the hypothetical PSMβ3 peptide does not appear to be produced. The quintuple (total) psm deletion mutants was then constructed by sequential deletion of the other psm loci, in the order as shown from top to bottom. Single psm deletion mutants were also constructed. In the case of hld (encoding δ-toxin) and psmα, start codon mutations were introduced instead of complete gene deletions, so as not to interfere with production of RNAIII or expression of psmδ, respectively. Deleted or replaced sections are shadowed in red. (c) Analysis of PSM production pattern by RP-HPLC/MS of all psm mutants used in thus study.
We previously described an isogenic deletion mutant of the psmβ operon, in which the psmβ operon was replaced with a spectinomycin resistance cassette [20]. We also described an hld (δ-toxin) mutant, in which the hld start codon was changed to abolish production of δ-toxin without affecting RNAIII function [34]. Here, a strategy was used to produce markerless deletions of all other single psm genes. We also used that strategy, as described in methods, to produce a sequential total PSM deletion mutant on the basis of the spectinomycin-resistant psmβ deletion mutant, in which all psm genes are completely deleted and hld expression is abolished (Fig. 1b). All mutants were verified for correct deletion by DNA sequencing as well as analysis of the PSM production pattern by reversed-phase high performance chromatography/electrospray ionization mass spectrometry (RP-HPLC/ESI-MS) (Fig. 1c). Mutations in agr that result in complete absence of PSM production happen frequently in the laboratory [35, 36]. Therefore, the correct PSM production pattern was verified at every step of the procedure. The total psm deletion strain (Δpsm), whose phenotype of complete PSM absence cannot be distinguished by this method from a spontaneous agr mutant, was verified as Agr-positive by analysis on milk agar plates, which assays for the maintained production of Agr-controlled proteases [37, 38].
Amyloid formation
We earlier described properties of pure S. epidermidis PSM peptides, such as capacities to lyse neutrophils [28]. We then did not analyze capacities to form amyloids, as amyloid formation by PSMs was only suggested and described later [26]. Therefore, we now subjected all pure S. epidermidis PSMs to the common thioflavin (ThT) test for amyloid formation. No PSM of S. epidermidis showed amyloid formation in this assay when tested at 0.1 mg/ml in physiological buffer conditions [10 mM sodium phosphate buffer (pH 8.0), 150 mM NaCl], whether diluted from DMSO stocks or pre-treated to destroy preformed amyloids, in contrast to amyloid-forming S. aureus PSMα3, which was used as control (Fig. 2a,b). As a further control, we treated all peptides with Tween 80, an amyloid-destroying agent, which led to abolishment of amyloid formation in the case of PSMα3, but did not alter fluorescence values, indicative of amyloid formation/destruction, with any S. epidermidis PSM. Thus, S. epidermidis PSMs do not form amyloids, at least under the tested conditions.
Fig. 2. Test for amyloid formation of S. epidermidis PSMs.
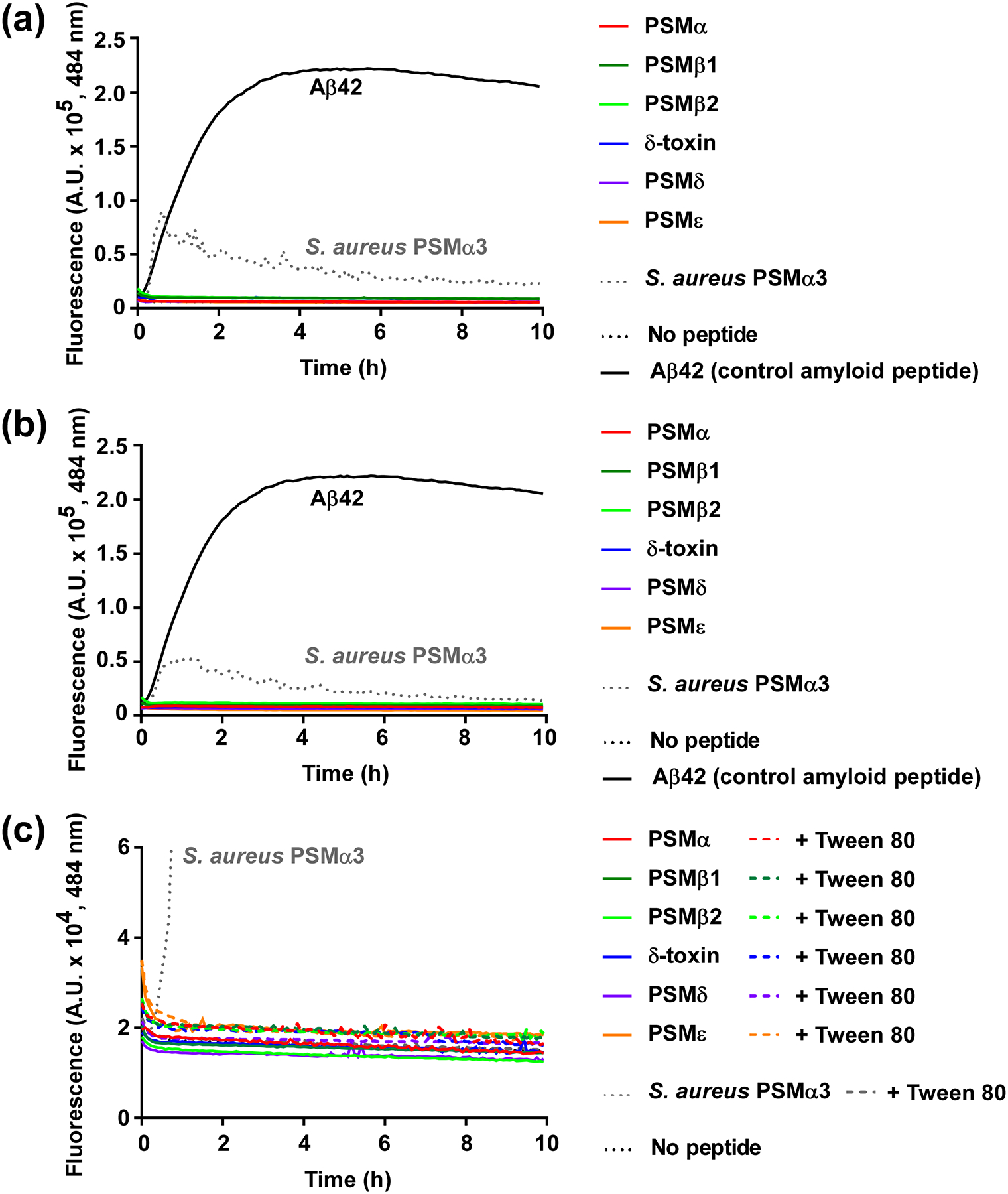
All S. epidermidis PSMs, as well as S. aureus PSMα3 and the control amyloid peptide Aβ42, were subjected to a ThT amyloid formation assay at a final concentration of 0.1 mg/ml in 10 mM sodium phosphate buffer (pH 8.0)/150 mM NaCl. The amyloid-indicative fluorescence at 484 nm was measured over 10 h. (a) Assay with PSM peptides diluted 1:100 from 10-mg/ml stocks in 100% DMSO. (b) Assay with PSM peptides pre-treated to abolish pre-formed amyloids using trifluoroacetic acid/hexafluoroisopropanol (1:1). (c) Assay comparing peptides pre-treated as in (b) with or without addition of Tween 80 to destroy pre-formed amyloids.
Role of S. epidermidis in in-vitro biofilms
There are many ways to analyze in-vitro biofilms. They can be categorized into two main setups: static and dynamic. In contrast to static models, dynamic models use flow, often applied in so-called flow cells, where fresh medium is run through the biofilm at a given flow rate [39]. Notably, no in-vitro model of biofilm formation can completely reflect the situation during infection, given the absence of host tissue formation, host matrix proteins, and immune defenses, to name but a few limitations. With this in mind, one could probably argue for either model, seen that device-associated infections occur in tissue or blood vessels, mimicking “static” or “dynamic” conditions, respectively. Thus, we used both a static and dynamic model to analyze the role of PSMs in in-vitro biofilms.
In the static model, we tested all single isogenic psm mutants as well as the total psm deletion strain (Δpsm). All showed significantly increased total and average biovolumes and biofilm thickness, and a reduced roughness coefficient (Fig. 3). Average biovolume is a readout for the degree of channel formation, thus for the structuring capacity associated with PSMs (with a higher value meaning less structuring). All S. epidermidis PSMs showed the same general impact on biofilm formation that has been described by us previously for S. aureus PSMs and the S. epidermidis PSMβ peptides [20, 21]. Interestingly, we detected average biovolume and thickness values similar to those of the total psm deletion mutant even for deletion mutants of PSMs that are only produced in comparatively low amounts, such as PSMδ or PSMε. Contrastingly, total biovolume and roughness coefficients showed some differences that seemed correlated with PSM production levels, with the highly produced PSMβ peptides and δ-toxin showing the most pronounced effects. In the dynamic flow-cell model, we compared the total psm mutant with the wild-type S. epidermidis strain. Total and average biovolume as well as thickness were increased in the Δpsm versus the wild-type strain, while roughness was decreased (Fig. 4). These results thus reflected those achieved in the static model.
Fig. 3. Static in-vitro biofilm formation by single and total S. epidermidis psm mutants.
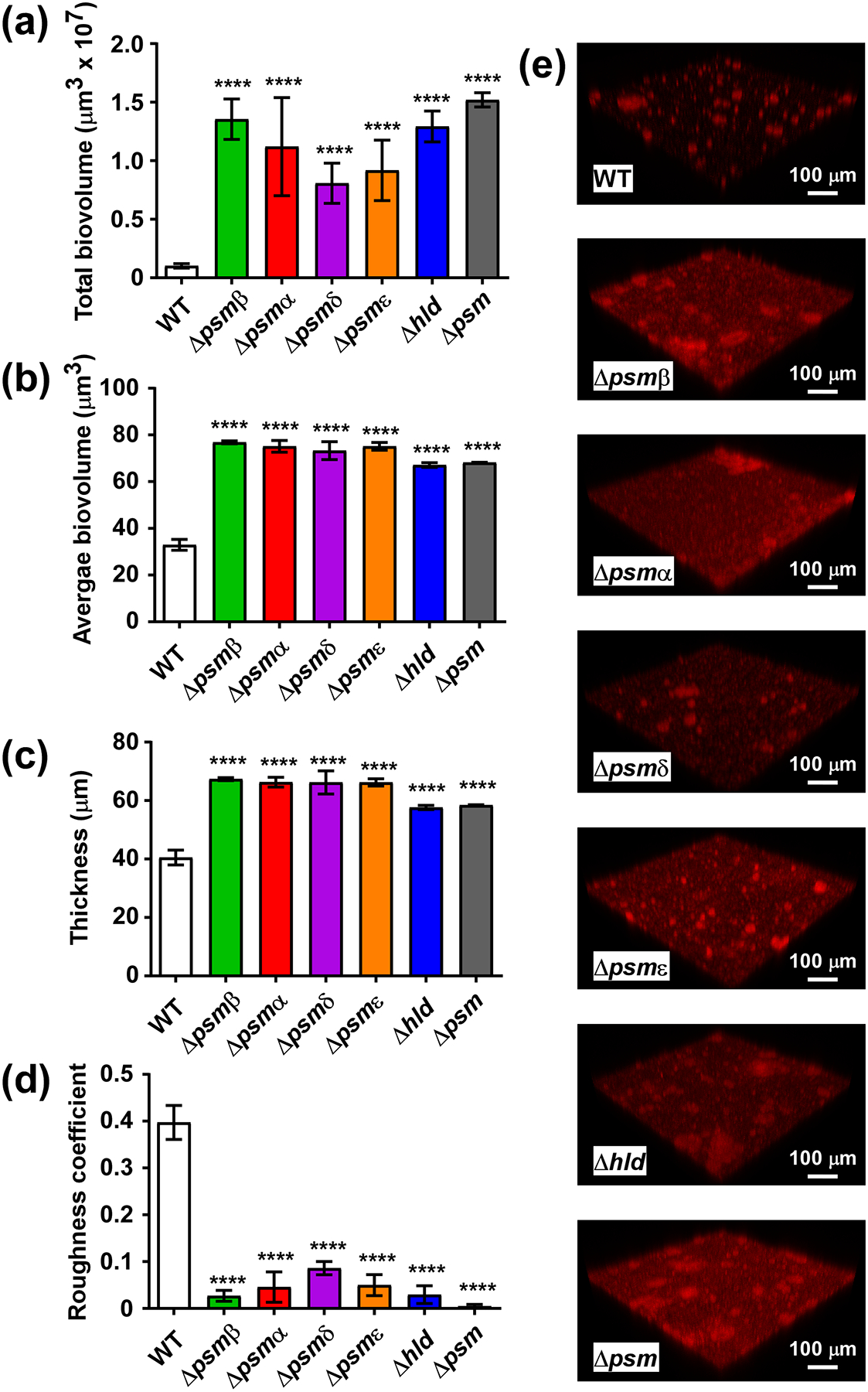
Biofilms were grown in TSBg in static mode for 48 h, measured by CLSM, and analyzed by Imaris or Comstat software. In addition to psm mutants, an isogenic mutant of the quorum-sensing psm control system agr was measured. (a) Total biovolumes (total biofilm mass). (b) Average biovolumes (More compact biofilm have higher; biofilms with more channel formation lower values.) (c) Thickness. (d) Roughness coefficient (Biofilms that are smoother have a lower value.) (e) Representative CLSM example pictures. (a-d) ****, p<0.0001 [1-way ANOVA with Dunnett’s post-test versus data obtained with the wild-type (WT) strain]. Data are from n=16 measurements of randomly selected areas in a sample. The experiment was repeated with similar results.
Fig. 4. Dynamic in-vitro biofilm formation by wild-type and isogenic Δpsm mutants strains.
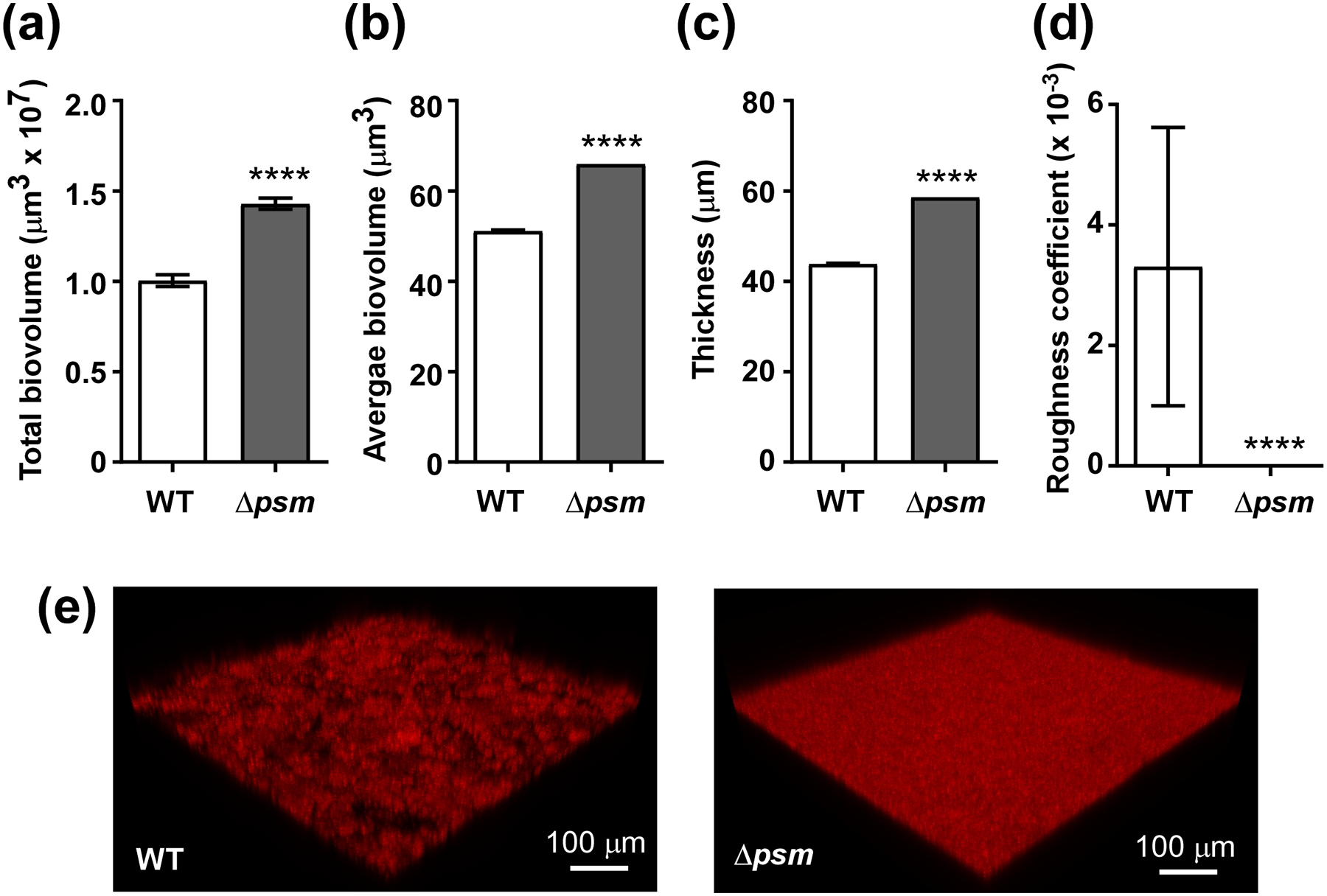
Biofilms were grown in TSBg in dynamic mode in flow cells for 48 h, measured by CLSM, and analyzed by Imaris or Comstat software. (a) Total biovolumes (total biofilm mass). (b) Average biovolumes (More compact biofilm have higher; biofilms with more channel formation lower values.) (c) Thickness. (d) Roughness coefficient (Biofilms that are smoother have a lower value.) (e) Representative CLSM example pictures. (a-d) ****, p<0.0001 (unpaired t-tests). Data are from n=15 measurements of randomly selected areas in a sample. The experiment was repeated with similar results.
Role of PSMs in S. epidermidis biofilm-associated infection
As mentioned already above, in-vitro biofilm models only poorly reflect the conditions of biofilm-associated infection. Thus, to investigate the role of PSMs in biofilm-associated infection, we employed a frequently used mouse infection model, in which catheter pieces with attached bacteria are placed under the skin of mice [11, 40]. Most device-associated infections with S. epidermidis occur in immune-compromised patients [2]. Using a similar model, we previously showed that S. epidermidis causes increased severity of device-associated infection in severe combined immunodeficiency (SCID) mouse strains as compared to immunocompetent mice [41]. These mice are impaired in B- and/or T-cell responses, thus primarily in adaptive immunity. However, innate host defenses, and especially neutrophils, are generally known to have the most important impact on staphylococcal infections [42]. Therefore, we here first tested whether mice deficient in leukocytes develop device-associated infections with increased severity. Leukocytes were destroyed by treatment with cyclophosphamide (CY), which affects all cell types of myeloid origin [43].
We measured bacterial CFU on the inserted catheter, in the catheter-surrounding tissue, in several organs (spleen, liver, kidney), and lymph nodes to test for development of device-associated infection as well as its systemic dissemination. Biofilms on the implanted device were significantly larger, and significantly more CFU were detected in the surrounding tissue and all tested organs, as compared to non-CY-treated, immunocompetent mice (Fig. 5). Thus, leukocyte function has a significant impact on biofilm-associated device infection and its dissemination in mice.
Fig. 5. Biofilm-associated device infection model.
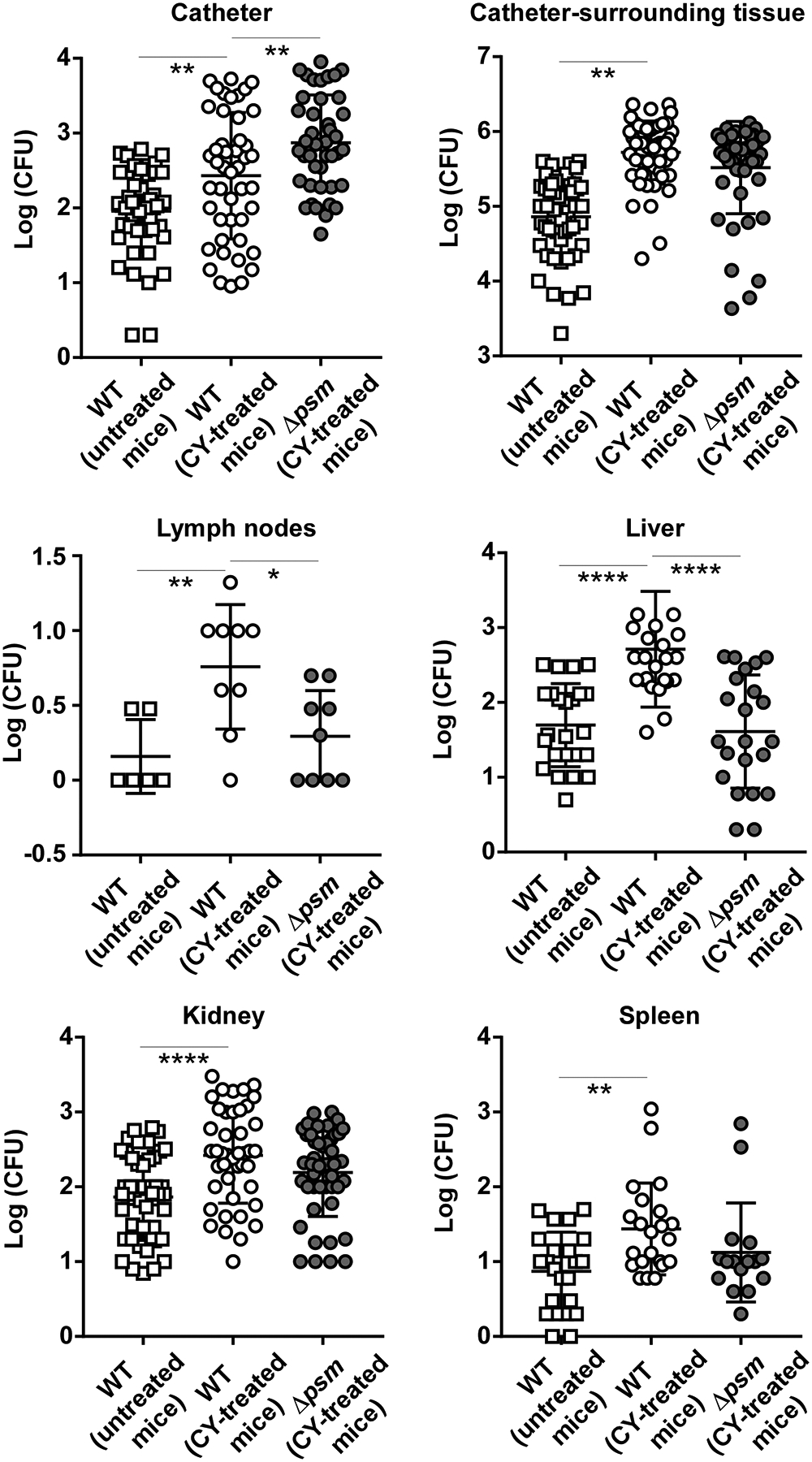
Catheter pieces coated with equal amounts of wild-type or Δpsm bacteria were inserted subcutaneously into the dorsa of mice, which were treated with cyclophosphamide to obtain leukocyte-deficient immune suppression. Another group of mice infected with wild-type S. epidermidis-coated catheters received saline as control to maintain immune competency. All groups consisted of n=26 mice with two inserted catheters each, resulting in n=52 data points. Occasionally, catheters fell out and were not included in the counting. For organs and lymph nodes, only data points are shown in case bacteria were detected. Especially in lymph nodes, detection of bacteria was rare. *, p<0.05; **, p<0.01; ****, p<0.0001 [1-way ANOVA with Tukey’s post-test. Only comparisons between untreated versus CY-treated, and CY-treated mice infected with wild-type (WT) versus Δpsm S. epidermidis are shown.]
We then used the same model under conditions of immune suppression to test for the role of PSMs using the total psm deletion mutant (Δpsm). Device-attached biofilms formed by the Δpsm mutant were significantly larger than those formed by wild-type S. epidermidis, while dissemination, as measured by CFU in the catheter-surrounding tissue, lymph nodes, and organs, was lower in mice infected with Δpsm bacteria. In the lymph nodes and liver, the differences were significant (Fig. 5).
Discussion
It is now well established that PSMs have a considerable impact on biofilm formation and represent major effectors of the previously described pronounced influence of Agr quorum-sensing regulation on staphylococcal biofilm development [17, 44, 45]. However, important questions have remained unanswered and there are contrasting schools of thought about how specific physico-chemical properties affect the biological functions of PSMs. First, except for our studies using a psmβ deletion mutant [20], the impact of PSMs on S. epidermidis biofilms and biofilm infection has remained undefined. Second, whether there is biological relevance of PSM amyloid formation, in particular for biofilm formation and biofilm infections, has remained a matter of debate [27]. Finally, in the present study we also addressed whether leukocyte depletion has a significant impact on the severity of S. epidermidis device-associated infection. This represents an important question given that S. epidermidis is the major cause of device-associated infections, as well as device infection-associated bacteremia; and many patients that suffer from S. epidermidis infections are immunocompromised.
We showed previously that in a device-associated S. epidermidis mouse infection model similar to that used in the present study, CBSCBG-MM and Nu/Nu SCID mice, deficient in B- and T-cells, had ~ 5 – 10 times higher average CFU on infected catheters and surrounding tissue than wild-type mice [41]. Here, mice deficient in leukocytes, a major arm of innate immunity, showed an at least similarly pronounced deficiency in clearing S. epidermidis biofilm infection. We also measured dissemination to lymph nodes and into organs, which was increased at comparable rates. Thus, our results show that deficiency in leukocytes as major cellular mediators of innate immunity leads to significantly increased severity of experimental device-associated biofilm infection.
As for the role of PSMs, our results are in accordance with the general principle indicated in our previous studies using S. aureus psm mutants [21] and the S. epidermidis psmβ mutant [20], inasmuch as dissemination into organs and establishment of second site infection was decreased in the absence of PSMs. However, we now also for the first time directly measured the extent of biofilm formation on the device. Our results show significantly increased biofilm formation on the device in the absence of PSMs, indicating in-vivo relevance of the model of PSM-mediated biofilm structuring and detachment in the used biofilm infection model.
While animal models or in-vitro flow-cell measurements of all single psm mutants would have required too large an amount of experiments, we tested all single psm mutants for their impact on static biofilm development. Some biofilm phenotypes showed a certain additivity of the effects of single PSMs; however, average biovolume and thickness values obtained for the total psm mutant were also reached by all single psm mutants. These observations reflect our previous findings using single S. aureus psm mutants [21]. They suggest that already the absence of PSMs that are expressed at comparatively low levels impacts biofilm phenotypes considerably, despite the continued presence of all other PSMs and an almost unchanged total PSM amount. We believe this may point to functional specification of PSMs with respect to their roles in biofilm structuring. Functional specification of PSMs is yet poorly understood and testing the hypothesis that different PSMs have different functions in biofilm structuring remains an important task for future in-depth mechanistic research.
Our previous in-vitro studies have consistently shown that PSMs structure biofilms and their absence leads to biofilms with enhanced thickness but less channel formation [20, 21, 23]. Others have shown that some S. aureus PSMs (PSMα1, PSMα2, PSMα4, PSMβ1, PSMβ2) are contained in amyloid-like fibrils that lead to enhanced resistance of biofilms toward enzymatic degradation [26]. While we previously confirmed in-vitro aggregation and potential amyloid formation by several S. aureus PSMs [27], both our in-vitro and in-vivo analyses using S. aureus psm mutants indicated that this capacity does not have significant relevance for in-vitro or in-vivo biofilm formation [20, 21, 27]. We also recently provided experimental evidence for the attachment of S. aureus PSMs to DNA [27], indicating that the enhanced resistance of PSM-positive biofilms to degradation can be explained without the involvement of PSM amyloids. Here, we analyzed S. epidermidis PSMs for amyloid formation. In contrast to amyloid-forming S. aureus PSMα3, which was used as a control, no S. epidermidis PSM showed significant amyloid formation in an in-vitro assay. Furthermore, our in-vitro data achieved with S. epidermidis psm mutants support the model of PSM-mediated biofilm structuring and detachment. Finally, our analysis of in-vivo biofilm formation on an implanted device indicate that this model is in accordance with in-vivo phenotypes, at least under the in-vivo infection conditions tested in this study.
Recently, PSMα3 of S. aureus was shown to adapt an exceptional amyloid fibril structure, which was claimed to be linked to the functional properties of that peptide [46], in particular its cytolytic capacity, which is higher than that of most other tested PSMs [47]. We previously demonstrated that PSMδ of S. epidermidis, a homologue of the S. aureus PSMα peptides, has cytolytic capacities at least at the same level as those exhibited by PSMα3 [28]. It is thus of interest that PSMδ, similar to all other tested S. epidermidis PSMs, did not show amyloid formation. This result adds to previous evidence we obtained using an alanine screen peptide bank of PSMα3, which showed that several PSMα3 derivatives with abolished or strongly reduced amyloid-forming capacities maintained cytolytic activity, indicating that amyloid formation of PSMα3 is not linked to its cytolytic capacity [27]. Altogether, our data show that amyloid formation is not a general PSM feature and lend further support to the notion that the occasionally observed amyloid-like aggregation of PSMs does not have biological relevance, at least not for the phenotypes that have so far been described for PSMs.
Blocking quorum-sensing is a frequently proposed strategy of modern antivirulence drug development [48]. In S. aureus, the Agr quorum-sensing system controls a plethora of toxins [49]. These include PSMs, which are under exceptionally direct and strict regulation by Agr [33]. As we verified in the present study, all S. epidermidis PSMs are also under strict Agr control. While targeting Agr certainly appears a valuable strategy to treat acute, non-biofilm-associated infections, we previously noted that caution should be applied when evaluating quorum-sensing blocking as a potential therapeutic option for biofilm-associated infections [50]. Our present results show that the same caution should be applied for S. epidermidis, which is of particular importance given that biofilm-associated infection of medical devices is by far the most frequent type of infection that this opportunistic pathogen causes. Then again, blocking Agr and thus, PSM production, according to our findings should diminish second-site infections. Future in-depth evaluation of quorum-sensing blockers for S. epidermidis infections should therefore include testing whether lowering the degree of biofilm formation on the device or the level of systemic dissemination is more important to achieve clinical improvement in such infections. Furthermore, S. epidermidis is a leading cause of nosocomial sepsis, an acute type of infection, for which quorum sensing blockers may be appropriate, a notion that remains to be tested.
Materials and methods
Bacterial strains and culture conditions
The S. epidermidis standard strain 1457 and its derivatives (see below) were used in all experiments (see Tab. 1 for a list of all strains). All strains were tested frequently, and always in pre-cultures of performed experiments, for the correct PSM production pattern and absence of Agr mutations (by RP-HPLC/MS or production of inhibition zones on proteolysis test plates). Cultures were grown in tryptic soy broth (TSB) or TSBg (TSB + 0.5% glucose), as indicated.
Table 1.
Strains used in this study
| Strains/plasmids | Relevant genotype and property | Source |
|---|---|---|
| S. aureus strains | ||
| RN4220 | Derived from NCTC8325–4; r−m+ | [52] |
| S. epidermidis strains | ||
| 1457 | Clinical biofilm-forming wild-type strain | [28] |
| 1457 Δpsmα | Isogenic start codon (ATG to ATA) psmα mutant of 1457 | This study |
| 1457 Δpsmβ | Isogenic psmβ deletion mutant of 1457; psmβ operon replaced with spectinomycin resistance cassette; specR | [17] |
| 1457 Δhld | Isogenic hld start codon mutant (ATG to ATA) of 1457; Hld (δ-toxin) protein production abolished without interfering with the function of RNAIII | [31] |
| 1457 Δpsmε | Isogenic markerless psmε deletion mutant of 1457 | This study |
| 1457 Δpsmδ | Isogenic markerless psmδ deletion mutant of 1457 | This study |
| 1457 Δpsm | Quintuple (total) isogenic psm mutant of 1457 (Δpsmβ/Δhld/Δpsmα/Δpsmδ/Δpsmε) | This study |
| 1457 Δagr | Isogenic agr deletion mutant of 1457 | [49] |
| E. coli strains | ||
| DH5α | endA1 recA1 gyrA96 thi-1 hsdR17(rκ− mκ+) relA1 supE44 (lacZYA-argF) U169 F- 80dlacZM15 deoR phoA | Invitrogen |
Gene deletion by allelic replacement
An allelic replacement procedure [51], was used to create markerless isogenic psmδ, and psmε mutants in S. epidermidis 1457, a biofilm-forming clinical isolate frequently used for biofilm research [31]. First, ~1-kb regions upstream and downstream of the psm genes were amplified from S. epidermidis 1457 genomic DNA using PCR oligonucleotide pairs, introducing attB1 and attB2 sites at the distal ends (Tab. 2). The creation of the isogenic psmβ mutant has been previously described [20]. Plasmids pKOR1-Δpsmδ and pKOR1-Δpsmε lack the coding sequences for psmα, psmδ, and psmε, respectively. Construction of the hld mutant (abolishing production of δ-toxin) has also been previously described [34]. In that strain, a start codon mutations was introduced (ATG to ATA) with pKOR1-Δhldepi, abolishing production of δ-toxin without interfering with the function of RNAIII, in whose gene it is embedded. The same strategy was used here to abolish production of PSMα (change of start codon of the psmα gene from ATG to ATA, using pKOR1-Δpsmα), as complete deletion of the gene proved to interfere with the production of PSMδ, whose gene is located downstream in the same operon.
Table 2.
Oligonucleotides used in this study
| Name | Sequence |
|---|---|
| For isogenic psmα/psmδ deletion mutant | |
| psmαδ-att1 | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCACAAATAATGTTGCACCCCAAAACATGGTCAT |
| psmαδ-rev1 | TGTCCCAGGCCCATGTAGAACCCTTACTAATTTGTTATCTATAGTGTAACTGGAAT |
| psmαδ-rev2 | TTCTACATGGGCCTGGGACATAATTCCTATC |
| psmαδ-att2 | GGGGACCACTTTGTACAAGAAAGCTGGGTTCATAAATGTGGCATCGTGATCTGTTTTAGAATAA |
| For isogenic psmε deletion mutant | |
| psmε-att1 | GGGGACAAGTTTGTACAAAAAAGCAGGCTTTTCAAAAAGATTTTTTACTACAAATATAATAGAATTAACA |
| psmε-rev1 | GAGGGGCGGAGTAGAGAATACACCTCCTATGTATTTGTTAACTAAATTATAGTATTGAAC |
| psmε-rev2 | TATTCTCTACTCCGCCCCTCCTGTTAGAGG |
| psmε-att1 | GGGGACCACTTTGTACAAGAAAGCTGGGTACCCTAGACCTGTTGAAAGTGGCATTATGA |
| For isogenic psmα start codon change mutant | |
| psmα-att1 | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCACAAATAATGTTGCACCCCAAAACATGGTCAT |
| psmα-rev1 | AGGGAGGTCATTTAAATAGCAGATGTAATC |
| psmα-rev2 | ATTACATCTGCTATTTAAATGACCTCCCTT |
| psmα-att2 | GGGGACCACTTTGTACAAGAAAGCTGGGTTCATAAATGTGGCATCGTGATCTGTTTTAGAATAA |
| For isogenic psmδ deletion mutant | |
| psmδ-att1 | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCACAAATAATGTTGCACCCCAAAACATGGTCAT |
| psmδ-rev1 | TGTCCCAGGCCCATGTAGAATCCTTTCAAAAGGGAAATGTATTTAGAGATG |
| psmδ-rev2 | TTCTACATGGGCCTGGGACATAATTCCTATC |
| psmδ-att2 | GGGGACCACTTTGTACAAGAAAGCTGGGTTCATAAATGTGGCATCGTGATCTGTTTTAGAATAA |
The same allelic replacement procedure and plasmids were used to produce an isogenic quintuple (total) psm mutant in S. epidermidis 1457, with the exception of the psmα and psmδ genes. Since the psmα and psmδ genes are found on the same operon, a separate allelic replacement plasmid (pKOR1-Δpsmα/Δpsmδ) was constructed, which allowed for the simultaneous deletion of both genes. The isogenic quintuple psm mutant was then created sequentially as follows; an hld mutation was first introduced into the previously described isogenic psmβ mutant [20] to generate an isogenic double psm mutant, Δpsmβ/Δhld. The genes for psmα and psmδ were then deleted with pKOR1-Δpsmα/Δpsmδ to generate a quadruple psm mutant (Δpsmβ/Δhld/Δpsmα/Δpsmδ). Finally, the quintuple isogenic psm mutant was created after allelic replacement with the pKOR1-Δpsmε. The S. epidermidis 1457 isogenic Δagr mutant used in this study has been described previously [52]. All mutants carry an introduced spectinomycin resistance gene.
PSM detection and relative quantification
PSMs were detected using reversed-phase high performance liquid chromatography/electrospray ionization mass spectrometry (RP-HPLC/ESI-MS) in principle as described [53] with some modifications, as described recently [54]. Obtained intensity values serve to quantify PSM production levels in a relative fashion. They can be used to directly compare the same PSM between different samples, and to a certain extent to compare with other PSMs of the same type, as intensity values for a given concentration are similar within PSMs of the α- and β-types.
Measurement of protease activity on proteolysis test plates
Protease activity was determined on skim-milk agar plates as described [37]. Two μl of bacterial cultures were dropped on the plates, which were incubated for 24 h at 37 °C.
Thioflavin T (ThT) assay for amyloid formation
The Thioflavin T (ThT) kit was purchased from AnaSpec [SensoLyte Thioflavin T β-Amyloid (1 – 42) Aggregation Kit]. S. epidermidis PSM peptides and S. aureus PSMα3 were prepared as 10 mg/ml stocks in DMSO and diluted for the ThT assay. Alternatively, they were subjected to pre-treatment with trifuoroacetic acid/hexafluoroisopropanol (1:1), as described [27] to abolish pre-formed amyloids. In each reaction, 0.1 mg/ml peptide was mixed with 200 μM ThT in 10 mM sodium phosphate buffer (pH 8.0) and 150 mM NaCl. For abolishment of amyloid formation, Tween 80 was added to a final concentration of 0.056%. The fluorescence of ThT was measured at 484 nm with excitation at 440 nm using a Tecan Spark multimode microplate reader every 5 min at 37 °C with 15 s shaking before each read.
In-vitro biofilm models
Wild-type and isogenic S. epidermidis psm deletion mutants were grown at 37 °C at 180 rotations per minute (rpm) to stationary growth phase. For static biofilm models, planktonic bacteria were vortexed and sonicated, and inoculated at an optical density (OD600) of 0.1 in TSBg in Lab-Tek chambered cover glass plates and incubated at 37 °C for 48 h. After 48 h, growth media were removed and biofilms were gently washed with phosphate buffered saline (PBS). Stationary biofilms were then stained for 15 min at 37 °C in 4 μM propidium iodide (PI). For flow biofilm systems, planktonic wildtype and isogenic psm mutants were inoculated at an OD600 of 0.1 in TSBg to Stovall flow-cells and allowed to adhere to the glass surface at 37 °C for 6 h. Adherent bacterial biofilms were grown in constant flow (0.5 ml/min) of TSB containing 4 μM PI. At the end of the respective incubation periods, biofilms were gently washed with PBS, then resuspended in PBS, and imaged using confocal laser scanning microscopy (CLSM) on a Zeiss LSM710 confocal microscope. Image analyses were performed using Imaris and Comstat.
Animal model of device-associated infection
In-vivo studies were approved by the Institutional Animal Care and Use Committee of the NIAID. Animal work was performed adhering to the institution’s guidelines for animal use, and followed the guidelines and basic principles in the United States Public Health Service Policy on Humane Care and Use of Laboratory Animals, and the Guide for the Care and Use of Laboratory Animals by certified staff in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International accredited facility.
All C57BL/6J mice were female and six to eight weeks of age at the time of use. Mice were injected intraperitoneally with a 2-ml loading dose of cyclophosphamide (CY, 150 mg/kg, (immunosuppressed model) or saline (immunocompetent model) on day 1, and additional maintenance-doses of 2 ml of CY (100 mg/kg) or saline, respectively, on days 3, 5, and 7. Wild-type and isogenic S. epidermidis psm deletion mutant strains were grown at 37 °C at 180 rpm to stationary growth phase in TSB on day 7, inoculated to an OD600 of 0.1 in TSB from these pre-cultures on day 8 and grown to exponential growth phase at 37 °C at 180 rpm. Then, bacteria were centrifuged in a Sorvall Legend RT centrifuge (4150 rpm, 25°C, 15 min), after which the supernatants were decanted and the bacterial pellets were resuspended each in 10 ml of TSBg, vortexed and sonicated, and added to 1-cm segments of sterilized catheters (Terumo Surflash 14 gauge IV catheters). Catheter segments were incubated within the bacterial suspension for 2 h at 37 °C. Then, biofilm-coated catheters were gently washed with PBS and allowed to air dry. Catheters were subcutaneously inserted on the dorsa of mice under general anesthesia. Mice were monitored for 48 h and euthanized on day 10. Catheters, peri-catheter skin and soft tissues, lymph nodes (inguinal, axillary, brachial, superficial cervical), as well as distal organs (liver, kidney, spleen) were collected for determination of bacterial CFU. To that end, catheters were suspended in PBS, vortexed and sonicated as described [20], and serially diluted. Peri-catheter skin and soft tissues, lymph nodes, and distal organs were homogenized in PBS suspension with 2-mm borosilicate beads in a FastPrep 96 (MP Bio) homogenizer and serially diluted. Bacterial CFU were determined from serially diluted samples.
Statistics
Statistical evaluation was performed using Graph Pad Prism Version 7.01. Data were analyzed using 1-way ANOVA with Tukey’s or Dunnett’s multiple comparison tests for more than two groups, and unpaired Student’s t-tests for the comparison of two groups. In-vivo data were subjected to Log10 transformation before analysis.
Acknowledgments
This study was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID), U.S. National Institutes of Health (NIH) (project number 1 ZIA AI000904, to M.O.), The National Natural Science Foundation of China (grant 81501804, to L.H.), and the General Program of Shanghai Municipal Commission of Health and Family Planning (grant 20154Y0014, to L.H.).
Abbreviations:
- Agr
accessory gene regulator
- CFU
colony-forming units
- CLSM
confocal laser scanning microscopy
- PIA/PNAG
polysaccharide intercellular adhesin/poly-N-acetylglucosamine
- PSM
phenol-soluble modulin
- RP-HPLC/ESI-MS
high performance chromatography/electrospray ionization mass spectrometry
- ThT
thioflavin
Footnotes
Declarations of interest: none
References
- [1].Darouiche RO. Device-associated infections: a macroproblem that starts with microadherence. Clin Infect Dis. 2001;33:1567–72. [DOI] [PubMed] [Google Scholar]
- [2].von Eiff C, Jansen B, Kohnen W, Becker K. Infections associated with medical devices: pathogenesis, management and prophylaxis. Drugs. 2005;65:179–214. [DOI] [PubMed] [Google Scholar]
- [3].Donlan RM, Costerton JW. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev. 2002;15:167–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Otto M Staphylococcus epidermidis--the ‘accidental’ pathogen. Nat Rev Microbiol. 2009;7:555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zheng Y, He L, Asiamah TK, Otto M. Colonization of medical devices by staphylococci. Environ Microbiol. 2018;20:3141–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kloos WE, Musselwhite MS. Distribution and persistence of Staphylococcus and Micrococcus species and other aerobic bacteria on human skin. Appl Microbiol. 1975;30:381–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Otto M Staphylococcal biofilms. Curr Top Microbiol Immunol. 2008;322:207–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mack D, Fischer W, Krokotsch A, Leopold K, Hartmann R, Egge H, et al. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear beta-1,6-linked glucosaminoglycan: purification and structural analysis. J Bacteriol. 1996;178:175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hussain M, Herrmann M, von Eiff C, Perdreau-Remington F, Peters G. A 140-kilodalton extracellular protein is essential for the accumulation of Staphylococcus epidermidis strains on surfaces. Infect Immun. 1997;65:519–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schaeffer CR, Woods KM, Longo GM, Kiedrowski MR, Paharik AE, Buttner H, et al. Accumulation-associated protein enhances Staphylococcus epidermidis biofilm formation under dynamic conditions and is required for infection in a rat catheter model. Infect Immun. 2015;83:214–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rupp ME, Ulphani JS, Fey PD, Bartscht K, Mack D. Characterization of the importance of polysaccharide intercellular adhesin/hemagglutinin of Staphylococcus epidermidis in the pathogenesis of biomaterial-based infection in a mouse foreign body infection model. Infect Immun. 1999;67:2627–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rupp ME, Ulphani JS, Fey PD, Mack D. Characterization of Staphylococcus epidermidis polysaccharide intercellular adhesin/hemagglutinin in the pathogenesis of intravascular catheter-associated infection in a rat model. Infect Immun. 1999;67:2656–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Conrady DG, Brescia CC, Horii K, Weiss AA, Hassett DJ, Herr AB. A zinc-dependent adhesion module is responsible for intercellular adhesion in staphylococcal biofilms. Proc Natl Acad Sci U S A. 2008;105:19456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Decker R, Burdelski C, Zobiak M, Buttner H, Franke G, Christner M, et al. An 18 kDa scaffold protein is critical for Staphylococcus epidermidis biofilm formation. PLoS Pathog. 2015;11:e1004735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang Y, Jiang J, Gao Y, Sun Y, Dai J, Wu Y, et al. Staphylococcus epidermidis small basic protein (Sbp) forms amyloid fibrils, consistent with its function as a scaffolding protein in biofilms. J Biol Chem. 2018;293:14296–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rohde H, Burandt EC, Siemssen N, Frommelt L, Burdelski C, Wurster S, et al. Polysaccharide intercellular adhesin or protein factors in biofilm accumulation of Staphylococcus epidermidis and Staphylococcus aureus isolated from prosthetic hip and knee joint infections. Biomaterials. 2007;28:1711–20. [DOI] [PubMed] [Google Scholar]
- [17].Otto M Staphylococcal infections: mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity. Annu Rev Med. 2013;64:175–88. [DOI] [PubMed] [Google Scholar]
- [18].Boles BR, Horswill AR. Staphylococcal biofilm disassembly. Trends Microbiol. 2011;19:449–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Loughran AJ, Atwood DN, Anthony AC, Harik NS, Spencer HJ, Beenken KE, et al. Impact of individual extracellular proteases on Staphylococcus aureus biofilm formation in diverse clinical isolates and their isogenic sarA mutants. Microbiologyopen. 2014;3:897–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang R, Khan BA, Cheung GY, Bach TH, Jameson-Lee M, Kong KF, et al. Staphylococcus epidermidis surfactant peptides promote biofilm maturation and dissemination of biofilm-associated infection in mice. J Clin Invest. 2011;121:238–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Periasamy S, Joo HS, Duong AC, Bach TH, Tan VY, Chatterjee SS, et al. How Staphylococcus aureus biofilms develop their characteristic structure. Proc Natl Acad Sci U S A. 2012;109:1281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Otto M Phenol-soluble modulins. Int J Med Microbiol. 2014;304:164–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dastgheyb SS, Villaruz AE, Le KY, Tan VY, Duong AC, Chatterjee SS, et al. Role of Phenol-Soluble Modulins in Formation of Staphylococcus aureus Biofilms in Synovial Fluid. Infect Immun. 2015;83:2966–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Peschel A, Otto M. Phenol-soluble modulins and staphylococcal infection. Nat Rev Microbiol. 2013;11:667–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cheung GY, Joo HS, Chatterjee SS, Otto M. Phenol-soluble modulins--critical determinants of staphylococcal virulence. FEMS Microbiol Rev. 2014;38:698–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schwartz K, Syed AK, Stephenson RE, Rickard AH, Boles BR. Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. 2012;8:e1002744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zheng Y, Joo HS, Nair V, Le KY, Otto M. Do amyloid structures formed by Staphylococcus aureus phenol-soluble modulins have a biological function? Int J Med Microbiol. 2018;308:675–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cheung GY, Rigby K, Wang R, Queck SY, Braughton KR, Whitney AR, et al. Staphylococcus epidermidis strategies to avoid killing by human neutrophils. PLoS Pathog. 2010;6:e1001133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mehlin C, Headley CM, Klebanoff SJ. An inflammatory polypeptide complex from Staphylococcus epidermidis: isolation and characterization. J Exp Med. 1999;189:907–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yao Y, Sturdevant DE, Otto M. Genomewide analysis of gene expression in Staphylococcus epidermidis biofilms: insights into the pathophysiology of S. epidermidis biofilms and the role of phenol-soluble modulins in formation of biofilms. J Infect Dis. 2005;191:289–98. [DOI] [PubMed] [Google Scholar]
- [31].Mack D, Siemssen N, Laufs R. Parallel induction by glucose of adherence and a polysaccharide antigen specific for plastic-adherent Staphylococcus epidermidis: evidence for functional relation to intercellular adhesion. Infect Immun. 1992;60:2048–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Novick RP, Ross HF, Projan SJ, Kornblum J, Kreiswirth B, Moghazeh S. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 1993;12:3967–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Queck SY, Jameson-Lee M, Villaruz AE, Bach TH, Khan BA, Sturdevant DE, et al. RNAIII-independent target gene control by the agr quorum-sensing system: insight into the evolution of virulence regulation in Staphylococcus aureus. Mol Cell. 2008;32:150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Munoz-Planillo R, Hasegawa M, et al. Staphylococcus delta-toxin induces allergic skin disease by activating mast cells. Nature. 2013;503:397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Somerville GA, Beres SB, Fitzgerald JR, DeLeo FR, Cole RL, Hoff JS, et al. In vitro serial passage of Staphylococcus aureus: changes in physiology, virulence factor production, and agr nucleotide sequence. J Bacteriol. 2002;184:1430–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Villaruz AE, Bubeck Wardenburg J, Khan BA, Whitney AR, Sturdevant DE, Gardner DJ, et al. A point mutation in the agr locus rather than expression of the Panton-Valentine leukocidin caused previously reported phenotypes in Staphylococcus aureus pneumonia and gene regulation. J Infect Dis. 2009;200:724–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Vuong C, Gotz F, Otto M. Construction and characterization of an agr deletion mutant of Staphylococcus epidermidis. Infect Immun. 2000;68:1048–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yao Y, Vuong C, Kocianova S, Villaruz AE, Lai Y, Sturdevant DE, et al. Characterization of the Staphylococcus epidermidis accessory-gene regulator response: quorum-sensing regulation of resistance to human innate host defense. J Infect Dis. 2006;193:841–8. [DOI] [PubMed] [Google Scholar]
- [39].Malone M, Goeres DM, Gosbell I, Vickery K, Jensen S, Stoodley P. Approaches to biofilm-associated infections: the need for standardized and relevant biofilm methods for clinical applications. Expert Rev Anti Infect Ther. 2017;15:147–56. [DOI] [PubMed] [Google Scholar]
- [40].Kadurugamuwa JL, Sin LV, Yu J, Francis KP, Purchio TF, Contag PR. Noninvasive optical imaging method to evaluate postantibiotic effects on biofilm infection in vivo. Antimicrob Agents Chemother. 2004;48:2283–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Vuong C, Kocianova S, Yu J, Kadurugamuwa JL, Otto M. Development of real-time in vivo imaging of device-related Staphylococcus epidermidis infection in mice and influence of animal immune status on susceptibility to infection. J Infect Dis. 2008;198:258–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rigby KM, DeLeo FR. Neutrophils in innate host defense against Staphylococcus aureus infections. Semin Immunopathol. 2012;34:237–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hall AG, Tilby MJ. Mechanisms of action of, and modes of resistance to, alkylating agents used in the treatment of haematological malignancies. Blood Rev. 1992;6:163–73. [DOI] [PubMed] [Google Scholar]
- [44].Joo HS, Otto M. Molecular basis of in vivo biofilm formation by bacterial pathogens. Chem Biol. 2012;19:1503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Le KY, Dastgheyb S, Ho TV, Otto M. Molecular determinants of staphylococcal biofilm dispersal and structuring. Front Cell Infect Microbiol. 2014;4:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tayeb-Fligelman E, Tabachnikov O, Moshe A, Goldshmidt-Tran O, Sawaya MR, Coquelle N, et al. The cytotoxic Staphylococcus aureus PSMalpha3 reveals a cross-alpha amyloid-like fibril. Science. 2017;355:831–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med. 2007;13:1510–4. [DOI] [PubMed] [Google Scholar]
- [48].Dickey SW, Cheung GYC, Otto M. Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance. Nat Rev Drug Discov. 2017;16:457–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Le KY, Otto M. Quorum-sensing regulation in staphylococci-an overview. Front Microbiol. 2015;6:1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kong KF, Vuong C, Otto M. Staphylococcus quorum sensing in biofilm formation and infection. Int J Med Microbiol. 2006;296:133–9. [DOI] [PubMed] [Google Scholar]
- [51].Bae T, Schneewind O. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid. 2006;55:58–63. [DOI] [PubMed] [Google Scholar]
- [52].Vuong C, Gerke C, Somerville GA, Fischer ER, Otto M. Quorum-sensing control of biofilm factors in Staphylococcus epidermidis. J Infect Dis. 2003;188:706–18. [DOI] [PubMed] [Google Scholar]
- [53].Joo HS, Otto M. The isolation and analysis of phenol-soluble modulins of Staphylococcus epidermidis. Methods Mol Biol. 2014;1106:93–100. [DOI] [PubMed] [Google Scholar]
- [54].Piewngam P, Zheng Y, Nguyen TH, Dickey SW, Joo HS, Villaruz AE, et al. Pathogen elimination by probiotic Bacillus via signalling interference. Nature. 2018;562:532–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Chatterjee SS, Joo HS, Duong AC, Dieringer TD, Tan VY, Song Y, et al. Essential Staphylococcus aureus toxin export system. Nat Med. 2013;19:364–7. [DOI] [PMC free article] [PubMed] [Google Scholar]