

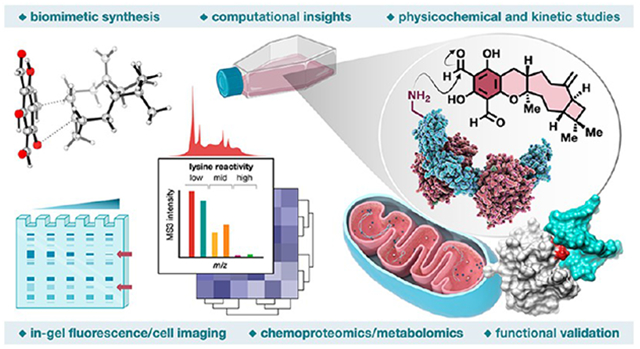
Abstract
Natural products perennially serve as prolific sources of drug leads and chemical probes, fueling the development of numerous therapeutics. Despite their scarcity, natural products that modulate protein function through covalent interactions with lysine residues hold immense potential to unlock new therapeutic interventions and advance our understanding of the biological processes governed by these modifications. Phloroglucinol meroterpenoids constitute one of the most expansive classes of natural products, displaying a plethora of biological activities. However, their mechanism of action and cellular targets have, until now, remained elusive. In this study, we detail the concise biomimetic synthesis, computational mechanistic insights, physicochemical attributes, kinetic parameters, molecular mechanism of action, and functional cellular targets of several phloroglucinol meroterpenoids. We harness synthetic clickable analogues of natural products to probe their disparate proteome-wide reactivity and subcellular localization through in-gel fluorescence scanning and cell imaging. By implementing sample multiplexing and a redesigned lysine-targeting probe, we streamline a quantitative activity-based protein profiling, enabling the direct mapping of global reactivity and ligandability of proteinaceous lysines in human cells. Leveraging this framework, we identify numerous lysine–meroterpenoid interactions in breast cancer cells at tractable protein sites across diverse structural and functional classes, including those historically deemed undruggable. We validate that phloroglucinol meroterpenoids perturb biochemical functions through stereoselective and site-specific modification of lysines in proteins vital for breast cancer metabolism, including lipid signaling, mitochondrial respiration, and glycolysis. These findings underscore the broad potential of phloroglucinol meroterpenoids for targeting functional lysines in the human proteome.
Graphical Abstract
INTRODUCTION
Chemical probes have emerged as indispensable tools in drug discovery and chemical biology due to their ability to elucidate the functions of proteins, thereby enabling in-depth investigations into associated biological mechanisms.1 Nonetheless, the paucity of selective chemical probes against a substantial fraction of human proteins persists, with numerous protein categories classified as undruggable.2 The traditional methods for discovering chemical probes, such as high-throughput screening of large compound libraries,3 have been recently complemented with more contemporary strategies like covalent ligand development,4 fragment-based5 and inverse6 drug discovery. These techniques have been systematically applied proteome-wide by leveraging reactive probes and quantitative mass spectrometry (MS) methods. Through judicious optimization of recognition scaffolds and reactive elements, electrophilic agents can engage shallow or dynamic protein binding pockets, thereby broadening the spectrum of druggable proteins. Notably, the irreversible nature of these interactions engenders sustained pharmacological effects, persisting until cellular protein turnover ensues.7
Despite the resurgence in covalent probes, initial efforts primarily focused on catalytic serine/threonine8 or cysteine9 residues within the active sites of enzymes. Recent studies, however, have broadened the scope of druggable sites to include non-catalytic cysteines in functional sites of proteins,10,11 noncatalytic tyrosine and phosphotyrosine sites,12,13 oncogenic variants of KRAS,14,15 and functionally diverse lysines involved in protein–protein and protein–RNA binding interfaces.13,16,17 Yet, most covalent drugs remain cysteine-targeting due to their predictable reactivity with acrylamides and haloacetamides.18 Expanding the scope of covalent probes hinges on a deep understanding of the reactivity and chemoselectivity of candidate electrophiles. This principle was exemplified by our in-depth chemical proteomic analysis of structurally diverse chemotypes, which, when integrated with human genetic information and cell-activation-state profiling, uncovered thousands of ligandable lysines, including those residing at functional sites on proteins historically deemed undruggable.17 Through these studies, we discovered that aldehydes exhibit disparate proteomic reactivities, spanning selective interactions with only a few lysines to more promiscuous covalent engagement with many lysines, with this divergent reactivity contingent upon their structural diversity.17 Aldehydes have long been viewed as a structural alert, leading to a historical dichotomy within the medicinal chemistry community and their dogmatic exclusion from drug discovery programs.19 However, recent FDA approvals of alcaftadine (lastacaft)20 and voxelotor (oxbryta)21 have challenged this dogma, suggesting that aldehydes might hold significant untapped potential in 21st-century drug discovery as a versatile modality to drug novel biological targets.19 As a prime example, voxelotor, an orally administered drug for the treatment of sickle cell disease, forms a reversible-covalent bond with the N-terminal amine of hemoglobin.22 This interaction enhances hemoglobin’s affinity for oxygen, preventing the polymerization that leads to the characteristic sickle shape of the affected cells.
Natural products and their structural analogues have been an enduring source of drug leads and chemical probes.23 Phloroglucinol meroterpenoids comprise one of the largest classes of natural products with >700 members displaying a vast array of biological activities (Figure 1A).24,25 Recent studies have identified several protein targets for a subset of phloroglucinol meroterpenoids, including ATP-citrate lyase (ACLY),26 phosphodiesterase-4 (PDE4D2),27 acetylcholinesterase (AChE), and α-glucosidase.28 Yet, elucidating their precise mechanism of action and systematically identifying their protein targets within native biological systems continue to pose significant challenges. The integration of natural product total synthesis and advanced chemoproteomic technologies has now emerged as a robust framework for unraveling complex mechanisms of action and identifying protein targets on a global scale within complex biological systems.29-34 Our investigation into phloroglucinol meroterpenoids is propelled by their elaborate terpenoid architecture featuring an electrophilic salicylaldehyde-based phloroglucinol core, reminiscent of voxelotor.21 This resemblance implies the potential for covalent interactions with proteinaceous amines. To date, only two natural products have been confirmed to impact protein function through irreversible lysine modification. These include Ophiobolin A, a mycotoxin, which disrupts the function of the protein calmodulin through the covalent modification of lysine K75,35 and Wortmannin, a fungal metabolite, which inhibits the function of phosphoinositide 3-kinase (PI3K) by covalently modifying the lysine K802 within the ATP catalytic site.36 Sonolisib (PX-866),37 an improved Wortmannin analogue, is an oral, irreversible, and potent PI3K inhibitor that has been subject to multiple clinical trials for glioblastoma,38 metastatic colorectal carcinoma,39 head and neck squamous cell cancer,40 non-small-cell lung cancer (NSCLC),41 prostate cancer,42 and advanced BRAF-mutant tumors.43 Despite their rarity, natural products that modulate protein function through covalent interactions with lysines hold immense potential to unlock new therapeutic modalities and advance our knowledge of the biological processes governed by these modifications.
Figure 1. Proposed mechanism of action and biomimetic synthesis of representative phloroglucinol meroterpenoids and analogues.

(A) Representative phloroglucinol meroterpenoids isolated from Eucalyptus globulus and Eucalyptus robusta. (B) Proposed reversible covalent reactivity of phloroglucinol meroterpenoids with proteinaceous lysines. (C) Plausible biosynthetic pathways to cattleianal, involving either an inverse electron-demand hetero-Diels–Alder, Michael addition/cyclization, or Alder-ene/cyclization sequence. (D) Biomimetic synthesis of cattleianal, euglobals G1–4, guadials B and C, grandinol, and jensenone. (E, F) Corresponding enantiomeric and clickable analogues as probes for in-gel fluorescence scanning, molecular imaging, and chemical proteomics. For a detailed description of reagents and reaction conditions, including stereochemical assignments by two-dimensional NMR, see the Supporting Information.
Motivated by these findings, we hypothesized that salicylaldehyde-based phloroglucinol meroterpenoids could engage in a reversible-covalent interaction with proteinaceous lysines, forming a Schiff base adduct that could be further stabilized by intramolecular resonance-assisted hydrogen bonding44 (Figure 1B). With this objective, we report the concise biomimetic syntheses of the recently isolated cattleianal45 and previously documented jensenone,46 grandinol,47 euglobals G1–4,48 and guadials B and C,49 together with synthetic clickable analogues and corresponding enantiomeric series. Through our advanced quantitative activity-based protein profiling platform, which incorporates sample multiplexing and features an optimized desthiobiotin-based probe, we identify numerous lysine–phloroglucinol meroterpenoid interactions in breast cancer cells that occur at tractable sites on proteins from diverse structural and functional classes. We demonstrate that phloroglucinol meroterpenoids perturb diverse biochemical functions through stereoselective and site-specific modification of lysines in proteins involved in lipid metabolism, mitochondrial respiration, and glycolysis. Collectively, these findings not only underscore the broad potential of phloroglucinol meroterpenoids for targeting functional lysines in the human proteome, but also vastly expand the toolbox available for studying lysine modifications.
RESULTS AND DISCUSSION
Biomimetic Synthesis of Representative Phloroglucinol Meroterpenoids and Clickable Analogues.
The Myrtaceae family, comprising dicotyledonous flora, constitutes approximately 5950 species dispersed across an estimated 132 genera.50 Cattleianal, first isolated in 2021 from the leaves of Psidium cattleianum, displays cytotoxicity against HT-29, Hep-2, MCF7, and MDA-MB-231 cancer cell lines, exerting an antiproliferative effect through the induction of G0/G1 cell cycle arrest.45 Biosynthetically, the sesquiterpenoid fragment of cattleianal can be traced to (−)-β-caryophyllene (1).51 In fact, several biomimetic approaches toward phloroglucinol meroterpenoids starting from β-caryophyllene have been reported.52-54 Inspired by these findings, we speculated three plausible mechanistic scenarios for the biosynthesis of cattleianal (Figure 1C). The first proposal incorporates a hetero-Diels–Alder reaction between 1 and an ortho-quinone methide 2, which may directly yield cattleianal. We envisaged that the relative stereochemistry of cattleianal could be partially controlled by the conformation of 1 during this reaction. This biosynthetic route was validated through the biomimetic synthesis of guajadial and psidial A in an aqueous three-component coupling reaction, involving 1, benzaldehyde, and diformylphloroglucinol.55 Alternatively, cattleianal may result from a Michael addition between 1 and 2, delivering intermediate 3. The intramolecular phenoxide cyclization of 3 would then establish the final stereocenter and complete the synthesis.51 Our third proposal pertains to the generation of an inconsequential mixture of diastereomers 4 through an Alder-ene reaction between 1 and 2. A subsequent tautomerization, followed by a proton transfer, would consequently intercept the intermediate 3.
Synthetic efforts toward cattleianal commenced with the Vilsmeier–Haack diformylation of commercial phloroglucinol, furnishing diformylphloroglucinol (S14) in 78% yield on a decagram scale (Figure 1D).55 This was followed by the C-methylation of S14 with iodomethane,56 which led to the formation of an ortho-quinone methide precursor (S18) in a 27% yield. Although the intermolecular cycloaddition of ortho-quinone methides has been shown to be difficult,57 the 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)-mediated oxidative cyclization of S18 in the presence of 1 in nitromethane afforded (−)-cattleianal in an 11% unoptimized yield with a 9:1 diastereomeric ratio. Next, our focus was shifted toward jensenone, a compound originally isolated in 1992 from the steam volatile leaf oil of Eucalyptus jensenii, a tree species indigenous to the northern regions of Australia.46 To date, no reported bioactivity has been attributed to jensenone. Initial attempts to reproduce the synthesis of jensenone under prior established conditions failed, resulting in complete decomposition.58 To mitigate this issue, we conducted the Friedel–Crafts acylation of phloroglucinol with isovaleryl chloride in the presence of methanesulfonic acid as catalyst and solvent,59 delivering isovalerylphloroglucinol (S1) in 53% yield. This was followed by a modified Duff formylation protocol using formamidine acetate,60 facilitating the isolation of jensenone in 41% yield without further purification. We next turned our attention toward guadials B and C, first isolated in 2015 from the leaves of Psidium guajava, commonly known as apple guava, an indigenous evergreen shrub prevalent in the Caribbean, Central America, and South America.49 The bioactivity of guadials B and C remains uncharacterized. The biomimetic synthesis of these two structurally isomeric natural products was achieved using established routes.53 In brief, methanolic solution of diformylphloroglucinol (S14) was initially treated with benzaldehyde in the presence of tetramethylethylenediamine (TMEDA) as a catalyst afforded a labile methyl ether 5 in a 31% yield. The methyl ether 5 was immediately heated at 90 °C with (+)-α-pinene under neat conditions to generate (+)-guadial B as the only product in 14% yield. The same reaction sequence was repeated with (−)-β-pinene to obtain (+)-guadial C in 16% yield. Proceeding further, our attention was drawn to grandinol and euglobals G1 –4, natural products isolated from the leaves of Eucalyptus grandis, commonly known as rose gum, in 1977 and 1994, respectively, a tall evergreen tree endemic to coastal areas stretching from Wales to Australia, known to exhibit mild to moderate antibacterial, antifungal, and antileishmanial activities.61 Grandinol and jensenone are postulated as the biosynthetic precursors to biologically active phloroglucinols, such as sideroxylonals, grandinal, euglobals, and macrocarpals.62 The synthesis of grandinol was achieved through an improved synthetic route that echoes a methodology described in 1985.56 Briefly, the Vilsmeier–Haack monoformylation of isovalerylphloroglucinol (S1) delivered aldehyde S2 in a 61% yield, which was then subjected to C-methylation with iodomethane56 to afford grandinol in a 28% yield. The biomimetic synthesis of euglobals G1–4, using grandinol as a biosynthetic precursor, was accomplished under modified conditions.57,63 DDQ-mediated oxidative cyclization of grandinol in the presence of (+)-α-pinene in nitromethane produced chromatographically separable regioisomeric (+)-euglobal-G1 and (+)-euglobal-G2 in a 21% unoptimized yield. Under identical reaction conditions, regioisomeric (−)-euglobal-G3 and (−)-euglobal-G4 were obtained from (−)-β-pinene as a terpene source.
Recent studies have underscored the integral function of stereochemistry in modulating small molecule–protein interactions, a principle which has been exploited in the discovery of electrophilic probes.11,13,64,65 With this in consideration, the enantiomeric counterparts of (+)-euglobal-G2 and (+)-guadial B were synthesized through an identical reaction sequence, however, with the use of (−)-α-pinene as the chiral terpene source to produce (−)-euglobal-G2 and (−)-guadial B (Figure 1E). The synthesis of clickable analogues derived from natural products has been demonstrated to be instrumental in quantitative proteomic experiments, facilitating the mapping of cellular protein targets and enabling rapid qualitative visualization of probe–protein interactions and their localization through in-gel fluorescence scanning and molecular imaging.30,34 Inspired by these studies, we embarked on the synthesis of alkyne- or azide-functionalized derivatives of selected phloroglucinol meroterpenoids. The rationale for the development of both alkyne- and azide-functionalized clickable probes was predominantly influenced by synthetic constraints and the chemical compatibility issues encountered during the modification of the phloroglucinol scaffold. The synthesis of alkyne-conjugated intermediates was marked by incompatibilities in key synthetic stages, resulting in decomposition and the formation of complex mixtures. In contrast, intermediates bearing azide functionalities demonstrated enhanced stability and predictability in their chemical behavior, thereby influencing the decision to utilize these tags in the probe design. Alkyne-bearing derivatives of jensenone (6) and grandinol (7) were synthesized by the C-alkylation of diformylphloroglucinol (S14) and isovaleryl formylphloroglucinol (S2) employing iodohexyne as the alkylating agent to afford alkynes 6 and 7 in 49% and 20% yield, respectively (Figure 1F). In pursuit of azide-functionalized derivatives of euglobals G1–4, we first engineered azide-tagged grandinol (8) through a four-step synthetic sequence. This involved the Friedel–Crafts acylation of phloroglucinol with 6-bromohexanoyl chloride, a displacement of the primary bromide using sodium azide, followed by the Vilsmeier–Haack monoformylation and C-alkylation using iodomethane to deliver 8 in a 36% overall yield. Subsequent DDQ-mediated oxidative cyclization of 8, in the presence of either (+)- or (−)-α-pinene in nitromethane, led to the formation of enantiomeric azide-tagged analogues of euglobal-G2, (+)-9 and (−)-9. Under identical reaction conditions, regioisomeric azide-tagged analogues of (−)-euglobal-G3 and (−)-euglobal-G4, (−)-10 and (−)-11, were acquired from (−)-β-pinene as a terpene source.
Combined Spectroscopic and Computational Studies into the Mechanism of the Formation of Cattleianal.
Next, we shifted our attention toward exploring the viability of our hetero-Diels–Alder proposal outlined in Figure 1C though additional spectroscopic and computational studies (Extended Data Figure S1A-C; see further discussion therein). Considering the inherent conformational flexibility of 1, it was imperative to determine how this dynamic nature might influence the facial bias in the hetero-Diels–Alder cycloaddition. Indeed, our 1H NMR analysis suggest that 1 predominantly exists as a 3:1 conformational mixture (Extended Data Figure S1A), aligning with the previously documented ratios of αα/βα (76%) and ββ (24%).66 Furthermore, under the reaction conditions, no conformational interconversion between αα/βα and ββ was observed. Examination of the crude reaction mixture revealed the initial formation of cattleianal as a 3:1 mixture of diastereomers (Extended Data Figure S1B). We ascertained that cattleianal and an unassigned minor diastereomer do not interconvert under the reaction conditions, confirming the irreversible nature of cycloadduct formation (Extended Data Figure S1C). Consequently, the observed ratio of cattleianal and an unassigned minor diastereomer from the crude reaction mixture aligns well with detected αα/βα and ββ conformational distributions.
To gain insights into the plausible mechanism for the formation of cattleianal through DDQ-mediated oxidative cyclization of 1 in the presence of 2, we conducted computational analysis to discern the inherent selectivity of the reaction between the conformers 1-βα and 1-ββ with an ortho-quinone methide 2 (Extended Data Figure S1D,E; see further discussion therein). The decision to compare the ββ to the βα conformer, rather than the αα conformer, was strategic, given their closely related spatial arrangement of the exocyclic methylene group. Despite our efforts, computational attempts to detect transition states consistent with a Michael addition or a concerted Alder-ene reaction between 1 and 2 proved unsuccessful. Notably, a high interconversion barrier (TSβα→ββ) between conformers 1-βα and 1-ββ, which are nearly equivalent in energy, aligns with our 1H NMR interconversion studies of 1 under the reaction conditions. Calculations further indicate that the 1-βα conformer reacts with 2 to afford cattleianal through a transition state (TS-βα) with an energy of 9.8 kcal·mol−1, while 1-ββ forms 4,10-epi-cattleianal through a TS-ββ with an energy of 11.4 kcal·mol−1 (Extended Data Figure S1D). The hetero-Diels–Alder pathway originating from 1-βα and 1-ββ is calculated to be exergonic by 28.8 and 23.5 kcal·mol−1, respectively. The results derived from the frontier molecular orbital analysis further underscore the inverse-electron-demand nature of the hetero-Diels–Alder cycloaddition with the HOMO–LUMO energy gap of 9.49 eV (Extended Data Figure S1E). Computational evaluations provide evidence that the final stoichiometry of the cycloadducts retains the inherent ratio of the conformational population of 1, corroborating the spectroscopic observations. Based on this, the synthesis of cattleianal is postulated to proceed through an inverse-electron-demand hetero-Diels–Alder mechanism involving the βα conformation of 1. Concurrently, the minor diastereomer 4,10-epi-cattleianal, arises from the less prevalent ββ conformer of 1. Collectively, these findings support the plausibility of the inverse-electron-demand hetero-Diels–Alder cycloaddition as a potential biosynthetic pathway.
Physicochemical and Kinetic Studies of Representative Phloroglucinol Meroterpenoids with a Model Amine Nucleophile.
The unique structural attributes inherent to the phloroglucinol core of meroterpenoids may hold the key to understanding their mechanisms of interaction with lysines in biological systems. We postulated that the presence of electron-withdrawing carbonyl groups on the phloroglucinol core may potentiate the acidity of phenolic moieties, subsequently resulting in the formation of anionic species at physiological pH. To validate this hypothesis, we embarked on the quantification of ionization constants (pKa) for select phloroglucinol meroterpenoids using UV spectrophotometric methods.67 The obtained pKa1 values for the representative phloroglucinol meroterpenoids were found to range from 5.04 to 5.56, indicating that these natural products may predominantly adopt anionic forms under physiological pH (Extended Data Figure S2A; see further discussion therein). The corresponding pKa2 values spanned between 8.01 and 9.45, while the pKa3 values for grandinol and jensenone were determined to be 10.86 and 10.47, respectively. The anionic nature of phloroglucinol meroterpenoids presents several biological implications. Primarily, the anionic electrophilic phloroglucinol core could potentially augment interactions with cationic lysine residues within proteins, likely mediated by proton relay and electrostatic mechanisms. Additionally, the juxtaposition of the anionic phloroglucinol core and lipophilic terpenoid framework may mimic lipid architectures, potentially facilitating interactions with proteins implicated in lipid metabolic pathways. In a concluding observation, the structural and physicochemical parallels between phloroglucinol meroterpenoids and ubiquinol (the reduced form of coenzyme Q10) are evident. Notably, ubiquinol is characterized by a hydrophobic isoprenyl chain and a richly oxygenated phenolic core, the latter exhibiting a calculated pKa1 value of 5.31 and a pKa2 value of 10.86.68 These structural analogies may serve as predictive indicators for the subcellular localization of these natural products within the mitochondrial electron transport chain.
At the outset, the synergistic interplay between electron-donating phenolic and electron-withdrawing carbonyl groups in modulating the inherent reactivity of the phloroglucinol scaffold toward lysine residues was ambiguous. To discern underlying kinetic trends that could be extrapolated to more complex structures, a focused array of benzaldehyde derivatives was selected, each varying in the number and spatial orientation of phenolic substituents relative to the aldehyde group. We postulated that observed rates would reflect the relative propensity of phloroglucinol meroterpenoids to associate with lysines in unstructured regions of proteins or from unfolded globular proteins prior to proteolytic degradation. In experiments conducted in the presence of an excess of butylamine as a prototypical amine nucleophile, salicylaldehyde and jensenone exhibited rapid imine adduct formation kinetics, with half-life times (t1/2) of 2.8 and 7.2 min, respectively (Extended Data Figure S2B). Comparatively, benzaldehyde displayed a decelerated rate of adduct formation, marked by a 10-fold increased half-life time (t1/2 = 28.9 min) in comparison to salicylaldehyde. Grandinol and phloroglucinaldehyde followed suit, showing 30-fold (t1/2 = 207.3 min) and 80-fold (t1/2 = 228.3 min) diminished rates, respectively, when compared to jensenone and salicylaldehyde. These kinetic observations substantiate that the rate of imine adduct formation is modulated by the balance between electron-donating phenolic and electron-withdrawing carbonyl groups present within the phloroglucinol core. Specifically, an increase in the electron-donating phenolic groups correlates inversely with the rate of imine adduct formation, thereby enabling the fine-tuning of intrinsic reactivity toward lysines in a predictable manner.
To investigate the structure–stability correlations governing imine adduct formation, we conducted dilution experiments to probe the dissociation kinetics through imine adduct hydrolysis.69,70 The results demonstrate that the initial imine adduct ratio was preserved post-dilution, thereby confirming the irreversible nature of the conjugation under physiologically relevant conditions (Extended Data Figure S2C). In contrast to the fast dissociation kinetics observed for iminoboronates71,72 and the tempered reversibility exhibited by diazaborines69,73 in reactions with amine nucleophiles, the conjugation of phloroglucinol meroterpenoids with a model amine nucleophile manifests in an essentially irreversible reaction. Encouraged by the robust stability of these adducts, our efforts focused on isolation of the natural product-butylamine adducts by column chromatography. To this end, methanolic solutions of grandinol and jensenone were subjected to an excess of butylamine, resulting in the production of monoadduct 12 and bisadduct 13 in 60% and 53% yields, respectively (Extended Data Figure S2D). Upon isolation, these complexes exhibited a noteworthy stability when stored neat under ambient temperature, preserving their structural integrity for several weeks without discernible degradation. When the isolated monoadduct 12 was subjected to the aforementioned dilution experiments, no evidence of reversibility was observed, further reaffirming the irreversible character of the conjugate under physiologically pertinent conditions.
To assess the stability of these putatively irreversible meroterpenoid–protein interactions in cells, we conducted a series of gel-based experiments using a synthesized clickable probe and hydroxylamine as an ambident α-effect nucleophile. The azide probe (−)-10 showed substantial labeling of proteins in both the soluble and insoluble fractions of MDA-MB-231 cells, confirming the irreversible probe–protein interactions (Extended Data Figure S2E). The resulting conjugates underwent hydroxylamine-mediated cleavage in a dose-dependent manner. Our data indicate that the cleavage of probe–protein conjugates occurs predominantly in the presence of hydroxylamine concentrations exceeding 10 mM. These observations collectively suggest that phloroglucinol meroterpenoids can form stable and irreversible adducts with lysine residues within proteins; these adducts not only withstand the cellular environment but are also impervious to the denaturing and reducing SDS-PAGE, succumbing only to cleavage under conditions marked by the abundant presence of a more potent nucleophile.
In-Gel Fluorescence Profiling of Clickable Phloroglucinol Meroterpenoid Probes in Breast Cancer Cells.
Prompted by the considerable intracellular stability exhibited by meroterpenoid–protein conjugates, we endeavored to assess their interaction profiles by qualitative in-gel fluorescence analysis using synthesized clickable probes (Figure 2A). Prior research has demonstrated that chemical proteomics platforms capable of broad-spectrum profiling serve as effective tools for elucidating ligandability events that are contingent upon biological state,11,17,74,75 thereby serving as a conceptual framework for the development of chemical probes capable of selectively modulating protein activity in a context-specific manner (e.g., pathological versus physiological states or wild-type versus mutant genotypes). To investigate this, experiments were performed using two triple-negative breast cancer (TNBC) cell lines,76 adherent mesenchymal MDA-MB-231 and MDA-MB-436 harboring wild-type and mutant forms of breast cancer susceptibility gene 1 (BRCA1), respectively (also see supplemental discussion on Supporting Information page S7).77
Figure 2. In-gel fluorescence profiling of clickable phloroglucinol meroterpenoid probes in breast cancer cells.

(A) Experimental workflow for detecting probe–protein interactions in cells by SDS-PAGE coupled with in-gel fluorescence scanning. (B) Time- and dose-dependent labeling by the clickable azide probe (−)-11. MDA-MB-231 cells were treated with (−)-11 for the indicated times or concentrations, harvested, lysed, and separated into soluble and insoluble fractions. Following CuAAC conjugation to BDP-TMR-alkyne (1 h, 20 °C), samples were resolved by SDS-PAGE and analyzed by in-gel fluorescence scanning. (C) Excess (−)-euglobal-G4 blocks probe (−)-11 labeling of several proteins in MDA-MB-231 cells (marked with red stars). (D) Comparative analysis of clickable phloroglucinol meroterpenoid probes shows differential protein targets across cell lines (marked with red stars) and enantiodivergent labeling within cell lines (marked by red boxes) in BRCA1-proficient and BRCA1-deficient triple-negative breast cancer cells. SDS-PAGE data shown are representative of three independent biological experiments (n = 3). For a detailed description of in-gel fluorescence studies, including compound treatment and CuAAC conjugation protocols, see the Supporting Information.
We conducted an initial evaluation of the time- and dose-dependent protein interaction landscape of the azide probe (−)-11 by gel-based analysis of treated cells (Figure 2B). In brief, MDA-MB-231 cells were exposed to various concentrations and durations of (−)-11 treatment, subsequently harvested, subjected to lysis, and separated into soluble and insoluble fractions. Following CuAAC conjugation to BDP-TMR-alkyne, samples were resolved by SDS-PAGE and analyzed by in-gel fluorescence scanning. The (−)-11 probe exhibited marked time- and concentration-dependent protein labeling in MDA-MB-231 cells, in contrast to negligible labeling observed in control cells treated with the vehicle, dimethyl sulfoxide (DMSO). This labeling profile was more conspicuous in the soluble fractions, particularly manifesting at or beyond the 8-h exposure. This pattern is plausibly attributable to the anionic nature of the phloroglucinol core, which may reduce transmembrane diffusion kinetics.78 In contrast, more immediate labeling was observed in the insoluble fractions, likely stemming from the combination of the anionic phloroglucinol core and the lipophilic terpenoid scaffold that mimics lipid architectures, potentially enhancing interactions with peripheral and integral proteins associated with biological membranes. To gauge the global selectivity profile of (−)-euglobal-G4, we implemented gel-based assays in a competitive format with the azide probe (−)-11 directly in MDA-MB-231 cells. Exposure to (−)-euglobal-G4 resulted in the blockade of probe (−)-11 labeling for a subset of protein targets in a dose-dependent manner (Figure 2C). This effect was further accentuated in the insoluble fractions. We next compared the protein interaction profiles of clickable alkyne and azide probes (6–11) across both MDA-MB-231 and MDA-MB-436 cell lines (Figure 2D). This comparative analysis revealed a spectrum of differentially engaged targets between the two cell lines and exhibited enantiodivergent labeling for (+)-9 and (−)-9 probes within these cell lines. Collectively, these results underscore the notion that phloroglucinol meroterpenoids can selectively interact with protein targets in a context-specific manner (e.g., BRCA1 wild-type versus mutant cells), thereby warranting further in-depth analysis to identify differential protein targets through quantitative MS-based chemical proteomics with the natural product library across both cell lines.
Multiplexed Mass Spectrometry-Based Quantification for Expedited Discovery of Lysine–Natural Product Interactions in Cells.
Our prior methodology, employing the isoTOP-ABPP (isotopic Tandem Orthogonal Proteolysis-Activity-Based Protein Profiling) platform as a quantitative MS-based measurement protocol for lysine interactions, exhibited commendable sensitivity and robustness.17 However, we acknowledge certain limitations. Notably, the restricted throughput hinders comprehensive investigation of structure–activity relationships (SARs) across the natural product library. Furthermore, this constraint poses challenges in comparing protein interaction profiles of multiple natural products under diverse conditions, such as varying cell types or concentrations. Finally, our previous chemical proteomic profiling of lysine interactions was conducted in cellular lysates,17 rather than in live cells, thereby limiting the exploration of lysine interactions that rely on an intact cellular environment or involve proteins prone to instability in cellular lysates. Addressing these limitations could significantly expand the coverage of lysine interactions and enhance the overall impact of this research.
We considered that the aforementioned challenges could be addressed by profiling natural products with a multiplexed approach for quantitative MS-based chemical proteomics that employs isobaric tandem mass tags (TMTs).11,64 Consequently, we developed the TMT18plex-based workflow, wherein up to 18 distinct populations of cells are each treated with a natural product (100 μM, 8 h) in live MDA-MB-231 cells and then chased with the redesigned desthiobiotin-tagged lysine-specific STP ester probe in cellular lysates, proteolytically digested by trypsin, and enriched by streptavidin (Figure 3A). The strategic selection of the MDA-MB-231 cell line was integral for a direct and consistent comparison with our prior work using the isoTOP-ABPP method,17 thereby highlighting the improvements in our current methodology with minimal biological variability. We designed and synthesized a novel desthiobiotin-tagged STP ester probe to supplant the conventionally used pentynoic acid STP ester,16,17 thereby obviating the need for CuAAC. Tryptic peptides from each treatment group are then labeled with a TMT of equivalent parent mass, but differentiable by MS3-derived fragmentation products, combined, and analyzed in a single MS experiment. To optimize both quantification and proteomic coverage without sacrificing the instrument speed, we implemented the real-time search (RTS) coupled with synchronous precursor selection (SPS) and MS3 fragmentation.79 Evaluation of MS2 spectra showed confident identification of all major y and b ions, including fragment ions that enabled identification of the probe-labeled lysines (Figure 3B). A median number of 6678 lysine residues was quantified per dataset, a considerable increase compared to the 2593 lysines quantified by the isoTOP-ABPP method.17 Cumulatively, these advancements represent a significant enhancement over existing lysine-targeted MS-based chemical proteomics methodologies, specifically tailored to increase experimental throughput.
Figure 3. Multiplexed mass spectrometry-based quantification for expedited discovery of lysine–natural product interactions in cells.

(A) Schematic of TMTpro-18plex-based workflow for mapping lysine–phloroglucinol meroterpenoid interactions in breast cancer cells. (B) MS/MS spectrum annotation of a desthiobiotin probe-modified tryptic peptide from PKFL. Covalent modification with desthiobiotin STP ester probe results in addition of +196.121 Da to the ε-amino groups of lysines (K677 from PFKL shown as a representative example). The TMTpro-18plex reagents are amine-reactive and modify the ε-amino groups of lysines and peptide N-termini by addition of +304.207 Da. Data shown are representative of three experiments (n = 3 biologically independent experiments). For a detailed description of the TMTpro-18plex-based protocol, see the Supporting Information.
Global Map of Phloroglucinol Meroterpenoid–Lysine Interactions in the Human Proteome.
Leveraging our innovative chemical proteomics platform, we conducted a comprehensive investigation of regioisomeric and enantiomeric phloroglucinol meroterpenoids across BRCA1-proficient MDA-MB-231 and BRCA1-deficient MDA-MB-436 cells. To evaluate the cytotoxic potential of phloroglucinol meroterpenoids under the experimental conditions used for mass spectrometry-based quantification, cell viability assays were conducted (Extended Data Figure S3A,B). These experiments revealed negligible cytotoxic effects under these conditions, supporting the notion that the alterations in protein target profiles observed in our chemical proteomics experiments are attributable to specific meroterpenoid–lysine interactions, rather than nonspecific cytotoxicity-induced alterations. For reliable assessment of natural product–lysine interactions, or “ligandability”, we imposed stringent criteria requiring that each lysine residue be quantified across all biological triplicates and TMT channels within independent datasets. Lysine residues were considered “liganded” if they manifested substantial reductions (≥60%) in enrichment by desthiobiotin-tagged STP ester probe in the presence of natural products when compared to DMSO (vehicle). The natural products were found to engage discrete sets of liganded lysines and displayed marked differences in their overall lysine reactivity (Figure 4A; also see Supplementary Dataset). A total of 159 lysines on 130 proteins were identified to be liganded by one or more natural products from the aggregate analysis of 9268 quantified lysines on 3172 unique proteins (Figure 4B; also see Extended Data Figure S3C). Most proteins contained only one liganded lysine, indicating site-specific ligandability within proteins (Figure 4C). Approximately 62% of the liganded lysines were targeted by a single natural product, while the remaining lysines exhibited distributed interaction profiles with 2 to many (≥5) natural products (Figure 4D). Notably, 47% of quantified lysines overlapped between MDA-MB-231 and MDA-MB-436 cells (Figure 4E; also see Extended Data Figure S3D), but only a small fraction (10%) of lysines were found to be liganded in both cells (Figure 4F; also see Extended Data Figure S3E-H), indicating that the natural products preferentially target unique lysines in cells with different BRCA1 mutational status.
Figure 4. Global map of phloroglucinol meroterpenoid–lysine interactions in the human proteome.

(A) Hierarchical clustering heatmap (Euclidean distance, complete-linkage method) of the liganded lysines and phloroglucinol meroterpenoids. Each row represents a liganded lysine and each column represents a phloroglucinol meroterpenoid. Dendrogram for liganded lysines is shown on the left side of the heatmap, and dendrogram for phloroglucinol meroterpenoids is shown below the heatmap. Dark blue to light gray color gradient denotes higher to lower ligandability (also see Supplementary Dataset). (B) Fraction of total quantified lysines liganded by natural products. (C) Number of liganded lysines per protein. (D) Number of natural product hits per liganded lysine. (E) Overlap of quantified lysines in MDA-MB-231 and MDA-MB-436 cells. (F) Overlap of liganded lysines in MDA-MB-231 and MDA-MB-436 cells. (G) Number of proteins harboring liganded lysines with reported probes. (H) Number of lysines bearing post-translational modifications (also see Extended Data Figure S3). (I) Functional class distribution of proteins with liganded lysines. (J) Sequence logo of the lysine-modified amino acid motif. Sequence logo of the probe lysine-modified amino acid motif depicting over- and underrepresented residues which are scaled to their log10 odds of the binomial probability, as a direct measure of a residue’s likelihood of being statistically significantly over- or underrepresented. The sequence plot was generated with the pLogo (http://plogo.uconn.edu).80 The horizontal red line indicates the threshold of the Bonferroni corrected p-value of p < 0.05. (K) Proportion of proteins harboring liganded lysines that are defined as Strongly Selective in the Cancer Dependency Map, reflecting a restricted dependency relationship with a subset of the cancer cell line panel, and as Common Essential to indicate their general requirement for the growth of most cancer cell lines. (L) Distribution of liganded lysines in proteins that have human-disease relevance (as assessed by pathogenic mutations that lead to monogenic disorders defined in the OMIM database). (M) Functional consequences of clinically relevant variants of the liganded lysine residues themselves (in cases where these mutations are associated with disease) as classified by ClinVar,81 gnomAD,82 and ClinGen83 databases. (N) Clusters of human diseases identified through DisGeNET84 term enrichment analysis on liganded proteins. Dark red represents breast carcinoma, and yellow highlights mitochondrial disorders. (O) Location of liganded lysines (dark red) mapped onto the crystal structures (gray) bound to metabolites or nucleic acids (blue). K469 of SHMT2 (PDB: 6QVG) bound to PLP; K72 of BANF1 (PDB: 2BZF) bound to DNA; K25 of RPS23 (PDB: 4CXG) bound to RNA; K299 of IDH2 mutant R172K (PDB: 5SVN) bound to NADPH. ND, not determined.
Phloroglucinol meroterpenoids showed disparate proteomic reactivities, spanning from promiscuous to site-specific and stereoselective interactions with lysine residues. Notable examples of lysine residues broadly engaged by phloroglucinol meroterpenoids include (1) K310 in cytochrome c1 (CYC1), a subunit of mitochondrial respiratory chain complex III, identified in insoluble fractions of MDA-MB-231 and MDA-MB-436 cells; (2) K99 in the ovarian carcinoma immunoreactive antigen-like protein (OCIAD2), a complex III-specific mitochondrial assembly factor, also identified in insoluble fractions of aforementioned cell lines; (3) K677 in transmembrane protein 168 (TMEM168), a putative nuclear transporter, identified in insoluble fractions of MDA-MB-231 cells; and (4) K121 in NADH-ubiquinone oxidoreductase subunit A6 (NDUFA6), an accessory subunit of mitochondrial membrane respiratory chain complex I, identified in soluble fractions of MDA-MB-436 cells. Upon further analysis of liganded lysines across protein targets, we found evidence for a substantial recognition component governing the meroterpenoid–lysine interactions, manifested through markedly distinct SARs for individual liganded lysines. Noteworthy examples of liganded lysines with highly stringent SAR targeted by a single phloroglucinol meroterpenoid include (1) K246 in annexin A4 (ANXA4), a phospholipid-binding protein implicated in membrane fusion and exocytosis, is exclusively targeted by (−)-euglobal-G4; (2) K35 in transthyretin (TTR), a thyroid hormone-binding protein responsible for the transport of thyroxine from the bloodstream to the brain, is selectively engaged by jensenone; (3) K197 in tyrosine-tRNA ligase (YARS), an enzyme that catalyzes the attachment of tyrosine to tRNA, is specifically liganded by (−)-cattleianal; and (4) K215 in glyceraldehyde-3-phosphate dehydrogenase (GAPDH), an enzyme that converts d-glyceraldehyde 3-phosphate to 3-phospho-d-glyceroyl phosphate, is solely targeted by (+)-guadial C. Moreover, the enantiomeric pairs of natural products, such as euglobal-G2 and guadial B, displayed pronounced enantiodivergent ligandability for a subset of lysines across the two breast cancer proteomes. Notable examples of lysines with enantiodivergent ligandability include (1) K330 in kynurenine-oxoglutarate transaminase 3 (KYAT3), an enzyme that catalyzes the irreversible transamination of the l-kynurenine, was preferentially targeted by (+)-guadial B with 74% ligandability, but only with 28% by (−)-guadial B; (2) K20 in nucleostemin (GNL3), a nucleolar GTP-binding protein that regulates the cell cycle, was selectively engaged by (−)-guadial B with 74% ligandability, in contrast to 46% by (+)-guadial B; (3) K24 in prohibitin-2 (PHB2), a chaperone that stabilizes mitochondrial respiratory enzymes, was liganded by (−)-euglobal-G2 with 61% ligandability, unlike 26% by (+)-euglobal-G2; and (4) K128 in stathmin (STMN1), a protein that regulates microtubule filament system by destabilizing microtubules, was preferentially engaged by (+)-euglobal-G2 with 67% ligandability, but only with 11% by (−)-euglobal-G2. We also identified instances of differential lysine ligandability in proteins with no discernible alterations in expression between BRCA1-proficient MDA-MB-231 and BRCA1-deficient MDA-MB-436 cells. Specifically, K99 in OCIAD2 exhibited marked engagement by (+)-euglobal-G2 with 92% ligandability in MDA-MB-231 cells, compared to 49% in MDA-MB-436 cells. Likewise, K24 in the mitochondrial 2-oxoglutarate/malate carrier protein (SLC25A11) displayed significant engagement by (−)-euglo-bal-G4 with 85% ligandability in MDA-MB-231 cells, in contrast to 33% in MDA-MB-436 cells. The observed differential lysine ligandability between BRCA1-proficient and BRCA1-deficient cells, despite comparable expression levels, suggests a complex interplay of biological mechanisms. These could include changes in post-translational modifications, protein interactions, structural conformations, compensatory cellular responses, microenvironmental factors, and protein stability. Future investigations in these areas could shed further light on this phenomenon and its relevance to BRCA1-related breast cancer pathology. A significant proportion (76%) of proteins possessing liganded lysines lacked reported chemical probes (Figure 4G), while a more modest fraction (31%) of liganded lysines are known to possess post-translational modifications (Figure 4H). The proteins with liganded lysines originated from diverse structural and functional classes (Figure 4I). Sequence logo analysis indicated no amino acid sequence conservation surrounding liganded lysines, suggesting that lysine ligandability is independent of protein sequence (Figure 4J). Among proteins harboring liganded lysines, 18% were defined as Strongly Selective in the Cancer Dependency Map, reflecting a restricted dependency relationship with a subset of the cancer cell line panel, and 32% were defined as Common Essential to indicate their general requirement for the growth of most cancer cell lines (Figure 4K). For instance, K121 in the NADH-ubiquinone oxidoreductase subunit A6 (NDUFA6), an assembly factor that exhibits a strongly selective dependency relationship with a subset of the cancer cell line panel, was selectively targeted with high ligandability in MDA-MB-436 cells by nearly all phloroglucinol meroterpenoids with the exception for jensenone.
In contrast, K76 in the mitochondrial citrate synthase (CS), an enzyme essential for the growth ofmost cancer cell lines, was exclusively targeted in MDA-MB-231 cells by (−)-euglobal-G3 and both enantiomers of euglobal-G2. A survey of the Online Mendelian Inheritance in Man (OMIM) database identified several human disease-relevant proteins (41%) harboring liganded lysines (Figure 4L). A noteworthy subset of these cases (45%) featured mutations with clinical ramifications in the liganded lysine residues themselves, as catalogued in the ClinVar, gnomAD, and ClinGen databases (Figure 4M). Notable examples include the pathogenic variant K524* (RCV001293010) in the syntaxin-binding protein 1 (STXBP1) in individuals diagnosed with developmental and epileptic encephalopathy, the likely pathogenic variant K398T (RCV000824963) in the α-actinin-1 (ACTN1) in individuals with platelet-type bleeding disorder 15, and the variant of uncertain significance K195E (RCV002355029) in the mitochondrial very long-chain specific acyl-CoA dehydrogenase (ACADVL) in individuals with inborn genetic diseases. Such convergence of ligandability and human genetic data point to functional lysines with the potential to be targeted by phloroglucinol meroterpenoid-based chemical probes. Term enrichment analysis from the gene–disease association database (DisGeNET) revealed a preferential ligandability of lysines within proteins implicated in mitochondrial pathologies (Figure 4N). This predilection is potentially attributable to the structural and physicochemical homology between phloroglucinol meroterpenoids and ubiquinol, which likely facilitates their subcellular trafficking to mitochondria and subsequent engagement with mitochondrial proteins. Furthermore, crystal structure analysis further revealed that various lysines targeted by natural products are near metabolite and nucleic acid binding sites (Figure 4O), thereby positing that meroterpenoid–lysine interactions could modulate the functional attributes of these proteins.
Phloroglucinol Meroterpenoids Target Key Metabolic Pathways in Breast Cancer Cells.
Subcellular distribution analysis revealed that of the protein targets harboring liganded lysines, 38% localized to the mitochondria, 35% to the cytoplasm, and the remainder were distributed among the endoplasmic reticulum, nucleus, Golgi apparatus, and extracellular space (Figure 5A). Gene ontology (GO) term analysis of diverse biological processes for proteins with liganded lysines confirmed an enrichment of proteins implicated in key mitochondrial metabolic pathways (Figure 5B). The subcellular localization of phloroglucinol meroterpenoids was validated through an in-cell treatment with clickable azide probe (−)-11, followed by fixed-cell CuAAC and fluorescence microscopy,85,86 which further underscored their mitochondrial targeting (Figure 5C).
Figure 5. Phloroglucinol meroterpenoids target key metabolic pathways in breast cancer cells.

(A) Subcellular distribution of proteins with liganded lysines. (B) Top-10 enriched clusters of biological processes from GO-term enrichment analysis of proteins harboring liganded lysines. (C) Fluorescence cell imaging of the clickable azide probe (−)-11 visualized by copper-catalyzed azide–alkyne cycloaddition (CuAAC). MDA-MB-231 cells were treated with (−)-11 at 100 μM for 8 h, fixed with 4% formaldehyde, permeabilized, and blocked (0.1% Triton-X, 5% BSA in PBS). Following CuAAC conjugation to BDP-TMR-alkyne (30 min, 20 °C), samples were counterstained with DAPI (nucleus, blue) and MitoView (mitochondria, green). (D) Hierarchical clustering heatmap (Euclidean distance, complete-linkage method) of identified metabolites in MDA-MB-231 cells treated with representative phloroglucinol meroterpenoids (also see Supplementary Dataset and Extended Data Figure S4). Shades of red and blue represent upregulation and downregulation of a metabolite, respectively. (E) Volcano plots depicting statistically significant (false discovery rate-corrected P value <0.05 and fold change >2.0) altered metabolites in MDA-MB-231 cells treated with jensenone and guadial B. P values were determined by Student’s t test (two-tailed, two-sample equal variance). Blue circles represent metabolites selectively downregulated by meroterpenoids, red circles represent metabolites selectively upregulated by meroterpenoids, and gray circles represent metabolites with no significant difference. Data represent average values ± SD, n = 6 per group from six biologically independent experiments. FC, fold change. (F) Top-25 metabolic pathway enrichment of metabolites significantly altered by representative phloroglucinol meroterpenoids. Colors represent the statistical significance (P value) of the enriched pathways, and point size represents the enrichment ratio of matched metabolites and total metabolites in the corresponding pathway. (G) Hierarchical clustering heatmap (Euclidean distance, complete-linkage method) of identified lipid species in MDA-MB-231 cells treated with representative phloroglucinol meroterpenoids (also see Supplementary Dataset and Extended Data Figure S4). Shades of red and blue represent upregulation and downregulation of a lipid, respectively. (H) Volcano plots depicting statistically significant (false discovery rate-corrected P value <0.05 and fold change >2.0) altered lipid species in MDA-MB-231 cells treated with jensenone and guadial B. P values were determined by Student’s t test (two-tailed, two-sample equal variance). Blue circles represent metabolites selectively downregulated by meroterpenoids, red circles represent metabolites selectively upregulated by meroterpenoids, gray circles represent metabolites with no significant difference. Data represent average values ± SD, n = 6 per group from six biologically independent experiments. FC, fold change. (I) Top-25 enriched lipid species significantly upregulated and downregulated by representative phloroglucinol meroterpenoids. Shades of red and blue represent upregulation and downregulation of a lipid, respectively. Point size represents the enrichment ratio of matched lipid species and total lipids in the corresponding pathway.
To corroborate the results of our chemoproteomics and cellular imaging studies, we applied an integrated untargeted metabolomics and lipidomics approach. This approach was instrumental in demonstrating that exposure of breast cancer cell lines to select phloroglucinol meroterpenoids modulates lipid, glucose, and mitochondrial metabolism. We directed our attention toward jensenone and the enantiomers of guadial B as representative phloroglucinol meroterpenoids for these studies on the basis of their disparate proteomic reactivity and enantiodivergent ligandability profiles. The untargeted metabolomics analysis revealed significant alterations across several interlinked metabolic pathways in breast cancer cells treated with jensenone and guadial B (Figure 5D; also see Extended Data Figure S4A). These natural products elicited unique patterns of metabolic modulation, characterized by specific upregulation and downregulation of metabolites, including distinct metabolic alterations induced by the enantiomers of guadial B (Figure 5E; also see Extended Data Figure S4B). Subsequent pathway enrichment analysis of the significantly altered metabolites by these meroterpenoids uncovered a profound influence on the metabolic networks within breast cancer cells, targeting multiple pathways concurrently (Figure 5F; also see Extended Data Figure S4C). Particularly, the analysis highlighted pronounced alterations in linoleic acid metabolism, glycolysis, the urea cycle, and gluconeogenesis, all of which are intrinsically linked to mitochondrial function. The observed modulation in linoleic acid metabolism, a crucial aspect of mitochondrial lipid metabolism, suggests a shift of mitochondrial fatty acid utilization. Alterations in glycolysis, closely linked to mitochondrial respiration, imply a rewriting in cellular energy production. In addition, changes in the urea cycle and gluconeogenesis, integral to mitochondrial amino acid and glucose metabolism, further underscore the influence of jensenone and guadial B on mitochondrial metabolic processes.
To gain further insight into the influence of these natural products on lipid metabolism, we conducted untargeted lipidomics studies. This investigation revealed significant alterations in the lipidomic profiles of breast cancer cells following treatment with jensenone and guadial B (Figure 5G; also see Extended Data Figure S4D). Each natural product elicited specific regulatory effects on lipid species, including differential alterations by the enantiomers of guadial B (Figure 5H; also see Extended Data Figure S4E).
The enrichment term analysis of lipids significantly affected by these meroterpenoids, underscored a substantial reprogramming of lipid profiles in the treated cells (Figure 5I; also see Extended Data Figure S4F). This analysis identified a pronounced upregulation of specific lipid categories, particularly monoacylglycerophosphocholines and acylcarnitines. The elevated acylcarnitine levels suggest a shift in mitochondrial fatty acid β-oxidation and a disruption in the balance of intracellular carbohydrate and lipid metabolism, reflecting a significant alteration in the cellular energy landscape. In contrast, a marked downregulation in diacylglycerols, diacylglycerophosphocholines, and diacylglycerophosphoethanolamines—key constituents of cellular membranes and signaling cascades—was observed, implying a disruption in membrane dynamics and intracellular signaling. Collectively, these findings underscore the potential of phloroglucinol meroterpenoids in orchestrating metabolic reprogramming in breast cancer cells, providing evidence for their polypharmacological mechanism of action under defined treatment conditions.
Mitochondrial metabolism plays a crucial role in cellular energy production and regulation of mitophagy, the targeted degradation of damaged or dysfunctional mitochondria.87 Prior investigations have elucidated that lysine residues exert regulatory control over proteins involved in mitochondrial metabolism and mitophagy.88 Lysine residues within mitochondrial proteins have been identified as key sites for modifications by phloroglucinol meroterpenoids in this study. Nevertheless, the functional implications of lysine–meroterpenoid interactions on target proteins integral to mitochondrial metabolism remain unexplored. Understanding the functional consequences of these modifications is imperative for uncovering their regulatory roles and underlying molecular mechanisms. Consequently, we next aimed to assess the functional impact of representative lysine–meroterpenoid interactions on target proteins. Proteins crucial to metabolic pathways that are essential for breast cancer progression—namely, lipid metabolism (ABHD12),89 mitochondrial respiration (IDH2),90 and glycolysis (PFKP, PFKM, PFKL),91,92—were chosen not only for their individual roles but also for their interplay with the mechanisms that regulate mitochondrial dynamics.
Functional Impact and Structure–Activity Relationship for Phloroglucinol Meroterpenoid Engagement of Lysine K373 in the Lysophosphatidylserine Lipase ABHD12.
The lipase ABHD12, which belongs to the α/β-hydrolase domain-containing protein family, is a serine hydrolase responsible for catalyzing the conversion of lysophosphatidylserine (LPS) into glycerophosphoserine,93 and is predominantly localized on the endoplasmic reticulum (ER) membrane.94 The recent development of a potent reversible thiourea-based inhibitor, DO264, and a covalent serine-reactive activity-based probe, JJH350, has significantly advanced our understanding of the pivotal role played by ABHD12 in lipid metabolism and disease.95,96 We postulate that exploration of inhibitory mechanisms through covalent modification of lysine residues might offer innovative avenues for the design of novel chemical probes targeting ABHD12.
In our chemical proteomics investigation, we identified a conserved lysine residue K373 in ABHD12 as a primary target showing moderate ligandability by meroterpenoids (+)-guadial C (64%), (−)-guadial B (66%), and jensenone (76%) in both MDA-MB-231 and MDA-MB-436 cell lines (Extended Data Figure S5A; see further discussion therein). Given that the crystallographic structure of ABHD12 is yet to be elucidated, we utilized an AlphaFold-generated three-dimensional structural model97—focusing on residues that exceeded a confidence threshold (pLDDT > 70)—for predictive analyses. The structural model revealed that K373 lines the cleft of the putative active site pocket, proximal to the catalytic triad residues S246, D333, and H372 (Figure 6A). Based on this spatial proximity to the catalytic site, we hypothesized that an initial qualitative assessment of proteome-wide reactivity and chemoselectivity for each meroterpenoid could be conducted using competitive gel-ABPP assays in conjunction with the fluorescent broad-spectrum serine-directed fluorophosphonate probe, TAMRA-FP.98 As anticipated, ABHD12 variants with K373R and K373M mutations maintained TAMRA-FP labeling, whereas the catalytic S246A mutant completely abrogated probe binding (Figure 6B). In agreement with our chemical proteomics data, phloroglucinol meroterpenoids possessing a dialdehyde core, selectively inhibited TAMRA-FP probe labeling in recombinantly expressed epitope-tagged WT-ABHD12, but not in the K373R-ABHD12 mutant (Extended Data Figure S5B). Intriguingly, immunoblot analysis revealed that jensenone treatment led to a marked reduction in the monomeric form of ABHD12 and a concomitant increase in high-molecular-weight ABHD12 aggregates. Notably, this effect was unique to jensenone and not observed with other dialdehyde-containing meroterpenoids, suggesting that the aggregation is governed not only by reactivity but also by a recognition motif specific to jensenone (Figure 6B).
Figure 6. Functional impact and structure–activity relationship for phloroglucinol meroterpenoid engagement of lysine K373 in the lysophosphatidylserine lipase ABHD12.

(A) The location of the liganded lysine K373 (dark red) is mapped onto the AlphaFold structure of the human ABHD12 (gray, AF-Q8N2K0-F1), with the catalytic triad (teal) residues (S246, D333, H372) positioned within the active site. Only the segment spanning residues 86–389 with high per-residue confidence score (pLDDT > 70) are displayed in the analysis. (B) Representative in-gel fluorescence data and immunoblot analysis showing SAR across phloroglucinol meroterpenoids for blockade of probe TAMRA-FP98 labeling and formation of high-molecular-weight aggregates of recombinantly expressed Flag-tagged WT-ABHD12 in HEK293T cell lysates. (C) SAR for phloroglucinol meroterpenoid inhibition (100 μM, 1 h, 37 °C) of LPS hydrolysis activity measured using recombinantly expressed WT-, S246A-ABHD12, and indicated K373 mutants in HEK293T cell lysates with 17:1 LPS substrate (100 μM, 20 min, 37 °C). Data represent average values ± SD, n = 3 per group from three biologically independent experiments. Statistical significance was calculated using a one-way ANOVA with Dunnett’s post hoc test: ****P < 0.0001. ns, not significant. (D) Fitted IC50 (95% CI) curves for the dose-dependent inhibition of the LPS hydrolysis activity by (+)-guadial B and (−)-guadial B measured using recombinantly expressed WT- and K373R-ABHD12 in HEK293T cell lysates. Data represent average values ± SD, n = 3 per group from three biologically independent experiments. (E) Fitted IC50 (95% CI) curves for the dose-dependent inhibition of the LPS hydrolysis activity by jensenone measured using recombinantly expressed WT- and K373R-ABHD12 in HEK293T cell lysates. Data represent average values ± SD, n = 3 per group from three biologically independent experiments. (F) Representative in-gel fluorescence data and Western blot analysis showing dose-dependent formation of high-molecular-weight aggregates and blockade of probe TAMRA-FP labeling of recombinantly expressed Flag-tagged WT and K373R mutant forms of ABHD12 in HEK293T cell lysates. (G) MDA-MB-231 cells treated with jensenone and (−)- and (+)-guadial B (100 μM, 8 h), show significantly elevated levels of monoacylglycerophosphoserine LPS(18:0) with a concurrent reduction in its hydrolytic degradation product, stearic acid FA(18:0). Data represent average values ± SD, n = 5 per group from five biologically independent experiments. Statistical significance was calculated using a one-way ANOVA with Dunnett’s post hoc test: ****P < 0.0001, ***P = 0.0003, **P = 0.0021. ns, not significant.
Next, using an LC/MS-based LPS hydrolysis assay,93 we observed that the K373R mutant elicited a marked attenuation in ABHD12 activity, with only 29% of residual activity, while the K373M-ABHD12 was essentially inactive, retaining merely 6% of the activity (Figure 6C). These findings accentuate the critical role of K373 in substrate binding. It is pertinent to highlight that K373R represents a variant of uncertain clinical significance, as denoted by the ClinVar (CA408453896). Subsequent screening of phloroglucinol meroterpenoids revealed that only those containing a dialdehyde core selectively inhibited LPS hydrolysis, congruent with ligandability values obtained from chemical proteomics and TAMRA-FP probe labeling blockade observed in gel-ABPP studies. For instance, guadials B and C led to a partial yet significant reduction in ABHD12 activity, retaining only 50% of residual activity, whereas jensenone elicited a more pronounced inhibition, with 23% residual activity. Dose–response profiles revealed apparent IC50 values of 18.9 μM for (+)-guadial B and 18.1 μM for (−)-guadial B against WT-ABHD12 (Figure 6D). In contrast, the K373R-ABHD12 mutant exhibited IC50 values exceeding 100 μM for both meroterpenoids, indicative of a K373-specific functional inhibition. Conversely, jensenone displayed comparable inhibitory efficacy against both WT and K373R mutant forms of ABHD12, with IC50 values of 32.6 μM and 45.7 μM, respectively, suggesting a K373-independent mechanism of LPS hydrolysis inhibition (Figure 6E). Subsequent gel-ABPP and immunoblot investigations confirmed that jensenone instigates the formation of high-molecular-weight ABHD12 aggregates from their monomeric counterparts in a dose-dependent manner (Figure 6F). Importantly, this aggregation effect was further validated with endogenous ABHD12 in MDA-MB-231 and HEK293T cell lines, characterized by a marked reduction in the monomeric form of ABHD12 and a concomitant increase in high-molecular-weight ABHD12 aggregates following dose-dependent administration of jensenone (Extended Data Figure S5D).
We then proceeded to validate the specific interaction between the clickable jensenone alkyne 6 and the targeted lysine K373 in ABHD12. This was accomplished through direct binding assays using CuAAC conjugation to BDP-TMR-azide, followed by in-gel fluorescence scanning of recombinantly expressed WT-ABHD12 and corresponding K373R mutant. The conducted experiments confirmed that probe 6 specifically binds to K373 in the WT-ABHD12, but not in the K373R mutant, further reinforcing the specificity of the K373–jensenone interaction (Extended Data Figure S5C). Furthermore, analogous to jensenone, probe 6 induced high-molecular-weight ABHD12 aggregation in a K373-independent manner. The amassed evidence currently suggests that although the binding of jensenone and its clickable analogue to K373 is specific, the resultant enzymatic inhibition (Figure 6E) and aggregation effects (Figure 6F and Extended Data Figure S5C) appear to be K373-independent. Moreover, our findings suggest that the principal mechanism by which jensenone impairs the enzymatic function of ABHD12 is through K373-independent aggregation. This raises the possibility that jensenone could promote aggregation through interactions with other lysine residues present in ABHD12. However, among the 21 lysine residues in ABHD12, only K373 was detected in our chemical proteomics, suggesting it might be the predominant lysine residue susceptible to covalent modification by the desthiobiotin STP ester probe.
We next sought to delineate the consequences of ABHD12 inhibition, specifically through aggregation by jensenone or targeted covalent modification at K373 by guadial B, on cellular glycerophosphoserine metabolism. Our untargeted lipidomics profiling revealed that jensenone and guadial B treatment significantly elevates levels of monoacylglycerophosphoserine LPS(18:0) concurrent with a reduction in its hydrolytic degradation product, stearic acid FA(18:0), a pattern consistent with genetic and pharmacological perturbation of ABHD12 (Figure 6H).94,95 These insights are of significant importance, as they bridge our chemoproteomic and lipidomic datasets with the outcomes from in vitro enzymatic assay, reinforcing the notion that inhibition of ABHD12, whether through aggregation or site-specific covalent modification of K373, plays a critical role in modulating glycerophosphoserine metabolism in breast cancer cells. Nonetheless, it is imperative to acknowledge that these effects may emanate from the polypharmacological mechanisms of phloroglucinol meroterpenoids under the specified experimental conditions, as suggested by the extensive alterations in the lipidome.
Structural and Functional Consequences of Phloroglucinol Meroterpenoid Engagement with Lysines K299 and K413 in the Mitochondrial Isocitrate Dehydrogenase IDH2 and Its Oncogenic Gain-of-Function R140Q Variant.
Mitochondrial isocitrate dehydrogenase 2 (IDH2) is an indispensable homodimeric enzyme that catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate within the tricarboxylic acid cycle and constitutes a pivotal element of the mitochondrial antioxidant pathway.100 IDH2 deficiency instigates the depolarization of the mitochondrial membrane potential, leading to mitochondrial dysfunction and triggering targeted mitochondrial degradation, known as mitophagy.101 The oncogenic R140Q variant of IDH2, which possesses neomorphic enzymatic activity, catalyzes the further reduction of α-ketoglutarate to the oncometabolite 2-hydroxyglutarate, thereby inducing cellular metabolic reprogramming (Figure 7B).102 Recognizing the consequential implications of this pathway, numerous inhibitors have been developed that predominantly target the IDH2 homodimer interface, displaying a broad spectrum of potencies against both the wild-type and oncogenic activity. A testament to this progress is the FDA’s recent approval of enasidenib, a compound that selectively obstructs the oncogenic activity of IDH2 variants through allosteric modulation of the homodimer interface.103 However, the emergence of resistance mutations against enasidenib necessitates continued research endeavors toward the identification of innovative chemical probes, aiming to target the oncogenic IDH2 variants by previously unexplored mechanisms of action.104
Figure 7. Structural and functional consequences for phloroglucinol meroterpenoid engagement with lysines K299 and K413 in the mitochondrial isocitrate dehydrogenase IDH2 and its oncogenic gain-of-function R140Q variant.

(A) SAR for phloroglucinol meroterpenoid inhibition (100 μM, 1 h, 37 °C) of α-KG production measured using recombinantly expressed WT-IDH2 and indicated K299 mutants in HEK293T cell lysates with d-(+)-threo-isocitrate substrate (1.3 mM, 1 h, 25 °C). The panel also includes corresponding representative immunoblot results. (B) IDH2 catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG); however, the hotspot mutation R140Q in IDH2 leads to neomorphic enzymatic activity that results in overproduction of the oncometabolite, d-2-hydroxyglutarate (2-HG). (C, D) Fitted IC50 (95% CI) curves for the dose-dependent inhibition of the oxidative IDH2 activity by (+)-guadial B and (−)-guadial B measured using recombinantly expressed WT- and K299R-IDH2 in HEK293T cell lysates. Data represent average values ± SD, n = 3 per group from three biologically independent experiments. Statistical significance was calculated using a one-way ANOVA with Dunnett’s post hoc test: ****P < 0.0001, ***P = 0.000315, *P = 0.027574. ns, not significant. (E) The location of liganded lysine K299 (dark red) and proximal lysine K413 (dark red) mapped onto the crystal structure of homodimer IDH2 (PDB ID: 5H3F) bound to NADPH (teal). Inset shows a magnified view of the pocket, detailing the spatial arrangement of K299 on chain A (gray) relative to K413 on chain B (tuscany) with an inter-residual distance of 8.6 Å. (F) Quantitative assessment of IDH2 oxidative activity inhibition by jensenone (100 μM, 1 h, 20 °C) in recombinantly expressed WT-IDH2 and indicated K299 mutants in HEK293T cell lysates, complemented by representative immunoblotting data. (G) Fitted IC50 (95% CI) curves for the dose-dependent inhibition of the oxidative IDH2 activity by jensenone measured using recombinantly expressed WT-, K299R-, K413R-, and double-mutant K299R/K413R-IDH2 in HEK293T cell lysates. Data represent average values ± SD, n = 3 per group from three biologically independent experiments. Statistical significance was calculated using a one-way ANOVA with Dunnett’s post hoc test: ****P < 0.0001. ns, not significant. (H) SAR for phloroglucinol meroterpenoid inhibition (100 μM, 2 h, 20 °C) of oncometabolite 2-HG production measured using recombinantly expressed R140Q-IDH2 and indicated K299 mutants in HEK293T cell lysates with α-KG substrate (0.6 mM, 1 h, 25 °C). Data represent average values ± SD, n = 3 per group from three biologically independent experiments. Statistical significance was calculated using a one-way ANOVA with Dunnett’s post hoc test: ****P < 0.0001. ns, not significant. (I) Representative immunoblot depicting jensenone-mediated IDH2 homodimerization of recombinantly expressed Flag-tagged WT- and R140Q-IDH2 with indicated K299 and K413 mutants in HEK293T cell lysates. (J) Fitted IC50 (95% CI) curves for the dose-dependent inhibition of reductive R140Q-IDH2 activity by jensenone measured using recombinantly expressed R140Q-, double mutant R140Q/K299R-, and triple mutant R140Q/K299R/K413R-IDH2 in HEK293T cell lysates. Data represent average values ± SD, n = 3 per group from three biologically independent experiments.
Our chemical proteomics data derived from MDA-MB-436 cells revealed that liganded lysine K299, which is evolutionarily conserved across eukaryotes, is situated on a cusp of the NADP+ binding site of IDH2 and forms a hydrogen bond with the phosphate group of NADP+ (Extended Data Figure S6). SAR analysis has uncovered a preferential engagement of K299 by phloroglucinol meroterpenoids bearing a dialdehyde core structure. The functional contributions of K299 to IDH2 catalytic activity have heretofore remained unexplored. Using a previously reported substrate assay,105 we found that the K299R-IDH2 mutant elicited only a marginal decrement in oxidative IDH2 activity, whereas the K299M-IDH2 mutant completely abolished enzymatic activity (Figure 7A). These observations imply that the binding of NADP+ and subsequent oxidative catalytic activity of IDH2 are modulated by the presence of a positively charged hydrogen bond donor at the K299 site, corroborating a direct interaction between K299 and the phosphate moiety of NADP+. In accordance with our chemical proteomics analysis, meroterpenoids bearing a dialdehyde phloroglucinol core exerted the most profound impact on IDH2 function. Specifically, guadial B induced a partial yet significant reduction of IDH2 oxidative activity, while jensenone treatment led to a complete functional inhibition. Dose–response evaluations indicated that both enantiomers of guadial B selectively inhibited the activity of WT-IDH2 with comparable IC50 values of approximately 24 μM (Figure 7C,D). Notably, these enantiomers exhibited an attenuated inhibitory effect on the K299R-IDH2 mutant (IC50 > 100 μM), thereby underscoring a K299-specific functional modulation. Subsequent immunoblotting analysis revealed an unexpected induction of IDH2 homodimerization by jensenone, with (+)-guadial C exhibiting a more subdued effect (Figure 7A).
Prompted by this observation, we revisited the crystallographic data for insights. Structural analysis of the murine isoform of IDH2, which exhibits >95% protein sequence homology with its human counterpart, and the human oncogenic variants R140Q- and R172K-IDH2, identified the K413 residue positioned opposite K299 on either flank of the NADP+ binding pocket, separated by an inter-residual distance of 8.6 Å and situated on distinct monomeric chains (Figure 7E). Previous studies indicate that endogenous acetylation at K413 due to SIRT3 protein depletion attenuates oxidative IDH2 enzymatic activity by impairing homodimer formation, consequently modulating mitochondrial redox status.106
Prior mutagenesis studies have documented a 44-fold reduction in oxidative IDH2 activity for the K413A variant, a 20-fold decrease in Vmax for the acetylation mimic K413Q, and no discernible difference in Km for K413R with respect to isocitrate and NADP+.107 Based on our structural examination and the functional role of K413, we posited that jensenone may engage with either of these lysine residues or induce IDH2 homodimerization through concurrent engagement of both lysines K299 and K413. This premise was inferred from our preceding observations involving a model amine nucleophile (Extended Data Figure S2D). To test this hypothesis, enzymatic assays measuring the oxidative activities of K413R-IDH2 and the double mutant K299R/K413R-IDH2 were conducted, and subsequently revealed that the K413R-IDH2 mutant maintained wild-type IDH2 activity, aligning with previous reports,107 whereas the activity of K299R/K413R-IDH2 mirrored that of K299R-IDH2 (Figure 7F). Following jensenone treatment, the double mutant K299R/K413R-IDH2 essentially preserved its oxidative activity, while the activity of all other IDH2 variants was substantially inhibited. Further immunoblotting affirmed jensenone-induced WT-IDH2 homodimerization, a phenomenon mitigated in the absence of one or both lysines. Moreover, jensenone exhibited a dose-dependent inhibitory effect on WT, K299R, and K413R mutant forms of IDH2, with respective IC50 values of 20.9 μM, 29.8 μM, and 21.4 μM, yet displayed negligible affinity (IC50 > 100 μM) toward the double mutant K299R/K413R-IDH2 (Figure 7G). These results substantiate the hypothesis that jensenone induces IDH2 homodimerization likely through the formation of a covalent bisadduct by concurrent engagement of lysines K299 and K413. In scenarios where either lysine is mutated to arginine, jensenone retains its inhibitory capacity through spatial proximity, potentially by binding to the remaining lysine residue.
Recognizing that phloroglucinol meroterpenoids operate through a distinct mechanism of action compared to known inhibitors targeting the dimeric interface, we sought to explore whether K299–meroterpenoid interaction could modulate the reductive activity of the oncogenic R140Q-IDH2 variant. Previous research has established that K413 acetylation commonly occurs in oncogenic R140-IDH2 and functions as a negative regulator of mutant IDH2 activity in human acute myeloid leukemia (AML) cells by attenuating dimer formation and inhibiting the binding of the substrate α-ketoglutarate and the cofactor NADPH.108 The K299R mutation was found to completely abrogate the reductive activity of R140Q-IDH2, possibly due to an augmented interaction between K299 and the cofactor NADPH within the R140Q-IDH2 variant (Figure 7H). Among the tested meroterpenoids, only jensenone fully abolished the reductive activity of the oncogenic R140Q-IDH2 variant with an IC50 of 26.2 μM, an inhibition potency comparable to that seen in WT-IDH2, but not against either the double mutant R140Q/K299- or the triple mutant R140Q/K299R/K413R-IDH2, further indicating K299-specific functional inhibition of R140Q-IDH2 (Figure 7J). This restrictive SAR among meroterpenoids further indicates a structural perturbation specific to the oncogenic R140Q-IDH2 variant, discernible through meroterpenoid–lysine interactions. Immunoblot analysis corroborated that jensenone promotes R140Q-IDH2 homodimerization through synchronous engagement of lysines K299 and K413, akin to its effect on WT-IDH2 (Figure 7I). Collectively, these findings substantiate a K299-dependent mechanism of IDH2 inhibition by meroterpenoids, effected through the obstruction of NADP+ binding, thereby presenting an alternative to existing compounds targeting the dimeric interface. Our data further underscore the potential of jensenone as a covalent chemical probe for elucidating K299/K413-mediated homodimerization mechanisms in both WT and oncogenic IDH2 variants.
Differential Ligandability and Functionality of Conserved Lysines in the Liver Isoform of Human Phosphofructokinase PFKL.
Phosphofructokinase-1 (PFK1) serves a critical function in the glycolytic pathway by catalyzing the phosphorylation of d-fructose-6-phosphate (F6P) into d-fructose-1,6-bisphosphate (FBP), the first committed step of glycolysis.109 At the transcriptional level, distinct PFK1 isoforms are differentially expressed across various human cell types; the platelet isoform (PFKP) is expressed in most cell types, while the muscle (PFKM) and liver (PFKL) isoforms are prevalent in muscle and immune cells, respectively.110 Considering that genes associated with glycolysis assume pivotal functions under mitochondrial damage,111 the role of PFK1 becomes particularly salient as glycolytic ATP production becomes a crucial cellular energy source. In addition, prior research has elucidated direct interactions between PFK1 and mitochondrial proteins, thereby linking glycolysis to mitochondrial metabolism.112 Such interactions underscore the indispensability of PFK1 in cellular metabolism under mitochondrial dysfunction.113 Consequently, PFK1 isoforms are likely instrumental in modulating glucose utilization and metabolic fate during mitophagy. Moreover, the expression profiles of PFK1 isoforms have been correlated with the mutational status of BRCA1; cells with BRCA1 deficiency upregulate PFKP compared to their wild-type counterparts.114 The development of covalent probes that exhibit subtype-specific selectivity for individual isoforms of PFK1 would enable precise elucidation of the functional interplay among these isoforms in relation to BRCA1 mutational status.
Our quantitative chemical proteomics investigations identified discernible enantioselective lysine–meroterpenoid interactions across all three isoforms of PFK1 in both MDA-MB-231 and MDA-MB-436 cell lines, with particularly notable events manifested by the enantiomers of guadial B (Figure 8A). In the liver isoform PFKL, three lysine residues—K315, K677, and K681—were found to be liganded, in contrast to the muscle (PFKM) and platelet (PFKP) isoforms, which exhibit ligandability solely at lysines K678 and K688, respectively. Intriguingly, the enantiomers of guadial B displayed differential ligandability for the three lysines located in the allosteric nucleotide effector binding site of PFKL. For instance, (+)-guadial B exclusively targeted K681 with a ligandability of 72%, while (−)-guadial B engaged lysines K677 and K681 with ligandabilities of 65% and 60%, respectively. Multiple sequence alignment analyses corroborated that these liganded lysine residues are phylogenetically conserved across diverse taxa, suggesting their functional relevance (Figure 8B). Comparative sequence alignment mapping further delineated that lysines K677 in PFKL, together with K678 in PFKM and K688 in PFKP, are analogous residues across the human PFK1 isoforms. Crystallographic analysis revealed that these liganded lysines are located in the allosteric nucleotide effector site >20 Å distal to the active catalytic site (Figure 8C).115 To dissect the functional implications of the meroterpenoid–lysine interactions identified through chemical proteomics, we generated a series of lysine-to-arginine mutants and evaluated their impact on the enzymatic activity of the PFKL isoform. By utilizing an enzyme-coupled assay that monitored the conversion of NAD+ to NADH by ultraviolet absorbance,116 we ascertained that the site-directed mutagenesis of key lysine residues in PFKL to arginines exhibited negligible to minimal perturbations on inherent enzymatic activity and protein expression levels (Figure 8D). Upon treatment with the phloroglucinol meroterpenoid library, we observed a stringent SAR pertaining to the inhibition of WT-PFKL activity, particularly with guadials B and C, recapitulating their ligandability for lysines K677 and K681, as determined by chemical proteomics (Figure 8E). To investigate the functional consequences of specific lysine residues and their interactions with enantiomers of guadial B, we conducted comparative analyses of their inhibitory dose–response profiles, assessing their impact on both the wild-type and a series of lysine-to-arginine PFKL variants.
Figure 8. Differential ligandability and functionality of conserved lysines in the liver isoform of human phosphofructokinase PFKL.

(A) Hierarchical clustering heatmap depicting TMT quantification of stereoselective meroterpenoid–lysine interactions on human phosphofructokinase isoforms in MDA-MB-231 and MDA-MB-436 breast cancer cells. (B) Multiple sequence alignment of phosphofructokinase orthologues across multiple vertebrate species. (C) Location of liganded lysines K315, K677, and K681 (dark red) mapped onto the Cryo-EM structure of the human PFKL (PDB ID: 7LW1) bound to ADP (teal; superimposed from PDB ID: 3O8N) in the nucleotide effector site. Catalytic and regulatory domains are colored light and dark gray, respectively. ADP (teal) and F6P (yellow) are bound to the catalytic site. FBP (green) is bound to the allosteric sugar effector site. (D) Quantitative assessment of enzymatic activity in recombinantly expressed WT-PFKL and indicated lysine mutants in HEK293T cell lysates, complemented by representative immunoblotting data. Data represent average values ± SD, n = 3 per group from three biologically independent experiments. Statistical significance was calculated using a one-way ANOVA with Dunnett’s post hoc test: *P = 0.0122. ns, not significant. (E) SAR for phloroglucinol meroterpenoid inhibition (100 μM, 1 h, 20 °C) of the F6P phosphorylation measured using recombinantly expressed epitope-tagged WT-PFKL in HEK293T cell lysates. Data represent average values ± SD, n = 3 per group from three biologically independent experiments. Statistical significance was calculated using a one-way ANOVA with Dunnett’s post hoc test: ****P < 0.0001, *P = 0.034. ns, not significant. (F, G) Fitted IC50 (95% CI) curves for the dose-dependent inhibition of PFKL activity by (+)-guadial B and (−)-guadial B measured using recombinantly expressed WT-PFKL and indicated lysine mutants in HEK293T cell lysates. Data represent average values ± SD, n = 3 per group from three biologically independent experiments.
Our results demonstrate that (+)-guadial B inhibited the WT-PFKL activity with an apparent IC50 value of 2.8 μM (Figure 8F), while the (−)-guadial B enantiomer displayed an IC50 of 4.6 μM (Figure 8G). Further examination revealed that the (+)-guadial B enantiomer inhibited K681R-PFKL with an IC50 of 6.8 μM, yet this inhibitory effect was completely abolished against K677R-PFKL, the K677R/K681R-PFKL double mutant, and the K677R/K681R/K315R-PFKL triple mutant (IC50 > 100 μM). These observations strongly suggest that the inhibitory effect of (+)-guadial B on PFKL activity is mediated through enantioselective, K677-specific covalent modification. In contrast, the (−)-guadial B enantiomer demonstrated inhibitory effects on both K677R-PFKL and K681R-PFKL mutants, with IC50 values of 8.6 μM and 7.0 μM, respectively. Notably, the suppression of PFKL activity by (−)-guadial B declined markedly for the K677R/K681R-PFKL double mutant (IC50 of 78.5 μM) and was completely abrogated in the K677R/K681R/K315R-PFKL triple mutant (IC50 > 100 μM). These findings indicate that (−)-guadial B modulates PFKL enzymatic activity predominantly through interactions with either K677 or K681 residues, while a subsidiary interaction with K315 may also contribute to its affinity for the allosteric nucleotide effector site. In summary, we uncovered SARs that point to unique and substantial contributions arising not only from the reactivity and recognition elements but also from the absolute stereochemical configuration of phloroglucinol meroterpenoids.
Enantiodivergent Ligandability and Functionality of Conserved Lysines in the Muscle and Platelet Isoforms of Human Phosphofructokinase PFKM and PFKP.
We next sought to elucidate whether the modulatory effects observed in meroterpenoid–lysine interactions were conserved in the muscle isoform of human phosphofructokinase PFKM. Through chemical proteomics analysis, we identified a conserved lysine residue K678 in PFKM as the primary binding site for both (+)-guadial C and (+)-guadial B, exclusively in the MDA-MB-436 cell line, exhibiting ligandabilities of 71% and 63% respectively. Subsequent functional assays with the K678R-PFKM mutant revealed an absence of discernible effects on inherent enzymatic activity or protein expression profiles. Among the meroterpenoids assessed, both (+)-guadial C and jensenone manifested the most pronounced inhibitory effects on catalytic activity of PFKM (Figure 9A). In contrast, the modulation of PFKM activity by (+)-guadial B was comparatively muted, with a residual activity of 85%, hence only partially mirroring our chemical proteomics data. Further comparison of their dose-dependent responses revealed apparent IC50 values of 10.8 μM for jensenone (Figure 9B) and 14.4 μM for (+)-guadial C (Figure 9C) against WT-PFKM. Notably, neither meroterpenoid exerted any inhibitory effect on the K678R-PFKM mutant (IC50 > 100 μM), indicating that lysine K678 is a requisite for the functional inhibition of PFKM by these meroterpenoids.
Figure 9. Enantiodivergent ligandability and functionality of conserved lysines in the muscle and platelet isoforms of human phosphofructokinase PFKM and PFKP.

(A) SAR for phloroglucinol meroterpenoid inhibition (100 μM, 2 h, 37 °C) of the F6P phosphorylation measured using recombinantly expressed epitope-tagged WT-PFKM in HEK293T cell lysates. Data represent average values ± SD, n = 3 per group from three biologically independent experiments. Statistical significance was calculated using a one-way ANOVA with Dunnett’s post hoc test: ****P < 0.0001. ns, not significant. (B, C) Fitted IC50 (95% CI) curves for the dose-dependent inhibition of PFKM activity by jensenone and (+)-guadial C measured using recombinantly expressed WT-PFKM and the corresponding K678R mutant in HEK293T cell lysates. Data represent average values ± SD, n = 3 per group from three biologically independent experiments. (D) HEK293T cell lysates recombinantly expressing epitope-tagged WT-PFKP and the corresponding K688R mutant were treated with the indicated phloroglucinol meroterpenoids (5 μM, 1 h, 20 °C) followed by treatment with the lysine-reactive probe P1 (1 μM, 1 h, 20 °C) and analysis by gel-ABPP and immunoblotting. (E) Representative gel-ABPP, immunoblotting, and densitometric analysis demonstrating dose-dependent blockade of probe P1 labeling of recombinant WT-PFKP in HEK293T cell lysates by (+)-guadial B and (−)-guadial B. Data represent average values ± SD, n = 3 per group from three biologically independent experiments. Statistical significance was calculated using a one-way ANOVA with Dunnett’s post hoc test: ****P < 0.0001, **P = 0.0011. ns, not significant. P1, NHS-fluorescein (carboxyfluorescein succinimidyl ester). (F) Fitted IC50 (95% CI) curves for the dose-dependent blockade of P1 probe labeling by (+)-guadial B and (−)-guadial B measured using recombinantly expressed WT-PFKP in HEK293T cell lysates. Data represent average values ± SD, n = 3 per group from three biologically independent experiments. (G) SAR for phloroglucinol meroterpenoid inhibition (1 μM, 1 h, 20 °C) of the F6P phosphorylation measured using recombinantly expressed epitope-tagged WT-PFKP in HEK293T cell lysates. Data represent average values ± SD, n = 3 per group from three biologically independent experiments. Statistical significance was calculated using a one-way ANOVA with Dunnett’s post hoc test: ****P < 0.0001. ns, not significant. (H) Fitted IC50 (95% CI) curves for the dose-dependent inhibition of PFKP activity by (+)-guadial B and (−)-guadial B measured using recombinantly expressed WT-PFKP and the corresponding K688R mutant in HEK293T cell lysates. Data represent average values ± SD, n = 3 per group from three biologically independent experiments. (I) Left: Volcano plot showing statistically significant (false discovery rate-corrected P value <0.05 and fold change >2.0) lysines liganded by (+)- and (−)-guadial B in MDA-MB-231 cells with log2(FC) on the x axis and −log10(P value) on the y axis (RTS-SPS-MS3 acquisition). P values were determined by Student’s t test (two-tailed, two-sample equal variance). Ligandable lysines are indicated as (+)-guadial B selective (red), (−)-guadial B selective (blue) and nonselective (gray). Data represent average values ± SD, n = 3 per group from three biologically independent experiments. Right: Volcano plot depicting statistically significant (false discovery rate-corrected P value <0.05 and fold change >2.0) altered metabolites in MDA-MB-231 cells treated with (+)- and (−)-guadial B. P values were determined by Student’s t test (two-tailed, two-sample equal variance). Blue circles represent metabolites selectively downregulated by (+)-guadial B treatment, red circles represent metabolites selectively upregulated by (+)-guadial B treatment, gray circles represent metabolites with no significant difference. Data represent average values ± SD, n = 6 per group from six biologically independent experiments. FC, fold change.
We subsequently centered our investigations on the platelet isoform of human phosphofructokinase PFKP. Our chemical proteomics data elucidated the enantioselective ligandability of lysine K688 in PFKP by two enantiomers of guadial B. Specifically, lysine residue K688 exhibited an 89% ligandability when targeted by (+)-guadial B, compared to a 60% ligandability when engaged by its enantiomer, (−)-guadial B. Among a panel of commercially available lysine-reactive fluorescent probes used for a convenient gel-ABPP analysis of ligandable lysines in recombinantly expressed proteins,16,17 we found that the fluorescein succinimidyl ester probe (P1) labeled WT-PFKP, but not K688R, in transfected HEK293T cell proteomes, and confirmed that (+)-guadial B, but not the (−)-guadial B enantiomer, blocked P1 probe labeling of WT-PFKP at 5 μM treatment (Figure 9D). To further elucidate the observed enantioselective ligandability, we quantitatively assessed the potency by dose-dependent blockade of probe labeling for each enantiomeric interaction with K688 in WT-PFKP (Figure 9E). We ascertained that (+)-guadial B blocked P1 probe labeling in WT-PFKP with an apparent IC50 of 1.5 μM, whereas the enantiomer (−)-guadial B manifested nearly a 10-fold reduction in potency, displaying an IC50 value of 13.8 μM (Figure 9F). Mutation of lysine K688 to arginine (K688R) in PFKP led to a partial but significant reduction in PFKP activity, indicating the importance of this lysine in the allosteric regulation of PFKP function (Figure 9G). It was next discerned that only (+)-guadial B, but not its enantiomer (−)-guadial B, elicited a K688-specific inhibition in WT-PFKP activity at 1.0 μM concentration. Comparative dose–response analyses revealed an IC50 of 0.41 μM for (+)-guadialB and 3.8 μM for (−)-guadial B against WT-PFKP (Figure 9H). Notably, the K688R-PFKP variant exhibited an IC50 > 100 μM for both enantiomers, suggesting a K688-centric inhibitory mechanism. Our subsequent investigation aimed to discern the correlation between the enantiodivergent ligandability of K688 in PFKP induced by the enantiomers of guadial B, and the concomitant alterations in cellular metabolite concentrations pertinent to PFKP inhibition. Our untargeted metabolomics analysis, although unable to detect F6P, revealed a notable downregulation in cellular FBP levels in cells treated with (+)-guadial B, a reduction that was more pronounced compared to (−)-guadial B (Figure 9I). These observations underscore a notable concordance between our chemoproteomic and metabolomic datasets, emphasizing the impact on key glycolytic intermediates and supporting the hypothesis that the influence of (+)-guadial B on PFKP extends beyond enzymatic inhibition to broader metabolic implications. However, it is imperative to approach these results with circumspection, as the effects noted are likely attributable to the polypharmacological mechanisms of these enantiomeric natural products under the specified experimental conditions and the array of identified targets. Collectively, our findings suggest that enantiomeric phloroglucinol meroterpenoids, despite their physicochemical indistinguishability, may serve as covalent probes capable of differentiating PFK1 isoforms through their enantioselective interactions with evolutionarily conserved lysine residues located within the allosteric nucleotide effector site.
CONCLUSIONS AND PERSPECTIVE
In this study, we elucidated a heretofore unrealized mechanism of action wherein phloroglucinol meroterpenoids modulate protein function through covalent interactions with lysine residues. Prior to this work, only two natural products—Ophiobolin A, a mycotoxin, and Wortmannin, a fungal secondary metabolite—had been validated to exert functional effects on proteins through irreversible lysine modification. Considering that phloroglucinol meroterpenoids constitute one of the largest classes of natural products, our study vastly expands the current repertoire of natural products capable of targeting proteinaceous lysines. Prospective synthetic and chemoproteomic investigations offer considerable promise for the development of innovative therapeutic modalities and advance our understanding of the intricate biological processes governed by lysine modifications.
This study has also mitigated multiple constraints inherent in our prior chemical proteomics platform, thereby significantly expanding the coverage of lysine interactions. We streamlined a quantitative activity-based protein profiling by introducing sample multiplexing and a redesigned lysine-targeting probe, optimized quantification and coverage with RTS-SPS-MS3 methodology, and conducted analyses in live cells to explore lysine interactions in an intact cellular environment. Cumulatively, these advancements represent a significant improvement over existing lysine-targeted MS-based chemical proteomics methodologies, specifically tailored to increase experimental throughput. Projecting forward, it remains imperative to optimize the protocol by reducing the requisite input proteome to enhance operational efficiency and resource conservation. It is, however, important to recognize that the total lysines quantified here constitute only a fraction of all lysine residues in the human proteome, and it is therefore possible that our ligandability estimates may not reflect the broader potential for phloroglucinol meroterpenoids to engage lysines across the entirety of human proteins. Nonetheless, we are encouraged by the discovery of liganded lysines in structurally and functionally diverse proteins, including those that lack chemical probes. This accentuates the potential of phloroglucinol meroterpenoids to expand the scope of the human proteome that can be targeted by natural products.
Projecting forward, we envision several exciting avenues that should address fundamental questions about the intricate biological processes governed by lysine modifications (also see supplemental discussion on Supporting Information page S14). Our study shows that meroterpenoids bearing dialdehyde phloroglucinol core may serve as covalent protein dimerizers and molecular glues through a unique mechanism of action that targets lysine residues within proteins. Given the burgeoning interest in chemical inducers of dimerization and molecular glues that modulate protein function through induced aggregation as a therapeutic strategy,99 these natural products could function as versatile tools for surveying protein aggregation and homo- and heterodimerization in the human proteome. Future efforts will leverage the chemical proteomics strategy coupled with size exclusion chromatography117 to assess the global impact of phloroglucinol meroterpenoids on protein complexes in cells, thereby elucidating the functional consequences and identifying potential neosubstrates.
In addition, the elucidation of the specific mass adducts of phloroglucinol meroterpenoids on their protein targets proved to be more complex than anticipated. Despite employing advanced LC-MS/MS techniques and targeted methodologies, we faced significant challenges in obtaining high-quality spectral data. We speculate that the challenges encountered in detecting these mass adducts may be attributed to several factors, including the inherent hydrophobicity of meroterpenoids, their ionization efficiency, characteristic fragmentation patterns, and the stability of the formed adducts. Lastly, the highly evolved terpenoid recognition architecture of phloroglucinols poses a consideration of reversible binding as a potential confounding factor in lysine reactivity within our chemical proteomics experiments. While we presently postulate that the inhibition of lysine reactivity is mediated predominantly through covalent modification by phloroglucinol meroterpenoids, it is plausible that reversible binding by these natural products could competitively interfere with probe–protein interactions. Such an occurrence, if substantiated, would extend the applicability of lysine reactivity profiling by enabling the identification of reversible meroterpenoid interactions that competitively perturb probe labeling of lysine residues within druggable protein pockets. In conclusion, our findings underscore the broad potential of phloroglucinol meroterpenoids for targeting functional lysines in the human proteome and vastly expand the toolbox available for studying lysine modifications.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NIH/NIGMS 1R35GM143027-01 (M.E.A.), Adam and Rachel Broder Fund for Cancer Research (M.E.A.), NIH/NIGMS T32GM138826 (A.K.B., C.E.G.), NSF/GRFP DGE-2139899 (A.K.B.), and NIH/SIG 1S10 OD017992-01 (S.Z.) for the Orbitrap Fusion mass spectrometer. We thank the Proteomic and Metabolomics Facility of Cornell University for generating the mass spectrometry data and the funding support from HHMI Transformative Technology 2019 program for the Orbitrap Eclipse system.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c10741.
Extended Data Figures S1–S6; supplemental discussion; biological protocols and synthetic procedures; compound characterization, including Supplementary Figures S1–S14 and Tables S1–S6; and additional references. (PDF)
Supplementary Dataset, including mass-spectrometry-based chemoproteomics datasets; untargeted metabolomics datasets; untargeted lipidomics datasets; and metanalysis (XLSX)
The authors declare the following competing financial interest(s): M.E.A. serves as a scientific consultant for AbbVie and Scorpion Therapeutics. Cornell University has filed provisional patent applications related to this work.
The raw mass spectrometry chemoproteomics data have been deposited to the ProteomeXchange Consortium through the PRIDE118 partner repository with the dataset identifier PXD045370.
Contributor Information
Amy K. Bracken, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States
Colby E. Gekko, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States
Nina O. Suss, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States
Emma E. Lueders, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States
Qi Cui, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States.
Qin Fu, Proteomics and Metabolomics Facility, Cornell University, Ithaca, New York 14853, United States.
Andy C. W. Lui, Proteomics and Metabolomics Facility, Cornell University, Ithaca, New York 14853, United States
Elizabeth T. Anderson, Proteomics and Metabolomics Facility, Cornell University, Ithaca, New York 14853, United States
Sheng Zhang, Proteomics and Metabolomics Facility, Cornell University, Ithaca, New York 14853, United States.
Mikail E. Abbasov, Department of Chemistry and Chemical Biology and Proteomics and Metabolomics Facility, Cornell University, Ithaca, New York 14853, United States
REFERENCES
- (1).Gerry CJ; Schreiber SL Chemical probes and drug leads from advances in synthetic planning and methodology. Nat. Rev. Drug Discovery 2018, 17 (5), 333–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hopkins AL; Groom CR The druggable genome. Nat. Rev. Drug Discovery 2002, 1 (9), 727–730. [DOI] [PubMed] [Google Scholar]
- (3).Macarron R; Banks MN; Bojanic D; Burns DJ; Cirovic DA; Garyantes T; Green DVS; Hertzberg RP; Janzen WP; Paslay JW; et al. Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discovery 2011, 10 (3), 188–195. [DOI] [PubMed] [Google Scholar]
- (4).Boike L; Henning NJ; Nomura DK Advances in covalent drug discovery. Nat. Rev. Drug Discovery 2022, 21 (12), 881–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Parker CG; Galmozzi A; Wang Y; Correia BE; Sasaki K; Joslyn CM; Kim AS; Cavallaro CL; Lawrence RM; Johnson SR; et al. Ligand and target discovery by fragment-based screening in human cells. Cell 2017, 168 (3), 527–541.e529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mortenson DE; Brighty GJ; Plate L; Bare G; Chen W; Li S; Wang H; Cravatt BF; Forli S; Powers ET; et al. Inverse drug discovery” strategy to identify proteins that are targeted by latent electrophiles as exemplified by aryl fluorosulfates. J. Am. Chem. Soc 2018, 140 (1), 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Johnson DS; Weerapana E; Cravatt BF Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med. Chem 2010, 2 (6), 949–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bachovchin DA; Cravatt BF The pharmacological landscape and therapeutic potential of serine hydrolases. Nat. Rev. Drug Discovery 2012, 11 (1), 52–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kato D; Boatright KM; Berger AB; Nazif T; Blum G; Ryan C; Chehade KAH; Salvesen GS; Bogyo M Activity-based probes that target diverse cysteine protease families. Nat. Chem. Biol 2005, 1 (1), 33–38. [DOI] [PubMed] [Google Scholar]
- (10).Backus KM; Correia BE; Lum KM; Forli S; Horning BD; Gonzalez-Paez GE; Chatterjee S; Lanning BR; Teijaro JR; Olson AJ; et al. Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534 (7608), 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Vinogradova EV; Zhang X; Remillard D; Lazar DC; Suciu RM; Wang Y; Bianco G; Yamashita Y; Crowley VM; Schafroth MA; et al. An activity-guided map of electrophile-cysteine interactions in primary human T cells. Cell 2020, 182 (4), 1009–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hahm HS; Toroitich EK; Borne AL; Brulet JW; Libby AH; Yuan K; Ware TB; McCloud RL; Ciancone AM; Hsu KL Global targeting of functional tyrosines using sulfur-triazole exchange chemistry. Nat. Chem. Biol 2020, 16 (2), 150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chen Y; Craven GB; Kamber RA; Cuesta A; Zhersh S; Moroz YS; Bassik MC; Taunton J Direct mapping of ligandable tyrosines and lysines in cells with chiral sulfonyl fluoride probes. Nat. Chem 2023, 15, 1616. [DOI] [PubMed] [Google Scholar]
- (14).Zhang ZY; Morstein J; Ecker AK; Guiley KZ; Shokat KM Chemoselective covalent modification of K-Ras(G12R) with a small molecule electrophile. J. Am. Chem. Soc 2022, 144 (35), 15916–15921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhang ZY; Guiley KZ; Shokat KM Chemical acylation of an acquired serine suppresses oncogenic signaling of K-Ras(G12S). Nat. Chem. Biol 2022, 18 (11), 1177–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hacker SM; Backus KM; Lazear MR; Forli S; Correia BE; Cravatt BF Global profiling of lysine reactivity and ligandability in the human proteome. Nat. Chem 2017, 9 (12), 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Abbasov ME; Kavanagh ME; Ichu TA; Lazear MR; Tao YF; Crowley VM; an Ende CW; Hacker SM; Ho J; Dix MM; et al. A proteome-wide atlas of lysine-reactive chemistry. Nat. Chem 2021, 13 (11), 1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Schaefer D; Cheng X Recent advances in covalent drug discovery. Pharmaceuticals (Basel) 2023, 16 (5), 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Gampe C; Verma VA Curse or cure? A perspective on the developability of aldehydes as active pharmaceutical ingredients. J. Med. Chem 2020, 63 (23), 14357–14381. [DOI] [PubMed] [Google Scholar]
- (20).Chigbu DI; Coyne AM Update and clinical utility of alcaftadine ophthalmic solution 0.25% in the treatment of allergic conjunctivitis. Clin. Ophthalmol 2015, 9, 1215–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Vichinsky E; Hoppe CC; Ataga KI; Ware RE; Nduba V; El-Beshlawy A; Hassab H; Achebe MM; Alkindi S; Brown RC; et al. A phase 3 randomized trial of voxelotor in sickle cell disease. New Engl. J. Med 2019, 381 (6), 509–519. [DOI] [PubMed] [Google Scholar]
- (22).Metcalf B; Chuang CH; Dufu K; Patel MP; Silva-Garcia A; Johnson C; Lu Q; Partridge JR; Patskovska L; Patskovsky Y; et al. Discovery of GBT440, an orally bioavailable R-state stabilizer of sickle cell hemoglobin. ACS Med. Chem. Lett 2017, 8 (3), 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Atanasov AG; Zotchev SB; Dirsch VM; Supuran CT Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discovery 2021, 20 (3), 200–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Nazir M; Saleem M; Tousif MI; Anwar MA; Surup F; Ali I; Wang DJ; Mamadalieva NZ; Alshammari E; Ashour ML Meroterpenoids: A comprehensive update insight on structural diversity and biology. Biomolecules 2021, 11 (7), 957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chen T; Ruan D.-d.; Zhang J.-h.; Wang H.-l.; Wu M; Wu Q.-y.; Zhang Y.-p.; Wu J.-b.; Ye Y; Chou G.-x.; et al. Eucarbwenstols A–H, eight novel compounds from Eucalyptus robusta prevents MPC-5 injury via ROS modulation and regulation of mitochondrial membrane potential. Bioorg. Chem 2022, 129, No. 106159. [DOI] [PubMed] [Google Scholar]
- (26).Li JS; Liu YN; Li JY; Lei C; Hou AJ Acylphloroglucinol-monoterpene meroterpenoids from Eucalyptus tereticornis and their inhibitory activity against ATP citrate lyase. Phytochemistry 2023, 207, 113565. [DOI] [PubMed] [Google Scholar]
- (27).Tang GH; Dong Z; Guo YQ; Cheng ZB; Zhou CJ; Yin S Psiguajadials A-K: Unusual psidium meroterpenoids as phosphodiesterase-4 inhibitors from the leaves of Psidium guajava. Sci. Rep 2017, 7 (1), No. 1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yu M-Y; Liu S-N; Luo EE; Jin Q; Liu H; Liu H-Y; Luo X-D; Qin X-J Phloroglucinols with hAChE and α-glucosidase inhibitory activities from the leaves of tropic Rhodomyrtus tomentosa. Phytochemistry 2022, 203, No. 113394. [DOI] [PubMed] [Google Scholar]
- (29).Cui C; Dwyer BG; Liu C; Abegg D; Cai Z-J; Hoch DG; Yin X; Qiu N; Liu J-Q; Adibekian A; et al. Total synthesis and target identification of the curcusone diterpenes. J. Am. Chem. Soc 2021, 143 (11), 4379–4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Davis DC; Hoch DG; Wu L; Abegg D; Martin BS; Zhang Z-Y; Adibekian A; Dai M Total synthesis, biological evaluation, and target identification of rare Abies sesquiterpenoids. J. Am. Chem. Soc 2018, 140 (50), 17465–17473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Isobe Y; Okumura M; McGregor LM; Brittain SM; Jones MD; Liang X; White R; Forrester W; McKenna JM; Tallarico JA; et al. Manumycin polyketides act as molecular glues between UBR7 and P53. Nat. Chem. Biol 2020, 16 (11), 1189–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Belcher BP; Machicao PA; Tong B; Ho E; Friedli J; So B; Bui H; Isobe Y; Maimone TJ; Nomura DK Chemoproteomic profiling reveals that anti-cancer natural product dankastatin B covalently targets mitochondrial VDAC3. bioRxiv 2023, DOI: 10.1101/2023.02.11.528139. [DOI] [PubMed] [Google Scholar]
- (33).Gowans FA; Thach DQ; Wang Y; Altamirano Poblano BE; Dovala D; Tallarico JA; McKenna JM; Schirle M; Maimone TJ; Nomura DK Ophiobolin A covalently targets complex IV leading to mitochondrial metabolic collapse in cancer cells. bioRxiv 2023, DOI: 10.1101/2023.03.09.531918. [DOI] [PubMed] [Google Scholar]
- (34).Tang J; Li W; Chiu TY; Martínez-Peña F; Luo Z; Chong CT; Wei Q; Gazaniga N; West TJ; See YY; Lairson LL; Parker CG; Baran PS Synthesis of portimines reveals the basis of their anti-cancer activity. Nature 2023, 622, 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kong Au T; Chow Leung P Identification of the binding and inhibition sites in the calmodulin molecule for ophiobolin A by site-directed mutagenesis. Plant Physiol. 1998, 118 (3), 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wymann MP; Bulgarelli-Leva G; Zvelebil MJ; Pirola L; Vanhaesebroeck B; Waterfield MD; Panayotou G Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol. Cell. Biol 1996, 16 (4), 1722–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hong DS; Bowles DW; Falchook GS; Messersmith WA; George GC; O’Bryant CL; Vo ACH; Klucher K; Herbst RS; Eckhardt SG; et al. A multicenter phase I trial of PX-866, an oral irreversible phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors. Clin. Cancer Res 2012, 18 (15), 4173–4182. [DOI] [PubMed] [Google Scholar]
- (38).Pitz MW; Eisenhauer EA; MacNeil MV; Thiessen B; Easaw JC; Macdonald DR; Eisenstat DD; Kakumanu AS; Salim M; Chalchal H; et al. Phase II study of PX-866 in recurrent glioblastoma. Neuro. Oncol 2015, 17 (9), 1270–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bowles DW; Kochenderfer M; Cohn A; Sideris L; Nguyen N; Cline-Burkhardt V; Schnadig I; Choi M; Nabell L; Chaudhry A; et al. A randomized, phase II trial of cetuximab with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with metastatic colorectal carcinoma. Clin. Colorectal Cancer 2016, 15 (4), 337–344. [DOI] [PubMed] [Google Scholar]
- (40).Jimeno A; Bauman JE; Weissman C; Adkins D; Schnadig I; Beauregard P; Bowles DW; Spira A; Levy B; Seetharamu N; et al. A randomized, phase II trial of docetaxel with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Oral. Oncol 2015, 51 (4), 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Levy B; Spira A; Becker D; Evans T; Schnadig I; Camidge DR; Bauman JE; Hausman D; Walker L; Nemunaitis J; et al. A randomized, phase II trial of docetaxel with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic non-small-cell lung cancer. J. Thorac. Oncol 2014, 9 (7), 1031–1035. [DOI] [PubMed] [Google Scholar]
- (42).Hotte SJ; Chi KN; Joshua AM; Tu D; Macfarlane RJ; Gregg RW; Ruether JD; Basappa NS; Finch D; Salim M; et al. A phase II study of PX-866 in patients with recurrent or metastatic castration-resistant prostate cancer: canadian cancer trials group study IND205. Clin. Genitourin. Cancer 2019, 17 (3), 201–208. [DOI] [PubMed] [Google Scholar]
- (43).Yam C; Xu X; Davies MA; Gimotty PA; Morrissette JJD; Tetzlaff MT; Wani KM; Liu S; Deng W; Buckley M; et al. A multicenter phase I study evaluating dual PI3K and BRAF inhibition with PX-866 and vemurafenib in patients with advanced BRAF V600-mutant solid tumors. Clin. Cancer Res 2018, 24 (1), 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Feng Z; Jia S; Chen H; You L Modulation of imine chemistry with intramolecular hydrogen bonding: Effects from ortho-OH to NH. Tetrahedron 2020, 76 (17), No. 131128. [Google Scholar]
- (45).Mahrous EA; Al-Abd AM; Salama MM; Fathy MM; Soliman FM; Saber FR Cattleianal and cattleianone: Two new meroterpenoids from Psidium cattleianum leaves and their selective antiproliferative action against human carcinoma cells. Molecules 2021, 26 (10), 2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Boland DJ; Brophy JJ; Fookest CJR Jensenone, a ketone from Eucalyptus-jensenii. Phytochemistry 1992, 31 (6), 2178–2179. [Google Scholar]
- (47).Crow WD; Osawa T; Paton DM; Willing RR Structure of grandinol – novel root inhibitor from Eucalyptus-grandis. Tetrahedron Lett. 1977, 18 (12), 1073–1074. [Google Scholar]
- (48).Takasaki M; Konoshima T; Kozuka M; Haruna M; Ito K; Shingu T Structures of Euglobal-G1, Euglobal-G2, Euglobal-G3, Euglobal-G4, and Euglobal-G5 from Eucalyptus-Grandis. Chem. Pharm. Bull 1994, 42 (12), 2591–2597. [Google Scholar]
- (49).Jian YQ; Huang XJ; Zhang DM; Jiang RW; Chen MF; Zhao BX; Wang Y; Ye WC Guapsidial A and guadials B and C: Three new meroterpenoids with unusual skeletons from the leaves of psidium guajava. Chemistry 2015, 21 (25), 9022–9027. [DOI] [PubMed] [Google Scholar]
- (50).Christenhusz MJM; Byng JW The number of known plants species in the world and its annual increase. Phytotaxa 2016, 261 (3), 201–217. [Google Scholar]
- (51).Shao M; Wang Y; Jian Y-Q; Huang X-J; Zhang D-M; Tang Q-F; Jiang R-W; Sun X-G; Lv Z-P; Zhang X-Q; et al. Guadial A and psiguadials C and D, three unusual meroterpenoids from Psidium guajava. Org. Lett 2012, 14 (20), 5262–5265. [DOI] [PubMed] [Google Scholar]
- (52).Newton CG; Tran DN; Wodrich MD; Cramer N One-step multigram-scale biomimetic synthesis of psiguadial B. Angew. Chem., Int. Ed 2017, 56 (44), 13776–13780. [DOI] [PubMed] [Google Scholar]
- (53).Dethe DH; B VK; Maiti R Biomimetic total syntheses of chromane meroterpenoids, guadials B and C, guapsidial A and psiguajadial D. Org. Biomol. Chem 2018, 16 (26), 4793–4796. [DOI] [PubMed] [Google Scholar]
- (54).Vieira de Castro T; Yahiaoui O; Peralta RA; Fallon T; Lee V; George JH Biomimetic synthesis enables the structure revision of littordials E and F and drychampone B. Org. Lett 2020, 22 (20), 8161–8166. [DOI] [PubMed] [Google Scholar]
- (55).Lawrence AL; Adlington RM; Baldwin JE; Lee V; Kershaw JA; Thompson AL A short biomimetic synthesis of the meroterpenoids guajadial and psidial A. Org. Lett 2010, 12 (8), 1676–1679. [DOI] [PubMed] [Google Scholar]
- (56).Yoshida S; Ujiie M; Takahashi N; Bolte ML; Crow WD Synthesis of deuterated grandinol. Agric. Biol. Chem 1985, 49 (1), 211–213. [Google Scholar]
- (57).Chiba K; Arakawa T; Tada M Synthesis of euglobal-G3 and -G4. Chem. Commun 1996, 15, 1763–1764. [Google Scholar]
- (58).Bharate SB; Chauthe SK; Bhutani KK; Singh IP An efficient two-step synthesis of jensenone isolated from Eucalyptus jensenii: Synthesis of analogues and evaluation as antioxidants. Aust. J. Chem 2005, 58 (7), 551–555. [Google Scholar]
- (59).Zhao Z; Li Z; Qiao W; Wang G; Cheng L An efficient method for the alkylation of α-methylnaphthalene with various alkylating agents using methanesulfonic acid as novel catalysts and solvents. Catal. Lett 2005, 102 (3), 219–222. [Google Scholar]
- (60).Mehr SHM; Depmeier H; Fukuyama K; Maghami M; MacLachlan MJ Formylation of phenols using formamidine acetate. Org. Biomol. Chem 2017, 15 (3), 581–583. [DOI] [PubMed] [Google Scholar]
- (61).Bharate SB; Khan SI; Yunus NA; Chauthe SK; Jacob MR; Tekwani BL; Khan IA; Singh IP Antiprotozoal and antimicrobial activities of O-alkylated and formylated acylphloroglucinols. Bioorg. Med. Chem 2007, 15 (1), 87–96. [DOI] [PubMed] [Google Scholar]
- (62).Ghisalberti EL Bioactive acylphloroglucinol derivatives from Eucalyptus species. Phytochemistry 1996, 41 (1), 7–22. [DOI] [PubMed] [Google Scholar]
- (63).Bharate SB; Bhutani KK; Khan SI; Tekwani BL; Jacob MR; Khan IA; Singh IP Biomimetic synthesis, antimicrobial, antileishmanial and antimalarial activities of euglobals and their analogues. Bioorg. Med. Chem 2006, 14 (6), 1750–1760. [DOI] [PubMed] [Google Scholar]
- (64).Wang Y; Dix MM; Bianco G; Remsberg JR; Lee H-Y; Kalocsay M; Gygi SP; Forli S; Vite G; Lawrence RM; et al. Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs. Nat. Chem 2019, 11 (12), 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Tao Y; Remillard D; Vinogradova EV; Yokoyama M; Banchenko S; Schwefel D; Melillo B; Schreiber SL; Zhang X; Cravatt BF Targeted protein degradation by electrophilic protacs that stereoselectively and site-specifically engage DCAF1. J. Am. Chem. Soc 2022, 144 (40), 18688–18699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Hübner M; Rissom B; Fitjer L Conformation and dynamics of (−)-β-caryophyllene. Helv. Chim. Acta 1997, 80 (6), 1972–1982. [Google Scholar]
- (67).Martínez CHR; Dardonville C Rapid determination of ionization constants (pKa) by UV spectroscopy using 96-well microtiter plates. ACS Med. Chem. Lett 2013, 4 (1), 142–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Hasegawa R; Saito K; Takaoka T; Ishikita H pKa of ubiquinone, menaquinone, phylloquinone, plastoquinone, and rhodoquinone in aqueous solution. Photosynth. Res 2017, 133 (1), 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Reja RM; Wang W; Lyu Y; Haeffner F; Gao J Lysine-targeting reversible covalent inhibitors with long residence time. J. Am. Chem.. Soc 2022, 144 (3), 1152–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Serafimova IM; Pufall MA; Krishnan S; Duda K; Cohen MS; Maglathlin RL; McFarland JM; Miller RM; Frödin M; Taunton J Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol 2012, 8 (5), 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Cal PMSD; Vicente JB; Pires E; Coelho AV; Veiros L. s. F.; Cordeiro C; Gois PMP Iminoboronates: A new strategy for reversible protein modification. J. Am. Chem. Soc 2012, 134 (24), 10299–10305. [DOI] [PubMed] [Google Scholar]
- (72).Quach D; Tang G; Anantharajan J; Baburajendran N; Poulsen A; Wee JLK; Retna P; Li R; Liu B; Tee DHY; et al. Strategic design of catalytic lysine-targeting reversible covalent BCR-ABL inhibitors. Angew. Chem., Int. Ed 2021, 60 (31), 17131–17137. [DOI] [PubMed] [Google Scholar]
- (73).António JPM; Gonçalves LM; Guedes RC; Moreira R; Gois PMP Diazaborines as new inhibitors of human neutrophil elastase. ACS Omega 2018, 3 (7), 7418–7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Bar-Peled L; Kemper EK; Suciu RM; Vinogradova EV; Backus KM; Horning BD; Paul TA; Ichu TA; Svensson RU; Olucha J; et al. Chemical proteomics identifies druggable vulnerabilities in a genetically defined cancer. Cell 2017, 171 (3), 696–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Kemper EK; Zhang Y; Dix MM; Cravatt BF Global profiling of phosphorylation-dependent changes in cysteine reactivity. Nat. Methods 2022, 19 (3), 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Chavez KJ; Garimella SV; Lipkowitz S Triple negative breast cancer cell lines: One tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2011, 32, 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Elstrodt F; Hollestelle A; Nagel JHA; Gorin M; Wasielewski M; van den Ouweland A; Merajver SD; Ethier SP; Schutte M BRCA1 mutation analysis of 41 human breast cancer cell lines reveals three new deleterious mutants. Cancer Res. 2006, 66 (1), 41–45. [DOI] [PubMed] [Google Scholar]
- (78).Fischer H; Gottschlich R; Seelig A Blood-brain barrier permeation: molecular parameters governing passive diffusion. J. Membr. Biol 1998, 165 (3), 201–211. [DOI] [PubMed] [Google Scholar]
- (79).Schweppe DK; Eng JK; Yu Q; Bailey D; Rad R; Navarrete-Perea J; Huttlin EL; Erickson BK; Paulo JA; Gygi SP Full-featured, real-time database searching platform enables fast and accurate multiplexed quantitative proteomics. J. Proteome Res 2020, 19 (5), 2026–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).O’Shea JP; Chou MF; Quader SA; Ryan JK; Church GM; Schwartz D pLogo: a probabilistic approach to visualizing sequence motifs. Nat. Methods 2013, 10 (12), 1211–1212. [DOI] [PubMed] [Google Scholar]
- (81).Landrum MJ; Lee JM; Riley GR; Jang W; Rubinstein WS; Church DM; Maglott DR ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42 (D1), D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Collins RL; Brand H; Karczewski KJ; Zhao X; Alföldi J; Francioli LC; Khera AV; Lowther C; Gauthier LD; Wang H; et al. A structural variation reference for medical and population genetics. Nature 2020, 581 (7809), 444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Rehm HL; Berg JS; Brooks LD; Bustamante CD; Evans JP; Landrum MJ; Ledbetter DH; Maglott DR; Martin CL; Nussbaum RL; et al. ClinGen – The clinical genome resource. New Engl. J. Med 2015, 372 (23), 2235–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Piñero J; Bravo A; Queralt-Rosinach N; Gutiérrez-Sacristán A; Deu-Pons J; Centeno E; García-García J; Sanz F; Furlong LI DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45 (D1), D833–D839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Hong V; Steinmetz NF; Manchester M; Finn MG Labeling live cells by copper-catalyzed alkyne–azide click chemistry. Bioconjugate Chem. 2010, 21 (10), 1912–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Uttamapinant C; Sanchez MI; Liu DS; Yao JZ; White KA; Grecian S; Clark S; Gee KR; Ting AY Site-specific protein labeling using PRIME and chelation-assisted click chemistry. Nat. Protocols 2013, 8 (8), 1620–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Melser S; Lavie J; Bénard G Mitochondrial degradation and energy metabolism. Biochim. Biophys. Acta 2015, 1853, 2812–2821. [DOI] [PubMed] [Google Scholar]
- (88).Shiba-Fukushima K; Inoshita T; Hattori N; Imai Y Lysine 63-linked polyubiquitination is dispensable for Parkin-mediated mitophagy. J. Biol. Chem 2014, 289 (48), 33131–33136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Jun S; Kim SW; Lim JY; Park SJ ABHD12 knockdown suppresses breast cancer cell proliferation, migration and invasion. Anticancer Res. 2020, 40 (5), 2601–2611. [DOI] [PubMed] [Google Scholar]
- (90).Aljohani AI; Toss MS; Kurozumi S; Joseph C; Aleskandarany MA; Miligy IM; Ansari RE; Mongan NP; Ellis IO; Green AR; et al. The prognostic significance of wild-type isocitrate dehydrogenase 2 (IDH2) in breast cancer. Breast Cancer Res. Treat 2020, 179 (1), 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Inaishi T; Shibata M; Ichikawa T; Kanda M; Hayashi M; Soeda I; Takeuchi D; Takano Y; Tsunoda N; Kodera Y; et al. Platelet isoform of phosphofructokinase accelerates malignant features in breast cancer. Oncol. Rep 2021, 47 (1), 8220. [DOI] [PubMed] [Google Scholar]
- (92).Ahsan H; Halpern J; Kibriya MG; Pierce BL; Tong L; Gamazon E; McGuire V; Felberg A; Shi JX; Jasmine F; et al. A genome-wide association study of early-onset breast cancer identifies PFKM as a novel breast cancer gene and supports a common genetic spectrum for breast cancer at any age. Cancer Epidemiol. Biomarkers Prev 2014, 23 (4), 658–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Blankman JL; Long JZ; Trauger SA; Siuzdak G; Cravatt BF ABHD12 controls brain lysophosphatidylserine pathways that are deregulated in a murine model of the neurodegenerative disease PHARC. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (4), 1500–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Joshi A; Shaikh M; Singh S; Rajendran A; Mhetre A; Kamat SS Biochemical characterization of the PHARC-associated serine hydrolase ABHD12 reveals its preference for very-long-chain lipids. J. Biol. Chem 2018, 293 (44), 16953–16963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Ogasawara D; Ichu TA; Vartabedian VF; Benthuysen J; Jing H; Reed A; Ulanovskaya OA; Hulce JJ; Roberts A; Brown S; et al. Selective blockade of the lyso-PS lipase ABHD12 stimulates immune responses in vivo. Nat. Chem. Biol 2018, 14 (12), 1099–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Ogasawara D; Ichu TA; Jing H; Hulce JJ; Reed A; Ulanovskaya OA; Cravatt BF Discovery and optimization of selective and in vivo active inhibitors of the lysophosphatidylserine lipase α/β-hydrolase domain-containing 12 (ABHD12). J. Med. Chem 2019, 62 (3), 1643–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Tunyasuvunakool K; Adler J; Wu Z; Green T; Zielinski M; Žídek A; Bridgland A; Cowie A; Meyer C; Laydon A; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596 (7873), 590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Patricelli MP; Giang DK; Stamp LM; Burbaum JJ Direct visualization of serine hydrolase activities in complex proteomes using fluorescent active site-directed probes. Proteomics 2001, 1 (9), 1067–1071. [DOI] [PubMed] [Google Scholar]
- (99).Wu H; Yao H; He C; Jia Y; Zhu Z; Xu S; Li D; Xu J Molecular glues modulate protein functions by inducing protein aggregation: A promising therapeutic strategy of small molecules for disease treatment. Acta Pharm. Sin. B 2022, 12 (9), 3548–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Jo SH; Son MK; Koh HJ; Lee SM; Song IH; Kim YO; Lee YS; Jeong KS; Kim WB; Park JW; et al. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP(+)-dependent isocitrate dehydrogenase. J. Biol. Chem 2001, 276 (19), 16168–16176. [DOI] [PubMed] [Google Scholar]
- (101).Han SJ; Jang HS; Noh MR; Kim J; Kong MJ; Kim JI; Park JW; Park KM Mitochondrial NADP(+)-dependent isocitrate dehydrogenase deficiency exacerbates mitochondrial and cell damage after kidney ischemia-reperfusion injury. J. Am. Soc. Nephrol 2017, 28 (4), 1200–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Chen JY; Lai YS; Tsai HJ; Kuo CC; Yen BLJ; Yeh SP; Sun HS; Hung WC The oncometabolite R-2-hydroxyglutarate activates NF-kappa B-dependent tumor-promoting stromal niche for acute myeloid leukemia cells. Sci. Rep 2016, 6, e32428 DOI: 10.1038/srep32428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Yen K; Travins J; Wang F; David MD; Artin E; Straley K; Padyana A; Gross S; DeLaBarre B; Tobin E; et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discovery 2017, 7 (5), 478–493. [DOI] [PubMed] [Google Scholar]
- (104).Zhuang XM; Pei HZ; Li TW; Huang JB; Guo Y; Zhao YM; Yang M; Zhang DY; Chang ZG; Zhang Q; et al. The molecular mechanisms of resistance to IDH inhibitors in acute myeloid leukemia. Front. Oncol 2022, 12, No. 931462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Dang L; White DW; Gross S; Bennett BD; Bittinger MA; Driggers EM; Fantin VR; Jang HG; Jin S; Keenan MC; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462 (7274), 739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Zou X; Zhu Y; Park SH; Liu G; O’Brien J; Jiang H; Gius D SIRT3-mediated dimerization of IDH2 directs cancer cell metabolism and tumor growth. Cancer Res. 2017, 77 (15), 3990–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Yu W; Dittenhafer-Reed KE; Denu JM SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem 2012, 287 (17), 14078–14086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Chen D; Xia S; Zhang R; Li Y; Famulare CA; Fan H; Wu R; Wang M; Zhu AC; Elf SE; et al. Lysine acetylation restricts mutant IDH2 activity to optimize transformation in AML cells. Mol. Cell 2021, 81 (18), 3833–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Schöneberg T; Kloos M; Brüser A; Kirchberger J; Sträter N Structure and allosteric regulation of eukaryotic 6-phosphofructokinases. Biol. Chem 2013, 394 (8), 977–993. [DOI] [PubMed] [Google Scholar]
- (110).Fernandes PM; Kinkead J; McNae I; Michels PAM; Walkinshaw MD Biochemical and transcript level differences between the three human phosphofructokinases show optimization of each isoform for specific metabolic niches. Biochem. J 2020, 477 (22), 4425–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Mor I; Cheung EC; Vousden KH Control of glycolysis through regulation of PFK1: Old friends and recent additions. Cold Spring Harb. Symp. Quant. Biol 2011, 76, 211–216. [DOI] [PubMed] [Google Scholar]
- (112).Zhou K; Yao Y-L; He Z-C; Chen C; Zhang X-N; Yang K-D; Liu Y-Q; Liu Q; Fu W-J; Chen Y-P; et al. VDAC2 interacts with PFKP to regulate glucose metabolism and phenotypic reprogramming of glioma stem cells. Cell Death Dis. 2018, 9 (10), No. 988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).To T-L; Cuadros AM; Shah H; Hung WHW; Li Y; Kim SH; Rubin DHF; Boe RH; Rath S; Eaton JK; et al. A compendium of genetic modifiers of mitochondrial dysfunction reveals intra-organelle buffering. Cell 2019, 179 (5), 1222–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Yeerken D; Hong R; Wang Y; Gong Y; Liu R; Yang D; Li J; Fan J; Chen J; Zhang W; et al. PFKP is transcriptionally repressed by BRCA1/ZBRK1 and predicts prognosis in breast cancer. PloS One 2020, 15 (5), No. e0233750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Brüser A; Kirchberger J; Kloos M; Sträter N; Schöneberg T Functional linkage of adenine nucleotide binding sites in mammalian muscle 6-phosphofructokinase. J. Biol. Chem 2012, 287 (21), 17546–17553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Amara N; Cooper MP; Voronkova MA; Webb BA; Lynch EM; Kollman JM; Ma T; Yu K; Lai Z; Sangaraju D; et al. Selective activation of PFKL suppresses the phagocytic oxidative burst. Cell 2021, 184 (17), 4480–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Lazear MR; Remsberg JR; Jaeger MG; Rothamel K; Her H.-l.; DeMeester KE; Njomen E; Hogg SJ; Rahman J; Whitby LR; et al. Proteomic discovery of chemical probes that perturb protein complexes in human cells. Mol. Cell 2023, 83 (10), 1725–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Perez-Riverol Y; Csordas A; Bai J; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47 (D1), D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.