

Abstract
Little is known about the extent to which species use homologous regulatory architectures to achieve phenotypic convergence. By characterizing chromatin accessibility and gene expression in developing wing tissues, we compared the regulatory architecture of convergence between a pair of mimetic butterfly species. Although a handful of color pattern genes are known to be involved in their convergence, our data suggest that different mutational paths underlie the integration of these genes into wing pattern development. This is supported by a large fraction of accessible chromatin being exclusive to each species, including the de novo lineage-specific evolution of a modular optix enhancer. These findings may be explained by a high level of developmental drift and evolutionary contingency that occurs during the independent evolution of mimicry.
As species diverge, mutations accumulate, and genes, regulatory elements, or pathways that are tightly regulated during development in one species may not be similarly constrained in the other. These genetic changes can generate different genomic environments that still underlie the same phenotypes, a process called developmental systems drift (1). Cases of convergent evolution allow us to study how natural selection can generate biological similarities in independent lineages despite their different genomic environments (2). This largely unanswered question has implications for understanding the molecular mechanisms that promote biological diversity.
Studying convergent evolution within Heliconius butterflies (3) and other adaptive radiations such as African cichlids (4) has provided insight into the link between natural selection and the genetic variation that has shaped the appearance of diverse morphologies. Recently, owing to technological advances in chromatin profiling, we can study the gene regulatory architecture of these morphological adaptations (5–8). Chromatin remodeling plays a key role in determining cellular identity by exposing cis-regulatory elements (CREs) to transcription factors (TFs) and regulating gene expression, thus presenting an important link between genetic mutations and developmental processes (9). In this work, we used this approach to study the degree of regulatory homology in a case of Müllerian mimicry between two pairs of Heliconius species and determine how mutational differences have affected the evolutionary trajectory toward convergence.
In Heliconius butterflies, divergence and convergence of wing color patterns has largely been assigned to allelic changes at only a few genes with major phenotypic effects (10–13). However, recent studies of accessible chromatin have revealed an intricate regulatory architecture near these genes that modulates their spatiotemporal expression patterns (14–17). Whereas one study revealed that independent modular CREs at the cortex gene control the mimetic yellow hindwing bar between Heliconius erato and Heliconius melpomene (16), a similar study on the optix gene proposed that conserved pleiotropic CREs underlie red color patterns between these comimics (17). Moreover, a third study on WntA suggested that divergent regulatory changes could explain the different melanic wing patterns induced by a CRISPR-Cas9 WntA gene deletion, or knockout (KO), across three pairs of Heliconius mimics (18, 19). Overall, these studies are suggesting a divergent regulation of mimetic wing patterning that has evolved from an ancestral developmental plan (20).
Our work focuses on a pair of comimetic Heliconius species from Panama that diverged ~11.1 million years ago and converged in forewing pattern (H. erato, geographic morphs demophoon and hydara, and H. melpomene, geographic morphs melpomene and rosina) (Fig. 1A). To understand the extent to which convergence in wing color patterns has occurred through a homologous or nonhomologous regulatory architecture, we combined differences in chromatin accessibility and gene expression data with a pangenome reference approach that accounted for genomic deletions and insertions (21). Using this strategy, we (i) investigated the level of chromatin similarities genome-wide, (ii) quantified and characterized differences in chromatin accessibility and gene expression in developing wings and sections of the forewing, and (iii) used CRISPR-Cas9 to validate a previously uncharacterized functional CRE near the red color pattern gene optix that underlies this convergent phenotype exclusive to the H. erato lineage.
Fig. 1. Sampling of chromatin accessibility (ATAC-seq) data and architecture of specific and shared chromatin landscape between H. erato and H. melpomene.
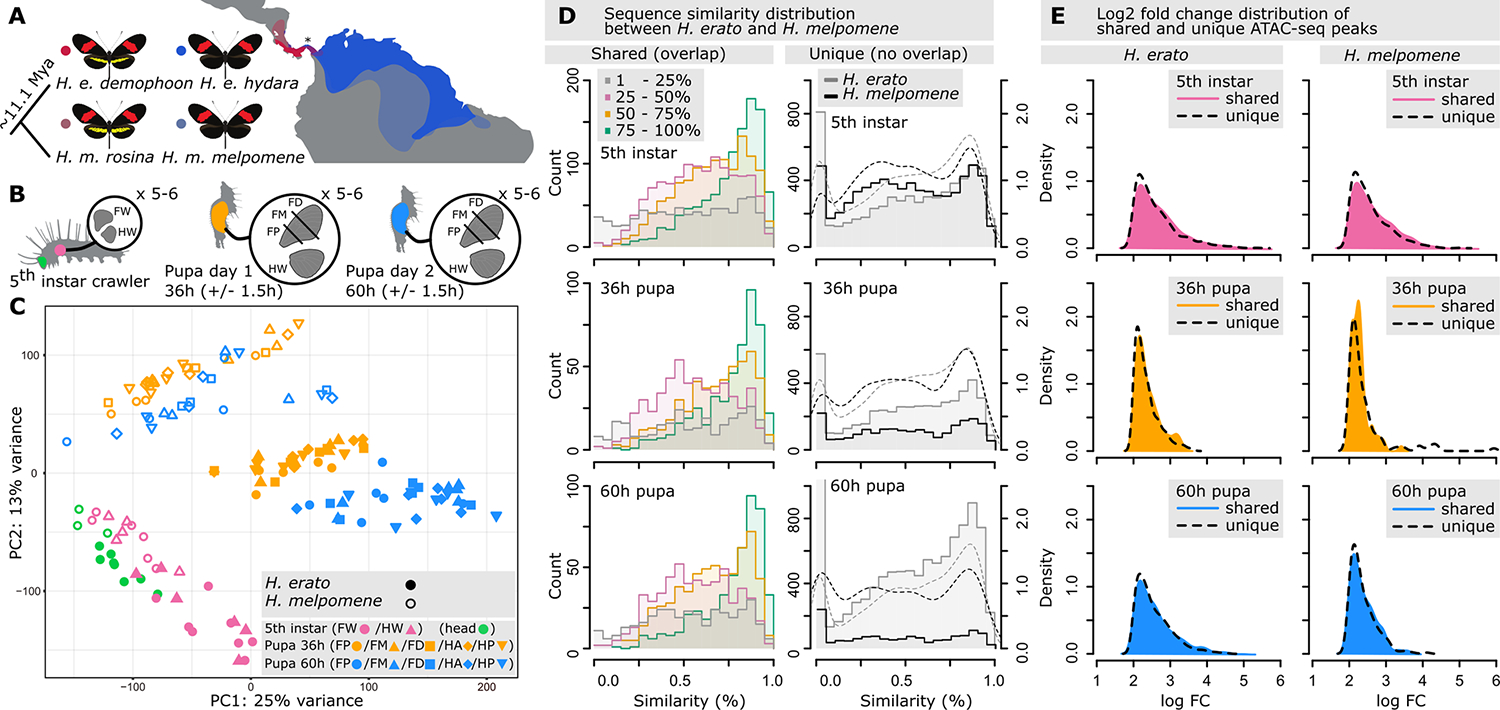
(A) Geographic distribution of red-banded H. erato and H. melpomene postman morphs used in this study. The populations of H. erato demophoon and H. melpomene rosina have a red forewing band and a yellow hindwing bar and admix with, respectively, H. erato hydara and H. melpomene melpomene that lacks the yellow hindwing bar. Samples come from reared morphs of Panama indicated with an asterisk (*). (B) Tissue sampling of fifth-instar head, forewing (FW), and hindwing (HW), and 36-hour pupal (day 1) and 60-hour pupal (day 2) FW sections (FP, FW posterior; FM, FW medial; FD, FW distal) and HW. (C) Principal components analysis (PCA) of ATAC-seq count values for peaks with at least 25% overlap between species. (D) Sequence similarity distribution between H. erato and H. melpomene for shared (left, ≥1-bp overlap, with overlapping ranges investigated at 25% intervals) and specific (right, 0-bp overlap) ATAC-seq peaks. Dashed lines indicate density distributions. (E) Log2-fold changes of shared (colored) and specific (dashed lines) ATAC-seq peaks that were differentially more accessible at a developmental time point in H. erato and H. melpomene.
Differences in chromatin accessibility suggest a divergent regulatory architecture
We quantified the magnitude of genome-wide changes in chromatin accessibility in the two butterfly species as a function of tissue and development (Fig. 1B). As expected, we observed highly dynamic chromatin remodeling over development (22), which represented the strongest predictor of chromatin accessibility within species (Fig. 1C). Out of the 152,897 ATAC-seq (assay for transposase-accessible chromatin with sequencing) peaks identified across the total dataset in H. erato (all tissues and time points), a total of 7.02, 4.51, and 7.92% were differentially more accessible (i.e., had significantly more ATAC-seq read counts), respectively, in fifth-instar larvae and 36- and 60-hour pupae. In H. melpomene, out of a total of 135,296 ATAC-seq peaks, 8.39, 3.08, and 2.55% were differentially more accessible, respectively in fifth-instar larvae and 36- and 60-hour pupae (fig. S1).
To explore the distinctness of the species’ chromatin landscapes, we compared the position and DNA sequence conservation of ATAC-seq peaks between H. erato and H. melpomene using a pangenome assembly. We tested for different overlap [1 base pair (bp) versus 50% reciprocal overlap] and replication criteria (i.e., peak present in at least two samples versus all samples, tissues, or time points) and consistently found a high number of species-specific open chromatin regions (table S1). For example, we found 57% of the total number of ATAC-seq peaks to be species-specific, with 7277 in H. erato and 10,762 in H. melpomene when we used our most conservative analyses (peak present in all samples for a tissue or time point within species and only 1-bp overlap between species) (table S1, panel iv). This level of distinctness was increased to 70.2% when we used a 50% reciprocal overlap between peaks from all samples and developmental time points, with 10,467 in H. erato and 13,952 in H. melpomene. Across all overlap criteria, the lowest proportion of specific ATAC-seq peaks observed was 26.1% in H. erato and 28.3% in H. melpomene (table S1). The distinctness in the chromatin landscape further increased when we accounted for differential accessibility among overlapping peaks (tables S2 and S3). For example, among the total of 33,678 ATAC-seq peaks that were identified as having 50% reciprocal overlap between the two species, 8.1% (2724) to 18.1% (6084) were significantly differentially accessible between the same tissues and time points [table S3, foldchange (FC) > 1, adjusted P < 0.05]. We found such a distinct chromatin architecture between H. erato and H. melpomene to be equally distributed across the 21 chromosomes (fig. S2).
Finally, we observed that ATAC-seq peaks with less overlap between species in the pangenome alignment generally occurred in less-conserved genomic regions (Fig. 1D, left column). We identified that for 11.7% (2347) and 7.9% (758) of the total ATAC-seq peaks identified, the sequence was only present (0% sequence similarity) in H. erato and H. melpomene, respectively, and up to 41.4% (8332) and 46.5% (4479) had less than 50% sequence conservation (Fig. 1F, right column). Specific ATAC-seq peaks (with 0-bp overlap between species) had similar fold changes when compared to shared peaks (Fig. 1E), which suggests that they have similar changes in accessibility (see supplemental text and figs. S3 to S5 for details on foldchange comparisons between species). Overall, these results highlight the existence of a widespread chromatin divergence, which isstrongly driven by genomic sequence evolution.
Dissimilarities in chromatin profiles of developing fore- and hindwings between comimics
To compare the chromatin landscape of developing wings between species, we first studied the differences between fore- and hindwing chromatin in each species-specific genomic background. Our analysis of ATAC-seq peak position and sequence similarity highlighted that highly overlapping peaks can have low sequence similarity (and vice versa) (Fig. 1D). For these analyses, we used a less-restrictive minimum of two samples within-species for the ATAC-seq peak to be retained in the analysis and a 50% reciprocal overlap between the species for the ATAC-seq peak to be considered “shared” (table S1, panel i). These criteria allowed us to also analyze the variable portion of the ATAC-seq signal within-species and enforced both physical overlap of ATAC-seq peaks and sequence similarity between species.
As expected from the shared ontogeny of wings (23), less than 0.5% of ATAC-seq peaks across development had significantly different chromatin accessibility between the fore- and hindwings in each of the two species (Fig. 2A). Out of 2535 ATAC-seq peaks subdivided into 1563 and 972 peaks that were significantly differentially more accessible in one of the two wings in H. erato and H. melpomene, respectively, only 7.2% (183 regions) were considered shared and had similar accessibility patterns in the two comimics. These included peaks near potentially important wing developmental genes such as distal-less (Dll), pangolin (pan), and dachsous (ds) in the forewing and Ultrabithorax (Ubx), aristaless (al), split ends (spen), winged eye (wge), and cubitus interruptus (ci) in the hindwing (Fig. 2A and table S4). Of the remaining peaks with a species-specific fore- or hindwing accessibility pattern, 58.8% (1490) were not identified (distinctly called peak) in the other species even at different time points, and 3.8% (96) were identified in both species but had significantly different accessibility (tables S2 and S3). The 183 shared wing identity peaks had an average sequence similarity of 80.7% (SD = 14.5), whereas the 2352 distinct wing identity peaks had an average sequence similarity of 66.6% (SD = 24.7), with 4.1% (97) being explained by 0% sequence conservation and 30.4% having less than 50% sequence similarity in the alternative species (tables S5 and S6).
Fig. 2. Forewing and hindwing identity observed from gene expression and chromatin landscape.
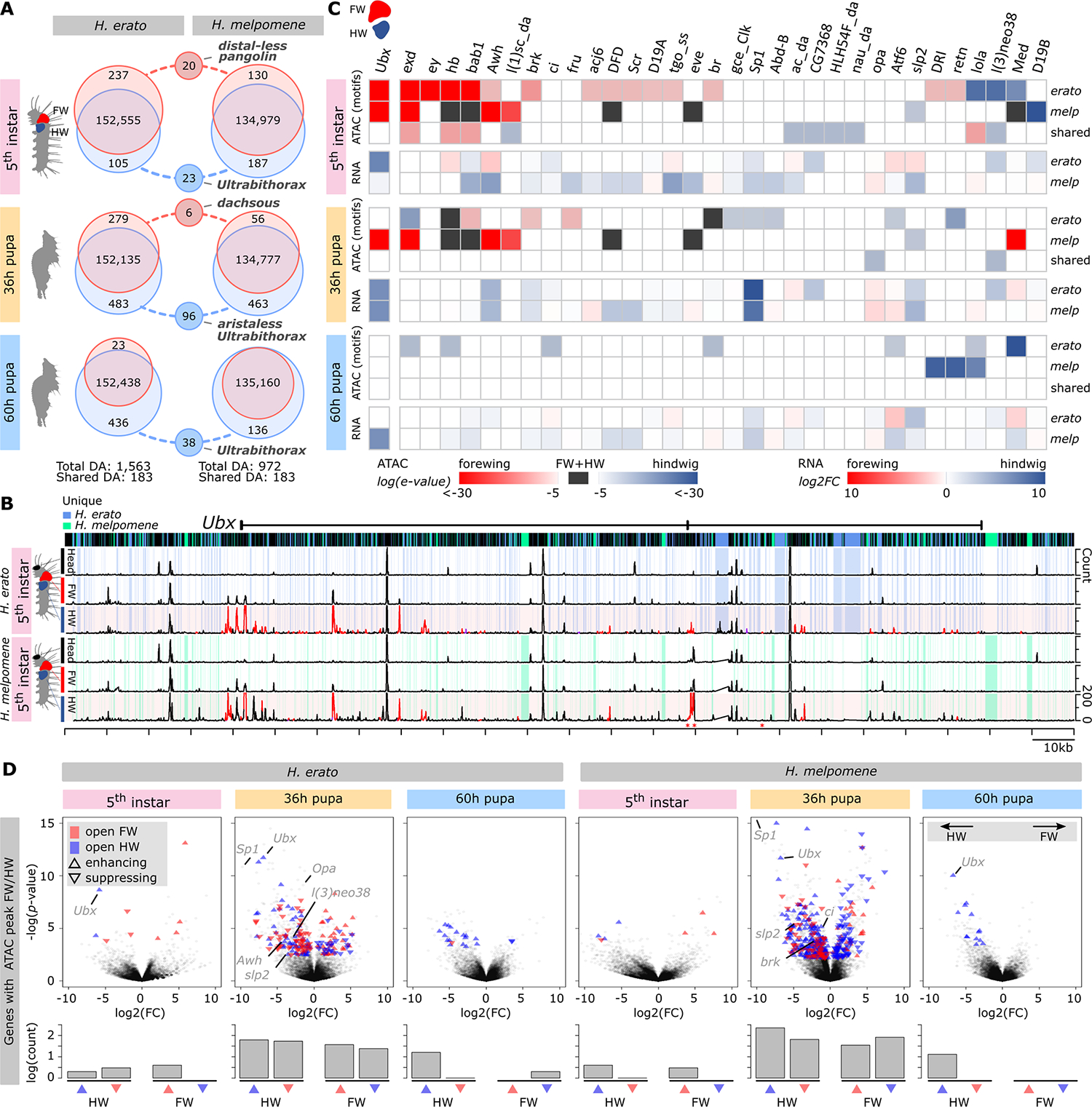
(A) Venn diagrams show the differentially accessible (DA) ATAC-seq peaks between the fore- and hindwings in H. erato and H. melpomene. Circles connected with dashed lines indicate how many of these wing-specific ATAC-seq peaks are shared between the two species (50% reciprocal overlap). (B) ATAC-seq profile near the Ubx gene in fifth-instar caterpillars. Blue and green shading indicate sequence that is specific to H. erato and H. melpomene, respectively. Peaks in red are significantly more accessible in the hindwing compared with forewing near Ubx and indicate the expected conserved homology at this gene. Asterisks (*) indicate peaks that are shared between species but significantly differentially accessible. (C) TF motifs enriched in differentially accessible ATAC-peaks between fore- and hindwing and their RNA expression levels. Log(e-value) indicates the significance level of the enrichment signal, with red and blue indicating higher enrichment in the fore- and hindwing, respectively, and black indicating enrichment in both fore- and hindwing. Log2FC indicates the expression level relative to the alternative wing. (D) Gene expression volcano plots with differentially expressed genes that have a differentially accessible ATAC-seq peak nearby. Red and blue indicate open ATAC-seq peak in fore- or hindwing, respectively. Upward and downward triangles indicate the enhancing or suppressing effect of the ATAC-seq peak. Significantly differentially expressed TFs with significant motif enrichment signal are indicated in gray. The bar plots show the counts of the enhancing and suppressing ATAC-seq peaks in fore- and hindwing.
Of the ATAC-seq peaks that were more accessible in the hindwing, many were concentrated within 100 kb of the Ubx gene [5.56% (82) and 2.04% (20) in H. erato and H. melpomene, respectively], which is known to be a key gene for insect hindwing specification (23, 24) (Fig. 2B and fig. S6). The Ubx gene was in the only genomic region where homologous ATAC-seq peaks were enriched in the hindwing between the species across all developmental time points investigated (fig. S7). Although most peaks had a similar accessibility pattern in both species, we also found 36 species-specific ATAC-seq peaks near Ubx. Sequence conservation was generally high at these chromatin regions (83.9%). Nevertheless, one peak at this genomic region was completely specific to H. erato and three peaks were shared between both species but significantly differentially accessible.
Enrichment analysis of TF-binding motifs in peaks differentially accessible between the fore- and hindwing also showed differences between H. erato and H. melpomene (Fig. 2C). At the fifth-instar stage, we found similarly enriched TF-binding motifs for Ubx, extradenticle (exd), hunchback (hb), bric a brac 1 (bab1), Arrowhead (Awh), and Deformed (DFD) in the forewing and Medea (Med) in the hindwing. Overrepresented TF-binding motifs specific to either H. erato or H. melpomene matched more than 26 additional TF-binding sites (Fig. 2C). The pattern of differentially accessible ATAC-seq peaks was corroborated by similarly highly divergent patterns of differential gene expression between the fore- and hindwings of H. erato and H. melpomene (Fig. 2D), including the TFs with enriched binding motifs. These genes showed patterns of activation or suppression by nearby CREs with a relative distribution that changes between wings, development, and species (Fig. 2D). Our ATAC-seq and gene expression data show conservation of chromatin accessibility at the Ubx locus but also a substantial number of distinct chromatin peaks between the fore- and hindwings of H. erato and H. melpomene. Our results thus suggest that regulatory divergence has evolved between the wings of these comimetic species, which may potentially include some functional changes at the Ubx locus itself.
Low conservation in forewing patterning between comimics
To study the developmental architecture (genes and CREs) of the comimetic red forewing band pattern, we collected and analyzed ATAC-seq and RNA sequencing (RNA-seq) data for forewing sections of 36-hour pupae of both species (Fig. 3). With this approach, we tested different possible combinations of wing patterning (wing section and gradient-like expressed ATAC-seq peaks and genes; Fig. 3A). As for the differences between whole wings, the results from the three forewing sections suggested a distinct architecture of patterning represented by the divergent chromatin landscape and gene expression between the comimics.
Fig. 3. Chromatin accessibility and gene expression in 36-hour pupa forewing sections.
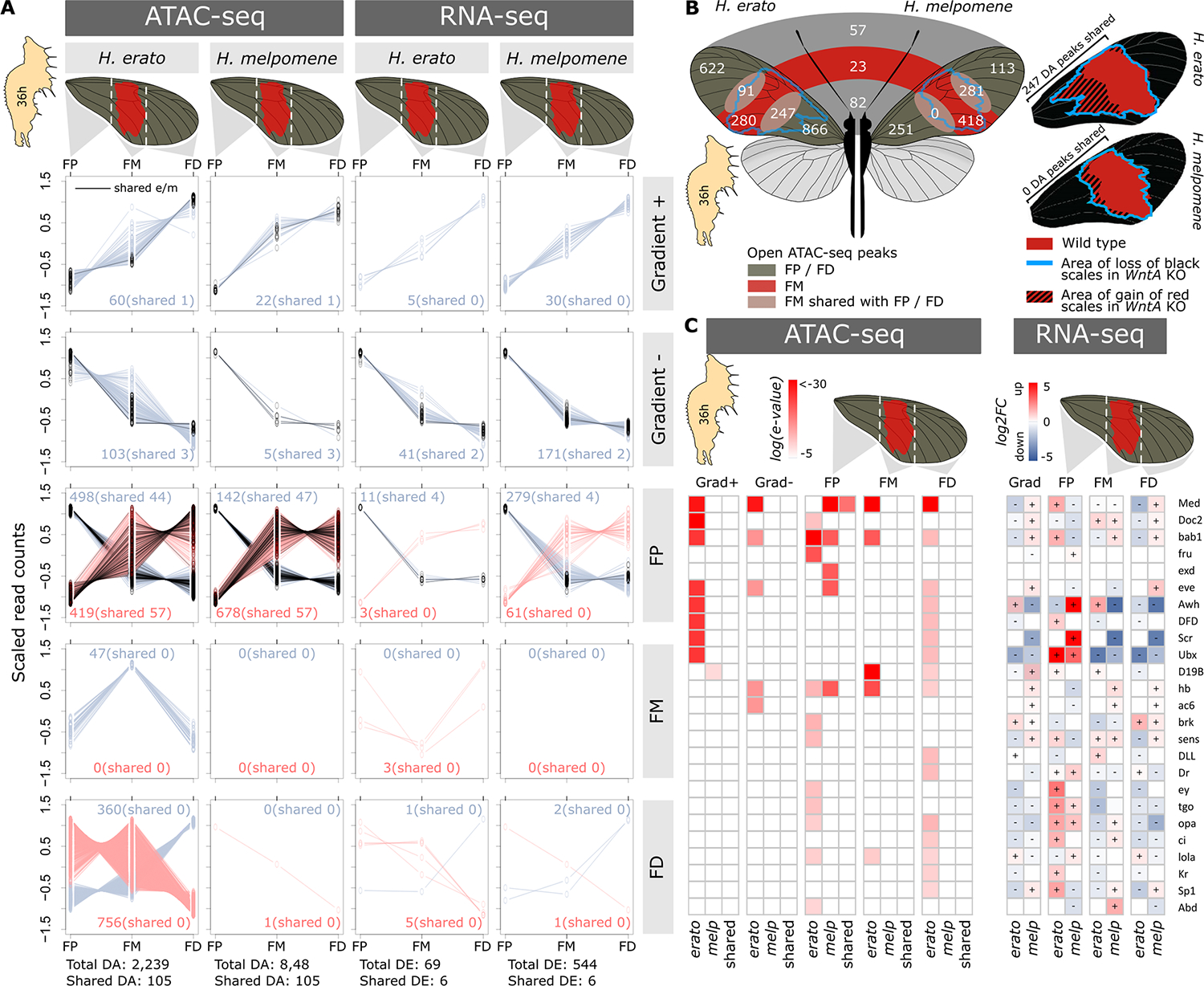
(A) Differentially accessible (DA) ATAC-seq peaks between forewing sections in H. erato and H. melpomene. ATAC-seq peaks are either significantly open (black lines) or closed (dark red lines) in FP, FM, FD, or a gradient + to − (increasing or decreasing accessibility from the proximal to distal wing section). Green lines indicate ATAC-seq peaks that are considered shared between H. erato and H. melpomene. For each comparison, we present the total and shared count numbers. (B) Numbers are differentially accessible ATAC-seq peaks in the wing sections. In contrast to (A), these numbers are obtained by pairwise comparisons between wing sections. Numbers at the boundaries of wing sections indicate peaks with shared differential accessibility compared to the other wing section. Numbers in the middle of the wings indicate peaks identified as shared between H. erato and H. melpomene (50% reciprocal overlap). Wings on the right show the wild-type phenotypes of H. erato and H. melpomene, with the blue lines indicating the extent of red scale development (and optix expression) in the WntA CRISPR-Cas9 KO. Numbers next to the wings represent DA peaks between FP or FM and FD in H. erato and H. melpomene, respectively. (C) TF motif enrichment (left) for differentially accessible ATAC-peaks between wing sections and expression of associated TFs (right). Log(e-value) indicates the significance level of the enrichment signal, and log2FC indicates the expression level relative to all other sections.
Using 50% reciprocal overlap, we identified, across all of the different patterning tests, a total of 2239 and 848 differentially accessible ATAC-seq peaks between sections of the forewing in H. erato and H. melpomene, respectively. Only 3.3% of these were shared between the two species. Similarly, when comparing gene expression across wing sections, we identified 69 and 544 differentially expressed genes in the forewing sections of H. erato and H. melpomene, respectively, of which only two (0.3%) had shared expression patterns (Fig. 3A). The shared ATAC-seq peaks had an average sequence similarity of 82.4% (SD = 12.6), whereas the total of 2871 differentially accessible ATAC-seq peaks specific to H. erato and H. melpomene had an average sequence similarity of 64.8% (SD = 25.1), with 3.0% explained by 0% sequence conservation and 30.2% having less than 50% sequence similarity in the alternative species (tables S5 and S6). Moreover, our forewing section data provided a molecular opportunity to investigate the distinct WntA KO behavior in the two comimics. Loss of function of the WntA morphogen resulted in the expansion of red scales and optix expression in the proximal part of the forewing only in H. erato and not in H. melpomene (Fig. 3B) (18). Differential accessibility analyses between forewing sections within each species resulted in 247 common chromatin peaks between the proximal and medial forewing sections in H. erato but zero between the proximal and medial forewing sections in H. melpomene. This result matches the different effect of WntA CRISPR-Cas9 Kos in H. erato and H. melpomene (fig. S8), thus reinforcing the existence of a distinct regulatory architecture of forewing proximal black in the two butterflies.
Apart from Med, bab1, and hb, we found no patterns of shared TF motif enrichment between H. erato and H. melpomene in wing sections (Fig. 3C). We identified 12 TFs and signaling molecules with nearby wing section–specific ATAC-seq peaks or differential expression patterns in both species (tables S7 and S8). These genes are known to be involved in developmental processes that include cell polarity, dorsoventral determination, and proximodistal axis identity and may represent important developmental building blocks around which gene regulatory networks have diverged. However, these TFs had distinct patterns of regulation or expression between the species because they were identified in different tissue comparisons. Genes such as engrailed and distal-less eyespots (25) represent additional genes, apart from the major known color pattern ones, that may be implicated in Heliconius wing pattern development and evolution. From these analyses, it emerges that a distinct regulatory architecture and gene expression of phenotypically identical wing patterning has evolved since their split ~11.1 million years ago.
A species-specific modular enhancer underlies the independent evolution of mimicry
To further investigate the implication of the observed widespread divergence in regulatory architectures between the comimetic butterflies on adaptive evolution (summarized in table S2), we studied the regulation of the “red” color pattern gene, optix. Our experimental design allowed us to study black and red sections of the wings during key developmental time points when optix expression is active [12 to 60 hours after pupation (10)]. Our expectation was thus to identify an open chromatin region that is significantly more accessible in the red medial forewing (FM) section and within the respective genomic association interval (13).
Within a 320-kb associated interval around the optix gene (13), we identified a total of 106 and 93 ATAC-seq peaks in H. erato and H. melpomene, respectively (Fig. 4A and fig. S9). Only one of these ATAC-seq peaks (155.5 kb downstream of the optix gene) was within a genetic yellow or red association interval hypothesized to be a candidate region for red forewing band regulation (13) and was significantly differentially accessible in the FM section in H. erato (Fig. 4B). Functional validation of this candidate CRE with CRISPR-Cas9 resulted in a mutant phenotype in which scale color–type changed from red to black or yellow in the FM of H. erato and did not affect red color patterns on the ventral side of the wings (efficiency equal to 65% of emerging adults) (Fig. 4C., fig. S10, and table S8; see fig. S11 for validation of excision mutations). Considering that these mutants may be mosaic because not all cells are being mutated in the wings, we generated a composite of the yellow-forewing mutant phenotypes, which resembled its sister species H. himera and similar yellow-forewing bands of other geographic H. erato morphs (Fig. 4D). Excising two additional candidate loci near optix, but outside the association interval, did not affect the red band phenotype (Fig. 4A and table S9). By contrast, a recent study proposed a pleiotropic architecture of the red hindwing rays and basal forewing pattern (referred to as “dennis”) and suggested that modular cis-acting enhancers of the gene optix that are sufficient to activate the presence of red rays and dennis patches likely do not exist (17, 26). Our data demonstrate that a modular CRE near optix is necessary to induce a red band phenotype.
Fig. 4. Key regulatory switch of red forewing band development.
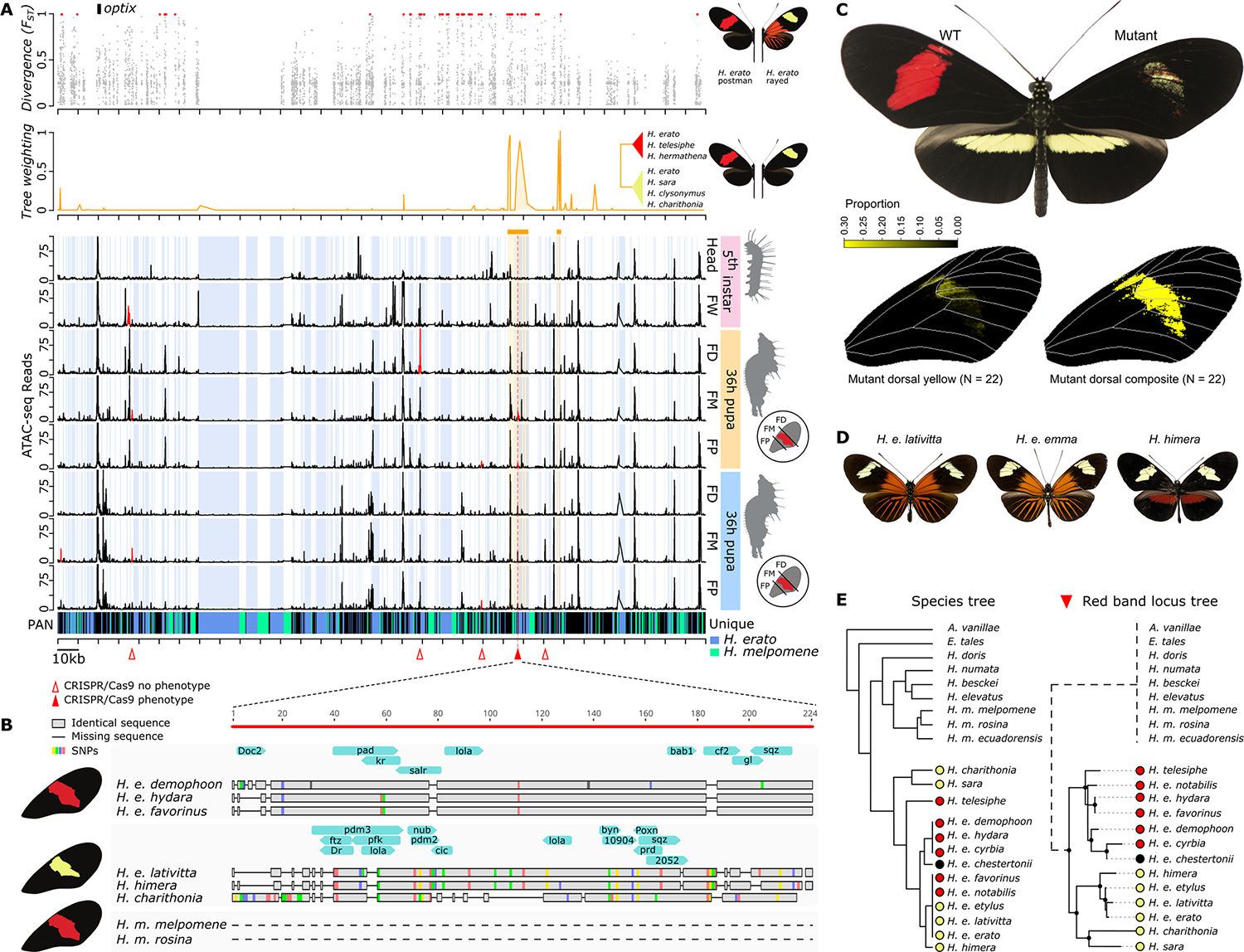
(A) Divergence [Fixation index (FST)], phylogenetic association (tree weighting), and ATAC-seq profile of red FW band near the optix gene. Blue shading indicates sequence that is specific to H. erato compared with H. melpomene. Red triangles indicate CRISPR-Cas9 excision targets. The solid red triangle indicates the target for which loss of red scales and gain of yellow scales in the FM section were observed. (B) Zoom-in on the only differentially accessible peak near optix associated with red forewing band. Gray bars and colors indicate aligned nucleotides and single-nucleotide polymorphisms (SNPs), respectively, whereas horizontal lines represent gaps. Blue arrows indicate in silico TF binding sites specific to each haplotype. The dashed lines indicate complete absence of homologous sequence. (C) CRISPR-Cas9 KO phenotype of key regulatory switch. Because of the mosaicism of CRISPR-Cas9 mutants, the complete color pattern transition is represented by the composite analysis of the individual mutant wing phenotypes. (D) Examples of geographic morphs with yellow forewing band phenotypes. (E) Detail of phylogeny of red (red circles) versus yellow (yellow circles) forewing band phenotypes at the key regulatory optix switch. The dashed branch for the outgroup species and H. melpomene indicates complete absence of homologous sequence.
Phylogenetic analysis of the H. erato optix CRE clustered H. erato populations or species within its lineage according to yellow or red color phenotypes (Fig. 4E). The sequence of this optix CRE was completely absent in H. melpomene and in butterfly species more distantly related to the Heliconius genus (Fig. 4E), thus suggesting its appearance at the origin of the H. erato clade. In silico identification of TF-binding sites, with the Drosophila database as a reference, identified up to nine potential TF-binding sites specific to the red band haplotype and 15 in the yellow haplotype (Fig. 4B). One of these TF-binding sites was for spalt-related (salr), a transcriptional repressor that, in Drosophila, mediates most decapentaplegic (dpp) functions during the development of the central part of the wing (27). These targets represent candidates for upstream regulation of optix and red pattern development in Heliconius. These results reveal the evolution of an adaptive optix CRE in H. erato, which demonstrates a distinct regulatory integration of a wing color pattern gene in the development of convergent morphologies.
Conclusion
Morphological characters of an individual require the organization of spatial and temporal gene expression (28). The integration of these genes and their products over the course of development defines a gene regulatory network (GRN) in which TFs interact with CREs of their target genes. There is a general consensus that gain and loss of CREs occurs at substantially higher rates than that of protein-coding genes (29). Despite the importance of CRE changes in the evolution of form and function in animals (30), the magnitude of CRE evolution and the context and evolutionary times necessary for CRE function to diverge or for new ones to evolve are not well understood and may be faster than generally described (31).
Convergent evolution provides an opportunity for comparative studies of CRE evolution and function during adaptive diversification. In Heliconius butterflies, mimetic species have independently evolved virtually identical wing color patterns through a shared set of color patterning genes. This has led us to assume that convergent wing color pattern evolution in Heliconius was achieved through a common developmental plan. However, this view has begun to shift recently (16, 18, 32). In this light, the highly divergent chromatin landscapes that we report for H. erato and H. melpomene suggest low conservation of CREs in the development of mimetic wing patterns. Aside from similarities at Ubx, many genes were distinctively expressed, regulated, or organized between H. erato and H. melpomene. Thus, our findings provide a contrast between the extremely conserved color pattern control at the level of protein-coding genes, with low similarity at the level of regulatory sequence. We show how these highly divergent regulatory architectures play out in the evolution of the red forewing band. A species-specific enhancer can switch red scales into yellow on the forewing of H. erato (Fig. 4C). The composite of the mosaic CRISPR-Cas9 mutants of the optix CRE in H. erato resembled H. erato’s sister species H. himera (33). This suggests that the modular regulatory changes that underlie wing color patterns also affect morphological diversification in the early stages of speciation. The lineage-specific nature of this CRE indicates that independent genetic changes are likely to be involved in species diversification of the erato and melpomene clades.
Over the ~11.1 million years since the H. erato and H. melpomene lineages split, they have retained a shared toolkit of genes involved in wing patterning (e.g., WntA, optix, cortex, aristaless, distal-less, engrailed, antennapedia). However, they evolved nonhomologous and quite distinct regulation of those genes throughout development. Although the wing patterns among mimics are highly similar, they are not identical, with consistent minor differences in pattern elements (fig. S12). These phenotypic differences may be a direct result of the fixation of independent developmental alterations (e.g., CRE changes) in the two butterfly lineages. Thus, since their split, H. erato and H. melpomene appear to have independently accumulated distinct genetic variations that modified an initially shared developmental system (20).
Our work highlights a high flexibility of evolutionary trajectories that could be a widespread property of any biological system. Whereas neutral and selected genetic changes in reproductively isolated species can create distinct genomic environments, a developmental system may thus be able to compensate for and accommodate these context-specific effects of genetic variation. This may, in turn, result in apparently similar but ultimately distinctive species-specific developmental solutions, as demonstrated by a high evolutionary turnover of CREs.
Supplementary Material
ACKNOWLEDGMENTS
We thank T. Mackay and F. Nijhout for providing valuable feedback on the manuscript.
Funding:
This work was funded by NSF EPSCoR RII Track-2 FEC (OIA 1736026) (R.P. and B.A.C.); NSF IOS 1656389 (R.P.); a Puerto Rico Science, Technology & Research Trust catalyzer award (2020–00142) (S.M.V.B. and R.P.); and a BBSRC grant (BB/R007500/1) (C.D.J.). S.M.V.B. and A.A.R. were also supported by a National Institutes of Health 4 NIGMS 604 COBRE Phase 2 Award from the Center for Neuroplasticity at the University of Puerto Rico (605 1P20GM103642). R.P. was also supported by the Hispanic Alliance for Clinical and Translational Research (Alliance) supported by the National Institute of General Medical Sciences (NIGMS), National Institutes of Health (U54GM133807). Additional funding to C.C. and W.O.M. was provided by the Smithsonian Tropical Research Institute and the Smithsonian’s Scholarly Studies program. For the support of sequencing and computational resources, we thank the University of Puerto Rico Sequencing and Genomics Facility INBRE Grant P20 GM103475 from NIGMS, a component of the NIH, and the Bioinformatics Research Core of the INBRE. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIGMS or NIH.
Footnotes
Competing interests: All authors declare no conflict of interest.
Data and materials availability: Custom codes, analyses pipelines, and images of CRISPR-Cas9 KO phenotypes are available through the archived Zenodo repository (34). ATAC- and RNA-seq sample GenBank accession numbers are available in the supplementary materials.
REFERENCES AND NOTES
- 1.True JR, Haag ES, Evol. Dev. 3, 109–119 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Blount ZD, Lenski RE, Losos JB, Science 362, eaam5979 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Van Belleghem SM, Lewis JJ, Rivera ES, Papa R, Curr. Opin. Genet. Dev. 69, 72–81 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kratochwil CF et al. , Science 362, 457–460 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Roscito JG et al. , Nat. Commun. 9, 4737 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo X et al. , Cell 184, 723–740.e21 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Uesaka M, Kuratani S, Takeda H, Irie N, Zoological Lett. 5, 33 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buenrostro J, Giresi P, Zaba L, Chang H, Greenleaf W, Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yadav T, Quivy JP, Almouzni G, Science 361, 1332–1336 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Reed RD et al. , Science 333, 1137–1141 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Martin A et al. , Proc. Natl. Acad. Sci. U.S.A. 109, 12632–12637 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nadeau NJ et al. , Nature 534, 106–110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Belleghem SM et al. , Nat. Ecol. Evol. 1, 52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Mazo-Vargas A, Reed RD, Proc. Natl. Acad. Sci. U.S.A. 114, 10707–10712 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mazo-Vargas A et al. , Proc. Natl. Acad. Sci. U.S.A. 114, 10701–10706 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Livraghi L et al. , eLife 10, e68549 (2021).34280087 [Google Scholar]
- 17.Lewis JJ et al. , Proc. Natl. Acad. Sci. U.S.A. 116, 24174–24183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Concha C et al. , Curr. Biol. 29, 3996–4009.e4 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Van Belleghem SM, Alicea Roman PA, Carbia Gutierrez H,Counterman BA, Papa R, Proc. Biol. Sci. 287, 20201267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mazo-Vargas A et al. , Science 378, 304–308 (2022). [DOI] [PubMed] [Google Scholar]
- 21.Ruggieri A et al. , Genome Res. 32, 1862–1875 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gorkin DU et al. , Nature 583, 744–751 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomoyasu Y, Curr. Opin. Insect Sci. 19, 8–15 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Tendolkar A et al. , Front. Ecol. Evol. 9, 643661 (2021). [Google Scholar]
- 25.Schachat SR, Oliver JC, Monteiro A, BMC Evol. Biol. 15, 20 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis JJ, Van Belleghem SM, Front. Ecol. Evol. 8, 261 (2020). [Google Scholar]
- 27.Sánchez J et al. , Biochem. J. 438, 437–445 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Hueber SD et al. , Development 134, 381–392 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Weirauch MT et al. , Cell 158, 1431–1443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wittkopp PJ, Kalay G, Nat. Rev. Genet. 13, 59–69 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Davidson PL et al. , Nat. Ecol. Evol. 6, 1907–1920 (2022). [DOI] [PubMed] [Google Scholar]
- 32.Bainbridge HE et al. , J. Evol. Biol. 33, 1516–1529 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Van Belleghem SM et al. , Evolution 75, 2251–2268 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Belleghem SM, StevenVB12/Genomics_ATAC: Genomics_ATAC, version 1.1, Zenodo (2022); 10.5281/zenodo.7368259. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.