

Abstract
Herein we report the first total synthesis of the indole diterpenoid natural product shearilicine by an 11-step sequence via a generalizable precursor to the highly oxidized subclass of indole diterpenoids. A native chiral auxiliary strategy was employed to access the target molecule in an enantiospecific fashion. The formation of the key carbazole substructure was achieved through a mild intramolecular Heck cyclization, wherein a computational study revealed noncovalent substrate–ligand and ligand–ligand interactions that promoted migratory insertion.
Carbazole is a heterocycle ubiquitously found in biologically active natural products and medically relevant compounds.1 Synthetic studies of diverse polycyclic scaffolds comprising carbazole alkaloids have enabled the development of strategies for rapid construction of conjugated aromatic systems embedded in topologically complex structures. For instance, the elegant synthesis of xiamycin (Figure 1A) from Li and co-workers featured an oxidative Heck cyclization to access the target heterocycle.2 This initial effort was followed by the first total synthesis of aflavazole, which leveraged an alkyne Prins cyclization and 6π-electrocyclization.3 In 2017, Garg and co-workers utilized a transient indolyne intermediate to install the terminal ring of the carbazole of tubingensin B.4
Figure 1.
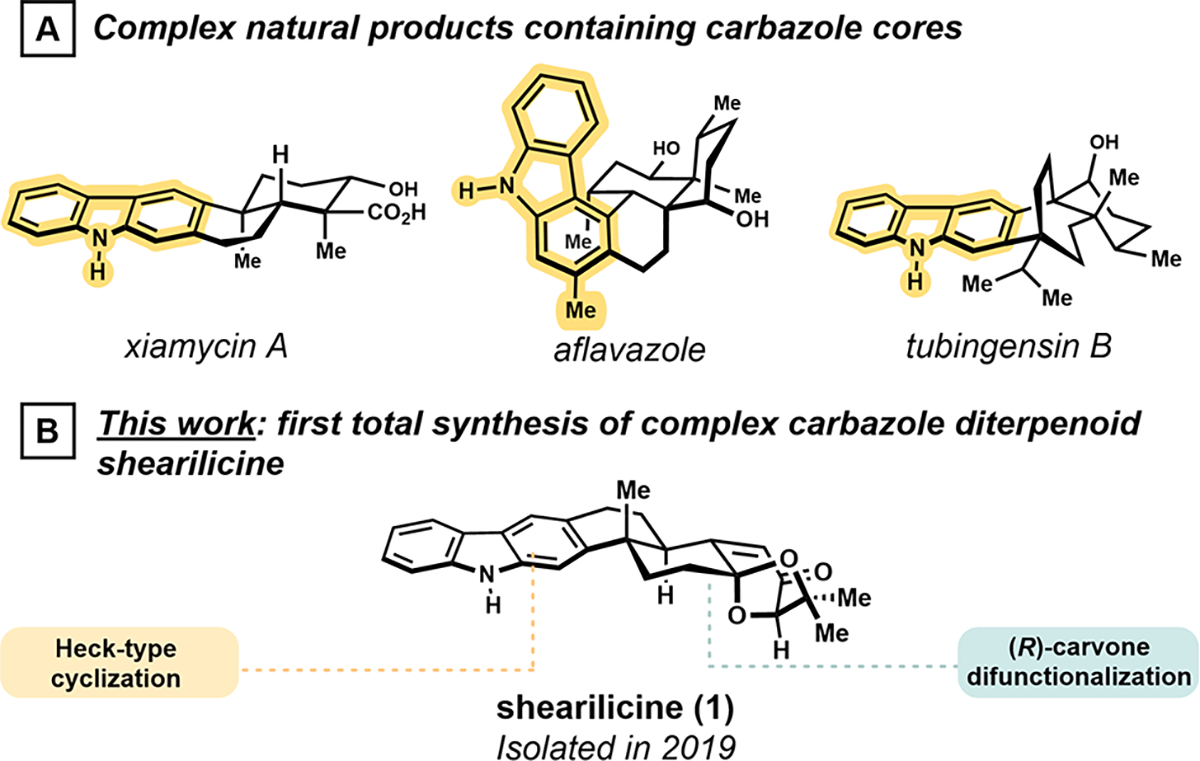
Complex carbazole-containing natural products.
In 2019, the isolation of the carbazole-containing indole diterpenoid shearilicine (1) from endophytic Penicillium sp. was reported (Figure 1B).5 Related indole diterpenoids exhibit potent and divergent mechanisms of action leading to the inhibition of BK ion channels,6 which has motivated many synthetic efforts within the class.7 Smith et al. pioneered and continues to advance the synthesis of indole diterpenoids, including the first total synthesis of paxilline-type indole diterpenoids,8,9 the only total synthesis of penitrem D,10 and more contemporary work on the nodulosporic acids.11 In these studies, Smith and co-workers employed the enantiopure Wieland–Miescher ketone to construct a 5,6,6-tricyclic intermediate, which enabled indole ring synthesis. Kuwahara and co-workers complemented these efforts in their strategy for paspalinine by realizing the reductive opening of a stereoselectively formed cyclopropane to install vicinal quaternary centers.12
In 2019, our group continued efforts in this area, leveraging a bioinspired strategy to the indole diterpenoids.13 Utilizing quantum chemical analysis enabled us to evaluate the relative energetics of two competing pathways for carbocation reactivity in the key step and design a versatile precursor that facilitated access to two skeletally distinct indole diterpenoids, paspaline A and emindole PB.
Shearilicine (1) incorporates a highly oxidized terminal ring, which is a structural feature associated with the most bioactive constituents of the class.7 Shearilicine (1) is the first carbazole-containing metabolite within the family. However, limited quantities have only allowed for evaluation against a few cancer cell lines; nonetheless compelling selective toxicity for L5178Y and A2780 was observed relative to J82 and HEK-293 at low micromolar levels (e.g., L5178Y, IC50 = 3.6 μM, SI = 11.3 compared to J82). Additionally, these combined structural features posed a unique synthetic challenge.
Herein we report the development of a strategy that utilizes two key bond formations to merge an indole synthon with a tricyclic terpenoid component via an alkylation and mild Heck cyclization reaction, which defines a broadly applicable approach to carbazole synthesis (Figure 2).
Figure 2.
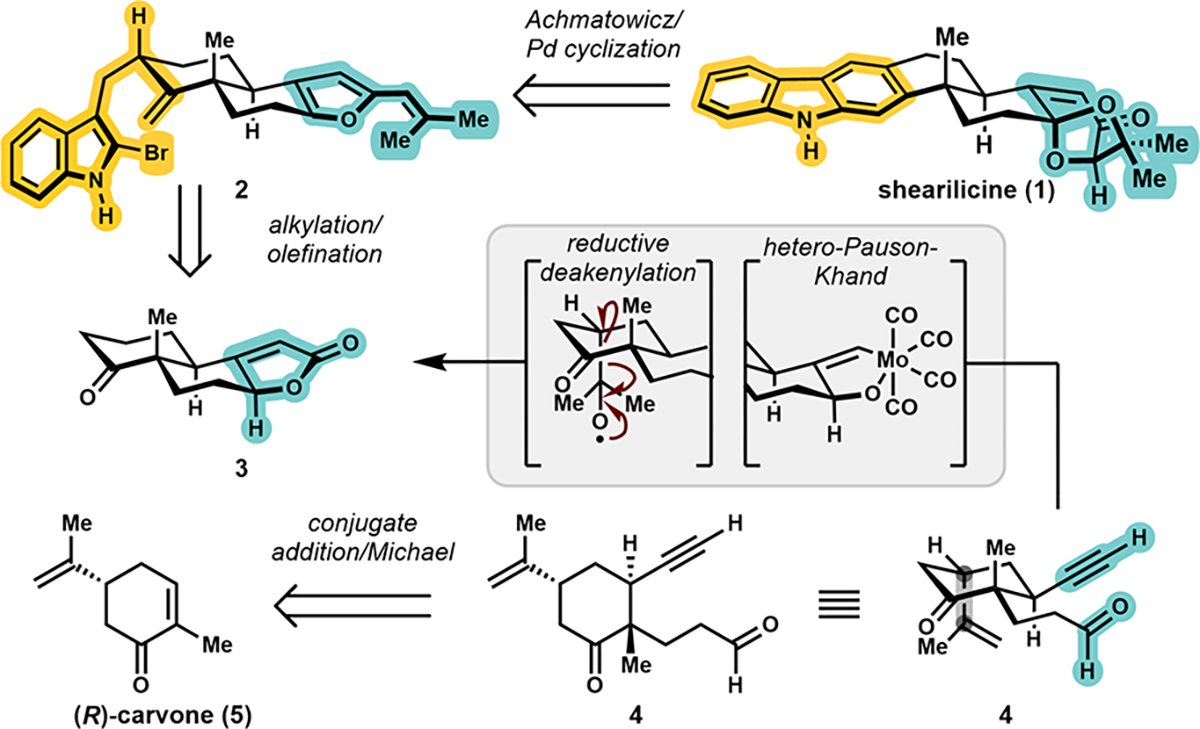
Retrosynthetic analysis of shearilicine (1).
Our retrosynthetic analysis of the oxidized indole diterpenoid framework was informed by two considerations: (1) topological and biosynthetic reasoning suggested synthesis through the ligation of an indole and terpene fragment; and (2) a starting material goal was defined as a chiral pool terpene for enantiospecific synthesis. The key aspects of our strategy hinged upon the Pd-catalyzed Heck cyclization and Achmatowicz rearrangement, the latter of which has been previously disclosed on related substrates by Saxton14 and Smith15 and recently applied by Carreira16 and Tong.17 The requisite vinylfuran (2) would be elaborated from a butenolide (3), an intermediate derived from intramolecular hetero-Pauson-Khand cycloaddition. This disconnection to 4 suggests a vicinal difuctionalization of a cyclohexanone.
Envisioning that a native chiral auxiliary would enable a diastereoselective conjugate addition, we elected to utilize (R)-carvone (5) as the starting material.18 Following the installation of the butenolide, a C–C bond cleavage was required to remove the isopropylidene unit. Although the invention of this strategy was previously disclosed,19 it had not been applied to complex molecule synthesis. Notably, this is distinct from other C–C bond cleavage strategies that classically target ring scission20 and other more recent developments in deconstructive functionalization.21
We began our synthesis with the conjugate addition of trimethylsilyl copper acetylide into (R)-carvone (5, Scheme 1). Subsequent trapping with TMSOTf resulted in the formation of enoxysilane 6 in 57% yield as a single diastereomer (d.r. > 20:1). The high level of diastereoinduction is attributed to the native chiral auxiliary function of the pendant isopropylidene group. The utility of the approach is generally encouraging, as asymmetric additions of alkynes into enones are a notable synthetic challenge.22 Enoxysilane (6) was cleaved with MeLi and treated with the masked acrolein equivalent (12)23 in the presence of Pd(PPh3)4 to afford the Tsuji–Trost allylation product 7 as an inconsequential mixture of E/Z isomers (5:1).
Scheme 1. Enantiospecific Synthesis of 10.
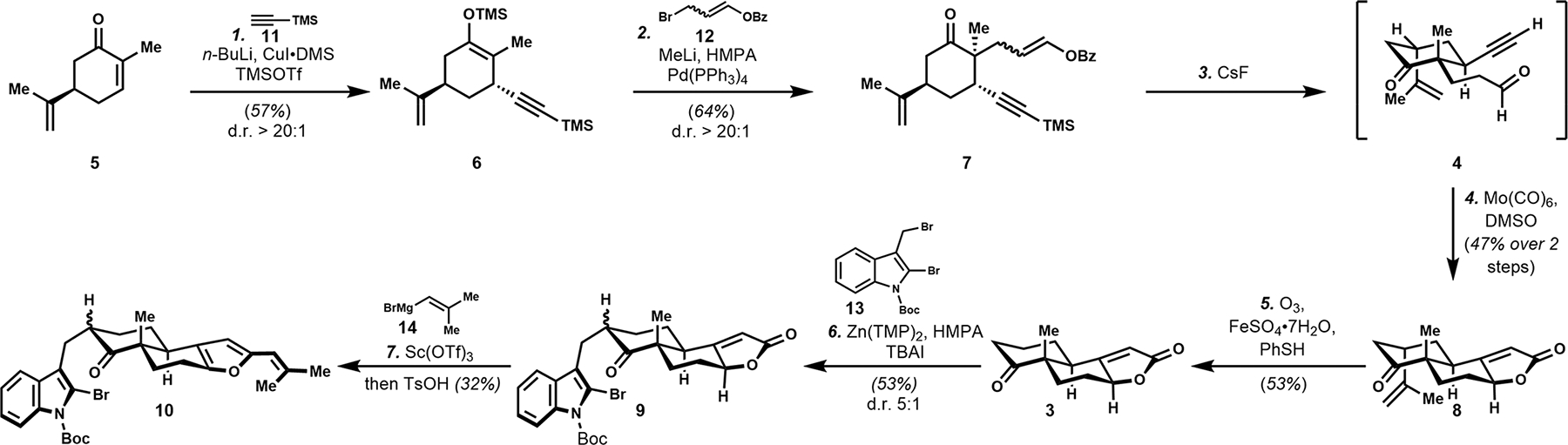
The aldehyde functionality was revealed, with concomitant desilylation by treatment with CsF to afford 4. The clean and reliable reactivity of CsF can be attributed to the mild basicity and high solubility of CsF in MeOH/THF. These conditions were uniquely effective, as more typical basic hydrolytic conditions promoted rapid decomposition of 4, likely via the engagement of the pendant aldehyde in an intramolecular aldol reaction. Additionally, it is noteworthy that the use of TBAF under analogous conditions was insufficient to effect Bz deprotection.
To avoid decomposition during chromatographic isolation, 4 was directly subjected to the hetero-Pauson–Khand cycloaddition. Treatment with Mo(CO)6 in the presence of a DMSO additive resulted in the formation of the tricycle 8 in 47% overall yield, producing a single diastereomer as observed by 1H NMR.24 At this stage, the isopropylidine moiety needed to be excised. While oxidative approaches to access the corresponding enone were considered,19,25 Kwon’s method was well-suited to provide the ketone at the desired oxidation state.26 With closely monitored and intermittent bubbling of O3 to avoid decomposition, enantioenriched 3 was obtained in 53% yield.
With tricycle 3 in hand, we proceeded to investigate chemoselective functionalization of the ketone and the butenolide substructures for the installation of the pendant dibrominated indole (13). This alkylation reaction required deprotonation of the ketone in the presence of the acidic butenolide. After extensive optimization, the selective alkylation of the ketone was achieved using Zn(TMP)2 in the presence of TBAI and the electrophile (13),27 producing 9 in 53% yield.
Due to competitive reactivity and acidity of the butenolide exacerbating the general challenge of electrophilic functionalization of neopentyl ketones, direct olefination of 9 proved to be ineffective including the “small” methylenation reagents such as Nysted’s reagent. We explored the possibility of converting the butenolide to the base-tolerant vinyl furan substructure (10). Given our hypothesis that steric hindrance present at the ketone would preference addition to the butenolide, we subjected 9 to the vinyl Grignard reagent. It was hypothesized that the Grignard addition could be promoted by treatment with a Lewis acid, and additionally, it was expected that this would help facilitate the aromatization process. Sc(OTf)328 was found to be the optimal additive for promoting selective reactivity, leading to the isolation of 10 in 32% overall yield.
With the reactivity of the butenolide attenuated through conversion to the furan (Scheme 2), the highly concentrated Wittig condition29 not only afforded the terminal olefin in 81% yield but also epimerized the homobenzylic position to the desired diastereomer (d.r. > 20:1). Having obtained the key cyclization precursor 2, we began to investigate the Pd-mediated bond formation.30 Ligandless conditions with Pd(OAc)2 in the presence of silver salts resulted in general decomposition (Entry 1). Encouragingly, the use of PPh3 allowed for 8% of the desired product to be obtained (Entry 2). The use of more electron rich but sterically bulky PBn(Ad)2 or bidentate ligands (Entries 3 and 4) were unsuccessful at improving the efficiency. Fortunately, a broad screen of Buchwald ligands identified XPhos as a promising lead, which afforded a yield of 28% (Entry 5). This result suggested testing other monodentate biaryl phosphines that preclude the formation of a bis-ligated complex. We expanded our search beyond the commonly used Buchwald-type ligands and ultimately identified CataCXium type ligand L1 as the most optimal ligand, affording 15 in 50% yield (Entry 6). The initial product of Heck cyclization (a dihydrocarbazole) was not observed, and instead, only the aromatized product 15 was identified.
Scheme 2. Synthesis of Shearilicine (1) and Ligand Studies for the Heck Cyclization.
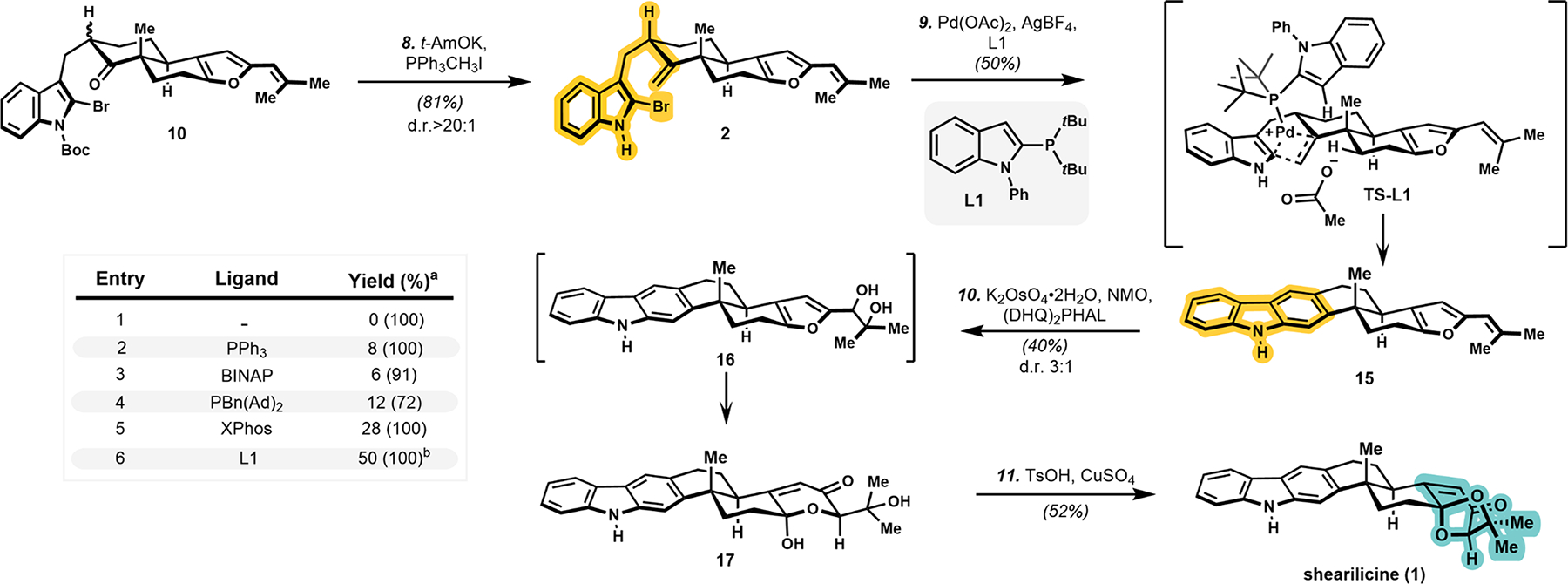
CataCXium ligand L1 has been utilized extensively for Suzuki,31 Buchwald–Hartwig,32 and Sonogashira coupling reactions.33 We were inspired to test L1 due to successful applications in Heck reactions between allylic alcohols and aryl halides.34 To understand the molecular basis for this ligand’s efficiency, we computed the full pathway and analyzed the stabilizing forces provided by L1 relative to PPh3. We first determined that the migratory insertion step has the highest transition state energy (see the Supporting Information (SI) for details).35
Consistent with experimental observation, the transition state for the migratory insertion step was computed to be lower in energy with L1 relative to PPh3 by 4.9 kcal/mol (Figure 3). We then evaluated differential weak noncovalent interactions in accordance with the method disclosed by Liu and co-workers,36 wherein we found favorable interactions between the apical methyl group on the trans-decalin and the indole unit of the CataCXium ligand (2.7 Å, 1.6 kcal/mol) and between the CataCXium tert-butyl groups and the indole unit of the substrate (3.2 Å, 2.3 kcal/mol). PPh3 exhibits analogous dispersive interactions with its phenyl substituents: between one phenyl group and the indole (2.5 Å, 1.9 kcal/mol) and between the other two phenyl groups of PPh3 and the apical methyl group (2.4 Å, 1.0 kcal/mol; 3.0 Å, 0.9 kcal/mol). Combined, the extended structure of CataCXium provides greater stabilization by 1.1 kcal/mol.
Figure 3.
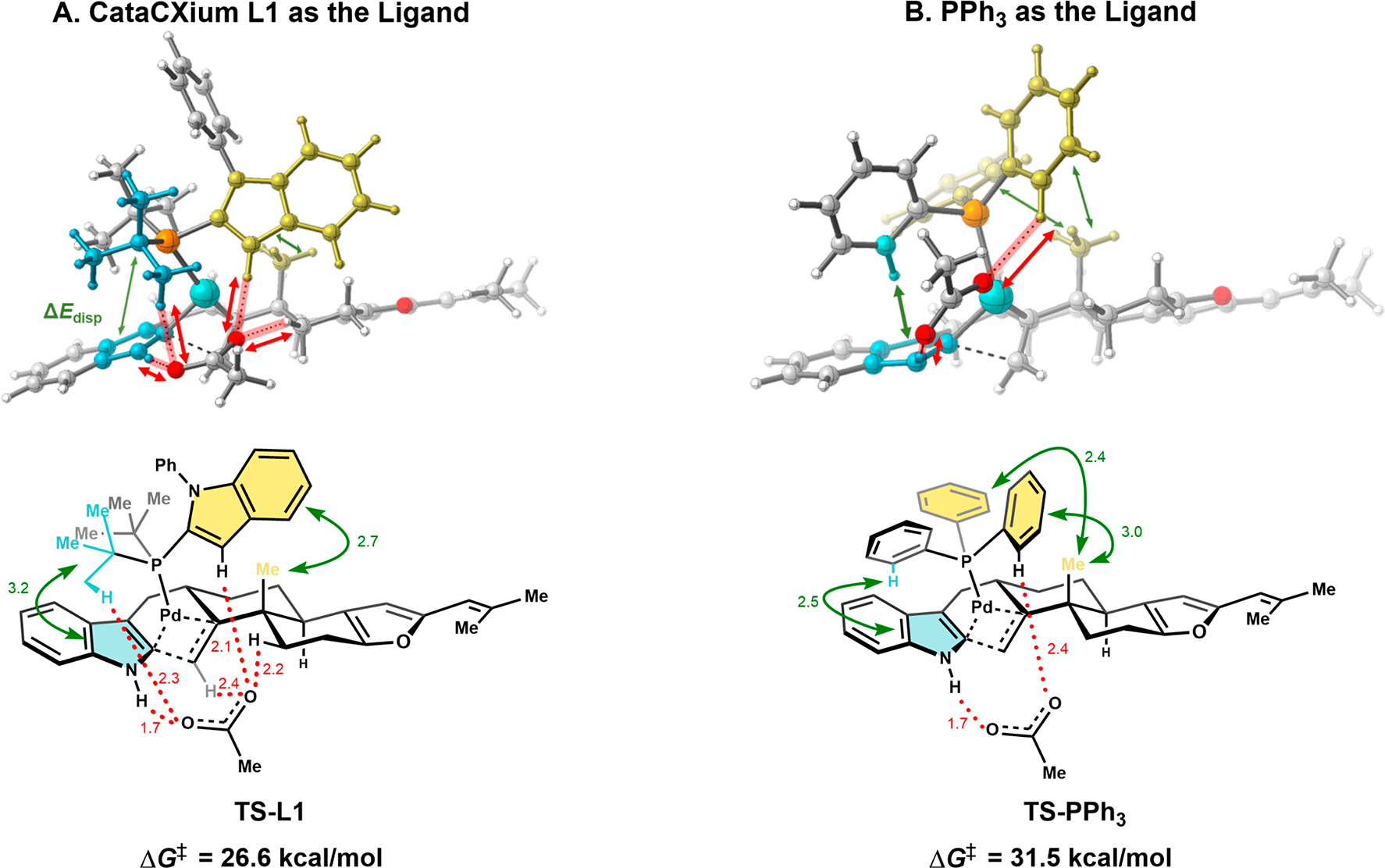
Significant differences between TS-L1 and TS-PPh3 in ligand–substrate interactions of the migratory insertion step of the key Heck reaction. Computed at the rωb97xd/SDD(Pd)/6–311+G**//rB3LYP/LANL2DZ (Pd)/6–31G* level of theory. Distances shown in Å.
In addition to these weak noncovalent interactions, the acetate anion coordinates more closely to the Pd-center for TS-PPh3 (2.2 vs 2.6 Å),37 because the steric bulk of the CataCXium ligand forces the acetate anion away from the Pd center in TS-L1, leading to contacts with the ligand and substrate. In TS-L1, one oxygen atom of the acetate anion displays interactions with two sites: (1) with the substrate’s indole N–H (1.7 Å) and (2) with the CataCXium t-Bu C–H (2.3 Å). The other oxygen atom of the acetate anion is involved in contacts with three sites on the substrate and the ligand: (1) with the vinylic C–H of the substrate (2.4 Å), (2) with the CataCXium indole C3–H (2.1 Å), and (3) with a proximal substrate methylene (2.2 Å). In TS-PPh3, there are only two such interactions: between one oxygen with the substrate’s indole N–H (1.7 Å) and the other oxygen of acetate anion and the PPh3 phenyl group (2.4 Å). The contacts with the proximal methylene and with the vinylic C–H are absent. While solvation likely plays a role and calculations with explicit solvation were not evaluated, this computational study shows striking differences in the transition states that provide a template for understanding mechanistic distinctions across ligands.
With a key step completed, carbazole 15 was subjected to Sharpless dihydroxylation conditions at low temperature. Extended reaction time allowed the intermediate furan diol 16 to further undergo Achmatowicz rearrangement in the same vessel, affording diol 17 in 40% yield with a d.r. of 3:1 favoring the desired diastereomer. The pendant alcohol was then ketalized through treatment with TsOH and CuSO4, leading to the formation of the natural product shearilicine (1).
In summary, we have completed the first total synthesis of shearilicine by an 11-step sequence, featuring: (1) a removable native chiral auxiliary to control conjugate addition facial selectivity and (2) a mild Heck reaction to form the carbazole core. The use and removal of native chiral auxiliaries to overcome selectivity challenges in multistep synthesis may be a broadly applicable strategy and compliments other developments in the use of C–C bond cleavage reactions.
The network of interactions about the Pd-center observed in this study highlights the ability of noncovalent bonding between substrates and ligands to influence selectivity in complex systems. In the context of natural product synthesis, the steric, electronic, and broader topological complexity of late-stage intermediates frequently induces deviations from selectivity and reactivity trends observed in simple substrates. Thus, obtaining a precise, molecular-level view of noncovalent bonding networks in key transition states could guide the design of specialized ligands for these complex high-value substrates and may represent a general and translatable mechanistic approach to improve catalysis.
Supplementary Material
ACKNOWLEDGMENTS
Financial support for this work was provided by Yale University, the NIH (GM118614), Digitalization-driven Transformative Organic Synthesis (22H05337), and Chiba University SEEDS Fund. The authors gratefully acknowledge the National Science Foundation for financial support in the establishment of the Yale University High Performance Computing (HPC) Center (CNS 08-21132). Dr. Fabian Menges is gratefully acknowledged for obtaining the high-resolution mass spectrometry data.
Footnotes
Notes
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.2c13584
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c13584.
Discussions of general experimental details, synthesis methods, computational details, computational methods for attractive dispersion, and determining anion-ligand/substrate interactions, figures of NMR spectra, Gibbs free-energy profiles, and method to evaluate attractive dispersion interactions, and tables of comparison of synthetic and natural 1H and 13C NMR, Cartesian coordinates, and associated distances (PDF)
Contributor Information
Daria E. Kim, Department of Chemistry, Yale University, New Haven, Connecticut 06520-8107, United States.
Yingchuan Zhu, Department of Chemistry, Yale University, New Haven, Connecticut 06520-8107, United States.
Shingo Harada, Department of Chemistry, Yale University, New Haven, Connecticut 06520-8107, United States; Graduate School of Pharmaceutical Sciences, Chiba University, Chiba 260-8675, Japan.
Isaiah Aguilar, Department of Chemistry, Yale University, New Haven, Connecticut 06520-8107, United States.
Abbigayle E. Cuomo, Department of Chemistry, Yale University, New Haven, Connecticut 06520-8107, United States
Minghao Wang, Department of Chemistry, Yale University, New Haven, Connecticut 06520-8107, United States.
Timothy R. Newhouse, Department of Chemistry, Yale University, New Haven, Connecticut 06520-8107, United States.
REFERENCES
- (1).(a) Knölker H-J; Reddy KR Isolation and Synthesis of Biologically Active Carbazole Alkaloids. Chem. Rev. 2002, 102, 4303–4427. [DOI] [PubMed] [Google Scholar]; (b) Schmidt AW; Reddy KR; Knölker H-J Occurrence, Biogenesis, and Synthesis of Biologically Active Carbazole Alkaloids. Chem. Rev. 2012, 112, 3193–3328. [DOI] [PubMed] [Google Scholar]
- (2).Meng Z; Yu H; Li L; Tao W; Chen H; Wan M; Yang P; Edmonds DJ; Zhong J; Li A Total Synthesis and Antiviral Activity of Indolosesquiterpenoids from the Xiamycin and Oridamycin Families. Nat. Commun. 2015, 6, 6096–6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Li H; Chen Q; Lu Z; Li A Total Syntheses of Aflavazole and 14-Hydroxyaflavinine. J. Am. Chem. Soc. 2016, 138, 15555–15558. [DOI] [PubMed] [Google Scholar]
- (4).Corsello MA; Kim J; Garg NK Total Synthesis of (−)-Tubingensin B Enabled by the Strategic Use of an Aryne Cyclization. Nat. Chem. 2017, 9, 944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ariantari NP; Ancheeva E; Wang C; Mandi A; Knedel TO; Kurtan T; Chaidir C; Muller WEG; Kassack MU; Janiak C; Daletos G; Proksch P Indole Diterpenoids from an Endophytic Penicillium sp. J. Nat. Prod. 2019, 82, 1412–1423. [DOI] [PubMed] [Google Scholar]
- (6).(a) Knaus H-G; McManus OB; Lee SH; Schmalhofer WA; Garcia-Calvo M; Helms LMH; Sanchez M; Giangiacomo K; Reuben JP Tremorgenic Indole Alkaloids Potently Inhibit Smooth Muscle High-Conductance Calcium-Activated Potassium Channels. Biochemistry 1994, 33, 5819–5828. [DOI] [PubMed] [Google Scholar]; (b) Zhou Y; Lingle CJ Paxilline Inhibits BK Channels by an Almost Exclusively Closed-Channel Block Mechanism. J. Gen. Physiol. 2014, 144, 415–440. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McManus OB; Rothberg BS An Old Probe Sheds New Light on BK Channel Pore Structure. J. Gen. Physiol. 2014, 144, 499–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Schatz DJ; Kuenstner EJ; George DT; Pronin SV Synthesis of Rearranged Indole Diterpenes of the Paxilline Type. Nat. Prod. Rep. 2022, 39, 946–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Smith AB; Mewshaw R Total Synthesis of (−)-Paspaline. J. Am. Chem. Soc. 1985, 107, 1769–1771. [Google Scholar]
- (9).Smith AB; Sunazuka T; Leenay TL; Kingerywood J Total Syntheses of (+)-Paspalicine and (+)-Paspalinine. J. Am. Chem. Soc. 1990, 112, 8197–8198. [Google Scholar]
- (10).(a) Smith AB; Kanoh N; Ishiyama H; Hartz RA Total Synthesis of (−)-Penitrem D. J. Am. Chem. Soc. 2000, 122, 11254–11255. [DOI] [PubMed] [Google Scholar]; (b) Smith AB; Kanoh N; Ishiyama H; Minakawa N; Rainier JD; Hartz RA; Cho YS; Cui HF; Moser WH Tremorgenic Indole Alkaloids. The Total Synthesis of (−)-Penitrem D. J. Am. Chem. Soc. 2003, 125, 8228–8237. [DOI] [PubMed] [Google Scholar]
- (11).Zou YK; Melvin JE; Gonzales SS; Spafford MJ; Smith AB Total Synthesis of (−)-Nodulisporic Acid D. J. Am. Chem. Soc. 2015, 137, 7095–7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Enomoto M; Morita A; Kuwahara S Total Synthesis of the Tremorgenic Indole Diterpene Paspalinine. Angew. Chem., Int. Ed. 2012, 51, 12833–12836. [DOI] [PubMed] [Google Scholar]
- (13).Kim DE; Zweig JE; Newhouse TR Total Synthesis of Paspaline A and Emindole PB Enabled by Computational Augmentation of a Transform-Guided Retrosynthetic Strategy. J. Am. Chem. Soc. 2019, 141, 1479–1483. [DOI] [PubMed] [Google Scholar]
- (14).Ali A; Guile SD; Saxton JE; Thornton-Pett M Synthetic Studies towards Paspalicine: Preliminary Investigations, and the Synthesis of 3′,4′,7′,7′a,9′,10′,11′,11′a-octahydro-4′,4′,7′a-trimethylspiro[1,3-dioxolane]-2,8′(6′H)-2′H-3′,5′a-epoxynaphth [2,1-b]oxepin-2′-one. Tetrahedron 1991, 47, 6407–6426. [Google Scholar]
- (15).(a) Smith AB; Minbiole KP; Verhoest PR; Schelhaas M Total Synthesis of (+)-Phorboxazole A Exploiting the Petasis-Ferrier Rearrangement. J. Am. Chem. Soc. 2001, 123, 10942–10953. [DOI] [PubMed] [Google Scholar]; (b) Smith AB; Safonov IG; Corbett RM Total Synthesis of (+)-Zampanolide and (+)-Dactylolide Exploiting a Unified Strategy. J. Am. Chem. Soc. 2002, 124, 11102–11113. [DOI] [PubMed] [Google Scholar]
- (16).Hauser N; Imhof MA; Eichenberger SS; Kündig T; Carreira, E. M. Total Synthesis of Shearinines D and G: A Convergent Approach to Indole Diterpenoids. Angew. Chem., Int. Ed. Engl. 2022, 61, No. e202112838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Guo L-D; Xu Z; Tong R Asymmetric Total Synthesis of Indole Diterpenes Paspalicine, Paspalinine, and Paspalinine-13-ene. Angew. Chem., Int. Ed. Engl. 2022, 61 (3), No. e202115384. [DOI] [PubMed] [Google Scholar]
- (18).Brill ZG; Condakes ML; Ting CP; Maimone TJ Navigating the Chiral Pool in the Total Synthesis of Complex Terpene Natural Products. Chem. Rev. 2017, 117, 11753–11795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Huang D; Schuppe AW; Liang MZ; Newhouse TR Scalable Procedure for the Fragmentation of Hydroperoxides Mediated by Copper and Iron Tetrafluoroborate Salts. Org. Biomol. Chem. 2016, 14, 6197–6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hoffmann RW SpringerLink. Elements of Synthesis Planning; Springer; Berlin Heidelberg, 2009; pp 106–117. [Google Scholar]
- (21).(a) Wang B; Perea MA; Sarpong R Transition-Metal-Mediated Cleavage of C-C Single Bonds: Making the Cut in Total Synthesis. Angew. Chem., Int. Ed. 2020, 59, 18898–18919. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lutz MDR; Morandi B Metal-Catalyzed Carbon–Carbon Bond Cleavage of Unstrained Alcohols. Chem. Rev. 2021, 121 (1), 300–326. [DOI] [PubMed] [Google Scholar]; (c) Lusi RF; Perea MA; Sarpong R C-C Bond Cleavage of α-Pinene Derivatives Prepared from Carvone as a General Strategy for Complex Molecule Synthesis. Acc. Chem. Res. 2022, 55, 746–758. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Xue Y; Dong G Deconstructive Synthesis of Bridged and Fused Rings via Transition Metal-Catalyzed “Cut-and-Sew” Reactions of Benzocyclobutenones and Cyclobutanones. Acc. Chem. Res. 2022, 55, 2341–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yu X; Chen J; Xiao W Visible Light-Driven Radical-Mediated C-C Bond Cleavage/Functionalization in Organic Synthesis. Chem. Rev. 2021, 121, 506–561. [DOI] [PubMed] [Google Scholar]
- (22).(a) Kwak YS; Corey EJ Catalytic Enantioselective Conjugate Addition of Trimethylsilylacetylene to 2-Cyclohexen-1-one. Org. Lett. 2004, 6, 3385–3388. [DOI] [PubMed] [Google Scholar]; (b) Knopfel TF; Zarotti P; Ichikawa T; Carreira EM Catalytic, Enantioselective, Conjugate Alkyne Addition. J. Am. Chem. Soc. 2005, 127 (27), 9682–9683. [DOI] [PubMed] [Google Scholar]; (c) Nishimura T; Guo XX; Uchiyama N; Katoh T; Hayashi T Steric Tuning of Silylacetylenes and Chiral Phosphine Ligands for Rhodium-Catalyzed Asymmetric Conjugate Alkynylation of Enones. J. Am. Chem. Soc. 2008, 130, 1576–1577. [DOI] [PubMed] [Google Scholar]; (d) Yazaki R; Kumagai N; Shibasaki M Direct Catalytic Asymmetric Conjugate Addition of Terminal Alkynes to α,β-Unsaturated Thioamides. J. Am. Chem. Soc. 2010, 132, 10275–10277. [DOI] [PubMed] [Google Scholar]; (e) Sanz-Marco A; Garcia-Ortiz A; Blay G; Pedro JR Catalytic Asymmetric Conjugate Addition of Terminal Alkynes to β-Trifluoromethyl α,β-Enones. Chem. Commun. 2014, 50, 2275–2278. [DOI] [PubMed] [Google Scholar]; (f) Dou XW; Huang YH; Hayashi T Asymmetric Conjugate Alkynylation of Cyclic α,β-Unsaturated Carbonyl Compounds with a Chiral Diene Rhodium Catalyst. Angew. Chem., Int. Ed. 2016, 55, 1133–1137. [DOI] [PubMed] [Google Scholar]; (g) Wang ZX; Li BJ Construction of Acyclic Quaternary Carbon Stereocenters by Catalytic Asymmetric Hydroalkynylation of Unactivated Alkenes. J. Am. Chem. Soc. 2019, 141, 9312–9320. [DOI] [PubMed] [Google Scholar]
- (23).Bottoni A; Lombardo M; Miscione GP; Algue JBP; Trombini C 3-Bromozinc Propenyl Esters: An Experimental and Theoretical Study of the Unique Stereocrossover Observed in Their Addition to Aromatic and Aliphatic Aldehydes. J. Org. Chem. 2008, 73 (2), 418–426. [DOI] [PubMed] [Google Scholar]
- (24).(a) Adrio J; Carretero JC Butenolide Synthesis by Molybdenum-Mediated Hetero-Pauson-Khand Reaction of Alkynyl Aldehydes. J. Am. Chem. Soc. 2007, 129, 778–779. [DOI] [PubMed] [Google Scholar]; (b) Gao P; Xu PF; Zhai HB Expeditious Construction of (+)-Mintlactone via Intramolecular Hetero-Pauson-Khand Reaction. J. Org. Chem. 2009, 74, 2592–2593. [DOI] [PubMed] [Google Scholar]; (c) Lu HH; Martinez MD; Shenvi RA An Eight-Step Gram-Scale Synthesis of (−)-Jiadifenolide. Nat. Chem. 2015, 7, 604–607. [DOI] [PubMed] [Google Scholar]; (d) Chen SJ; Jiang CG; Zheng N; Yang Z; Shi LL Evolution of Pauson-Khand Reaction: Strategic Applications in Total Syntheses of Architecturally Complex Natural Products (2016–2020). Catalysts 2020, 10, 1199–1223. [Google Scholar]
- (25).(a) Schreiber SL Fragmentation Reactions of α-Alkoxy Hydroperoxides and Application to the Synthesis of the Macrolide (±)-Recifeiolide. J. Am. Chem. Soc. 1980, 102, 6163–6165. [Google Scholar]; (b) Schreiber SL; Liew WF Iron Copper Promoted Fragmentation Reactions of α-Alkoxy Hydroperoxides - the Conversion of Octalins into 14-Membered Ring Macrolides. J. Am. Chem. Soc. 1985, 107, 2980–2982. [Google Scholar]
- (26).Smaligo AJ; Swain M; Quintana JC; Tan MF; Kim DA; Kwon O Hydrodealkenylative C(sp3)-C(sp2) Bond Fragmentation. Science 2019, 364, 681–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Liu R; Zhang P; Gan T; Cook JM Regiospecific Bromination of 3-Methylindoles with NBS and Its Application to the Concise Synthesis of Optically Active Unusual Tryptophans Present in Marine Cyclic Peptides. J. Org. Chem. 1997, 62, 7447–7456. [DOI] [PubMed] [Google Scholar]
- (28).Saito S; Hatanaka K; Yamamoto H Nucleophilic Addition of Organomagnesiums to Aldimines: Scandium Triflate (Sc(OTf)3) as an Effective Catalyst. Synlett 2001, 2001 (12), 1859–1861. [Google Scholar]
- (29).Shimokawa K; Smith AB Total Synthesis of the Pollen-Growth Inhibitor (−)-Emeniveol. Assignment of Absolute Stereochemistry. Tetrahedron Lett. 1993, 34, 7383–7384. [Google Scholar]
- (30).(a) Shibasaki M; Boden CDJ; Kojima A The Asymmetric Heck Reaction. Tetrahedron 1997, 53, 7371–7395. [Google Scholar]; (b) Amatore C; Jutand A Anionic Pd(0) and Pd(II) Intermediates in Palladium-Catalyzed Heck and Cross-Coupling Reactions. Acc. Chem. Res. 2000, 33 (5), 314–321. [DOI] [PubMed] [Google Scholar]; (c) Dounay AB; Overman LE The Asymmetric Intramolecular Heck Reaction in Natural Product Total Synthesis. Chem. Rev. 2003, 103, 2945–2964. [DOI] [PubMed] [Google Scholar]; (d) Amatore C; Jutand A; Khalil F Neutral Palladium(0) Complexes from Pd(OAc)2 and Tri-2-furylphosphine and Their Reactivity in Oxidative Addition of Iodobenzene. ARKIVOC 2006, 2006 (4), 38–48. [Google Scholar]; (e) Knowles JP; Whiting A The Heck-Mizoroki Cross-Coupling Reactions: A Mechanistic Perspective. Org. Biomol. Chem. 2007, 5, 31–44. [DOI] [PubMed] [Google Scholar]
- (31).(a) Choi YL; Yu CM; Kim BT; Heo JN Efficient Synthesis of Dibenzo[a,c]cyclohepten-5-ones via a Sequential Suzuki-Miyaura Coupling and Aldol Condensation Reaction. J. Org. Chem. 2009, 74, 3948–3951. [DOI] [PubMed] [Google Scholar]; (b) Hosoya T; Iimori R; Yoshida S; Sumida Y; Sahara-Miura Y; Sato J; Inouye S Concise Synthesis of v-Coelenterazines. Org. Lett. 2015, 17, 3888–3891. [DOI] [PubMed] [Google Scholar]
- (32).(a) Rataboul F; Zapf A; Jackstell R; Harkal S; Riermeier T; Monsees A; Dingerdissen U; Beller M New Ligands for a General Palladium-Catalyzed Amination of Aryl and Heteroaryl Chlorides. Chem. Eur. J. 2004, 10 (12), 2983–2990. [DOI] [PubMed] [Google Scholar]; (b) Diedrich CL; Haase D; Christoffers J New Octahydropyrido[3,4-b]acridine Scaffolds for Combinatorial Chemistry. Synthesis 2008, 2008, 2199–2210. [Google Scholar]
- (33).(a) Torborg C; Zapf A; Beller M Palladium catalysts for Highly Selective Sonogashira Reactions of Aryl and Heteroaryl Bromides. ChemSusChem 2008, 1, 91–96. [DOI] [PubMed] [Google Scholar]; (b) Prateeptongkum S; Driller KM; Jackstell R; Spannenberg A; Beller M Efficient Synthesis of Biologically Interesting 3,4-diaryl-substituted Succinimides and Maleimides: Application of Iron-Catalyzed Carbonylations. Chem. Eur. J. 2010, 16 (31), 9606–9615. [DOI] [PubMed] [Google Scholar]; (c) Ko E; Liu J; Perez LM; Lu GL; Schaefer A; Burgess K Universal Peptidomimetics. J. Am. Chem. Soc. 2011, 133, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Colbon P; Ruan J; Purdie M; Mulholland K; Xiao J Double Arylation of Allyl Alcohol via a One-Pot Heck Arylation–Isomerization–Acylation Cascade. Org. Lett. 2011, 13, 5456–5459. [DOI] [PubMed] [Google Scholar]
- (35).(a) Amatore C; Jutand A; Thuilliez A Formation of Palladium(0) Complexes from Pd(OAc)2 and a Bidentate Phosphine Ligand (dppp) and Their Reactivity in Oxidative Addition. Organometallics 2001, 20, 3241–3249. [Google Scholar]; (b) Johnson ER; Keinan S; Mori-Sánchez P; Contreras-García J; Cohen AJ; Yang W Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lu G; Liu RY; Yang Y; Fang C; Lambrecht DS; Buchwald SL; Liu P Ligand-Substrate Dispersion Facilitates the Copper-Catalyzed Hydroamination of Unactivated Olefins. J. Am. Chem. Soc. 2017, 139, 16548–16555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Amatore C; Carre E; Jutand A; M’Barki MA; Meyer G Evidence for the Ligation of Palladium(0) Complexes by Acetate Ions - Consequences on the Mechanism of Their Oxidative Addition with Phenyl Iodide and PhPd(OAc)(PPh3)2 as Intermediate in the Heck Reaction. Organometallics 1995, 14 (12), 5605–5614. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.