

ABSTRACT
Immunotherapy has revolutionized the treatment of cancers. Reinvigorating lymphocytes with checkpoint blockade has become a cornerstone of immunotherapy for multiple tumor types, but the treatment of glioblastoma has not yet shown clinical efficacy. A major hurdle to treat GBM with checkpoint blockade is the high degree of myeloid-mediated immunosuppression in brain tumors that limits CD8 T-cell activity. A potential strategy to improve anti-tumor efficacy against glioma is to use myeloid-modulating agents to target immunosuppressive cells, such as myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment. We found that the co-inhibition of the chemokine receptors CCR2 and CCR5 in murine model of glioma improves the survival and synergizes robustly with anti-PD-1 therapy. Moreover, the treatment specifically reduced the infiltration of monocytic-MDSCs (M-MDSCs) into brain tumors and increased lymphocyte abundance and cytokine secretion by tumor-infiltrating CD8 T cells. The depletion of T-cell subsets and myeloid cells abrogated the effects of CCR2 and CCR5 blockade, indicating that while broad depletion of myeloid cells does not improve survival, specific reduction in the infiltration of immunosuppressive myeloid cells, such as M-MDSCs, can boost the anti-tumor immune response of lymphocytes. Our study highlights the potential of CCR2/CCR5 co-inhibition in reducing myeloid-mediated immunosuppression in GBM patients.
KEYWORDS: Immunotherapy, MDSC, Glioma
Introduction
Chemokine receptors are G-protein-coupled receptors that promote migration of cells or chemotaxis toward their ligands known as chemokines. The activation of chemokine receptors is critical for cellular function and impacts paracrine signaling in the local tissue environment.1 Receptor and ligand expression are tightly regulated processes, with temporal, spatial, and etiological control.2 Pathological processes can dysregulate the expression of the receptors or ligands and modulate the inflammatory nature of the process in a detrimental manner.3 Therefore, targeting chemokine receptors and ligands have become an attractive target across multiple diseases.4
Chemokine receptors such as CCR2 and CCR5 have been observed to exert complementary effects in infectious diseases such as HIV, in autoimmune pathologies such as multiple sclerosis, and in cancers.5,6 The complimentary functions can be attributed to the high level of sequence homology between the two proteins.7 CCR5 is found in diverse subsets of immune cells, including macrophages, dendritic cells, T lymphocytes, neurons, and endothelial cells, while CCR2 expression is limited to NK cells, T cells, and macrophages.5 Previous studies in the context of tumors and infection, where the inhibition of CCR2 or CCR5 alone results in the modulation of immunosuppressive myeloid cells, such as tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), make the dual-inhibition of CCR2 and CCR5 an attractive strategy to further reduce immunosuppression in tumors such as GBM where immunosuppressive myeloid cells predominate.8
The deficiency of CCR2 has been shown to reduce the infiltration of MDSCs, but not DCs and resident tissue macrophages, to sites of infections.9 There is also evidence in cancers, suggesting that the infiltration of CCR2-expressing M-MDSCs into tumors is negatively correlated with the infiltration of CD8 TILs, thereby limiting the efficacy of immunotherapy.10 The study also found that CCR2 ablation did not change the intrinsic immunosuppressivity of M-MDSCs but reduced infiltration of M-MDSCs to tumors. In addition to MDSCs, TAMs exhibit CCR2-dependent recruitment to esophageal tumors, lymphoma, breast carcinoma, and prostate cancer.11–13 The ligand for CCR2, CCL2, is abundantly produced in tumors, promoting the infiltration of TAMs in glioma, renal tumors, and breast cancer among other tumors.14–16 CCL2 levels also correlate with TAM-infiltration, tumor vascularity, and early relapse in patients with gastric carcinoma and breast cancer.14,17
Similarly, CCR5-expressing MDSCs accumulate in melanoma and exhibit higher degree of immunosuppressivity compared to CCR5(-) MDSCs.18 Furthermore, anti-PD-1 therapy in gastric cancer has been shown to be improved with CCR5 blockade, as it reduces MDSC accumulation in tumors.19 Similar to CCL2, CCL5 (the ligand for CCR5) is also overexpressed in tumors and promotes infiltration of CCR5+ TAMs across multiple tumor types.20,21 The predilection of infiltrative MDSCs in localizing to GBM makes CCR2 and CCR5 viable therapeutic targets for co-inhibition. CCR2 inhibition as a monotherapy increased median survival in murine models of glioma and further increased the survival in combination with anti-PD-1.22 CCR5 inactivation with maraviroc has been shown to reduce M2 phenotype and to induce an M1 phenotype in microglia.23 While efforts to co-inhibit CCR2 and CCR5 have been tested in pre-clinical pancreatic adenocarcinoma model,24 it has not been tested in GBM therapy.
In this study, we combined the bioinformatic analysis of human GBM single-cell sequencing results to demonstrate the upregulation of these chemokine receptors on a specific myeloid population with in vivo dual CCR2/CCR5 blockade in a murine model of glioma, using a mouse surrogate for small-molecule dual antagonism of the receptors. We found that CCR2 and CCR5 are co-expressed among M-MDSCs in humans and murine models and co-inhibiting these markers synergizes with anti-PD-1 therapy in a glioma model by reducing the infiltration of M-MDSCs and thereby increasing CD8 TIL density and overall survival (OS).
Materials and methods
Animal model
Female C57BL/6J (Jackson Laboratory) mice (6–8 weeks) were housed in pathogen-free conditions under animal protocols approved by the Johns Hopkins University Institutional Animal Care and Use Committee. GL261-Luc2 cells were cultured with DMEM + 10% FBS + 1% penicillin/streptomycin media. To establish syngeneic murine glioma models, 1.3 × 105 GL261-Luc2 cells were injected with a mouse stereotaxic frame at coordinates 2 mm anterior and 2 mm lateral to lambda, 3 mm deep to the cortical surface as previously described.25 Mice were imaged for tumor burden 7 d post-implantation with IVIS®. Mice with tumors on day 7 were randomized and used for survival studies and treated with and without the inhibitors ± anti-PD-1 and followed until moribundity.
The CCR2/CCR5 inhibitor, BMS-687681, was generously provided to the laboratory of Dr. Michael Lim by Bristol Myers Squibb.26 It was administered via oral gavage twice (50 mg/kg) daily starting on day 8 after tumor implantation.24,27 Anti-PD-1 was administered via intraperitoneal (IP) injection on days 10, 12, and 14 per previous protocols.
Flow cytometry
On day 24, mice brains were harvested, mechanically dissociated, strained through a 70 µm filter (corning), and spun down in a 30%-70% Percoll® (Sigma-Aldrich) gradient at 2200 rpm for 20 min without brakes. Tumor-infiltrating lymphocytes and myeloid populations were extracted from the interface of 30% and 70% Percoll® and resuspended in phosphate buffered saline (Quality Biological, Gaithersburg, MD) for flow cytometric staining. Cells requiring IFN-γ staining of lymphocytes were activated in vitro with PMA/ionomycin for 6 h at 37°C prior to staining.
Tumor-derived cells were stained with LIVE/DEAD Fixable Cell Stain for 15 min (Thermo Fisher Scientific) for excluding dead cells. Cells were then stained for CD45 (BioLegend), CD3 (BD Bioscience), CD4 (Thermo Fischer), CD8 (BioLegend), CD11b (BioLegend), CD11c (BioLegend), F4/80 (BioLegend), Ly6C (BioLegend), Ly6G (BioLegend), IFN-γ (BioLegend), FoxP3 (BioLegend), PD-1 (BioLegend), LAG-3 (Thermo Fischer), and TIM-3 (BioLegend). After extracellular markers were stained for 15 min at room temperature, we fixed the cells for intracellular staining in a 1:3 mixture of fixation/permeabilization concentrate and diluent (eBioscence) for 30 min at room temperature. IFN-γ and FoxP3 were subsequently stained in permeabilization buffer (eBioscience) for 30 min on ice. Fluorescence minus one was used to gate IFN-γ+ and FoxP3+ cells, by controlling for data spread resulting from multiple fluorophores and nonbinary expression of the markers. Flow data was acquired using a FACSCelesta flow cytometer (BD) and analyzed using FlowJo (BD).
IVIS imaging
For imaging luminescent activity of luciferase protein in glioma to verify successful tumor implantation in vivo, GL261-luc2 bearing mice were injected with 200 μL IP D-luciferin on day 7 post-implantation. After 5 min, mice were anesthetized in an induction chamber with an isoflurane-O2 gas mixture at 2.5 L/min. After achieving adequate anesthesia as confirmed by the lack of response to toe pinch, mice were moved to the imaging chamber and remained anesthetized by continuous administration of isoflurane-O2 via nose cones. The images were captured by the IVIS® Spectrum in vivo imaging system.
Depletion of immune cells
C57BL/6J mice received IP injections of either isotype control, anti-CD4 (clone GK1.5, BioXCell, catalog#: BE0003–1), anti-CD8 (clone 2.43, BioXCell, catalog#: BE0004–1), or anti-CSF1R depletion antibodies (Clone AFS98, BioXCell, catalog#: BE0213), on days -1, 2, 5, 8, and 11 from GL261 intracranial tumor implantation. The depletion of CD4 and CD8 T-cell populations, as well as myeloid cell populations were confirmed by flow cytometry analysis of 2 mice harvested from each group. The depletion was approximately 90%+.
Bioinformatics
Single-cell RNA-sequence analysis
Our previously generated single-cell RNA-sequencing data from Jackson et al. was reanalyzed for this study.28 We analyzed all FACS-sorted CD45+/CD3- cells from 21 untreated Grade IV patients, yielding 96,132 cells. Clustering resolution and cell annotation methods were previously described in Jackson et al. Counts were normalized to the total UMI count by cell and log scaled using Seurat,29 and these normalized counts were used for CCR2 and CCR5 expression analysis.
For differential expression analysis (DEA) and gene set enrichment analysis (GSEA), all the cells in the M-MDSC cluster were separated into CCR2+ and CCR2- M-MDSCs. DEA between these two groups was performed using the MAST method,30 which uses a hurdle model that accounts for stochastic dropout and bimodal expression distributions that are characteristic of scRNA-seq data. The genes were deemed significantly differentially expressed if they had an absolute log2FC >0.5 and an FDR-adjusted p-value <0.05. The log2FC values from DEA were then used to generate ranked gene lists for GSEA,31 and the Hallmark and KEGG databases were analyzed for pathway enrichment, with pathways with FDR-adjusted p-values <0.05 considered significant.
TCGA and CGGA analysis
The GEPIA database (http://gepia.cancer-pku.cn), an interactive web server developed by Peking University for cancer expression profiling, was used to compare the expression of CCR2, CCR5, CCL2, and CCL5 signatures in TCGA tumors (glioblastoma, low-grade glioma, pancreatic adenocarcinoma, prostate adenocarcinoma, melanoma, breast carcinoma, and bladder carcinoma) and normal samples from TCGA and GTEX.32 The gene expression data for CCL2, CCL5, CCR2, and CCR5 along with corresponding clinical data from glioma patients (mRNAseq_325) were downloaded from CGGA (http://www.cgga.org.cn/) and expression of the genes compared among grades II-IV (regardless of IDH status of the tumors).33 The survival was also compared among primary GBM patients (regardless of IDH mutation status), exhibiting high expression (defined as greater than median expression) and low expression (lower than median expression) of CCR2 and CCR5.
Results
CCR2, CCR5, and their ligands are highly expressed in GBM
To gauge the role of CCR2-CCL2 and CCR5-CCL5 axes in GBM, we analyzed the bulk RNA-sequencing data from The Cancer Genome Atlas (TCGA) to compare the expression of CCR2, CCR5, CCL2 and CCL5 gene signatures between malignant and nonmalignant tissues (Figure 1a).32 Out of 31 cancers that are analyzed, only 9 had significant upregulation of the gene signature (|Log2FC| > 1 and p < 0.05), and these included GBM and low-grade glioma (Figure 1a). To further examine the expression of CCR2 and CCR5 across tumor grades, we analyzed bulk RNA-sequencing data from the Chinese Glioma Genome Atlas (CGGA)33 and observed that mRNA level of CCR2, CCR5, CCL2, and CCL5 incrementally increases with tumor grade (Figures 1b, S1a). As myeloid cell-mediated immunosuppression is higher in GBMs compared to low-grade glioma,28 we correlated the expression of CCR2, CCR5, and their ligands with signatures of MDSCs and observed a strong positive correlation (Figure S1b). There was also a strong correlation with T-cell exhaustion signatures, implying that CCR2/CCR5 signaling is associated with increased MDSC activity and CD8 T-cell exhaustion in GBMs (Figure S1c).
Figure 1.
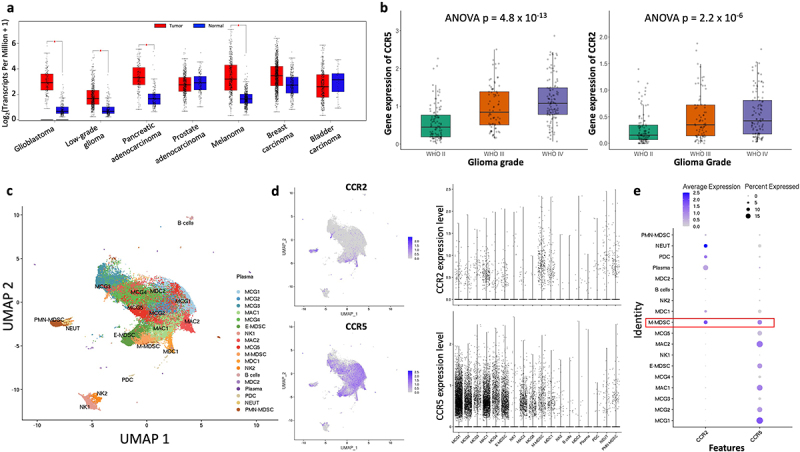
CCR2 and CCR5 are highly expressed in GBM. (a) Pan-cancer TCGA analysis comparing expression of CCR2, CCR5, CCL2 and CCL5 across multiple tumors. Red represents expression within tumors, and blue represents expression within normal tissue. (b) CGGA analysis comparing CCR2 and CCR5 expression across glioma grades. (c) UMAP of 96,132 CD45+/CD3- myeloid cells from 21 patients with Grade IV glioma (reanalysis of scRNA-Seq data from Jackson et al.). (d) CCR2 and CCR5 expression across all myeloid cell clusters. (e) Average expression and percent expressed of CCR2 and CCR5 in myeloid clusters, highlighting co-expression in M-MDSCs.
To characterize the different immune cell populations that may be responsible for increase in the expression of CCR2 and CCR5 in GBM, we analyzed our recently published single-cell RNA-sequencing data of 96,132 CD45+/CD3- myeloid cells from 21 patients with Grade IV glioma28 (Figure 1c). While CCR2 and CCR5 were heterogeneously expressed among various subsets of myeloid cells (Figure 1d), the co-expression of CCR2 and CCR5 was most strongly observed in monocytic-myeloid derived suppressor cells (M-MDSCs, sub-clustered as per Jackson et al.28) (Figure 1e). We also confirmed that M-MDSCs that infiltrated murine GL261 glioma exhibited a statistically significant and greater co-expression of CCR2 and CCR5 compared to tumor cells, whereas granulocytic-myeloid derived suppressor cells (G-MDSCs) did not (Figure S1d).
CCR2/CCR5 dual inhibitor improves survival in murine model of glioma
We hypothesized that CCR2 and CCR5 expression in tumors is detrimental to an effective anti-tumor response and is correlated with poor prognosis. Analyzing the survival of GBM patients from CGGA database based on the expression of receptors showed that patients with low expression of each of the two markers had a higher OS (Figure 2a). Patients with low CCR2 and CCR5 signature also had higher OS in the CGGA database (Figure S2a) and the TCGA database (Figure S2b). To test the therapeutic relevance of co-inhibiting CCR2 and CCR5 signaling in GBM, we used a GL261 murine glioma model to test anti-tumor efficacy of a dual inhibitor. The mice bearing orthotopic GL261 tumors were treated with dual CCR2/CCR5 inhibitor via oral gavage twice a day. The inhibitor significantly improved survival compared to untreated mice (Figure 2b) and did not elicit any adverse reactions. We also hypothesized that CCR2/CCR5 dual inhibition could synergize robustly with anti-PD-1 therapy to reduce glioma progression. Mice with GL261 tumors were either treated with isotype control, anti-PD-1 alone, CCR2/CCR5 co-inhibitor alone, or combination of anti-PD-1 and co-inhibitor. While anti-PD-1 treated mice had better survival than untreated mice, combination therapy elicited greater anti-tumor immune response compared to all other groups. (Figure 2c).
Figure 2.
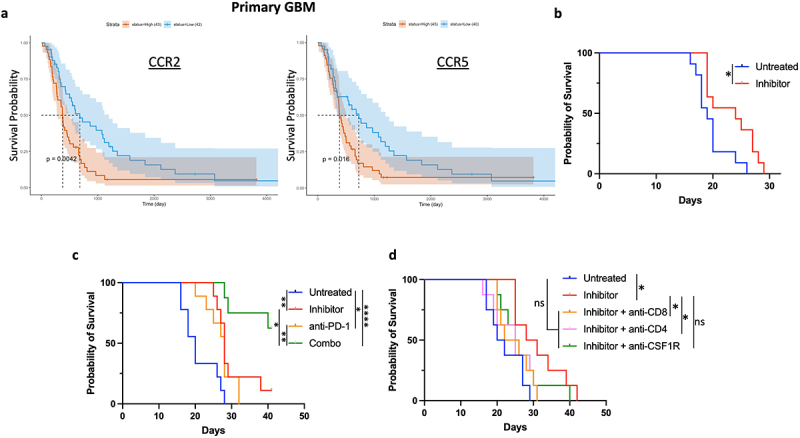
CCR2-CCR5 dual inhibitor improves survival in murine model of glioma. (a) Survival probability of GBM patients from CGGA database based on CCR2 and CCR5 expression. Blue represents low expression and red represents high expression. (b) Survival of C57BL/6 mice with intracranial orthotopic GL261 glioma (untreated n = 11, inhibitor n = 11). (c) Survival of C57BL/6 mice with intracranial orthotopic GL261 glioma (untreated n = 9, inhibitor n = 9, anti-PD-1 n = 9, anti-PD1 and inhibitor (combo) n = 8). (d) Survival of C57BL/6 mice with intracranial orthotopic GL261 glioma and depletion of CD4, CD8, and myeloid cells with anti-CSF1R antibodies (n = 8 for all groups). Differences in survival were calculated by the Mantel-Cox log-rank test. *p ≤ 0.05; **p ≤ 0.01; ****p ≤ 0.0001; ns, not significant. Survival experiments were repeated two times with similar results, and data from representative experiments are shown.
To delineate the contribution of myeloid and lymphoid compartments in the efficacy of CCR2/CCR5 co-inhibitor, we repeated administration of the inhibitor with anti-CSF1R, anti-CD4, and anti-CD8 antibodies. The depletion of all three immune subsets abrogated the efficacy of CCR2/CCR5 blockade (Figure 2d). Survival with inhibitors and anti-CSF1R antibodies was non-significantly different from untreated or inhibitor-treated groups, indicating that while the efficacy of CCR2/CCR5 co-inhibition could be attributed to our hypothesized targeting of immunosuppressive M-MDSCs with CCR2 and CCR5 expression, a broad depletion of myeloid cells with anti-CSF1R antibodies negatively impacted survival by depleting inflammatory myeloid subsets such as cross-presenting APCs and anti-tumor macrophages. We hypothesized that the survival pattern observed with lymphoid depletion indicates the role of CD8 and CD4 T cells as effector cells that mediate the efficacy of blocking M-MDSC infiltration with CCR2/CCR5 co-inhibition.
CCR2/CCR5 dual inhibitor reprograms immunosuppressive M-MDSC infiltration in glioma
To characterize changes in myeloid cell subsets in brain tumors with CCR2/CCR5 co-inhibition, we conducted flow cytometry with mice bearing orthotopic GL261 gliomas (Figure 3a). There were no significant differences in tumor-infiltration of CD11b+ myeloid cells, F4/80+ macrophages, and CD11c+ myeloid cell subsets between mice that were untreated or treated with the CCR2/CCR5 inhibitor (Figure 3b-d). Furthermore, anti-PD-1 or combination therapy also did not affect the infiltration of major myeloid subsets into brain tumors (Figure 3b-d). However, among CD11b+ myeloid cells, we observed a significantly lower mean fluorescence intensity of Ly6C (a marker for M-MDSCs) within glioma of mice treated with CCR2/CCR5 co-inhibitor, compared to untreated mice (Figure 3e). Quantifying the percentage of M-MDSCs among CD11b+ cells showed a robust reduction of M-MDSCs among myeloid cells with CCR2/CCR5 dual-inhibition (Figure 3f). However, G-MDSCs among CD11b+ cells were not significantly different among the groups, indicating M-MDSC infiltration was uniquely affected (Figure 3g). Quantifying M-MDSCs as a percentage of tumor cells also showed a lower infiltration of M-MDSCs into brain tumors with inhibitor and combination therapy. Notably, this reduction in M-MDSC infiltration was not affected with anti-PD1 alone (Figure 3h). Similarly, we did not see a difference in the proportion of G-MDSCs residing in the tumor across the four arms (Figure 3i). Combinedly, our results show that CCR2/CCR5 co-inhibition improves outcomes by reducing the tumor infiltration of CCR2 and CCR5 expressing M-MDSCs.
Figure 3.

CCR2-CCR5 co-inhibitor reprograms immunosuppressive myeloid infiltration into glioma. (a) Flow cytometry plots showing gating scheme of myeloid cells from tumor-infiltrating immune cells. Flow cytometry analysis measuring percent of (b) CD11b+ myeloid cells (c) CD11b+F4/80+ macrophages (d) CD11c+ DCs among CD45+ immune cells infiltrating the tumor in the untreated, anti-PD1, CCR2/CCR5 co-inhibitor, and anti-PD1 + CCR2/CCR5 co-inhibitor group (combo) (n = 5 each). (e) Histograms depicting Ly6C expression among myeloid cells from tumors of untreated, anti-PD1, CCR2/CCR5 co-inhibitor, and combo groups. Flow cytometry analysis measuring polarization of CD11b+ myeloid cells into (f) M-MDSCs and (g) G-MDSCs within tumors from the four groups (n = 5 each). Flow cytometry analysis comparing percent of (h) M-MDSCs and (i) G-MDSCs infiltrating brain tumors across the four groups. Graphs show mean ± SEM. Statistical significance was analyzed by one-way ANOVA with Sidak’s multiple-comparisons post hoc test. *p ≤ 0.05; ***p ≤ 0.001; ns, not significant.
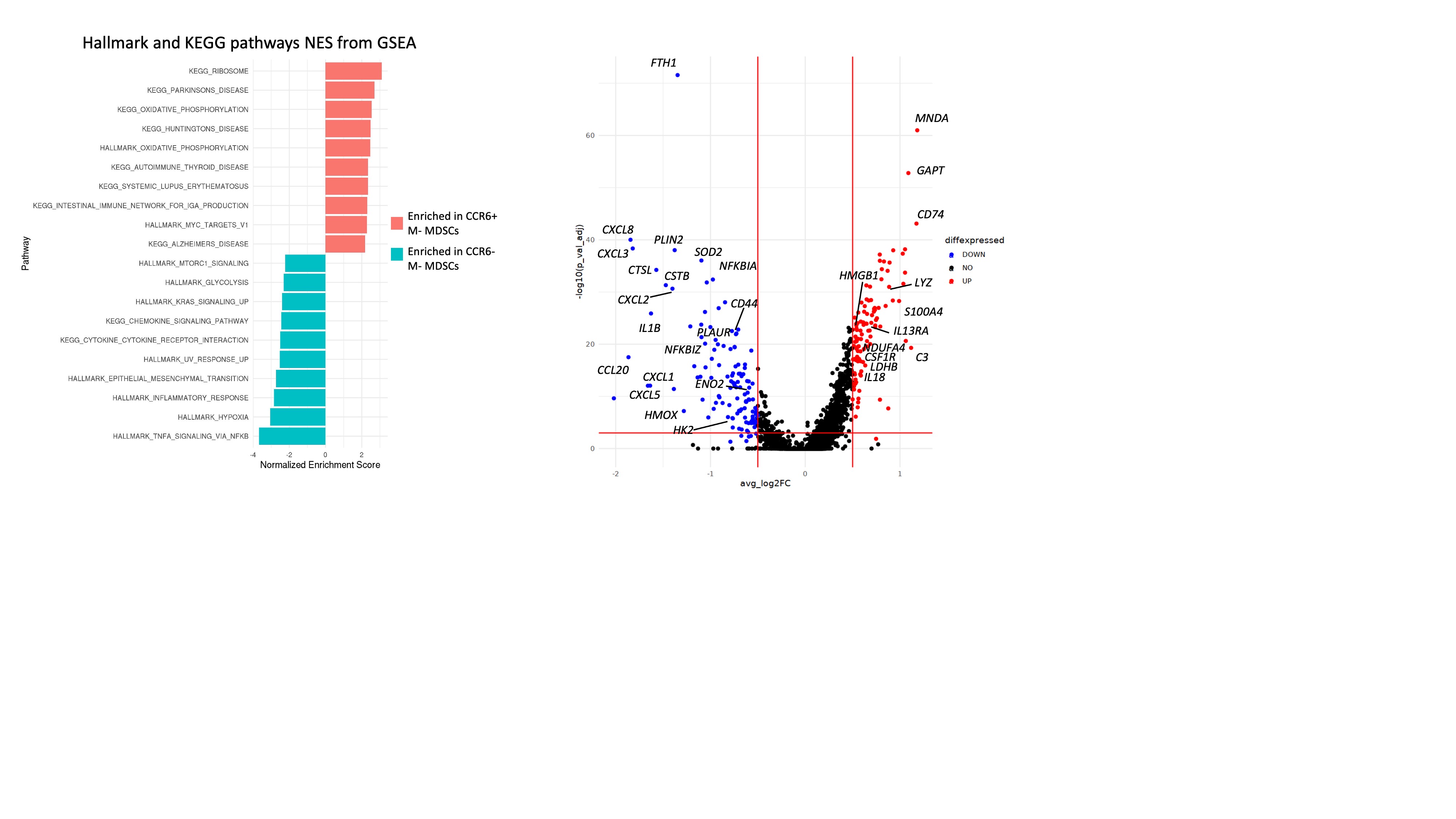
GSEA of Hallmark and KEGG showed CCR2 expressing M-MDSCs in GBM had positive enrichment of oxidative phosphorylation (OXPHOS) pathway, neurodegeneration pathologies (Alzheimer’s, Parkinson’s and Huntington’s diseases) and autoimmune pathologies (autoimmune thyroid and lupus erythematosus) but negative enrichment for glycolysis, TNF-α signaling via NF-kB, inflammatory response, hypoxia and chemokine signaling (Figure S3). Switch from glycolysis to OXPHOS has been previously reported to occur during immunosuppressive polarization of TAMs.34 CCR2-expressing M-MDSCs also downregulated genes related to chemokine signaling to other immune cells such as CXCL8, CXCL3, CXCL2, CCL20, CXCL5, and CXCL1. CCR2-expressing M-MDSCs upregulated markers of MDSC differentiation and immunosuppressivity including HMGB1, LYZ, and S100A4. Combinedly, these results indicate that CCR2/CCR5 co-inhibition reduces tumor infiltration of M-MDSCs that express the markers and are likely to play an anti-inflammatory and immunosuppressive role in the tumor microenvironment.
Inhibition of CCR2/CCR5 improves T-cell persistence and effector function in glioma
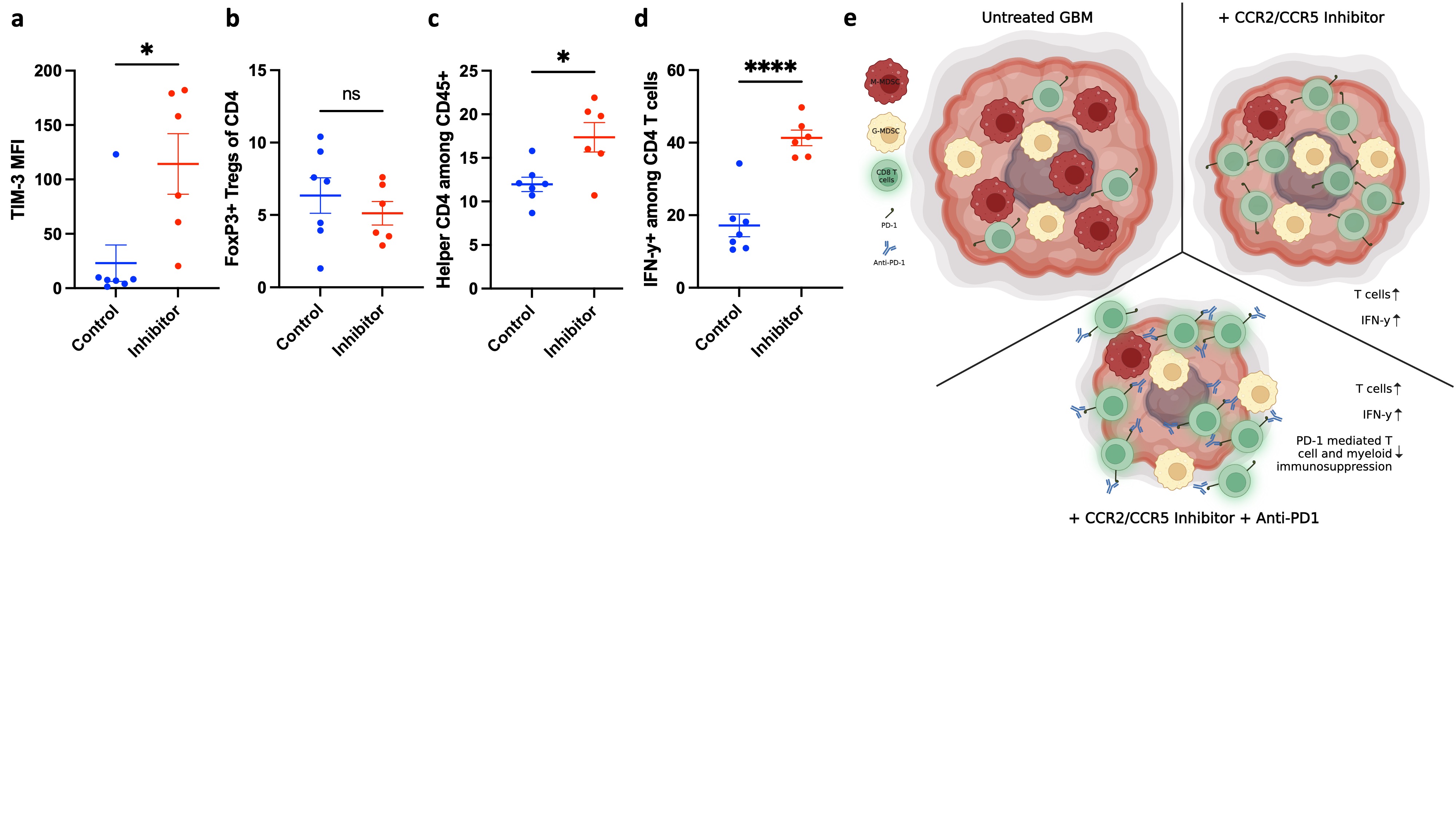
To characterize intratumoral lymphoid compartment following CCR2/CCR5 co-inhibition, we conducted flow cytometry analysis of lymphocyte abundance and effector phenotype in inhibitor-treated and untreated mice bearing GL261 orthotopic gliomas (Figure 4a). As anti-PD-1 alone did not elicit statistically significant differences in M-MDSC infiltration into glioma, we compared the effects of CCR2/CCR5 inhibition alone on lymphocyte compartment. Mice that received co-inhibitor had greater tumor abundance of CD3+, CD4+, and CD8+ T cells (Figure 4b-d). To delineate the anti-tumor efficacy of the CD8 T cells, we also assessed T-cell activation and exhaustion markers. In mice treated with the inhibitor, 60% of CD8 TILs expressed IFN-γ, while approximately 25% of CD8 T cells produced IFN-γ in untreated mice (Figure 4e). The mean fluorescent intensity of IFN-γ amongst CD8 T cells was also higher in the treated group, indicating greater T-cell activation capacity on a per cell basis (Figure 4f). Additionally, we measured checkpoint expression to assess if decreased expression of checkpoints was associated with the improved effector phenotype. However, we observed no significant difference in the expression of PD-1 and LAG-3, although there was an increase in TIM-3 expression with co-inhibitor therapy (Figures 4g,h and S4a). This suggests that while CCR2/CCR5 blockade does not directly decrease exhaustion phenotype of CD8 T cells, it increases effector function, indirectly through the effect on M-MDSC infiltration in the tumor. Although there were more CD4+ T cells in tumors of treated mice, there was no change in Tregs between mice treated with and without the CCR2/CCR5 inhibitor (Figure 4i and S4b). The increase in CD4+ T-cell infiltration was attributable to increase in helper CD4 T cells in the treated group (Figure S4c). Notably, inhibitor treatment also increased IFN-γ-expressing CD4 T cells in the tumor (Figure S4d).
Figure 4.
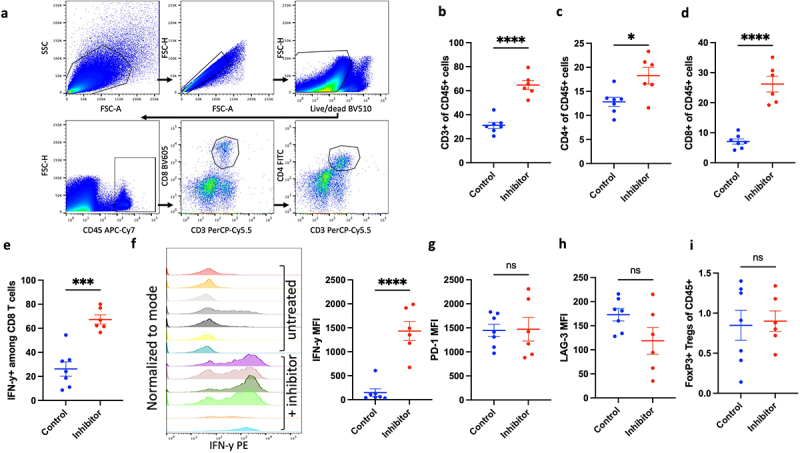
Inhibition of CCR2/CCR5 improves T-cell persistence and effector function in the tumors. (a) Flow cytometry plots showing gating scheme of lymphocytes from tumor-infiltrating immune cells. Flow cytometry analysis comparing abundance of (b) CD3+, (c) CD4+, (d) CD8+ TILs and (e) IFN-y+ CD8 TILs between control (n = 7) and inhibitor group (n = 6) in mice bearing GL261 orthotopic gliomas. (f) Mean fluorescence intensity (MFI) of IFN-y in CD8 TILs from untreated and inhibitor treated mice (g) Comparison of MFI of PD-1 and (h) LAG-3 between mice in control (n = 7) and inhibitor group (n = 6). (i) Comparison of FoxP3+ Tregs between control (n = 7) and inhibitor group (n = 6). Graphs show mean ± SEM. Statistical significance was analyzed by unpaired two-tailed Student’s t test (b-i), *p ≤ 0.05; ***p ≤ 0.001; ****p ≤ 0.0001; ns, not significant.
Discussion
Despite the advancements in immunotherapy, such as checkpoint blockade, GBM patients have not seen improvement in OS. Given the preponderance of immunosuppressive myeloid cells in the tumor microenvironment, we hypothesized that targeting the infiltration of immunosuppressive myeloid cells into glioma would reduce the dysfunction of lymphocytes and synergize with anti-PD-1 therapy in murine models. Our analysis of single-cell sequencing of GBM-derived immune cells showed co-expression of CCR2 and CCR5 on M-MDSCs, as has been reported by others across multiple tumor types. CCR2-expressing M-MDSCs also exhibited the upregulation of anti-inflammatory pathways, further supporting our hypothesis that CCR2/CCR5 expression on M-MDSCs can be targeted to prevent immunosuppression in glioma.
While previous attempts to ablate Ccr2 in murine models of glioma resulted in modest decrease in TAM infiltration but increase in tumor growth,35 co-blockade of CCR5 along with CCR2 with BMS-687681 increased survival as monotherapy and resulted in a robust decrease in M-MDSC infiltration into brain tumors (Figure S4e). It also elicited a significant increase in effector function of CD8 T cells, as evidenced by increased IFN-γ secretion from CD8 T cells. Quantifying myeloid cell infiltrates, we observed a strong reduction in infiltration of only M-MDSCs, whereas G-MDSCs, DCs, Macrophages and overall myeloid cell infiltration were not affected. Despite studies showing CCR5-mediated infiltration of Tregs into tumors,36 we did not observe any differences in tumor infiltration between untreated and CCR2/CCR5 dual-inhibitor treated mice. This shows the potential impact of CCR2/CCR5 co-inhibition in reducing M-MDSC infiltration into brain tumors and promoting CD8 activity in tumors. Our results show that targeting T lymphocytes with anti-PD-1 therapy while reducing immunosuppressive myeloid infiltration with CCR2/CCR5 co-inhibition together can promote maximum anti-tumor efficacy.
Our study provides preclinical evidence of CCR2/CCR5 co-blockade using orally administered agent, slowing glioma progression in murine models. Further studies are warranted to characterize the effects of CCR2/CCR5 co-inhibition on not just M-MDSC infiltration into tumors but also its immunosuppressivity toward lymphocytes. Moreover, single-cell sequencing data revealed an OXPHOS phenotype among CCR2 expressing M-MDSCs as opposed to greater reliance on glycolysis among CCR2(-) M-MDSCs. Further examination of the switch in phenotype between glycolytic to OXPHOS-reliant MDSCs and how this can be attenuated with CCR2/CCR5 co-inhibition can further enhance our understanding of the metabolic processes that can be disrupted in MDSCs to attenuate their immunosuppressivity.
Limitations
A limitation of our study is that using in vivo GL261 model does not recapitulate GBM progression in humans, and as such, examining the immunomodulatory effects of CCR2/CCR5 co-blockade in orthotopic glioma models is limiting. Our observation of changes in M-MDSC infiltration into GL261 glioma with CCR2/CCR5 co-blockade highlights the need for future studies that are specifically designed to study the mechanism of action by which CCR2/CCR5 blockade selectively modulates M-MDSCs, relative to other cell populations, despite CCR2 and CCR5 being present on multiple tumor-infiltrating immune cell types. This could reveal unique vulnerabilities of M-MDSCs that can be targeted by CCR2/CCR5 blockade. In addition, the analysis of clinical data supported our hypothesis that CCR2 and CCR5 expression in tumor can be prognostic for patients. However, survival studies examining the expression of CCR2 and CCR5 on specific target cells such as M-MDSCs and among patient samples that adhere to the current diagnosis of GBM (IDH-wildtype) will be better equipped to assess the contribution of CCR2/CCR5 on immunosuppressive myeloid cells in determining anti-tumor response. Although we did not find further changes in M-MDSC infiltration with the addition of anti-PD-1, we found an enhanced T-effector phenotype with inhibitor alone and a greater anti-tumor response in combination with anti-PD-1. As the role of PD-1 on lymphocytes and myeloid cells is an area of active interest in tumor immunology,37,38 future studies looking at the combined lymphoid- and myeloid-specific effects of both CCR2/CCR5 blockade and checkpoint blockade (such as anti-PD-1 or anti-TIM-3, based on increase in TIM-3 expression after CCR2/CCR5 co-blockade) could reveal previously unappreciated synergy between myeloid and lymphoid targeted therapies.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We would like to acknowledge Bristol Myers Squibb for providing the CCR2/CCR5 co-inhibitor BMS-687681.
Funding Statement
This work was completed with the help of private donors who gifted Michael Lim with research funds to explore treatments for GBM. This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Disclosure statement
M.L. has received research support from Arbor, BMS, Accuray, Tocagen, Biohaven, Kyrin-Kyowa and Biohaven, has been a consultant to Tocagen, VBI, InCephalo Therapeutics, Pyramid Bio, Merck, BMS, Insightec, Biohaven, Sanianoia, Hemispherian, Black Diamond Therapeutics, Novocure, is a shareholder of Egret Therapeutics and has patents for Focused radiation + checkpoint inhibitors, Local chemotherapy + checkpoint inhibitors and Checkpoint agonists for Neuro-Inflammation.
Data availability statement
The data that support the findings of this study stem from the pre-print: https://doi.org/10.1101/2023.03.26.534192
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2024.2338965
References
- 1.Hughes CE, Nibbs RJB.. A guide to chemokines and their receptors. FEBS J. 2018;285(16):2944–8. doi: 10.1111/febs.14466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bennett LD, Fox JM, Signoret N. Mechanisms regulating chemokine receptor activity. Immunology. 2011;134(3):246–256. doi: 10.1111/j.1365-2567.2011.03485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baggiolini M. Chemokines in pathology and medicine. J Intern Med. 2001;250(2):91–104. doi: 10.1046/j.1365-2796.2001.00867.x. [DOI] [PubMed] [Google Scholar]
- 4.Proudfoot AEI. Chemokine receptors: multifaceted therapeutic targets. Nat Rev Immunol. 2002;2(2):106–115. doi: 10.1038/nri722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fantuzzi L, Tagliamonte M, Gauzzi MC, Lopalco L. Dual CCR5/CCR2 targeting: opportunities for the cure of complex disorders. Cell Mol Life Sci. 2019;76(24):4869–4886. doi: 10.1007/s00018-019-03255-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kohli K, Pillarisetty VG, Kim TS. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 2022;29(1):10–21. doi: 10.1038/s41417-021-00303-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kothandan G, Gadhe CG, Cho SJ, Verma C. Structural insights from binding poses of CCR2 and CCR5 with clinically important antagonists: a combined in silico study. PloS One. 2012;7(3):e32864. doi: 10.1371/journal.pone.0032864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Groot J, Penas-Prado M, Alfaro-Munoz K, Hunter K, Pei BL, O’Brien B, Weathers S-P, Loghin M, Kamiya Matsouka C, Yung WKA, et al. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro Oncol. 2020;22(4):539–549. doi: 10.1093/neuonc/noz185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunay IR, Damatta RA, Fux B. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity. 2008;29(2):306–317. doi: 10.1016/j.immuni.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, Rizzuto GA, Lazarus JJ, Pamer EG, Houghton AN, et al. Monocytic CCR2+ myeloid derived suppressor cells promote immune escape by limiting activated CD8 T cell infiltration into the tumor microenvironment. Cancer Res. 2012;72(4):876–886. doi: 10.1158/0008-5472.CAN-11-1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loberg RD, Ying C, Craig M, Yan L, Snyder LA, Pienta KJ. CCL2 as an important mediator of prostate cancer growth in vivo through the regulation of macrophage infiltration. Neoplasia. 2007;9(7):556–562. doi: 10.1593/neo.07307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ren G, Zhao X, Wang Y, Zhang X, Chen X, Xu C, Yuan Z-R, Roberts A, Zhang L, Zheng B, et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell. 2012;11(6):812–824. doi: 10.1016/j.stem.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang H, Zhang Q, Xu M, Wang L, Chen X, Feng Y, Li Y, Zhang X, Cui W, Jia X, et al. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol Cancer. 2020;19(1):41. doi: 10.1186/s12943-020-01165-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heiskala M, Leidenius M, Joensuu K, Heikkilä P. High expression of CCL2 in tumor cells and abundant infiltration with CD14 positive macrophages predict early relapse in breast cancer. Virchows Arch. 2019;474(1):3–12. doi: 10.1007/s00428-018-2461-7. [DOI] [PubMed] [Google Scholar]
- 15.Boi SK, Orlandella RM, Gibson JT, Turbitt WJ, Wald G, Thomas L, Buchta Rosean C, Norris KE, Bing M, Bertrand L, et al. Obesity diminishes response to PD-1-based immunotherapies in renal cancer. J Immunother Cancer. 2020;8(2):e000725. doi: 10.1136/jitc-2020-000725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, Kanojia D, Pituch KC, Qiao J, Pytel P, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T Cells and myeloid-derived suppressor cells. Cancer Res. 2016;76(19):5671–5682. doi: 10.1158/0008-5472.CAN-16-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohta M, Kitadai Y, Tanaka S. Monocyte chemoattractant protein-1 expression correlates with macrophage infiltration and tumor vascularity in human gastric carcinomas. Int J Oncol. 2003;22:773–778. doi: 10.3892/ijo.22.4.773. [DOI] [PubMed] [Google Scholar]
- 18.Blattner C, Fleming V, Weber R, Himmelhan B, Altevogt P, Gebhardt C, Schulze TJ, Razon H, Hawila E, Wildbaum G, et al. CCR5+ myeloid-derived suppressor cells are enriched and activated in melanoma lesions. Cancer Res. 2018;78(1):157–167. doi: 10.1158/0008-5472.CAN-17-0348. [DOI] [PubMed] [Google Scholar]
- 19.Yang L, Wang B, Qin J, Zhou H, Majumdar APN, Peng F. Blockade of CCR5-mediated myeloid derived suppressor cell accumulation enhances anti-PD1 efficacy in gastric cancer. Immunopharmacol Immunotoxicol. 2018;40(1):91–97. doi: 10.1080/08923973.2017.1417997. [DOI] [PubMed] [Google Scholar]
- 20.Walens A, DiMarco AV, Lupo R, Kroger BR, Damrauer JS, Alvarez JV. CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. Elife. 2019;8:e43653. doi: 10.7554/eLife.43653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sima AR, Sima HR, Rafatpanah H, Hosseinnezhad H, Ghaffarzadehgan K, Valizadeh N, Mehrabi Bahar M, Hakimi HR, Masoom A, Noorbakhsh A, et al. Serum chemokine ligand 5 (CCL5/RANTES) level might be utilized as a predictive marker of tumor behavior and disease prognosis in patients with gastric adenocarcinoma. J Gastrointest Cancer. 2014;45(4):476–480. doi: 10.1007/s12029-014-9652-5. [DOI] [PubMed] [Google Scholar]
- 22.Flores-Toro JA, Luo D, Gopinath A, Sarkisian MR, Campbell JJ, Charo IF, Singh R, Schall TJ, Datta M, Jain RK. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proceedings of the National Academy of Sciences 2020; 117:1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laudati E, Currò D, Navarra P, Lisi L. Blockade of CCR5 receptor prevents M2 microglia phenotype in a microglia-glioma paradigm. Neurochem Int. 2017;108:100–108. doi: 10.1016/j.neuint.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Saung MT, Li K, Fu J, Fujiwara K, Niu N, Muth S, Wang J, Xu Y, Rozich N, et al. CCR2/CCR5 inhibitor permits the radiation-induced effector T cell infiltration in pancreatic adenocarcinoma. J Exp Med. 2022;219(5):e20211631. doi: 10.1084/jem.20211631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, Kim E, Wu A, Xia Y, Garzon-Muvdi T, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. 2018;7:e1466769. doi: 10.1080/2162402X.2018.1466769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carter PH, Brown GD, Cherney RJ, Batt DG, Chen J, Clark CM, Cvijic ME, Duncia JV, Ko SS, Mandlekar S, et al. Discovery of a potent and orally bioavailable dual antagonist of CC chemokine receptors 2 and 5. ACS Med Chem Lett. 2015;6(4):439–444. doi: 10.1021/ml500505q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puengel T, Lefere S, Hundertmark J, Kohlhepp M, Penners C, Van de Velde F, Lapauw B, Hoorens A, Devisscher L, Geerts A, et al. Combined therapy with a CCR2/CCR5 antagonist and FGF21 analogue synergizes in ameliorating steatohepatitis and fibrosis. Int J Mol Sci. 2022;23(12):6696. doi: 10.3390/ijms23126696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jackson C, Cherry C, Bom S, Dykema AG, Thompson E, Zheng M, Ji Z, Hou W, Li R, Zhang H, et al. Distinct Myeloid Derived Suppressor Cell Populations Promote Tumor Aggression in Glioblastoma. bioRxiv. 2023:2023.03.26.534192. [Google Scholar]
- 29.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16(1):278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sergushichev AA. GAM: a web-service for integrated transcriptional and metabolic network analysis. Nucleic Acids Res. 2016;44(W1):W194–200. doi: 10.1093/nar/gkw266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao Z, Zhang K-N, Wang Q, Li G, Zeng F, Zhang Y, Wu F, Chai R, Wang Z, Zhang C, et al. Chinese glioma genome atlas (CGGA): a comprehensive resource with functional genomic data from Chinese glioma patients. Genomics, Proteomics & Bioinformatics. 2021;19(1):1–12. doi: 10.1016/j.gpb.2020.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity. 2015;43(3):435–449. doi: 10.1016/j.immuni.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 35.Felsenstein M, Blank A, Bungert AD, Mueller A, Ghori A, Kremenetskaia I, Rung O, Broggini T, Turkowski K, Scherschinski L, et al. CCR2 of tumor microenvironmental cells is a relevant modulator of glioma biology. Cancers Basel. 2020;12(7):1882. doi: 10.3390/cancers12071882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tan MCB, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, Eberlein TJ, Hsieh CS, Linehan DC. Disruption of CCR5-dependent homing of regulatory T Cells Inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182(3):1746–1755. doi: 10.4049/jimmunol.182.3.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, Pal R, Yuan M, Asara J, Patsoukis N, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol. 2020;5(43):eaay1863. doi: 10.1126/sciimmunol.aay1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 pathway. Sci Adv. 2020;6(38):eabd2712. doi: 10.1126/sciadv.abd2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study stem from the pre-print: https://doi.org/10.1101/2023.03.26.534192