

Abstract
Friedreich’s ataxia is an inherited disorder caused by depletion of frataxin (FXN), a mitochondrial protein required for iron–sulfur cluster (ISC) biogenesis. Cardiac dysfunction is the main cause of death. Yet pathogenesis, and, more generally, how the heart adapts to FXN loss, remains poorly understood, though it is expected to be linked to an energy deficit. We modified a transgenic (TG) mouse model of inducible FXN depletion that permits phenotypic evaluation of the heart at different FXN levels and focused on substrate-specific bioenergetics and stress signaling. When FXN protein in the TG heart was 17% of normal, bioenergetics and signaling were not different from control. When, 8 weeks later, FXN was ~ 97% depleted in the heart, TG heart mass and cardiomyocyte cross-sectional area were less, without evidence of fibrosis or apoptosis. mTORC1 signaling was activated, as was the integrated stress response, evidenced by greater phosphorylation of eIF2α relative to total eIF2α, and decreased protein translation. We interpret these results to suggest that, in TG hearts, an anabolic stimulus was constrained by eIF2α phosphorylation. Cardiac contractility was maintained in the 97% FXN-depleted hearts, possibly contributed by an unexpected preservation of β-oxidation, though pyruvate oxidation was lower. Bioenergetics alterations were matched by changes in the mitochondrial proteome, including a non-uniform decrease in abundance of ISC-containing proteins. Altogether, these findings suggest that the FXN-depleted heart can suppress a major ATP-demanding process such as protein translation, which, together with some preservation of β-oxidation, could be adaptive, at least in the short term.
Introduction
Friedreich’s ataxia (FRDA) is caused by an expansion repeat of GAA between exons 1 and 2 of the gene encoding frataxin (FXN). This leads to depletion of FXN, which is part of the protein machinery responsible for iron–sulfur (Fe–S) cluster (ISC) biogenesis located in the mitochondrial matrix. It is generally accepted that FXN depletion below ~ 30% of normal levels leads to pathology, the severity of which is generally correlated with GAA expansion length [1]. FRDA is characterized by a fully penetrant ataxia, as well as other pathology, such as cardiomyopathy and progression to heart failure, which are less well predicted by GAA repeat length [2–5]; thus, cardiomyopathy in FRDA has been described as highly variable [5].
Cardiac dysfunction is the main cause of mortality in FRDA [3, 5]. Yet, its pathogenesis, including a variable presentation, is not well understood, though it might be expected that a major energy deficit, because of loss of oxidative phosphorylation (oxphos) capacity, is a driver of pathology [6–10]. Energy deficit seems logical as a source of pathology because the heart has an incessant requirement for ATP supplied by oxphos, and three of the electron transport chain (ETC) complexes require Fe–S centers for electron transfer, as do other enzymes of substrate metabolism within the mitochondrial matrix. Yet substrate oxidation pathways have not, to our knowledge, been studied in detail in cellular or animal models of FRDA.
An elegant time course study using the MCK (creatine kinase, muscle isoform) mouse model of FXN loss points to the activation of the integrated stress response (ISR) as an event concurrent with or preceding a putative energy deficit in the heart [11]. The ISR involves phosphorylation of eIF2α (p-eIF2α) that leads to decreased global protein translation on one hand and on the other hand, to the translation of the transcription factor ATF4 and genes induced by ATF4 [12, 13]. Induction of ATF4 targets can serve as readout of elevated ATF4 and is observed in many models of mitochondrial diseases [14–20]. Considering models of mitochondrial disease, a small number of cell models have reported a rise in p-eIF2α [18, 21], whereas most mouse models that investigated changes in signaling in response to mitochondrial dysfunction have focused on mTORC1 [17, 22–24] and AMPK signaling [19, 25]. AMPK and mTORC1 signaling have received less attention in FRDA models, including the MCK mouse. Of note, the MCK model features cardiac hypertrophy [7–9, 11], which is not easily explained by elevated p-eIF2α. The latter point, together with the mitochondrial disease literature, suggests that multiple signaling pathways can be altered to drive phenotypes. Thus, for any given model, it would be useful to more broadly understand nutrient and stress signaling. A broader understanding might also expand the therapeutic options and provide insight into variability in the cardiac status of FRDA patients.
The MCK mouse model has been used to evaluate potential therapies [7, 10, 11, 26, 27]. The model features a complete loss of FXN soon after birth and rapid progression to heart failure. Yet, given the variability of cardiomyopathy in FRDA, it can be valuable to have available and to characterize other mouse models in which FXN protein is depleted by >70%. Such a model was recently developed, with FXN depletion induced by doxycycline (Doxy) [28]. The mice developed a cardiomyopathy that was milder and slower in onset compared with the MCK mouse. Here we have used this newer genetic model to undertake a detailed study of substrate-specific bioenergetics in heart mitochondria and to broadly interrogate nutrient and stress signaling pathways. We used a modified Doxy dosing regimen that lowered mortality and allowed phenotypes to be evaluated with FXN protein just <20% of normal levels, and when FXN protein was almost fully depleted but before mice had undergone a major loss in body weight (BW). We show that ~20% FXN is sufficient to maintain heart function and oxphos capacity, with no evidence of altered signaling. When FXN was almost fully depleted, heart function and bioenergetics from β-oxidation were preserved, but hearts were smaller, AMPK and mTORC1 signaling were altered and ISR activation was associated with lower global protein translation. We interpret this response as protective for the heart, at least in the short term.
Results
Model of adult-onset Doxy-induced FXN depletion
We used a genetic mouse model to deplete FXN via Doxy induction of an shRNA targeting Fxn mRNA; this is the same genetic model used by Chandran et al. [28]. Wild-type (WT) mice and mice harboring one copy of the Doxy-inducible shRNA transgene against Fxn [transgenic (TG)] were generated from a WT × TG cross [28]. Doxy dosing was initiated when mice were ~9 weeks of age and was supplied in the chow (200 p.p.m.); TG mice and their WT littermates were fed the Doxy diet. Gross phenotypic changes were evident in TG mice after ~ 13 weeks of Doxy feeding. Notably, whereas BW of TG and WT mice was comparable during the first 13 weeks of Doxy, TG mice subsequently underwent a progressive weight loss that was pronounced by 20 weeks of Doxy (Supplementary Material, Fig. S1A). Mortality was evident only after ~16 weeks of Doxy feeding in TG mice, and by 18 weeks of Doxy, ~ 35% of the TG mice had died (Supplementary Material, Fig. S1B).
We focused our studies on the heart to two time points: mice fed with Doxy for 9–10 weeks (‘10 weeks’) as an early time point, and for 17.5–18.5 weeks (‘18 weeks’) (some BW loss was evident, but prior to major BW loss and mortality). After 10 weeks of Doxy feeding, FXN protein was 83% depleted in the heart of TG mice (Fig. 1B), and by 18 weeks of Doxy, FXN was 97% depleted in TG hearts, as assessed by immunoblot (Fig. 1B). Mass spectrometry analysis of isolated heart mitochondria from 18 weeks Doxy-fed mice failed to identify unique FXN peptides in heart mitochondria from TG mice, whereas multiple peptides were identified in WT heart mitochondria (Supplementary Material Table S1).
Figure 1 .
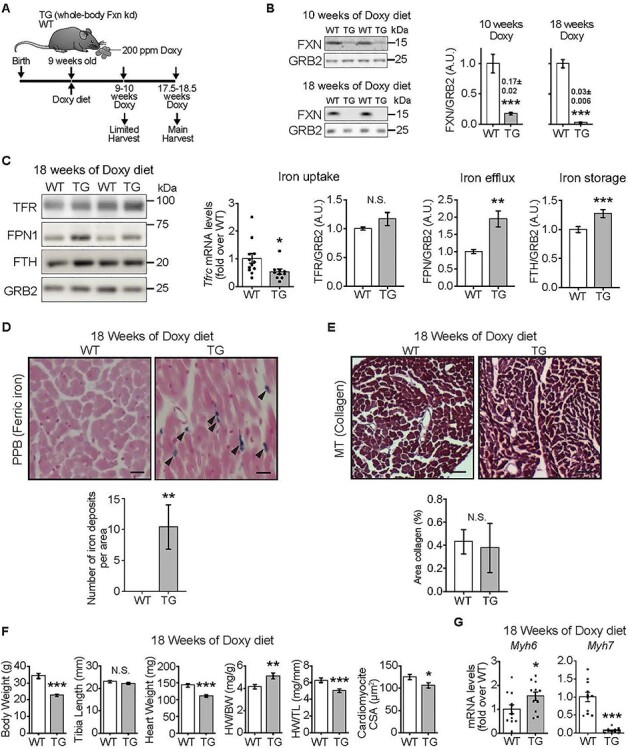
FXN-depleted hearts exhibit normal contractility but are smaller. (A) Experimental model. Mice harbored a whole body-wide Doxy-inducible shRNA construct targeting Fxn (TG mice). Doxy (200 p.p.m.) was delivered in the chow. Both TG and WT mice (WT littermates) received the Doxy-dosed diet. (B) Representative immunoblots of FXN and GRB2 (loading control) in heart lysates from WT and TG mice and quantification (n = 7/genotype). (C) Iron homeostasis in heart lysates, determined by immunoblot (left panels: TFR, FPN1, FTH, with GRB2 as loading control) and qPCR (Tfrc). Bar charts: mRNA levels of Tfrc, normalized to Actb (β-actin) and expressed relative to WT, value = 1 means no change, (n = 12 WT/10 TG) and protein levels of TFR, FPN1 and FTH (n = 4–7 WT/5 TG). (D) Upper: Sections from heart stained with PPB to reveal iron deposition. Lower: Quantification of number of iron deposits per area (353 μm × 264 μm), (n = 7 WT/4 TG). (E) Upper: Cross-sections from hearts stained with MT to detect collagen as a measure of fibrosis. Lower: Quantification of area of collagen (%) from heart cross-sections (n = 7 WT/4 TG). (F) BW, TL, HW, HW/TL (n = 7/genotype) and CSA of cardiomyocytes (n = 7 WT/4 TG). (G) Transcript levels [normalized to Actb (β-actin) and expressed relative to WT] of Myh6 (α-MHC) and Myh7 (β-MHC) (n = 11–12/genotype; points are values from each sample). (B–G): Values are mean ± standard error of mean (SEM). Statistical comparison was by unpaired t-test; *P < 0.05, **P < 0.01, ***P < 0.001. N.S.: not significant.
Lack of overt hypertrophy and evidence for compensated function
Next, we evaluated the levels of iron-handling proteins in the heart as well as iron deposition, because marked changes in these proteins occur in FXN-depleted mouse hearts, and iron deposits have been reported in FXN-depleted mouse and human hearts (reviewed in [29]). Hearts from TG mice fed Doxy for 18 weeks showed lower levels of transferrin receptor mRNA and unchanged protein levels, accompanied by higher levels of iron-handling proteins, ferroportin and ferritin (Fig. 1C). Perls staining revealed ferric iron deposits in TG but not WT hearts from 18-week Doxy-fed mice (Fig. 1D). Thus, TG hearts responded to FXN depletion with an iron homeostatic response consistent with elevated cellular iron [30].
The elevated ferric iron in TG hearts is abnormal and could be accompanied by pathological morphological and functional changes in the heart. Histological evaluation of WT and TG hearts from 18-week Doxy-fed mice revealed normal cardiac fiber structure and absence of fibrosis (Fig. 1E), and there was no evidence for apoptosis, on the basis of lack of caspase-3 cleavage and lack of BAX recruitment to mitochondria (Supplementary Material, Fig. S1D). Echocardiography to evaluate cardiac function revealed slightly higher fractional shortening in hearts from TG mice fed Doxy for 18 weeks, a change already evident in 10-week Doxy-fed TG mice (Table 1). The greater fractional shortening was driven primarily by a smaller left ventricular (LV) end-systolic diameter (Table 1), consistent with increased contractility.
Table 1.
Echocardiography.
| Before Doxy | 10 weeks of Doxy | 18 weeks of Doxy | ||||
|---|---|---|---|---|---|---|
| WT | TG | WT | TG | WT | TG | |
| HR, beats/min | 449 [11] | 444 [11] | 447 [15] | 428 [9] | 432 [12] | 398 [11] |
| IVS,d (mm) | 0.73 (0.05) | 0.76 (0.04) | 0.74 (0.04) | 0.75 (0.02) | 0.75 (0.03) | 0.79 (0.03) |
| IVS,s (mm) | 1.00 (0.03) | 1.05 (0.05) | 1.06 (0.03) | 1.11 (0.04) | 1.10 (0.04) | 1.23 (0.02)***, &&& |
| LVID,d (mm) | 4.23 (0.05) | 4.11 (0.16) | 4.21 (0.12) | 4.04 (0.11) | 4.12 (0.07) | 3.91 (0.14) |
| LVID,s (mm) | 3.16 (0.02) | 3.11 (0.18) | 3.2 (0.09) | 2.71 (0.13) | 3.08 (0.07) | 2.58 (0.08)***, &&& |
| LVPW,d (mm) | 0.66 (0.03) | 0.70 (0.03) | 0.75 (0.02) | 0.77 (0.03) | 0.79 (0.03) | 0.77 (0.03) |
| LVPW,s (mm) | 1.06 (0.04) | 1.00 (0.04) | 1.04 (1.2 | 1.05 (0.03 | 1.06 (0.03) | 1.11 (0.04) |
| EF (%) | 50 (1.2) | 49 (2.8) | 48.2 (1.2) | 62.1 (2.1)***, &&& | 50.2 (1.0) | 63.3 (2.0)***, &&& |
| FS (%) | 25.2 (0.8) | 24.6 (1.6) | 24.1 (0.8) | 33.1 (1.4)***, &&& | 25.2 (0.6) | 33.9 (1.6)***, &&& |
M mode cardiac echocardiography. HR: heart rate. Left ventricular inner diameter at end-diastole (LVID,d) and end-systole (LVID,s), left-ventricular posterior wall thickness at end-diastole (LVPW,d) and end-systole (LVPW,s), interseptal wall thickness at end-diastole (IVS,d) and end-systole (IVS,s), ejection fraction (EF) and fractional shortening (FS) were measured serially at indicated time points during doxy feeding. Values are mean ± SEM; n = 5/genotype; statistics: two-way ANOVA; P < 0.001 for time × genotype; P-values shown are from post hoc Tukey test: ***P < 0.001 for WT versus TG at a given time point, &&&P < 0.001 for TG time 0 versus TG 10 weeks and versus TG 18 weeks).
Cardiac hypertrophy is often a feature of the cardiomyopathy of FRDA [3]. Thus, we determined if cardiac hypertrophy was present in TG hearts. Morphometric evaluation of the heart histology revealed a smaller cardiomyocyte cross-sectional area (CSA) in TG hearts from 18-week Doxy-fed mice (Fig. 1F) and measurement of heart weight in 18-week Doxy-fed mice revealed a lower heart weight/tibia length ratio (HW/TL) in TG mice. Yet, HW relative to BW was higher (Fig. 1F), and LV posterior wall and septal wall thickness during diastole were unchanged in TG hearts (Table 1). To determine if smaller heart weight is intrinsic to the TG genotype, we measured HW/TL in mice that were not fed the Doxy diet and found no difference between WT and TG (Supplementary Material, Fig. S1C).
Lower levels of Myh6 (α-MHC; myosin heavy chain, α isoform) and higher levels of Myh7 (β-MHC) are often found in diseased hearts [31]. TG hearts from 18-week Doxy-fed mice showed slightly elevated Myh6 and lower Myh7 (Fig. 1G).
The expression pattern of the MHC isoforms, together with the functional and morphological data suggested that FXN-depleted hearts were not normal. We hypothesized that the status of TG hearts reflected adaptive changes. To address this hypothesis, we questioned how, energetically, the FXN-depleted hearts could sustain their function (Figs 2 and 3), and whether the smaller heart size could reflect altered stress and nutrient signaling (Figs 4–6).
Figure 2 .
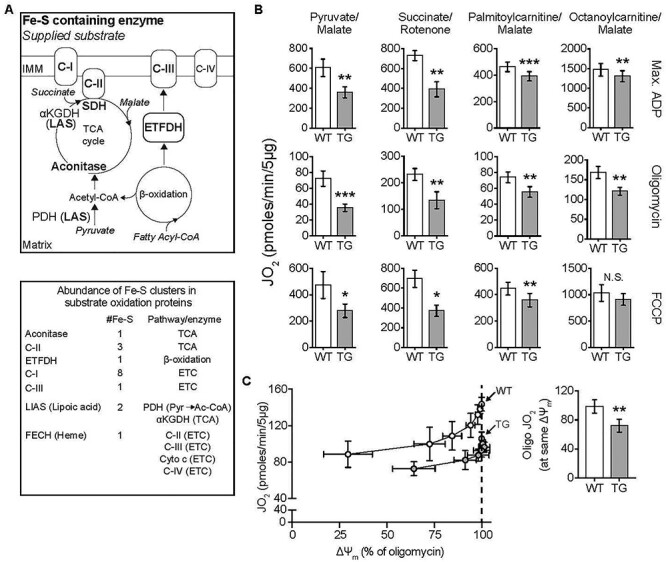
Substrate-dependent decrease in mitochondrial oxphos in TG hearts. All measurements were done in heart mitochondria from WT and TG mice fed Doxy for 18 weeks. (A) Upper: Diagram shows ISC-containing enzymes (bold font), and the substrates supplied in bioenergetics experiments (italics). Lower: List of ISC-containing complexes/enzymes with corresponding number of ISCs and associated metabolic pathway or enzyme. αKGDH: α-ketoglutarate dehydrogenase, Cyto c: Cytochrome c. C-I, C-II, CIII and C-IV are mitochondrial respiratory complexes I, II, III and IV. Pyr: pyruvate; Ac-CoA: acetyl-CoA. (B) Oxygen consumption rate (JO2) measured in isolated heart mitochondria supplied with pyruvate/malate (10 mM/5 mM), succinate (10 mM + 1 μM rotenone to prevent electron backflow through complex I), palmitoyl-L-carnitine plus malate (20 μM + 1 mM), or octanoyl-L-carnitine plus malate (200 μM + 1 mM). Max. ADP, saturating [ADP] (4 mM), was used to evaluate JO2 reflecting maximal oxphos. Oligomycin was used to evaluate JO2 that reflects maximal leak-dependent oxidation. The chemical uncoupler, FCCP (1 μM), 2.5 μg/ml) as used to evaluate JO2 that reflects maximal ETC capacity under the prevailing substrate conditions. N = 4–5/genotype except for octanoyl-L-carnitine (n = 4/genotype). (C) Proton leak was determined by measuring mitochondrial membrane potential (ΔΨm) and JO2 during antimycin (Complex III inhibitor) titration of octanoyl-L-carnitine oxidation, in the presence of oligomycin. Bar chart: JO2 values at the same ΔΨm (dashed line in the panel at left) (n = 3/genotype). B and C: Values are mean ± SEM. Statistical analysis was by unpaired t-test; *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 3 .
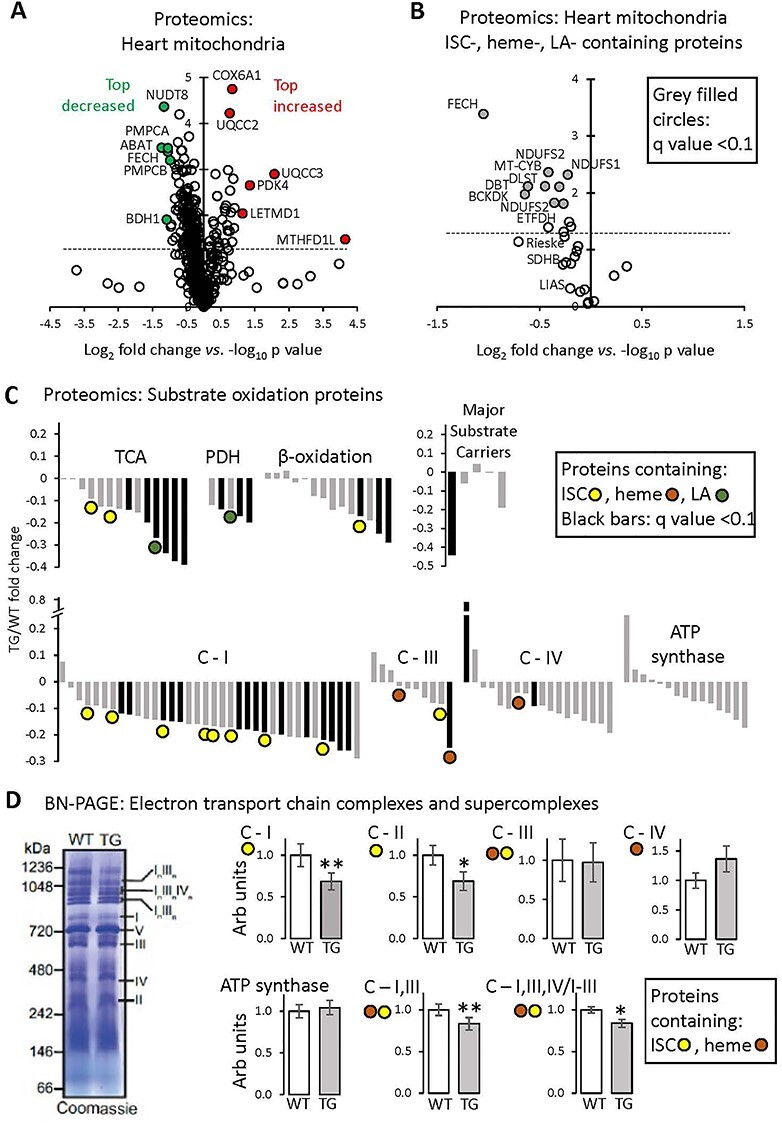
Proteomics analysis of heart mitochondria: variable effect of FXN depletion on ISC-containing proteins. All measurements were done in heart mitochondria from WT and TG mice fed Doxy diet for 18 weeks. (A) Volcano plot (log2 TG/WT-fold change versus log10 P-value, unpaired t-test) of heart mitochondrial proteins determined by mass spectroscopy (n = 3/genotype). Inclusion criteria: proteins identification on the basis of more than one peptide in at least one genotype. Dashed line: P = 0.05 (unpaired t-test). (B) Volcano plot (log2 TG/WT-fold change versus log10 P-value, unpaired t-test) of heart mitochondrial proteins containing ISCs or heme or LA moieties. Proteins determined by mass spectroscopy (n = 3/genotype). Dashed line: P = 0.05 (unpaired t-test). Black-filled circles: proteins with q-value (P-value corrected for FDR) < 0.10. (C) Fold change of substrate oxidation-related proteins, determined by mass spectroscopy (n = 3/genotype). Black bars: proteins with q-value < 0.10. C-I, III, IV: Complexes I, III, IV of the ETC. Complex II subunits are included in the ‘TCA’ category. (D) BN-PAGE gel electrophoresis of isolated heart mitochondria, to reveal the abundance of ETC complexes and SCs. Left: Example of Coomassie-stained gel, with ETC complexes and SCs shown by Roman numerals. Right: Quantification; values are mean ± SEM (n = 5/genotype, *P < 0.05, **P < 0.01 by unpaired t-test).
Figure 4 .
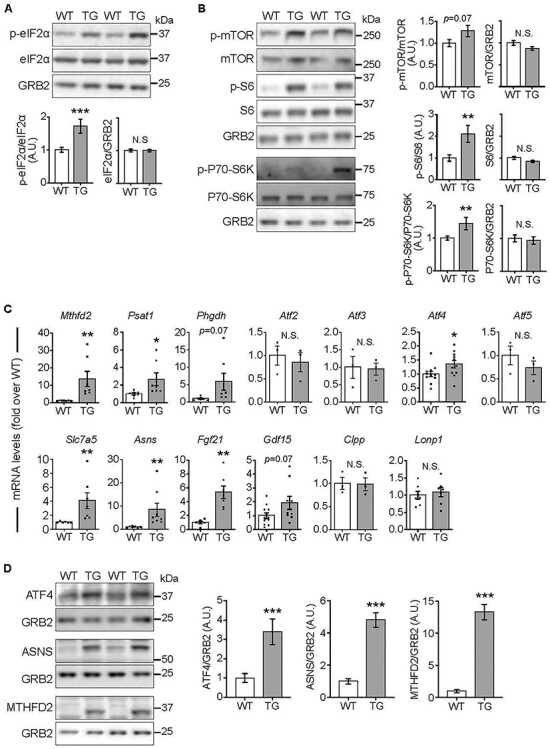
Elevated mTORC1 signaling and ISR activation in the heart after 18 weeks of FXN depletion. All measurements were done in heart lysates from WT and TG mice fed Doxy for 18 weeks. (A) Phosphorylation status of eIF2α. Top: Representative immunoblot of p-eIF2α(Ser52), total eIF2α and GRB2 from heart lysates of WT and TG mice. Lower: Quantification (n = 9 WT/7 TG). (B) mTORC1 signaling. Left: Representative immunoblots of p-mTORC1 (Ser2448), total mTOR, p-S6 (Ser235/236), total S6, p-P70-S6K (Thr389), total P70-S6K and GRB2 (loading control). Right: quantification (n = 9 WT/7 TG). (C) Transcripts levels [normalized to Actb (β-actin) and expressed relative to WT] of stress signaling targets. Points show values from each sample. Value = 1 means no change, (n = 3–11/genotype). (D) Protein levels of ATF4 and targets. Left: Representative immunoblots. GRB2 was used as loading control. Right: Quantification (n = 6/genotype). In all panels: bars represent mean ± SEM. Statistical comparison was by unpaired t-test, *P < 0.05, **P < 0.01, ***P < 0.01.
Figure 6 .
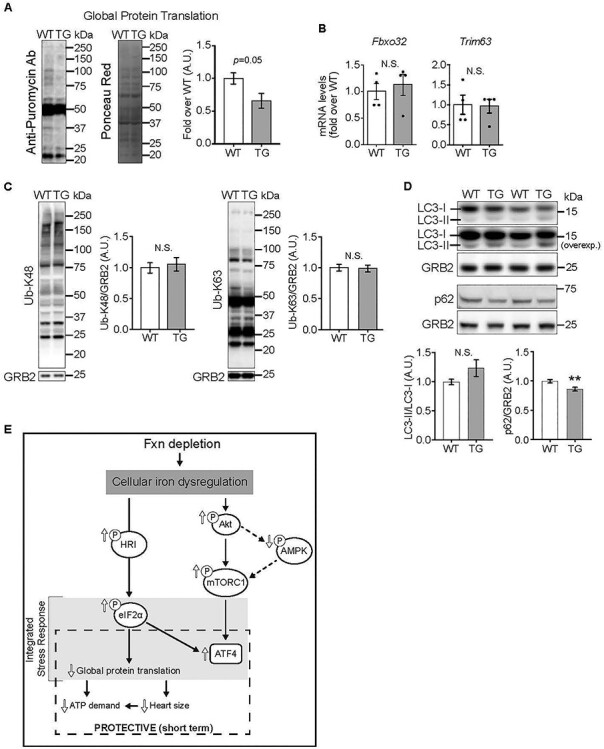
Decreased global protein translation in hearts depleted of FXN for 18 weeks (A) Global protein translation was determined using the SUnSET method. Left: Representative immunoblot using anti-puromycin antibody, with Ponceau Red staining (shown in gray scale) to estimate protein loading. Right: Quantification of puromycin signal (P = 0.05 by unpaired t-test, n = 4/genotype). (B, C) Protein degradation, estimated by relative mRNA levels of Fbxo32 and Trim63 (B, n = 4/genotype, not significant (N.S.), by unpaired t-test) and by K48- and K63-linked ubiquitination of proteins by immunoblot (C; N.S. by unpaired t-test, n = 6/genotype). (D) Autophagy, estimated by levels of LC3 and lipidated LC3 (n = 4/genotype, N.S.) and levels of p62 (n = 4/genotype, **P < 0.01), all by immunoblot. Statistical comparison was by unpaired t-test. (E) Model of altered signaling in FXN-depleted hearts. All values are the mean ± SEM.
The extent of oxphos suppression in TG heart mitochondria is substrate dependent
Many of the ISC-containing proteins within the mitochondrial matrix participate in substrate oxidation that is necessary for oxidative ATP synthesis, an absolute requirement for heart function. In particular, ISC-containing proteins are present in substrate oxidation pathways for both pyruvate and fatty acids (Fig. 2A). Thus, FXN depletion was expected to be accompanied by lower oxphos capacity in heart mitochondria, whether supplied with pyruvate or fatty acids. To this end, we performed a detailed bioenergetics analysis in isolated heart mitochondria supplied with pyruvate (with malate), succinate (with rotenone to inhibit backflow of electrons to Complex I) or one of two fatty acid substrates [palmitoyl-L-carnitine (PCarn) or octanoyl-L-carnitine (OCarn)]. Oxidation was evaluated as O2 consumption rate (JO2) under the following conditions: saturating concentrations of substrate and ADP were used to evaluate maximal oxphos; oligomycin, an ATP synthase inhibitor, was used to determine maximal leak-dependent JO2 and the chemical uncoupler, FCCP, was used to evaluate maximal ETC capacity.
In heart mitochondria from TG mice fed Doxy for 10 weeks (83% FXN loss), with pyruvate/malate as the substrate, ADP-stimulated oxphos tended to be lower and leak-dependent JO2 was significantly lower (Supplementary Material, Fig. S2). However, for succinate or PCarn/malate, maximal oxphos, leak and ETC capacity were similar between WT and TG mitochondria (Supplementary Material, Fig. S2). After 18 weeks of Doxy, (complete FXN loss in TG hearts) TG mitochondria supplied with pyruvate/malate or succinate showed a 40–50% deficit in maximal oxphos, leak-dependent JO2 and maximal ETC capacity (Fig. 2B). In contrast, TG mitochondria supplied with PCarn/malate showed a modest (~15%) decrease in these parameters (Fig. 2B).
We noted that maximal ADP-driven JO2 in WT mitochondria supplied with PCarn/malate was similar to the rate in TG mitochondria supplied with pyruvate/malate. Thus, we questioned if the modest changes in bioenergetics parameters in TG mitochondria oxidizing PCarn/malate reflected a limitation of the PCarn/malate reaction. To test this possibility, we used octanoyl-L-carnitine (OCarn) which, despite its shorter carbon chain length (C8:0), had higher maximal oxphos than PCarn (C16:0) likely because more substrate can be oxidized without causing uncoupling [32]; indeed, the ratio of maximal oxphos to maximal leak JO2 was ~ 5.5 for PCarn/malate and ~8.8 for OCarn/malate in WT mitochondria. Yet, even with OCarn/malate, there was only a modest decrease in JO2 in TG mitochondria (Fig. 2B).
We noted that maximal leak-dependent (oligomycin-insensitive) JO2 was lower in TG heart mitochondria supplied with any of the substrates tested (Fig. 2B). Leak-dependent JO2 can reflect supply of reducing equivalents to the ETC to generate the driving force for oxphos or can reflect proton leak back into the matrix by pathways other than the ATP synthase, lowering the efficiency of substrate oxidation to generate ATP. To discriminate between these possibilities, we evaluated leak-dependent JO2 as a function of driving force (membrane potential, ΔΨm), by titrating OCarn/malate oxidation with the Complex III inhibitor antimycin, in the presence of oligomycin, allowing leak-dependent JO2 to be compared between TG and WT at the same driving force (i.e. supply of reducing equivalents). Consistently, JO2 was lower at any given ΔΨm in TG versus WT heart mitochondria (Fig. 2C), indicating lower proton leak in TG mitochondria.
Altogether, the bioenergetics data indicate that, as expected, FXN loss leads to lower oxphos capacity, especially after 18 weeks of Doxy. The data also reveal a substrate dependence of the lower oxphos capacity, which, along with the lower proton leak in TG mitochondria, suggests that mitochondrial substrate metabolism in FXN-depleted mitochondria cannot simply be ascribed to changes in ISC-containing proteins.
Broad decreases in levels of complex I and tricarboxylic acid cycle enzymes in TG heart mitochondria
To address the differential impact of FXN loss on substrate oxidation, and to more broadly understand the impact of FXN depletion on heart mitochondria, we undertook a proteomics analysis of heart mitochondria isolated from 18-week Doxy-fed WT and TG mice. A total of 595 mitochondrial proteins were identified (Supplementary Material, Table S1). A volcano plot of TG protein abundance relative to WT levels revealed that many proteins were less expressed in TG mitochondria (Fig. 3A and B); these included some, but not all, ISC-, lipoic acid (LA)- or heme-containing proteins, as well as other proteins. Among the most substantially decreased non-ISC-containing proteins were PMPCA and PMPCB, the α and β subunits of the matrix-localized mitochondrial processing peptidase, a major protease that has FXN as a substrate [33]. Several proteins were increased in TG mitochondria (see below) (Fig. 3A).
Among the ISC-, LA- and heme-containing proteins, ferrochelatase (FECH), required for cellular heme synthesis, was one of the most substantially decreased proteins we detected (Fig. 3A and B). Lower FECH expression was confirmed by immunoblot in heart lysates and isolated mitochondria (Supplementary Material, Fig. S3). Heme-containing matrix proteins were, however, not uniformly decreased in abundance in TG mitochondria. Cytochrome b of Complex III was significantly decreased, whereas the cytochrome C1 subunits of Complex III and the mitochondrial-DNA-encoded mt-CO1 subunit of Complex IV were unchanged (Fig. 3C: brown dots). Cytochrome c was not detected by mass spectrometry; however, immunoblotting revealed a robust decrease in expression in TG mitochondria (Supplementary Material, Fig. S3A). Besides FECH, other substantially decreased (q < 0.1) ISC-containing proteins were subunits of Complex I (Fig. 3B). SDHB, aconitase and the Rieske protein of Complex III trended lower, whereas lipoic acid synthetase (LIAS) (required for LA synthesis) was clearly unchanged, as was ISC-containing NDUFS7 of Complex I (Fig. 3C). Immunoblotting of heart lysates and isolated heart mitochondria confirmed lower SDHB, whereas differences between WT and TG could not be detected in the abundance of aconitase, Rieske protein or LIAS (Supplementary Material, Fig. S3). Note that antibodies were first evaluated to determine conditions that yielded an approximately linear dynamic range (not shown). Thus, ISC-containing proteins were not uniformly downregulated in mitochondria from FXN-depleted hearts. Moreover, similar findings were obtained in isolated mitochondria and heart lysates; this indicates that the findings in isolated mitochondria do not reflect a subselection of the healthiest mitochondria.
We next focused on proteins of the main substrate oxidation pathways in the heart, particularly subunits of the ETC and ATP synthase, tricarboxylic acid (TCA) cycle and β-oxidation proteins, subunits of pyruvate dehydrogenase (PDH) and carriers for pyruvate (MPC1/2), activated fatty acids (CPT1a/b) and succinate and malate (SLC25A10). These are plotted in Figure 3C and include proteins containing ISCs (yellow dots), heme (brown dots) and LA (green). The most affected pathways or protein complexes were the TCA cycle (6/14 proteins decreased including SDHA, P < 0.05, q < 0.1), PDH (3/5 subunits decreased, P < 0.05, q < 0.1) and Complex I (15/36 subunits were decreased, P < 0.05, q < 0.1); immunoblotting of SDHA and NDUFA9 (Complex I subunit) also revealed significant decreases in these proteins in TG heart lysates and mitochondria (Supplementary Material, Fig. S3). Of the 24 members of these pathways determined by mass spectrometry to be decreased (q < 0.1) in TG mitochondria, four contained ISCs (out of the 12 ISC proteins in these pathways), one contained heme (out of three heme-containing proteins) and one contained LA (of two). Only one protein was increased (q < 0.1; a subunit of Complex IV). Levels of carriers of pyruvate and fatty acids were unchanged, though there was ~ 50% less SLC25A10 (Fig. 3C). The lower abundance of SLC25A10 did not reflect a broad decrease in SLC25A family members (Supplementary Material, Table S1).
In light of the decreased proton leak (Fig. 2C) and the recent report that proton leak is mediated by the adenine nucleotide translocase (ANT) [34], we noted the levels of ANT1 and ANT2 in the proteomics dataset (Supplementary Material, Table S1). ANT1, the major isoform in the heart, showed similar expression in TG and WT mitochondria (P = 0.6).
Because ETC subunits function in complexes and higher order supercomplexes (SC), we performed blue native polyacrylamide gel electrophoresis (BN-PAGE) analysis to determine complex and SC abundance and activity in heart mitochondria from mice fed Doxy for 18 weeks. The abundance of assembled Complexes III and IV and ATP synthase was similar between WT and TG mitochondria (Fig. 3D). However, TG mitochondria exhibited slightly less assembled Complex I and II (Fig. 3D) and their in-gel activity (Supplementary Material, Fig. S3D), and lower abundance of SCs containing Complex I, III and IV (Fig. 3D; Supplementary Material, Fig. S3D).
Finally, we determined if there was a substantial alteration in substrate oxidation in FXN-depleted hearts, evident as a switch from β-oxidation to glycolysis when oxphos is compromised [35]. Levels of mRNA for transporters and enzymes involved in glucose transport and metabolism (Slc2a4, Hk2, Pfkfb1) and fatty acid transport and metabolism (Cd36, Cpt1b, Acadm) were similar in hearts from WT and 18-week Doxy-fed mice (Supplementary Material, Fig. S4A). Furthermore, triglycerides levels, measured in whole hearts, were similar between WT and TG hearts (Supplementary Material, Fig. S4B).
Many mitochondrial proteins were expressed at lower abundance in hearts from 18-week Doxy-fed TG mice. Yet, a few proteins were increased. In particular, MTHFD1L (methylene tetrahydrofolate dehydrogenase 1 like, a matrix protein involved in folate metabolism) was barely detectable in WT mitochondria but readily detected after FXN loss (Fig. 3A). MTHFD1L is a target of the transcription factor ATF4 [36]. CYB5R1 was also substantially increased and is an ATF4 target [37]. ATF4 protein expression is normally low, but increases when translation initiation factor eIF2α is phosphorylated, under the activation of the ISR. ATF4 levels can also be induced via the mTORC1 pathway [38, 39]. Thus, we evaluated the status of the ISR and mTORC1 signaling.
Stress signaling and aberrant nutrient signaling in FXN-depleted hearts
We evaluated the phosphorylation status of eIF2α, the core event of ISR activation [13], in heart lysates. Compared with WT hearts, TG hearts from 18-week Doxy-fed mice showed substantially higher levels of phosphorylated eIF2α, with no changes in total eIF2α (Fig. 4A). Investigating the mTORC1 pathway, though phosphorylation of mTOR showed no difference between WT and TG hearts (Fig. 4B), downstream targets, namely p-S6 and p-P70-S6K, were more phosphorylated in TG hearts, consistent with increased activation of mTORC1 (Fig. 4B).
To evaluate downstream consequences of mTORC1/ISR activation, we measured levels of downstream transcripts and proteins (e.g. Mthfd2 and Fgf21 [18, 20]). After 10 weeks on Doxy diet, there were no differences between WT and TG hearts (Supplementary Material, Fig. S5). In contrast, TG hearts from 18-week Doxy-fed mice had higher mRNA levels of one-carbon metabolism enzymes, components of amino acid transport/metabolism, Fgf21 and Atf4 (Fig. 4C). Protein levels of ATF4, as well as MTHFD2 and ASNS (two ATF4 targets), were substantially elevated in TG heart lysates (Fig. 4D).
Insights into upstream regulators of eIF2α and mTORC1
To gain insight into drivers of the elevated p-eIF2α and mTORC1 signaling in hearts from 18-week Doxy-fed mice, we assessed the activation of AMPK and AKT, which, respectively, negatively and positively regulate mTORC1. AKT has also been reported to inhibit AMPK in the heart [40, 41]. Phosphorylation of AKT was increased at both phosphorylation sites (Thr308 and Ser473) in TG heart lysates, indicating increased AKT activation (Fig. 5A). Phosphorylation of AMPK was consistently less in FXN-depleted hearts, supporting suppressed AMPK signaling (Fig. 5B).
Figure 5 .
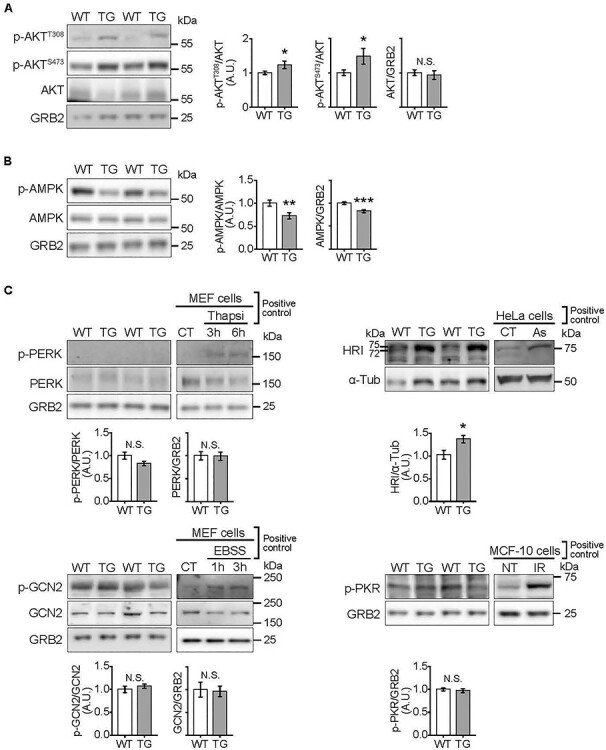
Altered activation of kinases upstream of eIF2α and mTORC1 in hearts depleted of FXN for 18 weeks. All measurements were done in heart lysates from WT and TG mice fed Doxy for 18 weeks. (A) AKT signaling. Left: Representative immunoblots of p-AKT (Ser473), p-AKT (Thr308), total AKT and GRB2 (loading control). Right: Quantification (n = 9 WT/7 TG). (B) AMPK signaling. Left: Representative immunoblots of p-AMPK (Thr302), total AMPK and GRB2 (loading control). Right panel: Averaged p-AMPK/AMPK, p-AMPK/GRB2 and AMPK/GRB2 values (n = 9 WT/7 TG). (C) Kinases upstream of eIF2α: PERK, GCN2, PKR and HRI. Representative immunoblots of p-PERK (Thr980), total PERK, p-GCN2 (Thr899) and total GCN2, HRI and p-PKR (Thr451), with GRB2 and α-Tubulin (α-Tub) as loading controls. Averaged p-PERK/PERK, p-GCN2/GCN2, HRI/(α-Tub) and pPKR/GRB2 values are shown below respective immunoblot. N = 4–9/genotype. Controls for antibodies: p-PERK: MEF treated with Thapsigargin 1 μM (Thapsi) to induce ER stress; p-GCN2: MEF cells cultured in EBSS to deprive cells of amino acids; p-PKR: MCF-10 cells were irradiated (20 Gy) to cause DNA damage to activate PKR (NT: non-treated, IR: irradiated); HRI: HeLa cells were incubated in 100 μM Na-Arsenite (As) for 1 h. All values are the mean ± SEM. Statistical comparison was by unpaired t-test, *P < 0.05, **P < 0.01, ***P < 0.001.
Four kinases phosphorylate eIF2α: PERK, GCN2, PKR and HRI [13]. Their activation was evaluated by phosphorylation status, by immunoblotting. Positive controls were generated to test the reliability of each antibody. Phospho-PERK was undetectable in heart lysates from both WT and TG mice (Fig. 5C). Additionally, BIP/GRP78 levels, usually increased with endoplasmic reticulum stress, were unchanged (Supplementary Material, Fig. S6A). Both p-GCN2 and p-PKR were detectable, but levels were similar in WT and TG hearts (Fig. 5C). HRI was evaluated using an antibody that detects total HRI, because an antibody against p-HRI is not available. When samples are run slowly on a 7% gel, activated HRI is seen as a doublet with a more intense higher molecular weight band or as a shift of a more intense band to a higher molecular weight (e.g. [11, 42, 43]). Using heart lysates, we detected a strong band at ~ 75 kDa in TG samples, whereas a less intense doublet was detected in WT samples (Fig. 5C, and see also Supplementary Material, Fig. S6B).
Finally, because mTORC1 and eIF2α activation have opposite efforts on protein synthesis, we evaluated global protein translation in vivo, by puromycin incorporation into elongating peptide chains [44]. A decrease in the signal from the anti-puromycin antibody in heart lysates from 18-week Doxy-fed TG mice indicated that global protein synthesis was lower (Fig. 6A). For a more complete picture of proteostasis, insight into protein breakdown was obtained. Transcript levels of E3 ubiquitin ligases, Atrogin1 (Fbxo32) and MuRF1 (Trim63), were similar in WT and TG hearts (Fig. 6B), as was protein polyubiquitination (Fig. 6C). Additionally, autophagy was estimated by measuring protein levels of LC3II, the lipidated form of LC3I, and p62. LC3II relative to LC3I was similar in WT and TG hearts, whereas p62 expression was lower in TG hearts from 18-week Doxy mice (Fig. 6D).
Discussion
The main findings and proposed mechanistic links are shown in Figure 6E. After 18 weeks on Doxy diet, and at least 8 weeks with FXN levels <20% of normal, TG hearts did not display overt hypertrophy. Furthermore, cardiac contractility was maintained, which we hypothesize is partly contributed by some preservation of β-oxidation, though pyruvate oxidation was lower. The bioenergetics changes generally matched the proteomics changes and were not entirely associated with a loss of ISC proteins. Global protein translation was lower in TG hearts from 18-week Doxy mice, whereas protein degradation by ubiquitination did not appear to be increased. In parallel, there were perturbations in AMPK and mTORC1 signaling and in AKT in TG hearts from 18-week Doxy-treated mice. Activated AKT could lead to the observed decrease in AMPK phosphorylation and independently drive the mTORC1 signaling. Lower AMPK activity would also be permissive for mTORC1 activation. The ISR was also activated, likely by HRI. We propose that the trophic state of TG hearts reflects an mTORC1-driven hypertrophic stimulus that is constrained by phosphorylation of eIF2α resulting in a net decrease in protein translation and, therefore, a decrease in the ATP demand of this energetically costly process. We speculate that this response is protective for the FXN-depleted heart, at least in the short term.
The TG model
The mouse model used here is the same genetic model as that used by Chandran et al. [28]. However, different Doxy regimens were used. Chandran supplied Doxy by intraperitoneal injection and in the drinking water, whereas we supplied Doxy in the chow. The time course and extent of FXN loss in the heart, and the BW loss, were similar for the two Doxy regimens. Yet, mortality of the mice used in the present study was lower; our mice showed ~ 35% mortality by 18 weeks of Doxy, whereas Chandran observed ~ 35% mortality by 10 weeks and >50% by 18 weeks of Doxy. Chandran tested several Doxy doses; higher dose associated with greater mortality. We propose that the dose used in the present study was lower and/or less toxic than the lowest dose used by Chandran (and on which they reported phenotypes).
Heart function
After 18 weeks on Doxy, hearts from TG mice showed no sign of fibrosis or apoptosis and showed signs of elevated contractility. This differs from the MCK model in which cardiac fibrosis, lower ejection fraction and markers of apoptosis were evident [7–11]. Chandran observed lower ejection fraction and fibrosis after 24 but not 12 weeks of Doxy [28]. Intermediates time points were not evaluated. We performed echocardiography on a small number of TG mice after 20 weeks of Doxy diet; ejection fraction continued to be above normal, whereas increased diastolic dimensions suggested the presence of hypertrophy (not shown). We did not pursue later time points because the condition of the mice deteriorated rapidly after 20 weeks of Doxy.
The heart from 18-week Doxy mice was not normal. Elevated septal wall thickness during systole in 18-week Doxy TG hearts, together with decreased LV lumen diameter in systole, suggests that TG hearts were hypercontractile. Thus, for the same heart rate, TG hearts worked harder to maintain cardiac output. Consistent with this possibility, there was a shift in MHC isoform, with increased α-MHC and decreased β-MHC [45, 46]. These changes in MHC isoform expression are opposite to what has been reported in failing hearts [31, 47], whereas a similar pattern of change was described in hibernating ground squirrels [48] and treadmill-trained rats [49].
Changes in transcript levels of glycolytic and β-oxidation enzymes that are associated with a cardiac energy crisis [35] were absent in TG hearts from 18-week Doxy-fed mice. Moreover, ejection fraction was similarly elevated in TG hearts at 10 and 18 weeks of Doxy, suggesting maintained contractility. These findings suggest that cardiac energy production was sufficient in TG hearts. It seems reasonable to suggest that β-oxidation contributed to the energy production in TG hearts, supported by the preservation of β-oxidation in isolated heart mitochondria, and the similar levels of triglycerides in those hearts. Greater oxphos efficiency, evidenced by lower proton leak, might also be a factor. Another consideration is that, though maximal oxphos capacity for pyruvate was lower, this maximal capacity includes a reserve, such that sufficient capacity might still be available for the pyruvate contribution to ATP production. Finally, we cannot rule out that glycolytic flux was elevated to support energy demand in TG hearts.
Mitochondrial bioenergetics and proteomics
A detailed bioenergetics analysis was performed in FXN-depleted heart mitochondria, which was lacking in other models of FRDA. This analysis yielded the novel observation that oxphos from fatty acids was less affected by FXN loss than oxphos derived from pyruvate. One explanation could be that, unlike pyruvate oxidation, oxphos from β-oxidation does not strictly depend on the TCA cycle and Complex I (Fig. 2A), the pathways with the greatest decrease in protein abundance in TG hearts. Furthermore, though the levels of electron transfer flavoprotein dehydrogenase (ETFDH) and short-chain specific acyl-CoA dehydrogenase were lower, the acyl-CoA dehydrogenases have overlap in substrate specificity, and Complex III, IV and ATP synthase were minimally changed with FXN loss. Thus, oxphos from β-oxidation would encounter fewer bottlenecks than pyruvate oxidation in TG hearts. That triglyceride levels in TG hearts were mostly within the WT range indirectly supports that β-oxidation was functional in TG hearts.
The heart mitochondrial proteome we obtained from 18-week Doxy-fed mice is, to our knowledge, the first broad proteomics analysis of FXN-depleted mitochondria. The closest comparison is with a proteomics analysis of heart mitochondria from mouse models of mtDNA depletion [37]. Similar to FXN-depleted hearts, mtDNA depletion activated the ISR. Important similarities between FXN- and mtDNA-depleted heart mitochondria are a broad decrease in oxphos proteins and ubiquinone biosynthetic enzymes (Supplementary Material, Table S1), combined with an upregulation of enzymes that promote the conversion of glutamate to proline (Supplementary Material, Table S1). A prominent difference in our proteomics data is the lack of change (or decrease) in the abundance of some heat shock proteins and proteases (e.g. CLPP and LONP1) that were strongly increased in mtDNA-depleted hearts [37]. This difference suggests that under stress conditions, mitochondrial proteases and chaperones are regulated by a mechanism other than the ISR.
Levels of ISC-containing proteins were expected to be lower (e.g. [9]). Indeed, FECH was substantially lower, and three Complex I subunits, SDHB and ETFDH were significantly, though modestly, decreased. Yet the other ISC-, heme- and LA-containing proteins were not significantly lower. A broad survey of the literature shows that ISC-containing proteins and their activity are not uniformly decreased when ISC biosynthetic machinery is lost. In particular, the abundance of FECH appears most sensitive to the loss of ISC biosynthetic proteins, followed by Complex I subunits/activity, then Complex II/SDHB and aconitase, and, finally, Complex III activity appears relatively insensitive to the loss of ISC-containing proteins [6, 7, 9, 11, 50–53]. An explanation for this unequal effect on ISC-containing proteins is not available, though mechanisms can be proposed. Longer half-life for Complex III and IV subunits compared with Complex I and SDH subunits (Supplementary Data 1 in [54]) suggests differential protein turnover could be a factor. Also, PO2 of cardiac mitochondria has been estimated to reach levels as low as 10 mmHg [55], raising the question of whether some FXN-independent ISC biogenesis shown to occur in hypoxia [56] is possible in cardiac mitochondria.
ISR and mTORC1 activation
We found that both the ISR and mTORC1 were activated in the heart of 18-week Doxy TG mice. Elevated p-AKT and lower p-AMPK could both have contributed to mTORC1 activation [57]. Elevated p-AKT has been linked to increased intracellular iron [58] and was evident in TG hearts. The lower p-AMPK was surprising, but has been reported in hearts depleted of acyl-CoA synthase-1 [59], and associated with higher p-AKT in the heart [40, 41], likely via phosphorylation of the AMPK-α1 subunit that prevents its activation [60]. We note that suppression of AMPK signaling and elevated mTORC1 would favor lower macroautophagy; however, this would need to be tested directly. Of the four kinases shown to phosphorylate eIF2α, only HRI showed evidence of activation in TG hearts. HRI activation was also found in the MCK mouse heart [11]. HRI was recently shown to be activated by DELE1 upon its cleavage and release of a soluble fragment from the inner mitochondrial membrane [61, 62]. DELE1 was not detected in our proteomics data and nor in transcriptomics data reported by Chandran et al. [28]. Thus, it seems more likely that HRI activation in TG (and MCK) hearts resulted from heme depletion (because of lower FECH), the canonical activator of HRI [63].
Elevated mTORC1 signaling would predict hypertrophy (e.g. [59]); however, there were no signs that cardiomyocyte size was increased. The phosphorylation of p-eIF2α relative to total eIF2α was also increased. Consistent with the latter, global protein translation was decreased, and cardiomyocyte CSA was smaller, possibly reflecting suppression of normal growth. Altogether, we interpret these results to suggest that, in TG hearts, an anabolic stimulus was constrained by eIF2α phosphorylation. MCK mice also showed elevated p-eIF2α [11]; however, cardiac hypertrophy was substantial [7, 9, 11]. The presence of hypertrophy in MCK mice suggests that mTORC1 signaling was elevated; however, mTORC1 signaling was not evaluated in that model. Interestingly, in the latter study, total eIF2α was increased (Fig. 4A in [11]); thus, the amount of non-phosphorylated eIF2α may have been maintained in MCK hearts, allowing for increased global protein translation and a substantial degree of hypertrophy. There was no sign of higher total eIF2α in TG hearts. Our study demonstrates that elevated mTORC1 signaling and the ISR can coexist and shows the importance of evaluating both pathways.
Phenotype versus FXN level
FXN abundance of 17% of the normal level was sufficient to preserve bioenergetics parameters, and was not associated with changes in the expression of ATF4 target genes and nor in FECH protein abundance. This raises the possibility that as little as ~ 20% of the normal FXN level may be enough to prevent pathology. Yet, a few points need to be considered. First, the bioenergetics profile at the 10-week Doxy time point may in part reflect the slow turnover of some mitochondrial proteins. Second, our study cannot rule out negative consequences of long-term expression of only 20% of the normal level of FXN in the heart. Concerning the 18-week Doxy time point, it should be noted that, although FXN protein was almost undetectable in TG hearts, substantial oxphos capacity remained. Even FECH was still present at 50% of the WT level, and LIAS expression was normal. Another consideration is the possibility that other factors could have interacted with FXN loss to generate the cardiac phenotype at the 18-week time point. For example, Doxy-treated mice were shown to have an altered microbiome [64], and the microbiome can influence cardiac pathology outcomes [65, 66]. Thus, discordance between FXN abundance and phenotypes needs to be considered here and likely in other FRDA models.
Implications
We show that several changes in stress signaling can occur in the FXN-depleted heart. The relative extent of these changes might differ among individuals with FRDA and contribute to the variability in cardiac phenotypes, including the degree of cardiac hypertrophy. We also show that the impact of FXN loss on substrate metabolism can be substrate dependent, raising the question of whether vulnerability to FXN depletion depends on metabolic phenotype; for example, neurons may be particularly vulnerable to FXN depletion because they rely on aerobic glucose oxidation. Finally, small molecules exist for the AMPK, mTORC1 and ISR pathways, raising the question of whether these pathways could be targeted to treat FRDA, as has been suggested for other mitochondrial diseases [17, 22, 25].
Materials and Methods
Mouse model
Mice were used in accordance with mandated standards of care, and use was approved by the Thomas Jefferson University Institutional Animal Care and Use Committee. Mice were housed at 22°C, under a 12-h light–dark cycle (lights on at 07:00). We used a Doxy-inducible model of FXN knockdown [28]. Here, TG mouse (on C56BL/6J background) contains a Tet-On FXN shRNA expression cassette. Littermates that do not contain the shRNA cassette were used as controls (WT). Doxy was administered in the chow (200 p.p.m. in Chow 5SHA, Animal Specialties) to WT and TG mice starting at 9 weeks of age. Only male mice were used for experiments.
Cell culture and stress conditions for antibody positive controls
Mouse embryonic fibroblast (MEF), HeLa and C2C12 cells were cultured in Dulbecco’s modified Eagle’s medium (11965-118, Invitrogen) supplemented with 10% fetal bovine serum, 2 mM glutamine and 100 U/ml penicillin and 100 mg/ml streptomycin and maintained at 37°C in a 5% CO2-humidified air. A series of positive controls were generated using stress conditions to validate the activation of the ISR kinases (PERK, GCN2, HRI and PKR) by their respective antibodies. For p-PERK antibody, MEF cells were treated with thapsigargin 1 μM for 3 and 6 h, to have a positive control for ER stress. For p-GCN2 antibody, MEF cells were cultured in Earle’s Balanced Salt Solution (EBSS; 5.33 mM KCl, 26.19 mM NaHCO3, 117.24 mM NaCl, 1.104 mM NaH2PO4 and 5.56 mM D-Glucose) for 3 and 6 h, to generate an amino acid deprivation condition. For HRI activation, HeLa cells were treated with Na-arsenite (100 μM AsNaO2) for 1 h. A positive control for p-PKR antibody was generated using MCF-10 cells irradiated with a CellRad350 at 0.8–0.9 Grey/min. Finally, a positive condition for apoptosis was generated by incubating C2C12 cells with 1 μM staurosporin for 1 and 3 h.
Echocardiography
Transthoracic two-dimensional M-mode echocardiography was performed using an echocardiographic imaging system (Vevo 2100, VisualSonic, Toronto, Canada) with a 40-MHz probe. Mice were anesthetized through the inhalation of isoflurane (∼1–2%); anesthesia was titrated with the goal of equalizing heart rate in all mice. M-mode interrogation was performed in the parasternal short-axis view at the level of the greatest LV end-diastolic dimension. LV wall thickness and internal dimensions were measured and used for calculation of fraction shortening and ejection fraction values.
Isolation of heart mitochondria
Heart mitochondria were isolated as described in [56]. All steps were performed on ice or at 4°C. The heart was dissected, washed in heart isolation buffer (HB: 225 mM mannitol, 75 mM sucrose, 20 mM HEPES, 0.1 mM EGTA, pH 7.4), minced with a razor blade, then suspended in HB + 0.5% defatted bovine serum albumin (BSA) in a glass/Teflon Potter-Elvehjem homogenizer and homogenized (350 rpm, 10 passes). Samples were centrifuged (5 min, 500 g); the supernatant was centrifuged (10 min, 9000 g), then the pellet was resuspended in HB devoid of BSA then centrifuged again (10 min, 9000 g). The final pellet was resuspended in HB (no BSA) in a volume that resulted in a protein concentration of ~ 20 mg/ml. Protein concentration was determined by bicinchoninic acid (BCA) assay (ThermoFisher Scientific 23 228, 1 859 078).
Bioenergetics analyses in isolated heart mitochondria
For most experiments, O2 consumption (JO2) was measured using the Seahorse XF24 Analyzer (Seahorse Bioscience, Billerica, MA). Isolated mitochondria were studied essentially as we have done previously [67]. Each well of the custom microplate contained 10 μg of mitochondria suspend in mitochondria assay medium (MAS; 70 mM sucrose, 22 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, 0.2% defatted BSA, pH 7.4 at 37°C). Amount of mitochondria had been optimized such that the O2 versus time signal was linear under all conditions. The microplate was centrifuged (2000 g, 20 min, 4°C) to promote adhesion of mitochondria to the plastic. Attachment was verified after centrifugation and again after experiments. Different substrates were tested: malate–pyruvate (5/10 mM), succinate (10 mM + 1 μM rotenone to inhibit Complex I and thereby prevent reverse electron flow through that complex), palmitoyl-L-carnitine–malate (20 μM/1 mM), octanoyl-L-carnitine (200 μM + 1 mM malate). Oligomycin (4 μg/ml) was used to measure non-phosphorylation ‘leak’ respiration JO2, and the uncoupler FCCP (6.7 μM) was used to measure maximal ETC activity. Antimycin titrations of octanoyl-L-carnitine (200 μM + 1 mM malate) were done in the presence of oligomycin (4 μg/ml) using the Oroboros O2k, with 200 μg of mitochondria per reaction, in MAS.
Fluorometric measurements in isolated mitochondria
Fluorometric measurements of mitochondrial membrane potential (ΔΨm) were performed using a multiwavelength-excitation dual-wavelength-emission fluorimeter (DeltaRAM, PTI). Briefly, isolated mitochondria (375 μg) were resuspended in 1.5 ml of intracellular medium containing 120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM Tris-HEPES at pH 7.2 and maintained in a stirred thermostated cuvette at 36°C. TMRM (1 μM) was added prior the experiment. Antimycin titrations of octanoyl-L-carnitine (200 μM μM + 1 mM malate) were done in the presence of oligomycin (4 μg/ml). FCCP (6.7 μM) was used to obtain maximal membrane depolarization.
Immunoblot analysis
For western blot analysis, heart was quickly frozen in liquid nitrogen. After that, tissue was homogenized with a lysis buffer containing: 150 mM NaCl, 25 mM HEPES, 2.5 mM EGTA, 1% Triton 100X, 1% Igepal (10%), 0.10% sodium dodecyl sulfate, 0.1% deoxycholate, 10% glycerol, protease inhibitor (Roche 11 873 580 001) and phosphatase inhibitor cocktail (200 mM sodium fluoride, 200 mM, imidazole, 115 mM sodium molybdate, 200 mM sodium orthovanadate, 400 mM sodium tartrate dihydrate, 100 mM sodium pyrophosphate and 100 mM β-glycerophosphate), using a glass/Teflon homogenizer at 300 rpm. After that, heart lysates were incubated at 4°C for 45 min, then centrifuged at 18000g for 20 min. From the supernatant, protein concentration was measured by BCA assay. Specifically, for LC3B detection in heart, we used T-PER tissue protein extraction reagent (ThermoFisher Scientific 78 510). Hearts were quickly pulverized over dry ice and then incubated for 1 h in T-PER at 4°C (shaking them at 1500 r.p.m.). After that, samples were centrifuged at 18000 g for 30 min at 4°C. From the supernatant, protein concentration was measured by BCA assay. For western blot analysis from cultured cells, cell lysis was obtained using RIPA buffer containing protease and phosphatase inhibitors. Cell lysates were spun at 12000 rpm for 10 min. From the supernatant, protein concentration was measured by BCA assay. For western blot analysis of mitochondrial proteins, isolated heart mitochondria were resuspended in 4× sample buffer. Primary antibodies were used for overnight incubation diluted in TBS-T 1% and are listed below.
Primary antibodies used in immunoblot analysis.
| Antibody | Catalog number | Dilution | Host |
|---|---|---|---|
| Phospho-mTOR (Ser2448) | Cell Signaling Technology (CST)-2971 | 1:1000 | Rabbit |
| mTOR | CST-2972 | 1:1000 | Rabbit |
| Phospho-P70-S6K (Thr389) | CST-9234 | 1:1000 | Rabbit |
| P70-S6K | CST-9202 | 1:1000 | Rabbit |
| Phospho-S6 (Ser235/236) | CST-2211 | 1:1000 | Rabbit |
| S6 | CST-2217 | 1:1000 | Rabbit |
| Phospho-AMPK (Thr172) | CST-2535 | 1:1000 | Rabbit |
| AMPKα | CST-2532 | 1:1000 | Rabbit |
| Phospho-AKT (Thr308) XP | CST-13038 | 1:1000 | Rabbit |
| Phospho-AKT (Ser473) XP | CST-4060 | 1:1000 | Rabbit |
| AKT | CST-9272 | 1:1000 | Rabbit |
| Phospho-GCN2 (Thr899) | Abcam (Ab)75 836 | 1:1000 | Rabbit |
| GCN2 | CST-3301S | 1:1000 | Rabbit |
| Phospho-PERK (Thr980) | CST-3179 | 1:1000 | Rabbit |
| PERK | CST-5683 | 1:1000 | Rabbit |
| Phospho-PKR (Thr451) | Invitrogen 44-668G | 1:1000 | Rabbit |
| HRI | EMD Millipore 07–728 | 1:5000 | Rabbit |
| Phospho-eIF2α (Ser51) | CST-3398 | 1:1000 | Rabbit |
| eIF2α | CST-5324 | 1:1000 | Rabbit |
| ATF4 | Novus Biologicals NB100852 | 1:500 | Goat |
| BIP | CST-3177 | 1:1000 | Rabbit |
| MTHFD2 | Proteintech 12 270-1-AP | 1:1000 | Rabbit |
| ASNS | Proteintech 14 681-1-AP | 1:1000 | Rabbit |
| FXN | Abcam (Ab)175 402 | 1:1000 | Rabbit |
| GRB2 | SC-8034 | 1:1000 | Rabbit |
| α-Tubulin | CST-2125S | 1:1000 | Rabbit |
| Transferrin receptor (TFR) | ThermoFisher Scientific H68.4 | 1:500 | Mouse |
| Ferroportin/FPN1/SLC40A1 | Novus Biological NBP1-21502SS | 1:1000 | Rabbit |
| FTH | CST-3998 | 1:1000 | Rabbit |
| FECH | SC-377377 | 1:1000 | Rabbit |
| LIAS | Proteintech 11 577-1-AP | 1:2500 | Rabbit |
| Ubiquitin | CST-3936 | 1:1000 | Mouse |
| K48-linkage-specific polyubiquitin | CST-8081 | 1:1000 | Rabbit |
| K63-linkage-specific polyubiquitin | CST-5621 | 1:1000 | Rabbit |
| Anti-puromycin | Millipore Sigma MABE343 | 1:5000 | Mouse |
| LC3B | CST-3868T | 1:1000 | Rabbit |
| SQSTM1/p62 | CST-5114S | 1:1000 | Rabbit |
| NDUFA9 | Ab14173 | 1:1000 | Mouse |
| SDHA | Ab14175 | 1:1000 | Mouse |
| SDHB | Novus Biologicals NBP1-54154SS | 1:1000 | Rabbit |
| UQCRC2 | Ab14745 | 1:1000 | Mouse |
| UQCRFS1 (Rieske) | Novus Biologicals NBP1-32367 | 1:1000 | Rabbit |
| ATP5A | Ab14748 | 1:1000 | Mouse |
| ETFA | Ab153722 | 1:1000 | Rabbit |
| ETFDH | Ab131376 | 1:1000 | Mouse |
| HSP70 | ThermoFisher Scientific MA3–008 | 1:1000 | Mouse |
| Cytochrome c | BD Pharmingen 556 433 | 1:1000 | Mouse |
| PDH-E1- α | Ab110330 | 1:1000 | Mouse |
| Aconitase 2 | Ab129069 | 1:1000 | Rabbit |
| Caspase-3 | CST-9665 | 1:1000 | Rabbit |
| BCL-2 | SC-7382 | 1:200 | Mouse |
| BAX | CST-2772S | 1:1000 | Rabbit |
Primers for qPCR (all are mouse specific).
| Primer name | Sequence | |
|---|---|---|
| Myh6 (α-MHC) | Forward | CGGAACAAGACAACCTCAAT |
| Reverse | TGGCAATGATTTCATCCAGC | |
| Myh7 (β-MHC) | Forward | TTGCTACCCTCAGGTGGCT |
| Reverse | CCTTCTCAGACTTCCGCAGG | |
| Cd36 | Forward | GGCCAAGCTATTGCGACAT |
| Reverse | CAGATCCGAACACAGCGTAGA | |
| Cpt1b | Forward | TGCCTTTACATCGTCTCCAA |
| Reverse | AGACCCCGTAGCCATCATC | |
| Acadm (MCAD) | Forward | CATTCCGGAAAGTTGCGGTGG |
| Reverse | TAATGGCCGCCACATCAGAG | |
| Slc2a4 (GLUT4) | Forward | TTATTGCAGCGCCTGAGTCT |
| Reverse | CAATCACCTTCTGTGGGGCAT | |
| Hk2 | Forward | CCGACTCGCCGCAACAAG |
| Reverse | ACTCGCCATGTTCTGTCCCAT | |
| Pfkfb1 | Forward | TTTCGCCCAGACAACATGGA |
| Reverse | TCAAAAACCGCAACGTGACC | |
| Mthfd2 | Forward | AGGTCCCAAGCCTTTGAGTT |
| Reverse | GTAAGGGAGTGCCGTTGAAA | |
| Psat1 | Forward | AGTGGAGCGCCAGAATAGAA |
| Reverse | CTTCGGTTGTGACAGCGTTA | |
| Phgdh | Forward | GACCCCATCATCTCTCCTGA |
| Reverse | GCACACCTTTCTTGCACTGA | |
| Atf2 | Forward | TGTCATTGTGGCTGATCAGACTC |
| Reverse | GTGTTGCAAGAGGGGACAAATC | |
| Atf3 | Forward | CCAGAATAAACACCTCTGCCATCG |
| Reverse | CTTCAGCTCAGCATTCACACTCTC | |
| Atf4 | Forward | CCACCATGGCGTATTAGAGG |
| Reverse | GTCCGTTACAGCAACACTGC | |
| Atf5 | Forward | CTGGGAACCCCTGTGGATTAAA |
| Reverse | GCAGCGTGGAAGATTGTTCA | |
| Slc7a5 | Forward | TGCAGCCATGACCCTAACAG |
| Reverse | AACAATGGGGACAGACCAGG | |
| Asns | Forward | TGGCTGCCTTTTATCAGGGG |
| Reverse | CAGATGCCCGAACTGTCGTA | |
| Fgf21 | Forward | ATGGAATGGATGAGATCTAGAGTTGG |
| Reverse | TCTTGGTGGTCATCTGTGTAGAGG | |
| Gdf15 | Forward | GAGCTACGGGGTCGCTTC |
| Reverse | GGGACCCCAATCTCACCT | |
| Clpp | Forward | CCAAGCACACCAAACAGAG |
| Reverse | GGACCAGAACCTTGTCTAAGAT | |
| Lonp1 | Forward | CTGTGTTCCCGCGCTTTATC |
| Reverse | GCCAGTGACAATCATTCGCAA | |
| Fbxo32 (Atrogin1) | Forward | ACCTGCTGGTGGAAAACATC |
| Reverse | CTTCGTGTTCCTTGCACATC | |
| Trim63 (MuRF1) | Forward | AAGGAGCGCCATGGGATACTG |
| Reverse | GTCACGCACGATTTCCC | |
| Tfrc | Forward | AGCCAGATCAGCATTCTCTAACT |
| Reverse | GCCTTCATGTTATTGTCGGCAT | |
| Actb (β-actin) | Forward | CAACACCCCAGCCATG |
| Reverse | GTCACGCACGATTTCCC |
BN electrophoresis and in-gel activity
Isolated heart mitochondria (100 μg) were lysed in 4% digitonin in extraction buffer [30 mM HEPES, 12% glycerol, 150 mM potassium acetate, 2 mM aminocaproic acid, 2 mM EDTA disodium salt, protease inhibitor tablet (Roche 11 873 580 001), pH 7.4], at 4°C for 30 min with constant shaking at 1500 rpm. The samples were centrifuged at 25000 g, 20 min, 4°C, then 1:400 diluted 5% Coomassie Blue G-250 was added to the supernatant. The samples were then loaded on a gradient gel (NativePAGE™ NovexR 3–12% Bis–Tris). The gel was run overnight in Native PAGE buffers (Invitrogen). The gel for Coomassie Blue staining was run in dark blue buffer until the dye front reached 1/3 of the gel (150 V) then in light blue buffer for the rest of the time up to 16 h (30 V). For gels used for in-gel activity, the gel was run first in light blue buffer then in clear buffer (Native PAGE Invitrogen). Following the electrophoresis, the gel was stained with Coomassie Blue R-250 for 30 min, then destained (40% MetOH, 10% acetic acid in water). In-gel activity of Complexes I, II and V was run as described [68].
Proteomics: LC–MS/MS analyses and data processing
Samples (25 μg each) were run into a NuPAGE 10% Bis–Tris gel (Thermo Scientific) for a short distance, and the entire gel lane was excised and digested with trypsin. Liquid chromatography tandem mass spectrometry (LC–MS/MS) analysis was performed using a Q Exactive HF mass spectrometer (Thermo Scientific) coupled with an UltiMate 3000 nano UPLC system (Thermo Scientific). Samples were injected onto a PepMap100 trap column (0.3 × 5 mm packed with 5 μm C18 resin; Thermo Scientific), and peptides were separated by reversed-phase HPLC on a BEH C18 nanocapillary analytical column (75 μm i.d. × 25 cm, 1.7 μm particle size; Waters) using a 4-h gradient formed by solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile). Eluted peptides were analyzed by the mass spectrometer set to repetitively scan m/z from 400 to 1800 in positive ion mode. The full MS scan was collected at 60 000 resolution followed by data-dependent MS/MS scans at 15 000 resolution on the 20 most abundant ions exceeding a minimum threshold of 20 000. Peptide match was set as preferred, exclude isotope option and charge-state screening were enabled to reject unassigned and single charged ions.
Peptide sequences were identified using MaxQuant 1.6.17.0 [69]. MS/MS spectra were searched against a UniProt mouse protein database (October 2020) and a common contaminants database using full tryptic specificity with up to two missed cleavages, static carboxamidomethylation of Cys, and variable Met oxidation, protein N-terminal acetylation and Asn deamidation. ‘Match between runs’ feature was used to help transfer identifications across experiments to minimize missing values. Consensus identification lists were generated with false discovery rates (FDRs) set at 1% for protein and peptide identifications. Statistical analyses were performed using Perseus 1.6.14.0 [70]. Missing values were imputed with a minimum value, and t-test P-values were adjusted (q-value) to account for multiple testing using Benjamini–Hochberg FDR function in Perseus.
The mitochondrial proteins were selected by matching gene names with a database of known and predicted mitochondrial genes using Python: the ‘Mitochondrial Part’ annotation from the Jensen Compartments database [https://doi.org/10.1093/database/bau012] downloaded from Enrichr (https://maayanlab.cloud/Enrichr/).
Quantitative polymerase chain reaction
Total RNA was extracted from tissues using Trizol® (Invitrogen, Carlsbad, CA) then treated with RQ1 DNase (Promega, Madison, WI) at 37°C for 30 min. RNA concentration was measured by Qubit® Fluorometer (Invitrogen, Carlsbad, CA). RNA was reverse transcribed using oligo(dT)20 primers and SuperScript III (Invitrogen, Carlsbad, CA). Primers were designed using Eurofins Primer Design Tool and checked for specificity and efficiency. Primers sequences are shown below. Quantitative polymerase chain reaction (qPCR) reactions were performed using ITaq SYBR green Supermix with ROX (BIO-RAD, Hercules, CA), with 20 ng cDNA/reactions, using an Eppendorf Mastercycler® ep realplex. The ∆∆Ct method was used to calculate mRNA levels relative to Actb (β-actin). Primers sequences used are listed below.
Global protein synthesis
To measure the rate of global protein synthesis, we used an in vivo SUnSET assay [44]. For this study, a sterilized puromycin (Sigma, 8833) solution in phosphate buffered saline (4 mg/ml) was injected intraperitoneally. The volume of the puromycin injection was calculated on the basis of the BW of the mouse, for a final concentration of 0.04 μmol/g. Thirty minutes after the injection, animals were sacrificed to collect tissue samples for biochemical detection of incorporated puromycin by western blot.
Tissue triglyceride detection
To measure triglyceride levels in tissue, hearts were digested overnight with an ethanolic KOH solution (100% ethanol and 30% KOH in a volume ratio of 2:1) at 60°C. After this, the digested sample was mixed thoroughly with a solution of 1 M MgCl2 and incubated at 4°C for 10 min. The samples were centrifuged at 14000 r.p.m. for 30 min. The supernatant was used for measurement using Stanbio Triglyceride Liquicolor® (Stanbio), following manufacturer’s instructions. Liver samples from fed and fasted (~16 h) mice were used as a positive control.
Histology
Hematoxylin/eosin (H&E), Masson’s Trichrome (MT) and Perls’ Prussian Blue (PPB) staining were performed at the translational research/pathology shared resource at Thomas Jefferson University. Image acquisition was performed using an Olympus CKX41 inverted microscope and cellSens Standard software. Images were analyzed using Image J. For H&E, the CSA of ~ 25 cardiomyocytes were measured per image (three 10× digital images per animal). For the quantification of iron deposits, five 20× digital images from each tissue section were analyzed. Images were converted to a RGB stack. PPB positive particles were identified and counted using Particle Analyzer plugin, establishing a threshold in the red channel. Only particles of area ≥ 1 μm2 were considered for the quantification. The size of the area analyzed was 353 μm × 264 μm (2048 × 1536 pixels). For area of collagen measurement, five 10× digital images from each tissue section were analyzed. Images were processed using Colour Deconvolution plugin. Collagen area was measured setting up a threshold in Colour_1 stack. Total tissue area was determined converting the original image to an RGB stack and measuring the area, setting up a threshold in the red channel.
Quantification and statistical analysis
Data analysis was performed using GraphPad Prism 8.2.1 Software. Unpaired Student’s t test for comparison of two means or one-way analysis of variance followed by Tukey post hoc comparisons, for multiple comparisons, were performed as appropriate. P-value < 0.05 was considered significant. Analysis of the proteomics data is described in the relevant section of Materials and Methods. In all cases, data not indicated as significant should be considered not statistically different. Details specific for a given measurement, including sample sizes, are provided in Results section and the figure legends.
Supplementary Material
Acknowledgements
The authors thank Dr George Porter (University of Rochester) for discussions. The authors thank Drs Vijay Chandran (University of Florida) and Daniel Geschwind (U.C.L.A.) for providing the mouse model, David Weaver (MitoCare, Thomas Jefferson University) for assistance with proteomics analysis, and Marco Tigano (NYU) for the irradiated MCF-10 cells.
Contributor Information
César Vásquez-Trincado, MitoCare Center for Mitochondrial Imaging Research and Diagnostics, Department of Pathology, Anatomy and Cell Biology, Thomas Jefferson University, Philadelphia, PA 19107, United States.
Monika Patel, MitoCare Center for Mitochondrial Imaging Research and Diagnostics, Department of Pathology, Anatomy and Cell Biology, Thomas Jefferson University, Philadelphia, PA 19107, United States.
Aishwarya Sivaramakrishnan, MitoCare Center for Mitochondrial Imaging Research and Diagnostics, Department of Pathology, Anatomy and Cell Biology, Thomas Jefferson University, Philadelphia, PA 19107, United States.
Carmen Bekeová, MitoCare Center for Mitochondrial Imaging Research and Diagnostics, Department of Pathology, Anatomy and Cell Biology, Thomas Jefferson University, Philadelphia, PA 19107, United States.
Lauren Anderson-Pullinger, MitoCare Center for Mitochondrial Imaging Research and Diagnostics, Department of Pathology, Anatomy and Cell Biology, Thomas Jefferson University, Philadelphia, PA 19107, United States.
Nadan Wang, Center for Translational Medicine, Department of Pathology, Anatomy and Cell Biology, Thomas Jefferson University, Philadelphia, PA 19107, United States.
Hsin-Yao Tang, Proteomics and Metabolomics Facility, The Wistar Institute, Philadelphia, PA 19104, United States.
Erin L Seifert, MitoCare Center for Mitochondrial Imaging Research and Diagnostics, Department of Pathology, Anatomy and Cell Biology, Thomas Jefferson University, Philadelphia, PA 19107, United States.
Conflict of Interest statement: None declared.
Funding
Friedreich’s Ataxia Research Alliance (to E.L.S.); National Institutes of Health (R01 GM123771 to E.L.S., P30 CA010815 to Wistar Institute Proteomics & Metabolomics Facility, R50 CA221838 to H.-Y.T).
References
- 1. Reetz K, Dogan I, Costa AS. et al. Biological and clinical characteristics of the European Friedreich's ataxia consortium for translational studies (EFACTS) cohort: a cross-sectional analysis of baseline data. Lancet Neurol 2015;14:174–12. [DOI] [PubMed] [Google Scholar]
- 2. Kipps A, Alexander M, Colan SD. et al. The longitudinal course of cardiomyopathy in Friedreich's ataxia during childhood. Pediatr Cardiol 2009;30:306–10. [DOI] [PubMed] [Google Scholar]
- 3. Tsou AY, Paulsen EK, Lagedrost SJ. et al. Mortality in Friedreich ataxia. J Neurol Sci 2011;307:46–9. [DOI] [PubMed] [Google Scholar]
- 4. St John Sutton M, Ky B, Regner SR. et al. Longitudinal strain in Friedreich ataxia: a potential marker for early left ventricular dysfunction. Echocardiography 2014;31:50–7. [DOI] [PubMed] [Google Scholar]
- 5. Legrand L, Diallo A, Monin ML. et al. Predictors of left ventricular dysfunction in Friedreich's ataxia in a 16-year observational study. Am J Cardiovasc Drugs 2020;20:209–16. [DOI] [PubMed] [Google Scholar]
- 6. Rotig A, de LonlayP, Chretien D. et al. Aconitase and mitochondrial iron-Sulphur protein deficiency in Friedreich ataxia. Nat Genet 1997;17:215–17. [DOI] [PubMed] [Google Scholar]
- 7. Seznec H, Simon D, Monassier L. et al. Idebenone delays the onset of cardiac functional alteration without correction of Fe-S enzymes deficit in a mouse model for Friedreich ataxia. Hum Mol Genet 2004;13:1017–24. [DOI] [PubMed] [Google Scholar]
- 8. Wagner GR, Pride PM, Babbey CM, Payne RM. Friedreich's ataxia reveals a mechanism for coordinate regulation of oxidative metabolism via feedback inhibition of the SIRT3 deacetylase. Hum Mol Genet 2012;21:2688–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stram AR, Wagner GR, Fogler BD. et al. Progressive mitochondrial protein lysine acetylation and heart failure in a model of Friedreich's ataxia cardiomyopathy. PLoS One 2017;12:e0178354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martin AS, Abraham DM, Hershberger KA. et al. Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich's ataxia cardiomyopathy model. JCI Insight 2017;2:e93885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang ML, Sivagurunathan S, Ting S. et al. Molecular and functional alterations in a mouse cardiac model of Friedreich ataxia: activation of the integrated stress response, eIF2alpha phosphorylation, and the induction of downstream targets. Am J Pathol 2013;183:745–57. [DOI] [PubMed] [Google Scholar]
- 12. Pakos-Zebrucka K, Koryga I, Mnich K. et al. The integrated stress response. EMBO Rep. 2016;17:1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Costa-Mattioli M, Walter P. The integrated stress response: from mechanism to disease. Science 2020;368:eaat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suomalainen A, Elo JM, Pietilainen KH. et al. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol 2011;10:806–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bao XR, Ong SE, Goldberger O. et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. elife 2016;5:e10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nikkanen J, Forsstrom S, Euro L. et al. Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell Metab 2016;23:635–48. [DOI] [PubMed] [Google Scholar]
- 17. Khan NA, Nikkanen J, Yatsuga S. et al. mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab 2017;26:419, e415–28. [DOI] [PubMed] [Google Scholar]
- 18. Quiros PM, Prado MA, Zamboni N. et al. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol 2017;216:2027–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dogan SA, Cerutti R, Beninca C. et al. Perturbed redox signaling exacerbates a mitochondrial myopathy. Cell Metab 2018;28:764, e765–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Forsstrom S, Jackson CB, Carroll CJ. et al. Fibroblast growth factor 21 drives dynamics of local and systemic stress responses in mitochondrial myopathy with mtDNA deletions. Cell Metab 2019;30:1040, e1047–54. [DOI] [PubMed] [Google Scholar]
- 21. Mick E, Titov DV, Skinner OS. et al. Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell. elife 2020;9:e49178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnson SC, Yanos ME, Kayser EB. et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 2013;342:1524–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Siegmund SE, Yang H, Sharma R. et al. Low-dose rapamycin extends lifespan in a mouse model of mtDNA depletion syndrome. Hum Mol Genet 2017;26:4588–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Civiletto G, Dogan SA, Cerutti R. et al. Rapamycin rescues mitochondrial myopathy via coordinated activation of autophagy and lysosomal biogenesis. EMBO Mol Med 2018;10:e8799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Viscomi C, Bottani E, Civiletto G. et al. In vivo correction of COX deficiency by activation of the AMPK/PGC-1alpha axis. Cell Metab 2011;14:80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Perdomini M, Belbellaa B, Monassier L. et al. Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich's ataxia. Nat Med 2014;20:542–47. [DOI] [PubMed] [Google Scholar]
- 27. Belbellaa B, Reutenauer L, Monassier L, Puccio H. Correction of half the cardiomyocytes fully rescue Friedreich ataxia mitochondrial cardiomyopathy through cell-autonomous mechanisms. Hum Mol Genet 2019;28:1274–85. [DOI] [PubMed] [Google Scholar]
- 28. Chandran V, Gao K, Swarup V. et al. Inducible and reversible phenotypes in a novel mouse model of Friedreich's ataxia. elife 2017;6:e30054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Llorens JV, Soriano S, Calap-Quintana P. et al. The role of iron in Friedreich's ataxia: insights from studies in human tissues and cellular and animal models. Front Neurosci 2019;13:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anderson CP, Shen M, Eisenstein RS, Leibold EA. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta 2012;1823:1468–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Razeghi P, Young ME, Alcorn JL. et al. Metabolic gene expression in fetal and failing human heart. Circulation 2001;104:2923–31. [DOI] [PubMed] [Google Scholar]
- 32. Wojtczak L, Schonfeld P. Effect of fatty acids on energy coupling processes in mitochondria. Biochim Biophys Acta 1993;1183:41–57. [DOI] [PubMed] [Google Scholar]
- 33. Branda SS, Cavadini P, Adamec J. et al. Yeast and human frataxin are processed to mature form in two sequential steps by the mitochondrial processing peptidase. J Biol Chem 1999;274:22763–69. [DOI] [PubMed] [Google Scholar]
- 34. Bertholet AM, Chouchani ET, Kazak L. et al. H(+) transport is an integral function of the mitochondrial ADP/ATP carrier. Nature 2019;571:515–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wende AR, Brahma MK, McGinnis GR, Young ME. Metabolic origins of heart failure. JACC Basic Transl Sci 2017;2:297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chung HK, Ryu D, Kim KS. et al. Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J Cell Biol 2017;216:149–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuhl I, Miranda M, Atanassov I. et al. Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. elife 2017;6:e30952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Park Y, Reyna-Neyra A, Philippe L, Thoreen CC. mTORC1 balances cellular amino acid supply with demand for protein synthesis through post-transcriptional control of ATF4. Cell Rep 2017;19:1083–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Selvarajah B, Azuelos I, Plate M. et al. mTORC1 amplifies the ATF4-dependent de novo serine-glycine pathway to supply glycine during TGF-beta1-induced collagen biosynthesis. Sci Signal 2019;12:eaav3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kovacic S, Soltys CL, Barr AJ. et al. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J Biol Chem 2003;278:39422–27. [DOI] [PubMed] [Google Scholar]
- 41. Soltys CL, Kovacic S, Dyck JR. Activation of cardiac AMP-activated protein kinase by LKB1 expression or chemical hypoxia is blunted by increased Akt activity. Am J Physiol Heart Circ Physiol 2006;290:H2472–79. [DOI] [PubMed] [Google Scholar]
- 42. Suragani RN, Zachariah RS, Velazquez JG. et al. Heme-regulated eIF2alpha kinase activated Atf4 signaling pathway in oxidative stress and erythropoiesis. Blood 2012;119:5276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang S, Macias-Garcia A, Velazquez J. et al. HRI coordinates translation by eIF2alphaP and mTORC1 to mitigate ineffective erythropoiesis in mice during iron deficiency. Blood 2018;131:450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 2009;6:275–77. [DOI] [PubMed] [Google Scholar]
- 45. Herron TJ, McDonald KS. Small amounts of alpha-myosin heavy chain isoform expression significantly increase power output of rat cardiac myocyte fragments. Circ Res 2002;90:1150–52. [DOI] [PubMed] [Google Scholar]
- 46. Rundell VL, Manaves V, Martin AF, de TombePP. Impact of beta-myosin heavy chain isoform expression on cross-bridge cycling kinetics. Am J Physiol Heart Circ Physiol 2005;288:H896–903. [DOI] [PubMed] [Google Scholar]
- 47. Gupta MP. Factors controlling cardiac myosin-isoform shift during hypertrophy and heart failure. J Mol Cell Cardiol 2007;43:388–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nelson OL, Rourke BC. Increase in cardiac myosin heavy-chain (MyHC) alpha protein isoform in hibernating ground squirrels, with echocardiographic visualization of ventricular wall hypertrophy and prolonged contraction. J Exp Biol 2013;216:4678–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Al-Horani RA, Mohammad MA, Haifawi S, Ihsan M. Changes in myocardial myosin heavy chain isoform composition with exercise and post-exercise cold-water immersion. J Muscle Res Cell Motil 2021;42:182–91. [DOI] [PubMed] [Google Scholar]
- 50. Mochel F, Knight MA, Tong WH. et al. Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am J Hum Genet 2008;82:652–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Navarro-Sastre A, Tort F, Stehling O. et al. A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am J Hum Genet 2011;89:656–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lim SC, Friemel M, Marum JE. et al. Mutations in LYRM4, encoding iron-sulfur cluster biogenesis factor ISD11, cause deficiency of multiple respiratory chain complexes. Hum Mol Genet 2013;22:4460–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Crooks DR, Maio N, Lane AN. et al. Acute loss of iron-sulfur clusters results in metabolic reprogramming and generation of lipid droplets in mammalian cells. J Biol Chem 2018;293:8297–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fornasiero EF, Mandad S, Wildhagen H. et al. Precisely measured protein lifetimes in the mouse brain reveal differences across tissues and subcellular fractions. Nat Commun 2018;9:4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mik EG, Ince C, Eerbeek O. et al. Mitochondrial oxygen tension within the heart. J Mol Cell Cardiol 2009;46:943–51. [DOI] [PubMed] [Google Scholar]
- 56. Ast T, Meisel JD, Patra S. et al. Hypoxia rescues frataxin loss by restoring iron sulfur cluster biogenesis. Cell 2019;177:1507, e1516–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol 2020;21:183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Varghese J, James J, Vaulont S. et al. Increased intracellular iron in mouse primary hepatocytes in vitro causes activation of the Akt pathway but decreases its response to insulin. Biochim Biophys Acta Gen Subj 2018;1862:1870–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ellis JM, Mentock SM, Depetrillo MA. et al. Mouse cardiac acyl coenzyme a synthetase 1 deficiency impairs fatty acid oxidation and induces cardiac hypertrophy. Mol Cell Biol 2011;31:1252–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hawley SA, Ross FA, Gowans GJ. et al. Phosphorylation by Akt within the ST loop of AMPK-alpha1 down-regulates its activation in tumour cells. Biochem J 2014;459:275–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Guo X, Aviles G, Liu Y. et al. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 2020;579:427–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fessler E, Eckl EM, Schmitt S. et al. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 2020;579:433–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Han AP, Yu C, Lu L. et al. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J 2001;20:6909–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cabral DJ, Penumutchu S, Reinhart EM. et al. Microbial metabolism modulates antibiotic susceptibility within the murine gut microbiome. Cell Metab 2019;30:800, e807–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mistry P, Reitz CJ, Khatua TN. et al. Circadian influence on the microbiome improves heart failure outcomes. J Mol Cell Cardiol 2020;149:54–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Troseid M, Andersen GO, Broch K, Hov JR. The gut microbiome in coronary artery disease and heart failure: current knowledge and future directions. EBioMedicine 2020;52:102649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Moffat C, Bhatia L, Nguyen T. et al. Acyl-CoA thioesterase-2 facilitates mitochondrial fatty acid oxidation in the liver. J Lipid Res 2014;55:2458–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jha P, Wang X, Auwerx J. Analysis of mitochondrial respiratory chain supercomplexes using blue native polyacrylamide gel electrophoresis (BN-PAGE). Curr Protoc Mouse Biol 2016;6:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 2008;26:1367–72. [DOI] [PubMed] [Google Scholar]
- 70. Tyanova S, Temu T, Sinitcyn P. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 2016;13:731–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.