

Abstract
Chronic hepatitis B (CHB) is a major global health challenge. CHB can be controlled by antivirals but a therapeutic cure is lacking. CHB is characterized by limited HBV-specific T cell reactivity and functionality and expression of inhibitory receptors. The mechanisms driving these T cell phenotypes are only partially understood. Here, we created a single-cell RNA-sequencing dataset of HBV immune responses in patients to contribute to a better understanding of the dysregulated immunity. Blood samples of a well-defined cohort of 21 CHB and 10 healthy controls, including a subset of 5 matched liver biopsies, were collected. scRNA-seq data of total immune cells (55,825) plus sorted HBV-specific (1,963), non-naive (32,773) and PD1+ T cells (96,631) was generated using the 10X Genomics platform (186,123 cells) or the full-length Smart-seq2 protocol (1,069 cells). The shared transcript count matrices of single-cells serve as a valuable resource describing transcriptional changes underlying dysfunctional HBV-related T cell responses in blood and liver tissue and offers the opportunity to identify targets or biomarkers for HBV-related immune exhaustion.
Subject terms: Hepatitis B, Transcriptomics, T cells, Data integration
Background & Summary
Background
Hepatitis B virus (HBV) infection is a major global public health problem despite the existence of safe and effective preventative vaccines. More than 250 million people worldwide are living with chronic HBV. Patients carry a high risk of cirrhosis and hepatocellular carcinoma (HCC)1. Current treatments are based on direct antiviral agents which can only limit HBV replication without achieving a long-term HBV cure2. Ineffective immune responses are a key feature enabling chronic HBV infection but also contributing to hepatocytes injury and liver inflammation. Understanding the mechanisms behind this dysfunctional immune response and the switches towards a functional response resulting in viral cure will be key for the development of new HBV treatments such as immunotherapies.
During acute infection a robust immune response involving HBV-specific antiviral CD8+ effector cells, CD4+ helper T cells and B cells producing HBV-specific antibodies leads to viral clearance and resolution of infection3–6.
However, during chronic HBV infection, immune responses show a number of dysregulated features including atypical B cells producing reduced antibody levels and T cell exhaustion, characterized by low levels of HBV-specific T cells with a state of low functionality7–10. T cell exhaustion is defined by restricted proliferation, progressively reduced cytotoxicity and cytokine production and increased expression of inhibitory receptors9,11–14.
Chronic exposure to high antigen levels, extended duration of disease as well as suboptimal T cell priming and activation have been identified as some of the factors driving this dysfunctional immune response14–18.
Recently, it has become clear that exhausted T cells in chronic HBV infection are not representing a homogeneous cell population19–21.
Data from a small cohort of patients achieving HBV functional cure either spontaneously or after cessation of NUC treatment shows that induction of a sufficient immune response is possible even in a setting of chronic HBV infection22,23.
Many studies on immune responses in HBV patients have focused on the analysis of peripheral blood immune cells due to easier sample accessibility9,11,13,16. However, HBV is a purely hepatotropic virus and it has become clear that the cell composition and functional features of intrahepatic immune cells can differ substantially from the ones in peripheral blood24–28.
Single-cell RNA-sequencing (scRNA-seq) is a breakthrough technology allowing for transcriptome analysis cell by cell. This technique has been widely used to assess immune cell profiles in chronic infectious diseases or cancers related to an immune exhaustion phenotype29–32. ScRNA-seq, as an unbiased transcriptome analysis, provides also a great opportunity to discover the underlying cellular and molecular mechanisms contributing to the dysfunctional immune response in chronic HBV infection.
So far, only few studies have explored the HBV immune landscape with scRNA-seq in human samples. Zheng et al. explored sorted T cells infiltrating HCC tumor tissue on an HBV background33. Zhang and colleagues focused on the liver immune cell infiltration during different phases of chronic HBV infection in an Asian patient cohort34. A pilot study from Genshaft et al. explored the technological feasibility of using fine needle aspirates for scRNA-seq comparing two different methods35 and the latest sc-RNAseq study in the HBV field investigated changes in the immune cell landscape during the progression from HBV infection to HBV cirrhosis and HBV-associated HCC36.
In this study, we established a scRNA-seq dataset of total immune cells and HBV-specific T cells from blood and liver of non-cirrhotic HBV patients without HCC (Fig. 1). We used both a droplet-based microfluidic scRNA-seq system (10X Genomics) and full-length scRNA-seq by Smart-seq2 to analyze total immune cells and HBV-specific T cells.
Fig. 1.

Schematic overview of the study workflow. 1. Blood samples from patients with chronic hepatitis B infection were collected. Matched liver biopsies were available for a subgroup of patients. Blood from healthy donors was used as control. 2. For liver biopsies, single cell suspensions were prepared by enzymatic digestion. PBMCs were isolated by density centrifugation. A subsample of whole blood was used in parallel with the liver cell suspension for magnetic bead isolation. 3. CD45+ cells were isolated using magnetic bead enrichment from both whole blood and liver cell suspensions. Specific T cell populations (Non-naive, PD1+ or HBV-specific T cells) were sorted from PBMCs via FACS sorting either in plates as single cells per well or as bulk populations in tubes. 4. Libraries for 3′ scRNA-seq were prepared from CD45+ cells and sorted T cell populations using the 10X Genomics Chromium platform. Libraries for full-length RNA-sequencing using the Smart-seq2 protocol were prepared from plate-sorted T cells. 5. scRNA-seq was performed using the Illumina NovaSeq instrument. 6. Sequencing reads were mapped to the human genome and a gene-by-cell count matrix was generated. 7. Sample quality was assessed and cell barcodes were filtered based on number of genes, read or UMI counts, and mitochondrial content. The gene-by-cell count matrix was normalized to counts per 10’000 (cp10k). 8. Highly variable genes were selected, principal components analysis was performed, nearest neighbors were identified, Leiden clustering and uniform manifold approximation projection (UMAP) were performed. 9. Signature enrichment scores were calculated to identify cell types.
To the best of our knowledge, this dataset represents very comprehensive scRNA-seq data focusing on the complete immune compartments in blood and liver of non-cirrhotic HBV patients without HCC and is the only published dataset including full-length sequencing of sorted HBV-specific T cells.
As such our data is a valuable resource for the scientific community to better understand the mechanisms underlying the dysfunctional immune response during chronic HBV infection contributing to the development of new treatment strategies for this disease.
Methods
Human clinical samples
Samples from two different groups of patients were included in the study. From 15 NUC treated CHB patients (HBeAg negative, HBsAg positive, HBV DNA < 25 IU/mL, ALT < 1.5 × UNL) 100 mL of blood were taken. From 6 untreated CHB patients (HBeAg negative, HBsAg positive, HBV DNA > 2000 IU/mL, ALT < 2x UNL) 100 mL blood as well as one matched liver biopsy for 5 of them was obtained. All patients were treated and followed at the Strasbourg University Hospital, France. Patient characteristics are shown in Table 1.
Table 1.
Characteristics of the study cohort.
| Patient ID | Age | Sex | Biopsy | Fibrosis | ALT (IU/mL) | AST (IU/mL) | Total bilirubin (µmol/L) | INR | Albumin (g/L) | platelets (G/L) | Disease duration (years) | HBV genotype | HBsAg (IU/mL) | HBV DNA (IU/ml) | Antiviral treatment | Duration antiviral treatment (years) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 65 | F | no | F0-F1 | 27 | 19 | NA | NA | NA | 242 | 7 | NA | 351.31 | <10 | TDF | 7 |
| 2 | 44 | M | no | F0-F1 | 96 | 60 | 16 | 1 | 45 | 251 | 15 | NA | 23547.07 | 265210 | No | NA |
| 3 | 55 | M | no | F0-F1 | 24 | 15 | 7.5 | 1 | 54 | 219 | 15 | B | 405.32 | <10 | ETV | 13 |
| 4 | 26 | M | no | F0-F1 | 32 | 23 | 18.7 | 1 | 54 | 203 | 25 | NA | 43715.09 | <10 | Yes, NA | 6 |
| 6 | 39 | M | no | F1-F2 | 26 | 26 | 12.5 | NA | 45 | 211 | 10 | E | 17581.84 | <10 | ETV | 5 |
| 7 | 39 | M | no | F0-F1 | 24 | 19 | 24.2 | 1.1 | 42 | 196 | 9 | E | 3448.37 | <10 | TDV | 8 |
| 8 | 39 | F | no | F0-F1 | 17 | 18 | 13.3 | NA | 44 | 285 | 18 | NA | 8836.84 | <10 | TDV | 16 |
| 9 | 68 | M | no | F0-F1 | 16 | 26 | 8.5 | NA | 45 | 251 | 23 | NA | 970 | <10 | TDV | 19 |
| 10 | 40 | F | no | F0-F2 | 29 | 41 | NA | NA | NA | 248 | 4 | NA | 24015.82 | <10 | TDV | 1 |
| 11 | 35 | M | no | F0-F1 | 19 | 19 | 7.2 | 1 | 40 | ND | 7 | D | 9717 | <10 | TDV | 6 |
| 12 | 45 | F | no | F0-F1 | 29 | 19 | 8.2 | 1 | NA | 384 | 24 | NA | 1980.26 | <10 | TDV | 7 |
| 14 | 57 | M | no | F2 | 82 | 47 | NA | NA | NA | ND | 6 | NA | 256 | <11 | TDV | 7 |
| 16 | 39 | F | no | F0-F1 | 29 | 29 | 2.2 | 1 | 46 | 255 | 11 | D | 20991 | <10 | TDV | 7 |
| 21 | 44 | F | yes | F0 | 31 | 26 | 10.2 | 1 | 45 | 261 | 27 | NA | 1608 | 899100 | No | NA |
| 26 | 21 | M | yes | 0 | 58 | 42 | 14 | 1.2 | NA | 219 | 13 | E | 2896.87 | 11457 | No | NA |
| 27 | 31 | M | no | NA | 33 | 31 | 10.2 | NA | 43 | 156 | NA | NA | 1540.41 | <10 | ETV | 10 |
| 28 | 54 | M | no | F0-F1 | 21 | 21 | 14.3 | 1.2 | 46 | 194 | 33 | D | 2122 | <10 | ETV | 11 |
| 34 | 23 | M | yes | F0 | 84 | 24 | 14.9 | 1 | 51 | 211 | 2 | NA | 1109.93 | 11162 | No | NA |
| 35 | 39 | M | yes | F0 | 43 | 26 | 32.3 | 1 | 47 | 239 | 5 | E | 27138.56 | 40773 | No | NA |
| 36 | 69 | M | no | F0-F1 | 19 | 27 | 13.9 | 1.03 | 44 | 266 | 36 | NA | 2605 | <10 | ETV | 7 |
| 37 | 24 | M | yes | F0 | 224 | 114 | 16 | 1 | 46 | 306 | 4 | NA | 22740.11 | 349793 | No | NA |
Abbreviations: ALT - alanine aminotransferase, ETV – entecavir, F - female, M - male, NA - not available, TDF – tenofovir. Fibrosis data were obtained by liver biopsy or transient elastography.
Exclusion criteria to the study were defined as (i) use of steroids or other immunosuppressive agent in the past 4 weeks; (ii) any disease or clinical test indicating the possibility of a disease or condition that could confound the study results (including, but not limited to: cancer, systemic lupus erythematosus, rheumatoid arthritis or other autoimmune disease); (iii) major surgery or traumatic injury (including blood transfusion) within the past 4 weeks; (iv) use of an investigational drug in the past 12 weeks; (v) HCV, HIV, HDV or HAV coinfection; (vi) significant acute infection such as influenza or other within the past 2 weeks; (vii) history of drug abuse in the past year; (viii) pregnancy or breastfeeding; (ix) patients with: (a) either a medical history or signs of cirrhosis demonstrated by a biopsy result or any other validated non-invasive test revealing cirrhosis, recorded in the patient’s medical file or (b) during the screening visit: a transient elastography value ≥ 10.5 kPa OR a Fibrotest®/Fibrosure® score ≥ 0.48 and an APRI (aspartate aminotransferase platelet ratio Index) score ≥ 1 at screening; history of ascites, gastrointestinal bleeding and/or encephalopathy; any comorbidity likely to lead to liver damage (excessive alcohol consumption; hemochromatosis; Wilson’s disease; autoimmune hepatitis; inflammatory colitis, etc.).
All patients who participated in the study provided prior written informed consent. The study was approved by the French national ethics committee, Comité de Protection des Personnes Ile-de-France.
Blood samples from 10 healthy controls were provided by the Roche medical service. Patients gave informed consent and the blood collection was approved by the Ethikkommission Nordwest- und Zentralschweiz.
Sample collection and processing
Sample collection
Blood samples were collected in Vacutainer Sodium Heparin Tubes. Freshly taken liver biopsies were immediately stored in 5 mL of MACS® Tissue Storage Solution (Miltenyi Biotec). Shipment of samples was temperature controlled with blood samples shipped at room temperature (15–25 °C) and biopsies shipped cooled (2–8 °C). Samples were processed freshly on the same day.
PBMC isolation
Isolation of PBMCs from fresh whole blood was performed through density centrifugation. In short, blood was diluted 1:3 with 1X PBS containing 2% heat inactivated FBS. In a 50 mL Leucosep tube (greiner, 227290) 30 mL of diluted blood were layered onto 15 mL of Ficoll-Paque PLUS (GE Healthcare, 17-1440-03) and centrifuged at 800 g for 30′ at room temperature without brake. PBMCs were collected at the interphase, washed with 50 mL isolation buffer and counted for further processing. Freshly isolated PBMCs were either used directly for sorting or frozen in Cryostor CS10 (Stemcell, 07930) for later use.
Generally, all samples used for 10X processing were used freshly. Additionally, a few samples were processed from frozen PBMCs to match exactly the samples used for Smart-seq2 processing and facilitate data integration. Furthermore, in order to increase cell numbers for a few donors (VHB11, VHB34, HC8 and 9) frozen samples were added. The information on fresh vs. frozen processing is available in the metadata of the dataset.
Biopsies processing
For preparation of single cell suspensions from biopsy, the biopsy was cut in small pieces with a sterile scalpel and digested in RPMI medium containing 0.25 mg/mL collagenase (Sigma-Aldrich, C6885) and 0.2 mg/mL DNAse (Roche, 1010415900) at 37 °C for 40′ with constant slow shaking. Suspensions were filtered through a 70 μm filter and leftover pieces ground through the filter. The filter was then rinsed with RPMi + 10% FCS. Another filtering step through a 40 μm filter was performed and the filter rinsed with RPMI + 10% FCS.
Magnetic bead isolation
Magnetic bead isolation was used to isolate CD45+ immune cells from fresh biopsy-derived cell suspensions. The cell suspension was first centrifuged at 300 g for 10′. The cell pellet was then washed in 5 mL MACS buffer and pelleted again. CD45+ cells were isolated using human CD45 MicroBeads (Miltenyi, 130-045-801) for positive selection according to the manufacturer protocol using MS columns (Miltenyi Biotec, 130-042-201). After magnetic bead isolation cells were pelleted for 10′ at 300 g, supernatant was discarded and cells were resuspended in 40 μL PBS with 0.04% BSA and counted. All biopsy samples were used freshly.
Matched blood samples from biopsy donors were split into two parts with 80 mL used for standard PBMC isolation (see above) and 20 mL used for CD45+ magnetic bead isolation to match the isolation procedure of biopsy-derived immune cells. For bead isolation from whole blood StraightFrom Whole Blood CD45 MicroBeads (Miltenyi Biotec, 130-090-872) and the Whole Blood Column Kit (Miltenyi Biotec, 130-093-545) were used according to manufacturer instructions.
Fluorescence-activated cell sorting (FACS)
For cell staining 1 × 106 fresh or frozen PBMCs were used. First, viability staining was performed using 200 μL of a 1:1000 dilution of the Zombie NIR™ Fixable Viability dye (Biolegend, 423105) for 15′ at room temperature in the dark. Cells were washed with staining buffer (BD Pharmingen) and resuspended in 200 μL Fc-receptor blocking solution, after 5′ the staining mix of antibodies was added and cells were incubated for 20′ at 4 °C in the dark. Cells were then washed twice with CSB and filtered before resuspension to a maximum of 30 × 106/mL for FACS sorting.
For cell staining intended for HBV-specific T cell sorting 2 × 106 PBMCs from HLA-A*02:01+ donors were used and stained with 10 μL tetramer (PE-labelled, iTAG MHC Tetramer, FLPSDFFPSV, MBL International Corporation, #TB-0018-1) in 100 μL CSB for 30′ at room temperature in the dark prior to viability and antibody staining described above.
The used antibodies were BUV395-labelled anti-CD4 (BD Biosciences, SK3, #563550), BUV737-labelled anti-CD8 (BD Biosciences, SK1, #564629), BV421-labelled anti-PD-1 (Biolegend, EH12.2H7, #329920), BV510-labelled anti-CD3 (BD Biosciences, UCHT1, #563109), BV711-labelled anti-CD19 (Biolegend, HIB19, #302246), BV711-labelled anti-CD14 (Biolegend, M5E2, #301838), BV711-labelled anti-CD16 (Biolegend, 3G8, #302044), BV711-labelled anti-CD11B (Biolegend, ICRF44, #301344), FITC-labelled anti-CCR7 (Biolegend, G043H7, #353216), BB700-labelled anti-CD45 (BD Biosciences, HI30, #746090), PE-CF594-labelled anti-PD-1, BD Biosciences, EH12.1, #565024), PE-Cy7-labelled, anti-CD45RA (Biolegend, HI100, #304126), APC-labelled anti-CD45RA (Biolegend, HI100, #304112).
FACS sorting was performed on a BD Fusion instrument using BD FACSDiva software version 8.0.1.
Library preparation and sequencing (10X Genomics)
For scRNA-seq employing the 10X Genomics Chromium platform cell suspensions were used for library preparation following the manufacturer instructions (Chromium Single Cell 3′ Reagent Kits v3 User Guide CG000183 Rev A). A cell recovery of 8000 cells was targeted.
Smart-seq2
For full-length scRNA-seq of T cells from three HLA-A*02:01+ donors, cells were sorted into 96-well plates with one cell per well directly into lysis buffer containing dNTPs and oligo-dT primers. Smart-seq2 library preparation was performed as originally published following the protocol from Picelli et al.37 using the Nextera XT DNA Library Preparation Kit (96 samples), Illumina, FC-131-1096. For all Smart-seq2 experiments frozen PBMCs were used.
Sequencing of 10X libraries
10X Libraries were quantified using the Qubit dsDNA HS assay and average library size was calculated running Bioanalyzer DNA High Sensitivity protocol. Libraries were pooled in an equimolar manner and pools were diluted to 2.5 nM before loading into the sequencer. Illumina NovaSeq 6000 instrument was used for sequencing using single-indexed paired-end parameters (28 cycles - 8 cycles - 91 cycles).
Sequencing of Smart-seq2 libraries
Dual-indexed Smart-seq2 libraries were pooled by equal volumes of library. Each library pool was quantified on a Qubit Fluorometer using the Qubit™ dsDNA HS kit (Thermo Fischer Scientific®). Library quality was assessed on a Bioanalyzer using the Agilent High Sensitivity DNA kit (Agilent Technologies®). Library pools were diluted to 2 nM and sequenced for 2 × 101 cycles on a NovaSeq 6000 instrument (Illumina Inc.).
Single cell RNA-sequencing data processing
In total, we sequenced 58 experiments on the 10X platform (excluding feature barcoding experiments) and 3 experiments using the Smart-seq2 protocol. We excluded 5 experiments:
One experiment due to inconclusive FACS staining for cell sorting (VHB5_PD1_10X_3p_blood), one experiment because of high mitochondrial gene count, low gene count and low UMI count (VHB17_CD45_10X_3p_blood), three experiments that were re-sequenced because they contained very few cells (VHB34_CD45_10X_3p_blood, VHB27_PD1_10X_3p_blood, and VHB27_NN_10X_3p_blood).
10X data preprocessing
FASTQ files were generated using 10X Genomics cellranger 5.5.0 mkfastq. In order to estimate UMI counts and gene expression levels, reads were mapped to the human genome (hg38) utilizing 10X Genomics cellranger 5.5.0 count. The gene-by-cell count matrix was further processed using Besca38 and Scanpy39.
10X data filtering
In order to achieve high quality data, only cells that expressed at least 800 and not more than 6.000 genes; included at least 2.500 and not more than 50.000 UMIs; had not more than 15% of UMIs mapping to mitochondrial genes were kept. This resulted in 186,123 total cells.
10X data normalization
Normalization was performed using count depth scaling to 10,000 total counts per cell, resulting in the cp10k (counts per 10,000) unit. Count values were log-transformed using natural logarithm: ln(cp10k + 1).
Smart-seq2 experiments
In total, we sequenced 1,713 cells using the Smart-seq2 protocol37 from 9 experiments (3 donors × 3 conditions). Two cells were excluded, because the raw sequencing data (FASTQ files) were missing or corrupted (cells 771 and 1320).
Smart-seq2 data preprocessing
Base calling was performed with BCL to FASTQ file converter bcl2fastq v2.17.1.14 from Illumina (https://support.illumina.com/downloads.html). In order to estimate gene expression levels, paired-end RNA-Seq reads were mapped to the human genome (hg38) with STAR aligner version 2.5.2a using default mapping parameters40. Numbers of mapped reads for all Ensembl transcript variants of a gene (counts) were combined into a single value by featureCounts software41 and normalized as TPM (transcripts per million). The gene-by-count matrix was further processed using Besca38 and Scanpy39.
Smart-seq2 data filtering
In order to achieve high quality data, we kept only those cells that expressed at least 800 and not more than 6,000 genes (same as for 10X data); had a percentage of UMIs mapping to mitochondrial genes not more than 15% (same as for 10X data). This resulted in 1,069 total cells.
Smart-seq2 data normalization
Normalization was performed using count depth scaling to 10,000 total counts per cell, resulting in the cp10k (counts per 10’000) unit. Count values were log-transformed using natural logarithm: ln(cp10k + 1).
Data Records
10X data
The gene-by-cell raw UMI count matrix and the processed data for the 10X single cell RNA-sequencing experiments are available from Zenodo record 839940942. The raw count matrix is available in the MTX format (barcodes.tsv, genes.tsv, matrix.mtx) together with the corresponding metadata in the TSV format (metadata.tsv) within the gzip archive: raw.tar.gz. The matrix can be processed using Besca38. The processed data files are available from the gzip archive standard_workflow_besca2.tar.gz. It contains mainly human-readable text-files or tab-separated files, which can be opened by any text editor or spreadsheet software. The processed data is also available as an AnnData object in the h5ad format: standard_workflow_besca2.h5ad. It can be loaded by Scanpy39 for further analyses, by the cellxgene visualization tool43, or by other compatible toolkits.
Smart-seq2 data
The gene-by-cell raw and tpm-normalized count matrices and the processed data for the Smart-seq2 experiments are available from Zenodo record 839945844. The tpm (transcripts per million) normalized count matrix is available in the MTX format (barcodes.tsv, genes.tsv, matrix.mtx) together with the corresponding metadata in the TSV format (metadata.tsv) within the gzip archive: raw.tar.gz. The matrices can be processed using Besca38. The processed data files are available from the gzip archive: standard_workflow_besca2.tar.gz. It contains mainly human-readable text-files or tab-separated files, which can be opened by any text editor or spreadsheet software. The processed data is also available as an AnnData object in the h5ad format: standard_workflow_besca2.h5ad. It can be loaded by Scanpy39 for further analyses, by the cellxgene visualization tool43, or by other compatible toolkits.
10X and Smart-seq2 integrated data
The gene-by-cell raw UMI count matrix and the processed data for the integrated data are available from Zenodo record 839947545. The raw count matrix is available in the MTX format (barcodes.tsv, genes.tsv, matrix.mtx) together with the corresponding metadata in the TSV format (metadata.tsv) within the gzip archive: raw.tar.gz. The matrix can be processed using Besca38. The processed data files are available from the gzip archive: integrated_10X_SS2.tar.gz. It contains mainly human-readable text-files or tab-separated files, which can be opened by any text editor or spreadsheet software. The processed data is also available as an AnnData object in the h5ad format: integrated_10X_SS2.h5ad. It can be loaded by Scanpy39 for further analyses, by the cellxgene visualization tool43, or by other compatible toolkits.
Technical Validation
Cell sorting
For subsequent scRNA-seq cells were sorted either as bulk populations when used for sequencing with the 10X Genomics platform or directly into lysis buffer of 96-well plates when used for sequencing by the Smart-seq2 protocol. The general gating strategy for these two approaches was identical. Cells were identified as lymphocytes and doublets excluded by forward and side scatter. Viable CD45+ cells were selected and CD3+ T cells identified.
In the following steps either total non-naive T cells by gating out CD45RA/CCR7 double-positive naive cells, PD1+ T cells or HBV specific CD8+ T cells were chosen for sorting (Fig. 2). For the sorting of HBV-specific T cells from HLA-A*02:01+ donors, cells double-positive for CD8 and the MHC-core-peptide (FLPSDFFPSV) tetramer complex were selected.
Fig. 2.

Gating strategy for T cell populations of interest for subsequent scRNA-seq. Representative image of the gating strategy used for cell sorting of non-naive T cells, PD1+ T cells and HBV-specific T cells using antibodies against surface markers and fluorescently labeled MHC tetramers against the core protein as indicated on the plot axes.
For the plate based Smart-seq2 protocol HBV-specific T cells from 3 different donors were included. These cells are assumed to display an exhausted phenotype, indicated by high PD1 expression. Figure 3 shows PD1 expression on HBV-specific cells for each of the donors displayed versus the expression in total T cells. The HBV-specific cell population shows a higher expression of PD1 versus total T cells.
Fig. 3.
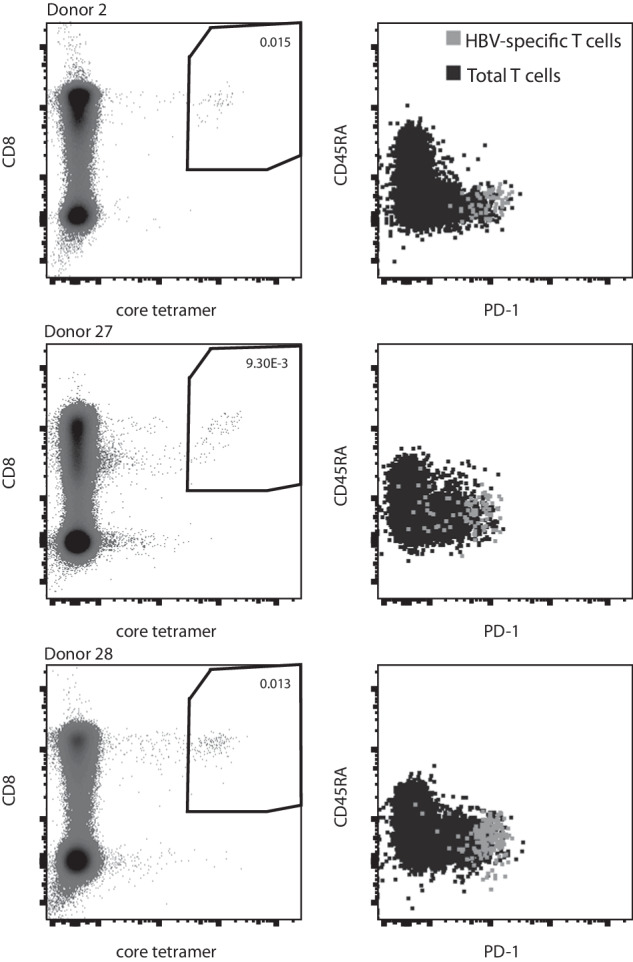
FACS staining and PD1 expression of HBV core-specific CD8 T cells from three donors used for scRNA-seq with Smart-seq2.
10X cell clustering
The steps in this paragraph were done to evaluate the 10X data alone and are not relevant for the integration of the 10X and Smart-seq2 data.
To reduce dataset dimensionality before clustering, the highly variable genes within the dataset were selected. Genes were defined as being highly variable when they have a minimum mean expression of 0.0125, a maximum mean expression of 3 and a minimum dispersion of 0.5.
Technical variance was removed by regressing out the effects of count depth and mitochondrial gene content and the gene expression values were scaled to a mean of 0 and variance of 1 with a maximum value of 10.
The first 50 principal components were calculated and used as input for calculation of the 10 nearest neighbours. The neighbourhood graph was then embedded into two-dimensional space using the Uniform Manifold Approximation and Projection (UMAP) algorithm)46. Cell communities are detected using the Leiden algorithm47 at a resolution of 1 (Fig. 4).
Fig. 4.
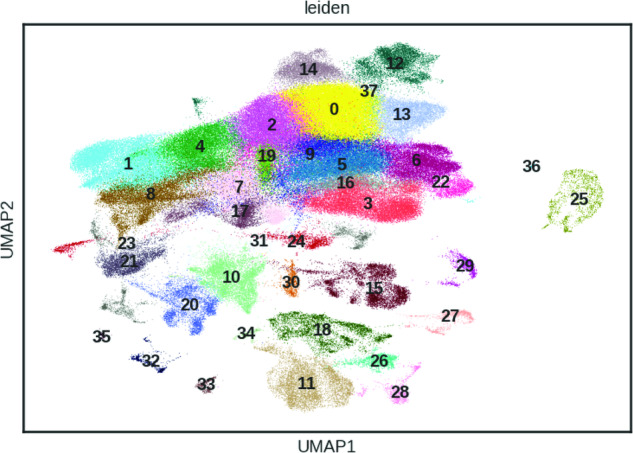
UMAP of 186,123 cells from the 10X platform coloured by 38 Leiden clusters.
Smart-seq2 cell clustering
The steps in this paragraph were done to evaluate the Smart-seq2 data alone and are not relevant for the integration of the 10X and Smart-seq2 data (see paragraph thereafter).
To reduce dataset dimensionality before clustering, the highly variable genes within the dataset were selected. Genes were defined as being highly variable when they have a minimum mean expression of 0.0125, a maximum mean expression of 3 and a minimum dispersion of 0.5.
Technical variance was removed by regressing out the effects of count depth and mitochondrial gene content and the gene expression values were scaled to a mean of 0 and variance of 1 with a maximum value of 10.
The first 50 principal components were calculated and used as input for calculation of the 10 nearest neighbours. The neighbourhood graph was then embedded into two-dimensional space using the UMAP algorithm46. Cell communities were detected using the Leiden algorithm47 at a resolution of 1 (Fig. 5).
Fig. 5.
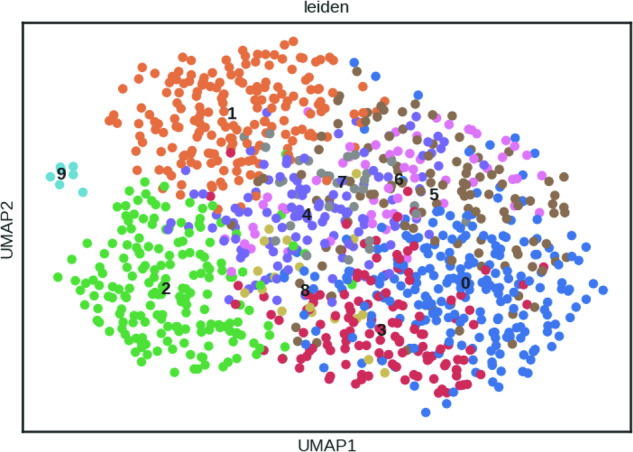
UMAP of 1,069 cells from the Smart-seq2 protocol coloured by 10 Leiden clusters.
Integration of 10X and Smart-seq2 data
The raw UMI count matrix (10X data) and TPM count matrix (Smart-seq2 data) were concatenated resulting in 187,192 cells. Normalization was performed using count depth scaling to 10,000 total counts per cell, resulting in the cp10k (counts per 10,000) unit for both protocols. Count values were log-transformed using natural logarithm: ln(cp10k + 1). We did not apply any advanced integration method and therefore cells cluster by protocol (see Fig. 7).
Fig. 7.

UMAP of 187,192 cells from the integrated 10X and Smart-seq2 dataset coloured by organ (left) and protocol (right).
Integrated cell clustering
The clustering performed on the integrated dataset is independent from the previous clusterings on the individual datasets (see previous paragraphs).
To reduce dataset dimensionality before clustering, the highly variable genes within the dataset were selected. Genes were defined as being highly variable when they have a minimum mean expression of 0.0125, a maximum mean expression of 3 and a minimum dispersion of 0.5.
Technical variance was removed by regressing out the effects of count depth and mitochondrial gene content and the gene expression values are scaled to a mean of 0 and variance of 1 with a maximum value of 10.
The first 50 principal components were calculated and used as input for calculation of the 10 nearest neighbours. The neighbourhood graph was then embedded into two-dimensional space using the UMAP algorithm46. Cell communities were detected using the Leiden algorithm47 at a resolution of 1 (Figs. 6–8).
Fig. 6.

UMAP of 187,192 cells from the integrated 10X and Smart-seq2 dataset coloured by 38 Leiden clusters (left) and experiment condition (right).
Fig. 8.

UMAP of 187,192 cells from the integrated 10X and Smart-seq2 dataset coloured by disease (left) and Leiden (right).
We assessed the cell types of all cells by calculating a signature scores for all signatures provided by Besca (https://github.com/bedapub/besca/blob/master/besca/datasets/genesets/CellNames_scseqCMs6_sigs.gmt)38. The score is the average expression of a set of genes subtracted with the average expression of a reference set of genes, calculated by Scanpy’s score_genes function (https://scanpy.readthedocs.io/en/stable/generated/scanpy.tl.score_genes.html)39.
These signatures were selected for the cell type annotation (Figs. 9, 10):
Hematopoietic signature genes: PTPRC, CORO1A, RAC2, CD53, LAPTM5, CXCR4, LCP1
Myeloid signature genes: CSF3R, MS4A6A, MS4A7, MNDA, C5AR1, FCGR2A, C3AR1, FPR1, LILRB2, HDC, FCGR3B, CCL22
B cell signature genes: CD19, MS4A1, TNFRSF13C, VPREB3, PAX5, CR2
T cell signature genes: CD3E, CD3D, CD3G, TRAC, BCL11B, TRAT1, CD2
NK cell signature genes: NCR1, LIM2, KIR2DL4, KLRC1, IL18RAP, KLRF1,
Endothelial signature genes: CDH5, ECSCR, CCL14, KDR, TIE1, PCAT19, MYCT1, FLT4
Fig. 9.

UMAP of 187,192 cells from the integrated 10X and Smart-seq2 dataset coloured by signature score from Scanpy’s score_genes function.
Fig. 10.

UMAP of 187,192 cells from the integrated 10X and Smart-seq2 dataset coloured by cell type annotation.
Acknowledgements
We thank Dr. Simona Tripon (Strasbourg University Hospital) for her help with patient inclusion. We thank colleagues at Roche who supported the project: Asja Praetor, Guillaume Marc Daniel and Volker Stucke for contracting and external alliance support, Petra Schwalie and Jitao David Zhang for bioinformatic discussions, Thomas Racek and Claudia Bossen for administrative support, Vera Griesser, Fabian Koechl and Alexia Phedonos for sequencing support.
Author contributions
K.H. performed bioinformatic data analysis, interpreted data, wrote the manuscript. T.K.T. contributed to bioinformatic data analysis. N.G. performed library preparation, sequencing and contributed to manuscript preparation. A.S. selected and enrolled patients and edited the manuscript. P.S. selected and enrolled patients and assured biological sample collection. N.K. helped with sequencing and contributed to manuscript preparation. T.N. gave substantial intellectual input for study design and analysis. F.H. selected and enrolled patients and reviewed the manuscript. T.F.B. initiated and designed the study and edited the manuscript. N.P. initiated and designed the study, interpreted data and gave substantial intellectual input. M.F. designed the study, performed experiments, interpreted data and wrote the manuscript.
Code availability
The Besca38 toolkit was used to process the data. The corresponding Jupyter notebooks are available from the Zenodo together with the data. The processing workflow for the 10X data is available from record 839940942 standard_workflow_besca2.ipynb or standard_workflow_besca2.html and the processing workflow for the Smart-seq2 data is available from record 839945844 standard_workflow_besca2.ipynb or standard_workflow_besca2.html. The Jupyter notebook to integrate both datasets is available from record 839947545 integrate_10x_smartseq2.ipynb or integrate_10x_smartseq2.html.
Competing interests
Authors previously or currently employed by F. Hoffmann-La Roche Ltd. might hold stocks. The authors declare that they have no other competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Klas Hatje, Email: klas.hatje@roche.com.
Antonio Saviano, Email: saviano@unistra.fr.
Marianne Forkel, Email: marianne.forkel@roche.com.
References
- 1.WHO. GLOBAL HEPATITIS REPORT, 2017. https://www.who.int/publications/i/item/9789241565455 (2017).
- 2.Lazarus JV, et al. The hepatitis B epidemic and the urgent need for cure preparedness. Nat Rev Gastroenterol. 2018;15:517–518. doi: 10.1038/s41575-018-0041-6. [DOI] [PubMed] [Google Scholar]
- 3.Maini MK, et al. Direct ex vivo analysis of hepatitis B virus-specific CD8 + T cells associated with the control of infection. Gastroenterology. 1999;117:1386–1396. doi: 10.1016/S0016-5085(99)70289-1. [DOI] [PubMed] [Google Scholar]
- 4.Webster GJM, et al. Incubation phase of acute hepatitis B in man: Dynamic of cellular immune mechanisms. Hepatology. 2000;32:1117–1124. doi: 10.1053/jhep.2000.19324. [DOI] [PubMed] [Google Scholar]
- 5.Dunn C, et al. Temporal Analysis of Early Immune Responses in Patients With Acute Hepatitis B Virus Infection. Gastroenterology. 2009;137:1289–1300. doi: 10.1053/j.gastro.2009.06.054. [DOI] [PubMed] [Google Scholar]
- 6.BÖCHER WO, et al. Regulation of the neutralizing anti‐hepatitis B surface (HBs) antibody response in vitro in HBs vaccine recipients and patients with acute or chronic hepatitis B virus (HBV) infection. Clin Exp Immunol. 1996;105:52–58. doi: 10.1046/j.1365-2249.1996.d01-732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burton AR, et al. Circulating and intrahepatic antiviral B cells are defective in hepatitis B. J Clin Invest. 2018;128:4588–4603. doi: 10.1172/JCI121960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salimzadeh L, et al. PD-1 blockade partially recovers dysfunctional virus-specific B cells in chronic hepatitis B infection. J Clin Invest. 2018;128:4573–4587. doi: 10.1172/JCI121957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boni C, et al. Characterization of Hepatitis B Virus (HBV)-Specific T-Cell Dysfunction in Chronic HBV Infection. J Virol. 2007;81:4215–4225. doi: 10.1128/JVI.02844-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. J Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoogeveen RC, et al. Phenotype and function of HBV-specific T cells is determined by the targeted epitope in addition to the stage of infection. Gut. 2018;68:893–904. doi: 10.1136/gutjnl-2018-316644. [DOI] [PubMed] [Google Scholar]
- 12.Ye B, et al. T-cell exhaustion in chronic hepatitis B infection: current knowledge and clinical significance. Cell Death Dis. 2015;6:e1694–e1694. doi: 10.1038/cddis.2015.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schuch A, et al. Phenotypic and functional differences of HBV core-specific versus HBV polymerase-specific CD8 + T cells in chronically HBV-infected patients with low viral load. Gut. 2019;68:905–915. doi: 10.1136/gutjnl-2018-316641. [DOI] [PubMed] [Google Scholar]
- 14.Boni C, et al. Restored Function of HBV-Specific T Cells After Long-term Effective Therapy With Nucleos(t)ide Analogues. Gastroenterology. 2012;143:963–973.e9. doi: 10.1053/j.gastro.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 15.Bénéchet AP, et al. Dynamics and genomic landscape of CD8 + T cells undergoing hepatic priming. Nature. 2019;574:200–205. doi: 10.1038/s41586-019-1620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bert NL, et al. Effects of Hepatitis B Surface Antigen on Virus-Specific and Global T Cells in Patients With Chronic Hepatitis B Virus infection. Gastroenterology. 2020;159:652–664. doi: 10.1053/j.gastro.2020.04.019. [DOI] [PubMed] [Google Scholar]
- 17.Montali I, et al. Antigen Load and T Cell Function: A Challenging Interaction in HBV Infection. Biomed. 2022;10:1224. doi: 10.3390/biomedicines10061224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raziorrouh B, et al. Inhibitory Phenotype of HBV-Specific CD4 + T-Cells Is Characterized by High PD-1 Expression but Absent Coregulation of Multiple Inhibitory Molecules. Plos One. 2014;9:e105703. doi: 10.1371/journal.pone.0105703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, et al. CXCL13-mediated recruitment of intrahepatic CXCR5 + CD8 + T cells favors viral control in chronic HBV infection. J Hepatol. 2020;72:420–430. doi: 10.1016/j.jhep.2019.09.031. [DOI] [PubMed] [Google Scholar]
- 20.Cheng, Y. et al. Multifactorial heterogeneity of virus-specific T cells and association with the progression of human chronic hepatitis B infection. Sci Immunol4, (2019). [DOI] [PubMed]
- 21.Cheng Y, et al. Non-terminally exhausted tumor-resident memory HBV-specific T cell responses correlate with relapse-free survival in hepatocellular carcinoma. Immunity. 2021;54:1825–1840.e7. doi: 10.1016/j.immuni.2021.06.013. [DOI] [PubMed] [Google Scholar]
- 22.Yuen M, et al. HBsAg seroclearance in chronic hepatitis B in the Chinese: Virological, histological, and clinical aspects. Hepatology. 2004;39:1694–1701. doi: 10.1002/hep.20240. [DOI] [PubMed] [Google Scholar]
- 23.Ferreira SC, et al. Factors associated with spontaneous HBsAg clearance in chronic hepatitis B patients followed at a university hospital. Ann Hepatol. 2014;13:762–70. doi: 10.1016/S1665-2681(19)30978-0. [DOI] [PubMed] [Google Scholar]
- 24.Nkongolo S, et al. Longitudinal liver sampling in patients with chronic hepatitis B starting antiviral therapy reveals hepatotoxic CD8 + T cells. J Clin Investigation. 2023;133:e158903. doi: 10.1172/JCI158903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maini MK, et al. The Role of Virus-Specific Cd8 + Cells in Liver Damage and Viral Control during Persistent Hepatitis B Virus Infection. J Exp Medicine. 2000;191:1269–1280. doi: 10.1084/jem.191.8.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pallett LJ, et al. IL-2high tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J Exp Med. 2017;214:1567–1580. doi: 10.1084/jem.20162115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Isogawa M, Kakimi K, Kamamoto H, Protzer U, Chisari FV. Differential dynamics of the peripheral and intrahepatic cytotoxic T lymphocyte response to hepatitis B surface antigen. Virology. 2005;333:293–300. doi: 10.1016/j.virol.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 28.Ouaguia L, et al. Circulating and Hepatic BDCA1+, BDCA2+, and BDCA3 + Dendritic Cells Are Differentially Subverted in Patients With Chronic HBV Infection. Front Immunol. 2019;10:112. doi: 10.3389/fimmu.2019.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yao C, et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8 + T cell persistence in chronic infection. Nat Immunol. 2019;20:890–901. doi: 10.1038/s41590-019-0403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pritykin Y, et al. A unified atlas of CD8 T cell dysfunctional states in cancer and infection. Mol Cell. 2021;81:2477–2493.e10. doi: 10.1016/j.molcel.2021.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang S, et al. An atlas of immune cell exhaustion in HIV-infected individuals revealed by single-cell transcriptomics. Emerg Microbes Infect. 2020;9:2333–2347. doi: 10.1080/22221751.2020.1826361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aizarani N, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature. 2019;572:199–204. doi: 10.1038/s41586-019-1373-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng C, et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell. 2017;169:1342–1356.e16. doi: 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 34.Zhang C, et al. Single-cell RNA sequencing reveals intrahepatic and peripheral immune characteristics related to disease phases in HBV-infected patients. Gut. 2023;72:153–167. doi: 10.1136/gutjnl-2021-325915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Genshaft, A. S. et al. Single-cell RNA sequencing of liver fine-needle aspirates captures immune diversity in the blood and liver in chronic hepatitis B patients. Hepatology Publish Ahead of Print (2023). [DOI] [PMC free article] [PubMed]
- 36.Bai Q, et al. Single-cell landscape of immune cells during the progression from HBV infection to HBV cirrhosis and HBV-associated hepatocellular carcinoma. Front. Immunol. 2023;14:1320414. doi: 10.3389/fimmu.2023.1320414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Picelli S, et al. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 38.Mädler SC, et al. Besca, a single-cell transcriptomics analysis toolkit to accelerate translational research. NAR Genom. Bioinform. 2021;3:lqab102-. doi: 10.1093/nargab/lqab102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 42.Hatje K, 2023. HBV Immunomics 10X data. Zenodo. [DOI]
- 43.Megill, C. et al. cellxgene: a performant, scalable exploration platform for high dimensional sparse matrices. bioRxiv10.1101/2021.04.05.438318 (2021).
- 44.Hatje K, 2023. HBV Immunomics Smart-seq. 2 data. Zenodo. [DOI]
- 45.Hatje K, 2023. HBV Immunomics 10X and Smart-seq. 2 integrated data. Zenodo. [DOI]
- 46.McInnes L, Healy J, Saul N, Großberger L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018;3:861. doi: 10.21105/joss.00861. [DOI] [Google Scholar]
- 47.Traag VA, Waltman L, van Eck NJ. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 2019;9:5233. doi: 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
Data Availability Statement
The Besca38 toolkit was used to process the data. The corresponding Jupyter notebooks are available from the Zenodo together with the data. The processing workflow for the 10X data is available from record 839940942 standard_workflow_besca2.ipynb or standard_workflow_besca2.html and the processing workflow for the Smart-seq2 data is available from record 839945844 standard_workflow_besca2.ipynb or standard_workflow_besca2.html. The Jupyter notebook to integrate both datasets is available from record 839947545 integrate_10x_smartseq2.ipynb or integrate_10x_smartseq2.html.