

Abstract
Chemical probes that specifically modulate the activity of heterotrimeric G proteins provide excellent tools for investigating G protein-mediated cell signaling. Herein we report a family of selective peptidyl Gαi/s modulators derived from peptide library screening and optimization. Conjugation to a cell-penetrating peptide rendered the peptides cell-permeable and biologically active in cell-based assays. The peptides exhibit potent guanine nucleotide exchange modulator-like activity towards Gαi and Gαs. Molecular docking and dynamic simulations revealed the molecular basis of the protein-ligand interactions and their effects on GDP binding. This study demonstrates the feasibility of developing direct Gαi/s modulators and provides a novel chemical probe for investigating cell signaling through GPCRs/G proteins.
Keywords: G proteins, G protein-coupled receptors, cell signaling, G protein modulator, peptidyl ligands
Graphical Abstract
INTRODUCTION
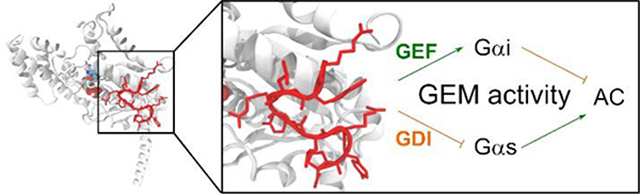
G protein-coupled receptors (GPCRs) transmit signals into the cell via membrane-associated heterotrimeric G proteins (Gαβγ), which are classified into the four subfamilies Gαi/o, Gαs, Gαq/11, and Gα12/13.1 G proteins function as molecular switches by alternating between inactive guanine nucleotide diphosphate (GDP)–bound and active guanine nucleotide triphosphate (GTP)–bound states and interacting with different downstream effectors, e.g., adenylyl cyclase (AC). Generally, Gαs stimulates whereas Gαi inhibits the activity of AC, thereby modulating the intracellular cyclic adenosine monophosphate (cAMP) level.2,3 Malfunction of GPCRs leads to a multitude of human diseases including cancer; consequently, GPCRs have been a fertile ground for drug discovery.4,5 Approximately 30% of FDA-approved drugs target more than 100 GPCRs.4 However, targeting GPCRs has its limitations, as some diseases involve the malfunctioning of several GPCRs, each of which mediates a specific G protein signaling,2,3,6 while in other cases, G proteins may initiate signaling pathways independent of GPCRs.2,3,5–7 Directly targeting intracellular G proteins thus represents an attractive alternative to GPCR–directed therapeutics, but remains a major challenge for several reasons. First, G proteins do not have well-defined pockets on their surfaces and are difficult to target with conventional small molecules. Second, different G protein subfamilies share a high degree of sequence and structure similarity, making it difficult to specifically target a given G protein.2,3,6 Finally, G proteins reside inside the cytosol necessitating cell permeability of any G protein modulator. To date, only a few specific G protein modulators are known, including FR900359 (FR) and YM–254890 (YM), both of which are natural products and target Gαq.2,5–9 No such specific and selective small- or medium-sized modulators exist for targeting the intracellular Gαi and Gαs subfamilies, although protein modulators are known, e.g., cholera toxin (CTX, which is a Gαs activator) and pertussis toxin (PTX, which is a Gαi inhibitor).3,10,11 Mastoparan and suramin compounds are notable outliers, which broad application is limited due to their lack of specificity12,+ and selectivity2, respectively.
Inspired by the fact that both FR900359 (FR) and YM–254890 (YM) are cyclic peptides, researchers have begun to explore linear and cyclic peptides as potential G protein modulators, often by screening combinatorial peptide libraries. For example, screening of mRNA display libraries has identified linear peptides R6A14, R6A-114,15, AR6–0516 and GSP17, as well as more proteolytically stable macrocyclic peptides cycGiBP18, cycPRP–119, cycPRP-319 and Gα SUPR20 as high-affinity binders for Gαi1·GDP (Fig. 1d).3,14–20 A GDP–selective ligand, KB-752,21 was obtained from a phage display library and acts as guanine-nucleotide exchange factor (GEF) for Gαi1 and as guanine-nucleotide dissociation inhibitor (GDI) for Gαs.21–23 This bifunctional activity is referred to as the guanine-nucleotide exchange modulator (GEM) activity, which was originally described for GIV and other proteins containing a GEM motif (ΦTΦX[D/E]FΦ, where Φ is a hydrophobic residue and X is any residue.24 The GEM motif has previously been referred to as the Gα-binding and activating (GBA) motif.25–27 Structural analyses of the Gαi1·GDP/KB–752 and Gαi3·GDP/GIV–GEM complexes revealed that both peptides bind to a region between the Switch II motif and α3 helix of Gαi.21,28 In this study, we discovered a relatively potent modulator against Gαi1·GDP, GPM-1, by screening a one-bead-one-compound (OBOC) peptide library. Subsequent optimization of GPM-1 and conjugation with a cell-penetrating peptide (CPP) improved its binding affinity, proteolytic stability, and cell permeability. Biomolecular simulations provided insights into the molecular basis of the underlying peptide-protein interactions. Our results show that the cell-permeable variants of GPM–1 (GPM–1c and GPM-1d) modulate the Gαi/s activity in cell culture in a GEM–like manner and thus provide a novel lead for further development into highly specific and potent Gα modulators in the future.
Figure 1.
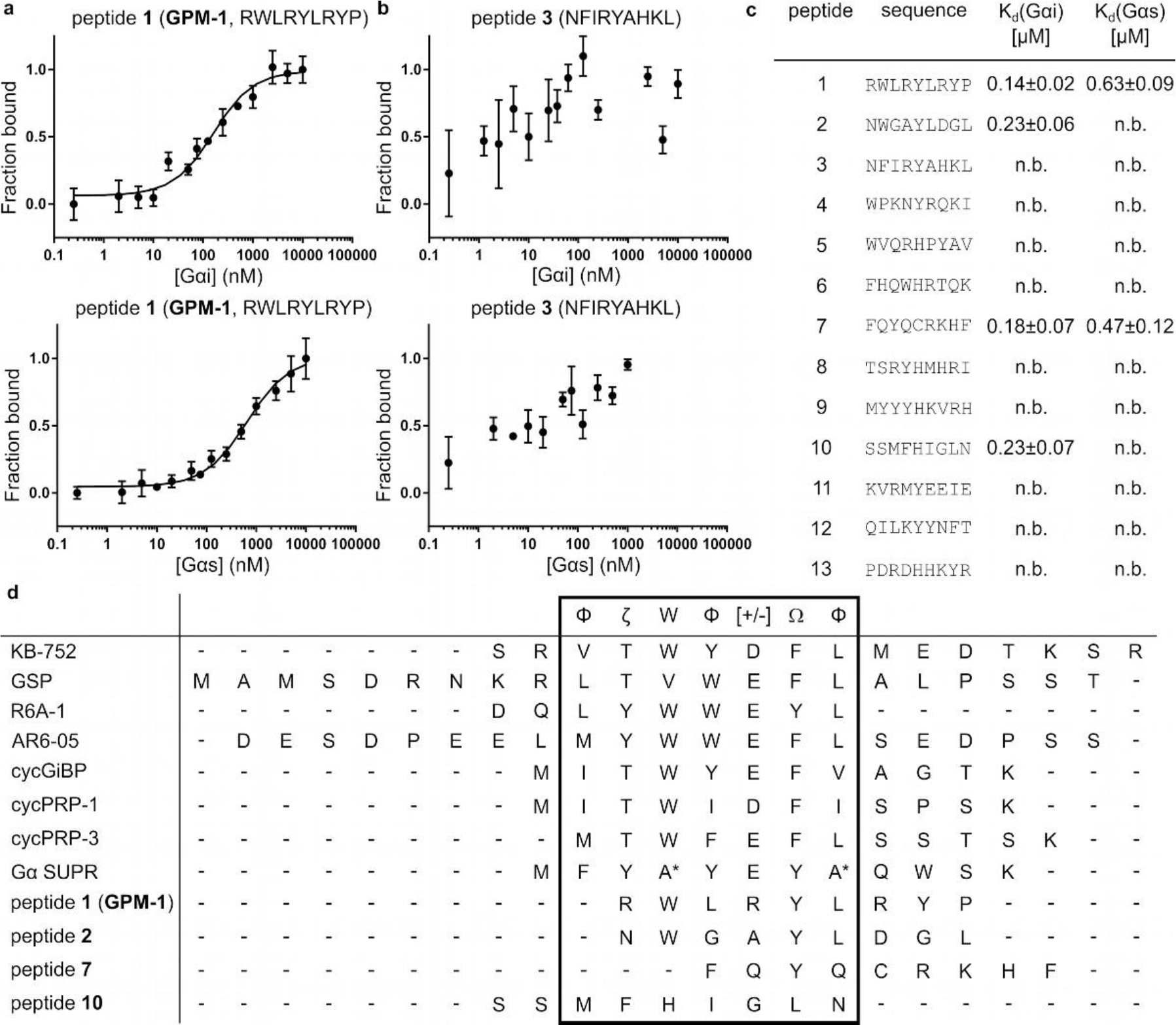
MST results and sequence alignment of the best Gαi1·GDP binders. (a) Binding curves of 5(6)-carboxyfluorescein-labeled peptide 1 (GPM–1) with Gαi1/s, error bars represent standard deviation (SD) for n = 3; (b) Peptide 3 as non–binding representative for Gαi1/s, n = 2; (c) MST results of all hits. (n.b.: no binding (< 1 μM or not saturated); M = Nle). (d) Sequence alignment of KB-75221, GSP17, R6A-114, AR6–0516, cycGiBP18, cycPRP-1/319, Gα SUPR20, and peptides 1 (GPM-1), 2, 7, 10. M = Nle29 for peptide 10, * = N-methylated amino acids, Φ: hydrophobic (V, I, L, F, W, Y, M), Ω: aromatic (F, W, Y), ζ: uncharged hydrophilic (N, Q, S, T), [+]: basic (H, K, R) and [−]: acidic (D, E) amino acids.24
RESULTS AND DISCUSSION
Identification of Gαi1 Binders.
The lengths/sizes of GEM motifs in proteins (7 aa),23,25 FR/YM (eight building blocks),9 and the high-affinity Gαi1·GDP ligand R6A–1 (9 aa)14,15 suggest that a peptide sequence of seven to nine residues should be sufficient for high-affinity binding to Gα proteins. We therefore screened a previously reported nonapeptide OBOC library29 for binding to biotinylated Gαi1·GDP by following a well-established library screening protocol (Figure S1 and text in SI).30,31 A total of 101 hit sequences (Table S1) were obtained and analyzed for recurring motifs to establish any consensus sequence(s) (Figure S1). Thirteen representative peptides (1–13) were selected from different consensus groups for resynthesis and binding studies (Figures 1c and S1, Tables S2, S3).
Specificity for Gα Binding of Selected Hits.
The binding affinity of the selected hits for Gαi/s was determined by microscale thermophoresis (MST, Figures 1a–c and S3) employing fluorescein-labeled peptides and varying concentrations of recombinant Gαi1·GDP or Gαs·GDP (Table S4, Figure S2). In addition, fluorescein-labeled KB-752 was used as a positive control (Tables S3, S4).21 Four of the peptides (1, 2, 7, and 10) showed potent binding to Gαi1·GDP (Kd = 140–230 nM) but no or only weak binding to Gαs·GDP (Figures 1a–d and S3). Under the same condition, KB–752 showed a Kd value of 345 ± 40 nM for Gαi and no binding to Gαs, which is somewhat different from the previously reported Kd values of 3900 nM (for Gαi) and 5100 nM (for Gαs) as determined by surface plasmon resonance (SPR, Figure S3).21 Compared to the previously reported peptide ligands,14–21 our peptides contain a similar number of hydrophobic aromatic amino acids but a higher percentage of basic (and less acidic) residues (Figure 1d). Peptides 1 and 2 contain a ζWΦ[+/−]ΩΦ-motif (ζ: polar; Φ: hydrophobic; Ω: aromatic amino acid24), which is also shared by KB–752, GSP, R6A and R6A–1, AR6–05, cycGiBP, cycPRP–1, cycPRP–3 and Gα SUPR (Figure 1d, Table S2).3,12–19 Moreover, the binding motif exhibits similarities to the GEM motif in proteins (ΦTΦX[D/E]FΦ).25–27 In comparison to the alignment of the library-derived peptides (ΦζWΦ[+/−]ΩΦ–motif), the alignment of peptide 1 with the GEM motif can be slightly adjusted to a ΦζΦX[+/−]ΩL–consensus. The sequence similarity suggests that like the previously described peptides and GEM proteins, peptides 1 and 2 may also bind to the Switch II/α3 region of Gαi1.18,19,21,26,28,32 To test this hypothesis, we examined the ability of peptide 1 to compete with fluorescein-labeled KB–752 for binding to Gαi1 by MST (Figure S4). Peptide 1 indeed inhibited KB–752 for binding to Gαi1 with an IC50 value of 155 ± 10 nM. Since Phe8 of KB-75221 and Phe168 of the GIV–GEM motif28,33 are crucial for Gαi binding and the GEM activity, we next replaced the corresponding residue of peptide 1 (Tyr5) with an alanine and tested the resulting variant (1Y5A) for binding to Gαi1 and Gαs (Tables S3, S4). 1Y5A exhibited ~2–fold lower affinity for Gαi1 than peptide 1 (Kd = 285 ± 40 nM) and no binding to Gαs (Figure S5). Taken together, our data suggest that peptide 1 binds to the Switch II/α3 region of Gαi1 and may function similarly to KB–752 and GEM proteins.21,28,33 On the other hand, peptides 7 and 10 have lower sequence similarity to the ζWΦ[+/−]ΩΦ–motif in peptides 1 and 2 or to the previously reported peptides and their binding sites on Gαi1 are currently unclear (Figure 1d, Table S2).
Optimization of Hit Peptide.
Linear peptide 1, referred to as GPM-1 (for “G protein modulator–1”, Figures 1, 2, and S6) hereafter, is likely proteolytically unstable and lacks membrane permeability. We thus undertook a limited medicinal chemistry campaign to improve its “drug-like” properties.
Figure 2.
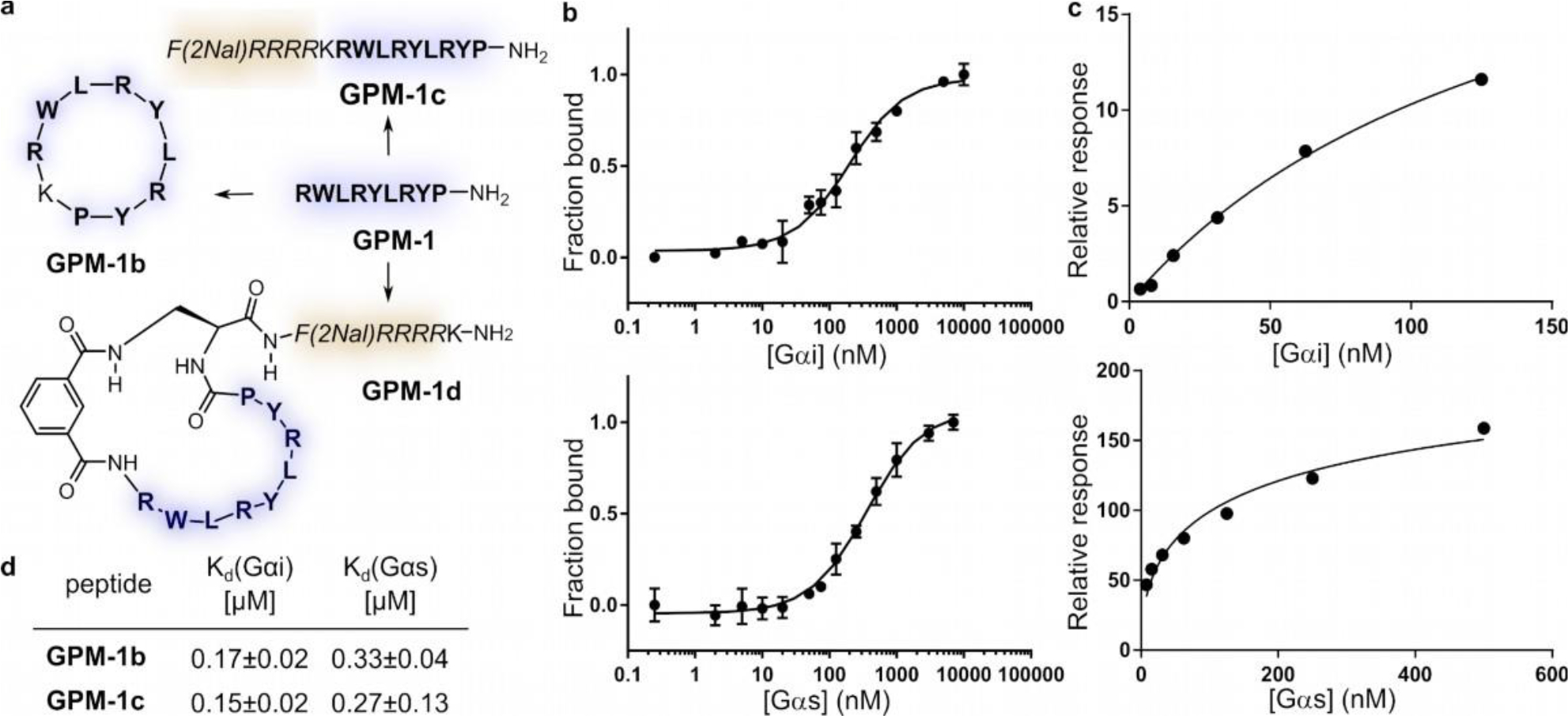
Optimization of GPM–1 and binding results for the derived peptides. (a) GPM–1b is the head-to-tail cyclized GPM–1 (H–RWLRYLRYP–NH2, blue); GPM–1c comprises GPM–1 and the CPP moiety [F(2Nal)RRRR, yellow]; GPM-1d includes the cyclic GPM-1(b) and the CPP. (b) MST data of FITC–labeled GPM–1b with Gαi1/s, error bars represent SD for n = 3. (c) SPR data of immobilized biotinylated GPM–1c with Gαi1/s, n = 1. (d) obtained Kd values of GPM–1b and GPM–1c.
First, head-to-tail cyclization of GPM–1 resulted in GPM–1b, which is expected to have improved metabolic stability (Figures 2a and S6, Table S3). Next, a CPP sequence, F(2Nal)RRRR (where 2Nal is L–2–naphthylalanine), and a lysine were added to the N-terminus of GPM-1 to facilitate cellular entry and fluorescent labeling, respectively, yielding linear peptide GPM–1c (Figures 2a and S6, Table S3). This CPP sequence has previously been shown to effectively deliver biologically active peptidyl cargos, e.g., inhibitors against the monomeric G protein K–Ras, into mammalian cells.34 Its small size also helps to keep the molecular weight of the final construct to a minimum. Finally, GPM–1 was extended at the C-terminus with a (S)-2,3-diaminopropionyl (Dap) unit followed by the CPP motif and subsequently cyclized between its N-terminus and the sidechain amine of the Dap residue by using a bifunctional linker, isophthalic acid (Ipa), to give cyclic peptide GPM–1d (Figures 2a and S6, Table S3).
Two control peptides were also prepared. Peptide 14 (H– KRWLRYLRYP–NH2) serves as a control to assess the effect of the inserted Lys on target binding and the biological activity of GPM–1, while peptide 15 reveals whether the CPP sequence alone has any biological activity (Figures 2a and S6, Table S3).
Peptides GPM–1b–d, 14 and 15 were fluorescently labeled and tested for binding to Gαi1/s by MST (Figures 2b and S3, Table S4). As expected, peptide 14 exhibited a similar binding affinity for Gαi (Kd = 140 ± 30 nM) to GPM–1, but a lower binding affinity for Gαs (Kd = 1010 ± 160 nM). Cyclization of GPM–1 slightly decreased its binding affinity for Gαi (Kd = 170 ± 20 nM for GPM–1b, Fig. 2b, d), but unexpectedly increased the affinity for Gαs (Kd = 330 ± 40 nM for GPM–1b). The CPP-containing peptides (GPM–1c, GPM–1d, and 15) could not be analyzed by MST, presumably because the positively charged sequences resulted in strong binding of the peptides to the negatively charged glass wall of the capillaries.35 We therefore labeled peptides GPM–1, GPM–1c, GPM–1d, and 15 with a biotin (for surface immobilization, Table S4) and analyzed them for binding to Gαi1·GDP and Gαs·GDP by surface plasmon resonance (SPR) (Figures 2c and S7). Peptides GPM–1, GPM–1c, and 15 bound to Gαi1 with Kd values of 170 ± 50 nM, 150 ± 20 nM, and 530 ± 70 nM, respectively (Figures 2c–d and S7). Thus, for GPM-1, there is good agreement between the Kd values derived from MST and SPR assays. As expected, GPM–1c retained the binding affinity of GPM–1 for Gαi1·GDP. Surprisingly, peptide 15 (the CPP moiety alone) showed significant binding affinity for Gαi1, although the affinity is considerably lower than that of GPM–1 and GPM–1c. Note that peptides 15 and GPM–1 share substantial structural similarities, in that they are both rich in arginine and aromatic hydrophobic residues. We were not able to reliably determine the binding affinity of GPM–1d by SPR, because protein binding to the immobilized peptide resulted in only small response unit (RU) changes. GPM–1d was directly used in the activity studies, as we expected it to have similar binding affinity to GPM–1b and GPM–1c, which are both high-affinity Gαi binders. Importantly, GPM–1 (Kd = 560 ± 60 nM) and GPM–1c (Kd = 270 ± 130 nM) showed weaker binding to Gαs than Gαi1, in agreement with the MST data for GPM–1 (Kd = 630 ± 90 nM). Peptide 15 displayed substantially lower affinity for Gαs (Kd = 2000 ± 720 nM, Figure S7).
Biological Activity in Cell Culture.
The biological activity of GPM–1 and its derivatives was assessed by two different assays. In a cell-free ELISA-based assay,36 membrane-bound inhibitory (δ–opioid) and stimulatory (β2–adrenergic) GPCRs converge at the level of AC, allowing the use of cAMP production as a common readout (Figure 3a–b). Regulation of the Gs function can be assessed under basal, isoproterenol (Iso; receptor activation)- and forskolin (Fsk; direct activation of AC)-stimulated conditions, whereas any regulatory effect on Gi is gleaned from the inhibition of Fsk-stimulated cAMP production by [Tyr–D–Ala2–Gly–Phe–D–Leu5]Enkephalin (DADLE). A whole-cell assay was also established with HEK293 cells overexpressing the SNAP- β2 adrenergic receptor to specifically monitor the Gs signaling pathway after stimulation with Iso in a concentration-dependent manner (Figure 3c). It should be noted that since the change in cAMP levels represent the sum of all G protein activities, these assays only detect major regulatory effects on dominant signaling cascades.
Figure 3.
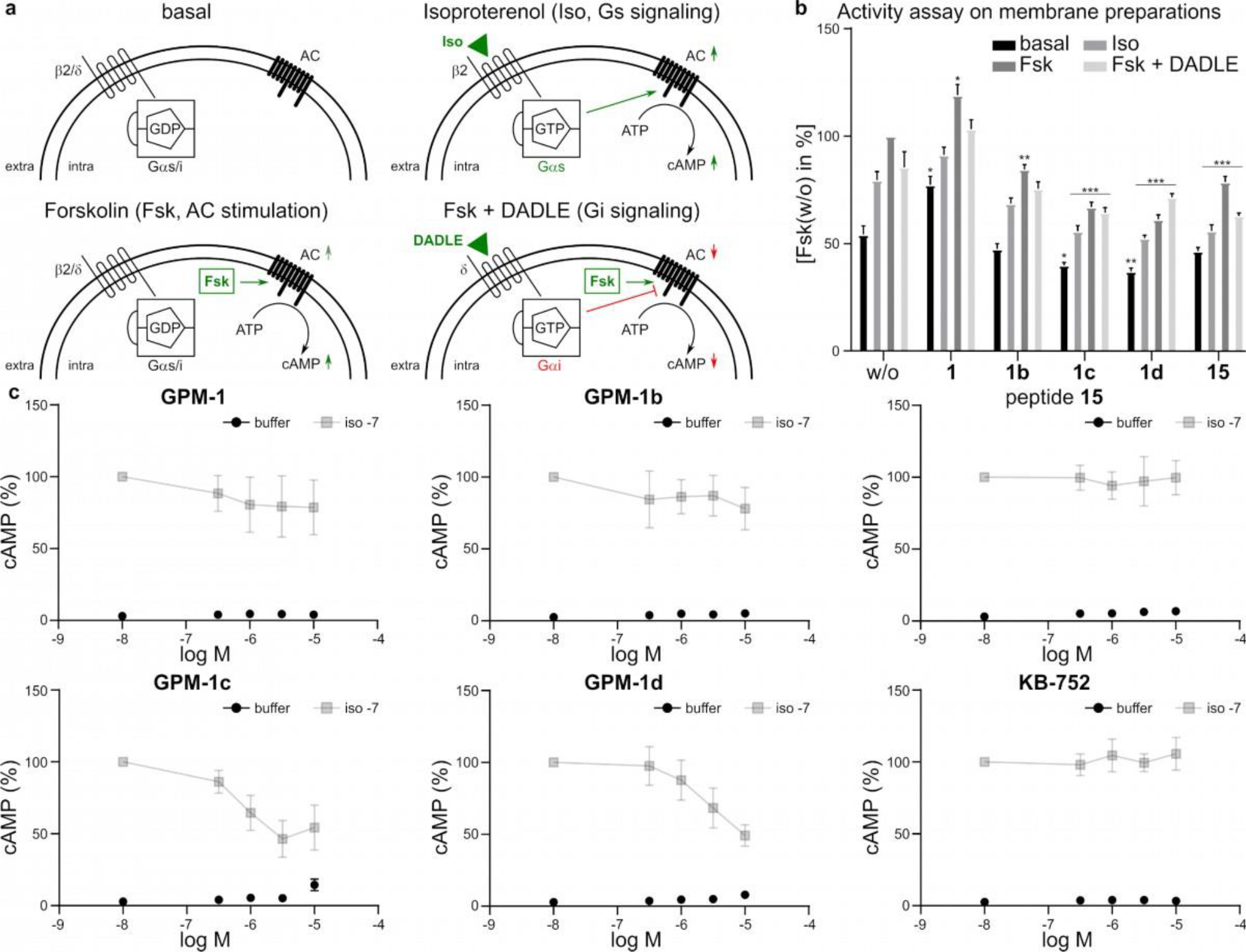
Functional studies of GPM-1 and its derivatives. (a) Schematic representation of the individual states in the activity studies on NG108–15 membrane preparations. Basal: GPCR (β2 or δ) available for ligand binding, Gα activity is measured independently of the GPCRs. Isoproterenol (Iso) binds to the β2–adrenergic receptor and activates the Gs signaling. Activated Gs stimulates AC leading to an increased cAMP level. Forskolin (Fsk) stimulates AC and increases the cAMP level. Fsk+DADLE: Fsk stimulates cAMP production by direct activation of AC. DADLE ([Tyr–D–Ala2–Gly–Phe–D–Leu5]Enkephalin) binds to the δ-opioid receptor and activates the Gi signaling. Activated Gi binds to AC and inhibits the Fsk-stimulated cAMP production. (b) Relative cAMP levels on differently induced membrane preparations incubated with or without the modulators (GPM–1, GPM–1b–d, 15) in the presence of a phosphodiesterase inhibitor. cAMP levels were normalized to Fsk or w/o. Shown are percentage values of membranes incubated in the absence (basal), or in the presence of Iso, Fsk and Fsk+DADLE. Error bars represent SD for n = 3. Statistical analysis was performed using the One–Way ANOVA Test Dunnet corrected, with *p<0.05, **p<0.01, and ***p<0.0001 for comparisons with the control (w/o). (c) Iso-induced cAMP accumulation in HEK293 cells. Depicted are the buffer controls (black) and the Iso values (grey) for GPM–1, GPM–1b–d, 15 and KB–752 in % cAMP. Error bars represent SD for n = 3. None of the peptides was found to influence the vitality of the cells in an MTT assay on HEK293 cells at a maximum concentration of 10 μM.
Among the peptides that bound to Gαi1 or Gαs in the biochemical tests (peptides GPM–1, 2, 7, and 10), only GPM–1 and its derivatives exhibited significant biological activity in the cell-free or whole-cell assay. In addition to our peptides, KB–752 was included for comparisons as no data from whole cell assays could be retrieved from the literature.21–23
In the cell-free assay, GPM–1 resulted in an overall increase in the basal, Iso- and Fsk-stimulated cAMP production, relative to the vehicle control (w/o) (Figure 3b). At the meantime, GPM–1 reduced the stimulatory effect of Iso by ~10%. In the cell-based assay, GPM-1 also decreased the Iso-mediated cAMP production in a concentration-dependent manner, by ~20% at 10 μM peptide concentration (Figure 3c). The cellular activity of GPM–1 may seem surprising, because it is not conjugated to any CPP; however, GPM–1 contains three arginine residues as well as several hydrophobic residues and likely possesses significant cell-penetrating activity. Interestingly, KB–752 has previously been reported to inhibit Gs signaling in a membrane preparation assay,22 which was confirmed in our ELISA experiment (Figure S8). However, KB–752 has no effect on Gαs in the whole cell assay (Figure 3c). The simplest explanation of these observations is that both KB–752 and GPM–1 act as GDI toward Gαs.22,27 Indeed, since GPM–1 shares a high sequence similarity to KB–752 within the Gαi/s-binding motif, a similar mechanism of action is not unexpected. Lack of activity in the whole-cell assay by KB–752 is likely because the latter contains multiple negatively charged residues in its sequence and is impermeable to the cell membrane. GPM–1 Y5A showed no effect in the ELISA (Figure S8), which further corroborates the KB–752–like effect of GPM-1, as Phe8 in KB–752 is critical for the GEM activity.21,28,33 As expected, peptide 14 behaved very similar to GPM-1 (Figure S9), whereas GPM–1b showed no functional regulation of Gαs (Figure 3b). The latter is in good agreement with the result of the whole cell assay (Figure 3c) and could be a consequence of the higher rigidity of the cyclic structure compared to GPM-1 despite of its binding affinity.
In the cell-free assay, the CPP-linked peptides GPM–1c and GPM–1d produced an overall reduction in the basal, Iso- and Fsk-stimulated cAMP levels (Figure 3b). This effect was much greater in the cell-based assay; both GPM–1c and GPM–1d dose-dependently reduced Iso-stimulated cAMP production, causing ≥50% inhibition at 10 μM (Figure 3c). The CPP alone (peptide 15) was inactive in the whole cell assay, suggesting that the observed effect in the ELISA-based assay is likely caused by non-specific interactions (Figure 3b–c). These data indicate that GPM–1c and GPM–1d are more effective GDIs than GPM–1.
With respect to Gi signaling, while GPM–1, GPM–1b, GPM–1, Y5A (as well as peptide 15 and KB–752) showed no effect on the Fsk-stimulated AC activity, GPM-1c and GPM–1d increased Gi signaling by 19% and 22%, respectively, relative to the control (w/o) (Figure 3b). Additionally, while GPM–1, GPM–1b, GPM–1 Y5A, KB–752 and peptide 15 had no effect on DADLE-mediated inhibition of the AC activity, GPM–1c almost completely abolished the effect of DADLE and GPM–1d increased the AC activity by ~10% after activation of inhibitory δ-opioid receptors with DADLE (Figure 3b). These results suggest that GPM–1c and GPM–1d function as Gαi specific GEFs resulting in permanent activation of Gαi as described previously for KB–752.21,22
Taken together, the above data demonstrate that GPM–1c and GPM–1d bind to and regulate the function of both Gi (as GEF) and Gs (as GDI) thus acting as bifunctional GEMs, as previously demonstrated for KB–75221,22,27 and GEM proteins.25–27 However, while KB–752 and GEM proteins are impermeable to the cell membrane, GPM–1c and GPM–1d are cell permeable and biologically active in whole-cell assays. Among the two peptides, GPM–1d is the preferred ligand, because of its higher potency in the cellular assay as well as greater proteolytic stability (thanks to its cyclic structure).
Computational Analysis of Gαi1/s-Peptide Complexes.
To gain insight into the structural basis for the observed biological activities, we carried out a series of computational analyses for the interaction of Gαi/s with the peptide modulators experimentally deemed to be active (GPM–1c, GPM–1d), slightly active (GPM–1), and inactive (GPM–1b, GPM–1 Y5A, 15). We describe briefly below the Gαi/s–peptide interactions observed from 50 ns MD trajectories of docked Gαi/s–peptide complexes and the structural and energetic implications. Additional details such as the Gαi homology model, determination of Gαi/s binding site, and molecular docking to Gαi/s are provided in SI (Figures 4, S10–S17, Tables S5–S10).
Figure 4.
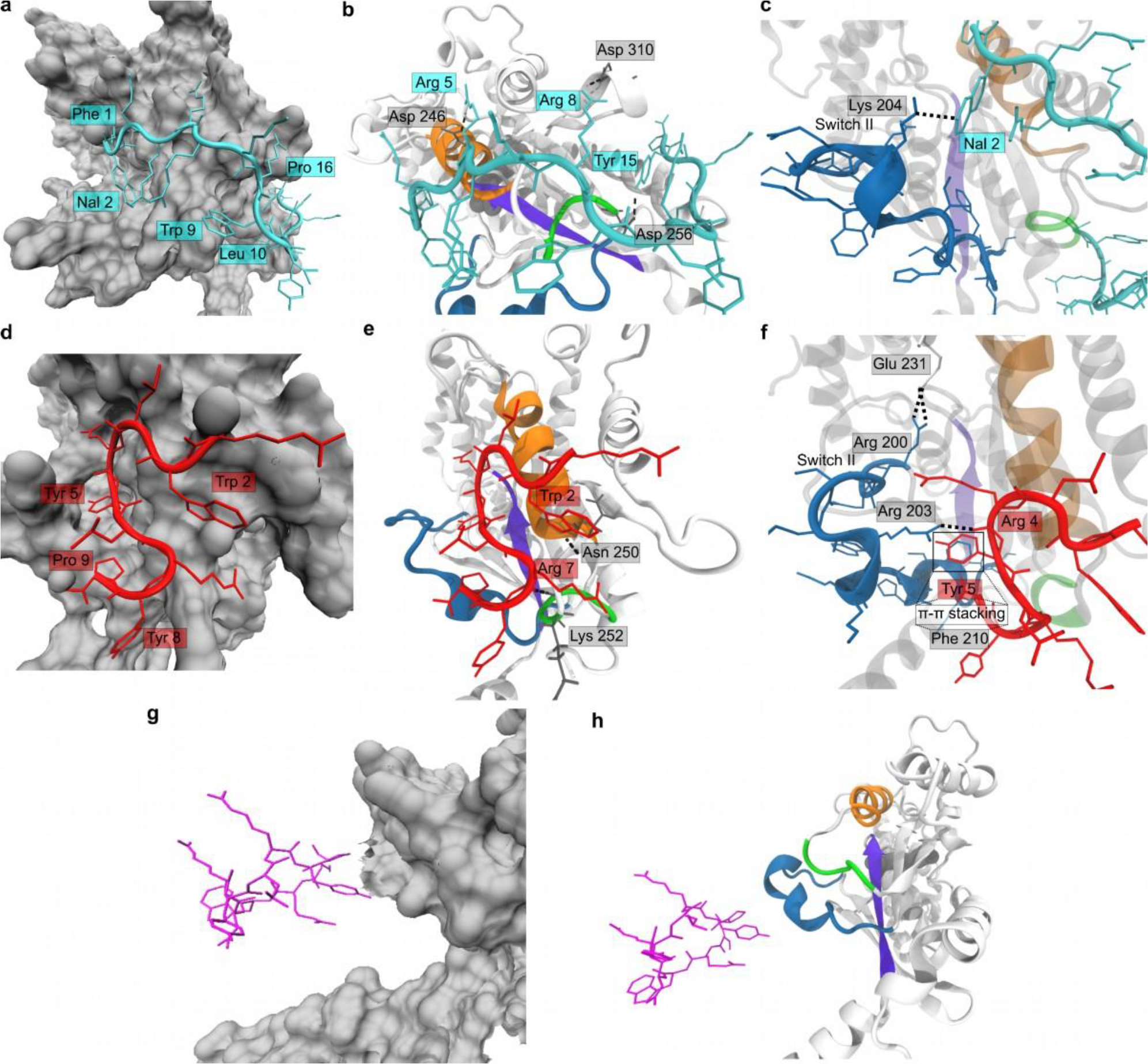
Computational studies of Gαi-peptide interactions. (a), (d), (g) Molecular surface (gray) of Gαi on which GPM–1c (cyan), GPM–1 (red), and GPM–1b (magenta) are bound. In (a) and (d), the side chains involved in hydrophobic interactions with Gαi are labeled. (b), (e), (h) Gαi structure (white cartoon) with Switch II (blue), α3 (orange), β1 (violet), and α3–β5 loop (green) depicted. The bound conformation of GPM–1c (cyan), GPM–1 (red), and GPM–1b (magenta) are presented, with the H-bonding (black dotted lines) partners labeled (for GPM–1c, GPM–1). (c), (f) Closer look at the interactions between Switch II (blue) of Gαi and GPM–1c (cyan) and GPM–1 (red). H-bonding interactions are shown as black dotted lines, the residues involved are labeled. In (f), the π-π stacking interactions between Tyr5 of GPM–1 and Phe210 of Switch II are highlighted in a square.
GPM-1c binds to a region around Switch II, α3, and β1 of Gαi (Figures 4a–c and S10, Video S3) through hydrophobic interactions between the side chains of Phe1, Nal2, Trp9, Leu10, and Pro16 of GPM–1c and binding sites 2 and 3 (Figure S12) on Gαi. Additional binding energy as well as specificity are derived from persistent GPM–1c–Gαi hydrogen (H)–bonding interactions between Arg5-Asp246, Arg8-Asp310, Tyr15–Asp256, and 2Nal–Lys204 (Switch II), which were observed for >80% of the simulation (Figure 4c). This renders the structure of the bound peptide stable upon Gαi binding with a backbone RMSD of 3.21 ± 0.81 Å relative to its initial conformation (Table S10).
GPM-1d and Gαi engage in a network of H-bonding interactions between Arg14–Asp246, Arg14–Asn250, Arg16-Asn251, Arg14–Lys252, and Arg4–Glu211 (Switch II) in >70% of the simulation. The complex is also stabilized by hydrophobic interactions betweenTrp2 of GPM–1d and binding site 3 on the Gαi surface (Figures S12 and S16) as well as an intramolecular stacking interaction between the side chain of Tyr8 and N-terminal Ipa. It appears that cyclization of GPM–1d enhances protein binding by constraining it into the binding conformation, (Table S10). On the other hand, cyclization prevented the peptide side chains from adopting optimal interactions with the Gαi surface, resulting in a reduction of its computed binding affinity.
GPM–1 showed a much lower binding affinity (−300.23 kJ mol−1, Table S11) than GPM–1c and GPM–1d. GPM–1 bound similarly to KB–75221 and the GIV GEM motif26,28 around α3 and the α3- β5 loop of Gαi (Figure 4d–f, S10, S11, Video S1). The side chains of Trp2, Tyr5, Tyr8 and Pro9 are largely buried into the hydrophobic groves of the binding site (Figure 4d), with GPM–1–Gαi H-bonding interactions between Trp2–Asn250, Arg7-Lys252 and Arg4–Arg203 (Switch II, Fig. 4e–f) providing additional stabilization. A π-π stacking interaction between Phe210 (Switch II) and Tyr5 of GPM–1 add to the stability as previously described for GEMs at the respective position (Figures 4f and 5).28 Switch II residues (including Phe210) are involved in Gβγ binding, explaining why the binding of GPM–1 and Gβγ to Gαi might be mutually exclusive.28,33
Figure 5.
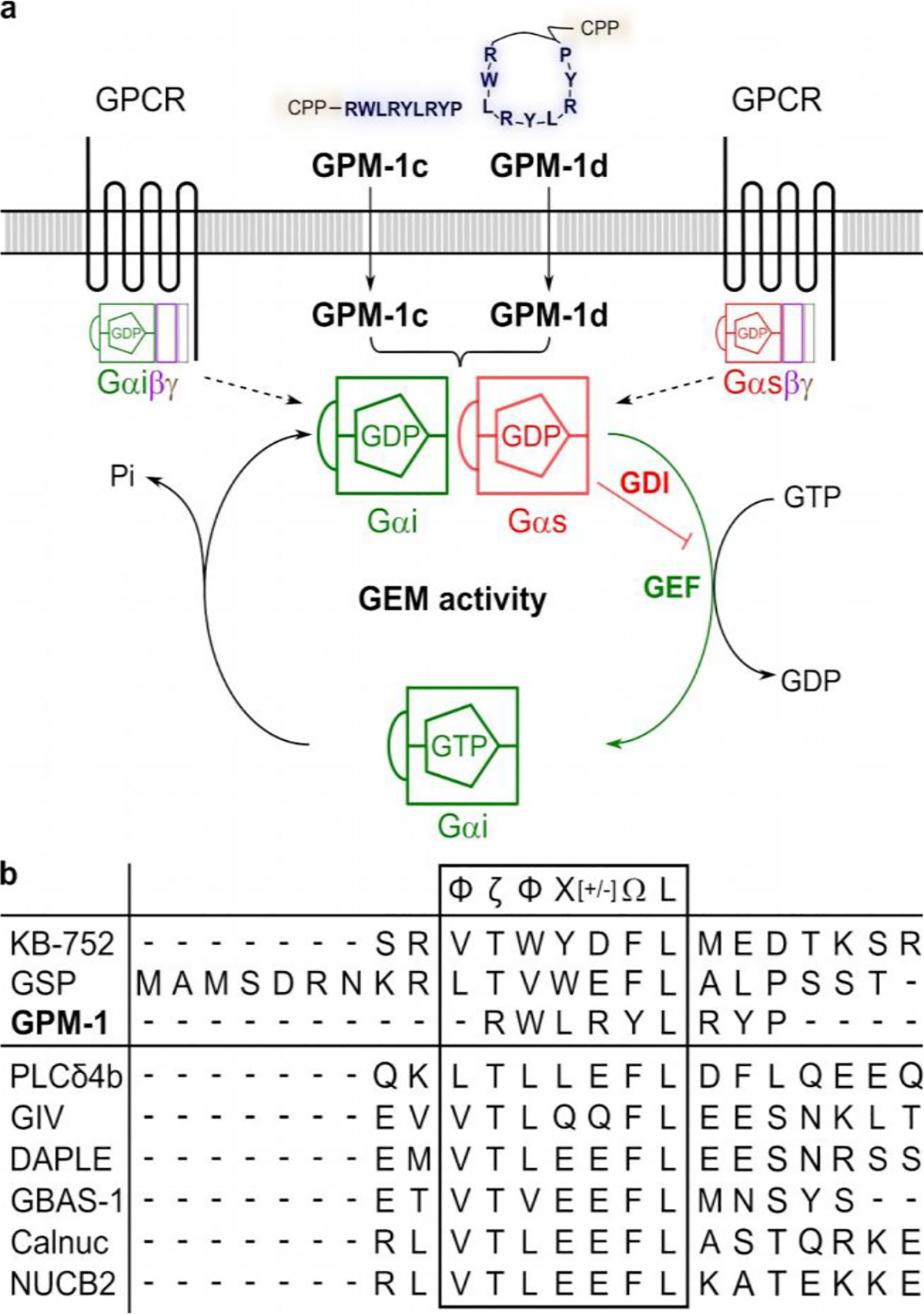
GEM-like effect of GPM–1c and GPM–1d. a: Schematic representation of the GEM activity of GPM–1c and GPM–1d which are able to penetrate the cell membrane. Inside the cell, both can cause a GEF effect on Gαi and a GDI effect on Gαs, thus reducing the overall cAMP production. b: Sequence alignment of GPM–1 with KB–75221, GSP17 and the GEM-motifs of the GEM-proteins25–27. Symbols as follows24: Φ: hydrophobic (V, I, L, F, W, Y, M), Ω: aromatic (F, W, Y), ζ: uncharged hydrophilic (N, Q, S, T), [+]: basic (H, K, R) and [–]: acidic (D, E) amino acids.
The inactivity of GPM–1b, GPM–1 Y5A and peptide 15 can be explained from the MD simulations of their bound conformations on Gαi. GPM–1 Y5A, adopting almost a cyclic conformation upon folding (Figure S15), bound poorly to Gαi (Figure S16, Table S11), reflecting its inactive nature in the experiments. This indicates that the interaction of Tyr5–Phe210 is important for the Gαi binding of GPM–1, which was also described for GEMs.23,28 The cyclic GPM–1b moves away from its bound conformation into solution during the simulation and is held in the vicinity of the protein only via long-range non-bonded interactions (Figure 4g–h, Video S2). Peptide 15 also moved away from its Gαi–bound conformation and remained in this unbound state throughout, i.e., peptide 15 also had a poor binding energy (Table S11).
An interesting observation was made with the binding energies computed between the Gαi·GDP protein with and without the peptides. It appears that the quality of peptide binding to Gαi is anticorrelated with the binding of GDP. Gαi·GDP had a MMPBSA binding energy of –306.23 kJ mol−1, which was considered as the reference value for the GDP association. With GPM–1c and GPM–1d bound, the average binding energy of GDP to Gαi decreased to –436.38 kJ mol−1 and –433.67 kJ mol−1, respectively (Table S11, Fig. S18). With the moderately active GPM–1 bound, the binding energy of GDP was 286.27 kJ mol−1, while with GPM–1 Y5A, GPM–1b and peptide 15, the GDP binding energies were 158.65 kJ mol−1, 318.34 kJ mol−1, and 309.67 kJ mol−1, respectively (Table S11). This observation suggests a tantalizing possibility that binding by the peptides may promote GDP release from Gαi (i.e., nucleotide exchange). This is consistent with the experimental results which suggest GPM–1c and GPM–1d as GEF for Gαi.28
Conversely, the binding of GPM–1, GPM–1c, and GPM–1d on Gαs (Figures S17 and S18) enhances GDP binding to Gαs. All three peptides bound on the predicted binding site 2 (Figures S13 and S17) between Switch II and α3 and provide a direct cover for the GDP molecule. Direct H-bonded interactions between the peptides and GDP were observed. The peptides exhibited higher MMPBSA binding energies in comparison to their interactions with Gαi, and improved binding of GDP to Gαs, indicating a GDI–like action (Table S12). GPM–1 Y5A, which bound to a different region between binding site 2 and 3, had a poor binding affinity.
Advantages over current Gαi/s modulators.
Direct targeting of G proteins provides an attractive alternative to GPCR modulators for treating many human diseases, such as cancer.2,3,5–7 However, modulators that are capable of binding reversibly and specifically to Gαi/s have been challenging to develop.2,3 The widely used protein modulators, such as CTX and PTX, covalently and irreversibly modify the G proteins,3,10,11 while KB–752 is impermeable to the cell membrane and cannot be used in cellular or in vivo assays. GPM–1c and GPM–1d have a high potential of pharmacological significance, because they possess GIV–like GEM activity, are able to occupy the Gαi–GIV interface and thus might influence the (GIV-mediated) Gαi activity.3,28 Furthermore, the peptides can affect the G protein activity independently of the GPCRs, which can provide insights into the G protein–mediated signaling and related diseases and bypasses the need to address individual receptors in disorders directed by multiple GPCRs.
CONCLUSION
In this work we discovered linear (GPM–1c) and cyclic peptides (GPM–1d) as a novel class of cell-permeable, Gαi/s–selective, and reversible modulators of Gα protein activity (Figure 5). These Gαi/s GEMs appear to bind to the Switch II/α3 region, as do KB–752 and the GEM proteins,25–28 and may thus affect Gαi/s downstream signaling and the resulting cellular response. Since G proteins are generally “undruggable”3,38 and have a crucial role in the pathogenesis of cancer, GPM–1c and GPM–1d should serve as invaluable chemical tools in pharmacological research and potential leads for further development into therapeutic agents to finally achieve druggability for Gs and Gi.3,5,38 This study also demonstrates that a combination of peptide library screening and medicinal chemistry offers a viable approach to developing novel Gα modulators.
METHODS
Expression and Purification of Gαi1/s Protein.
Transformed E. coli BL21 (DE3) cells were used that contained a vector construct of the plasmid pET28a (+) and the DNA sequence encoding for a hexahistidine tag and an enterokinase cleavage site [Met-Gly-Ser-Ser-(His)6-Ser-Ser-Gly-Leu-Val-Pro-Arg-Gly-Ser-His-Met-Ala-Ser-Met-Thr-Gly-Gly-Gln-Gln-Met-Gly-Arg-Ser-(Asp)4-Lys] followed by the sequence encoding for Gαi1 (Uniprot ID: P63096), where the initial methionine was deleted according to Suzuki et al.39 The same procedure was followed for Gαs (short isoform, Uniprot ID: P63092–2).40 Expression of Gαi1 was modified from Chen et al.41 The His-tagged protein was purified in a slightly modified approach according to Tesmer et al.42, whereas Gαs protein preparation was performed as described earlier introducing modifications to these protocols.40–44 Protein samples (Gαi1 in 20 mM HEPES pH 8.0, 100 mM NaCl, 1 mM MgCl2, 250 mM imidazole, 10 mM β-mercaptoethanol, 50 μM GDP, 10% (v/v) glycerol and Gαs in20 mM HEPES pH 8.0, 100 mM NaCl, 1 mM MgCl2, 2 mM dithiothreitol, 50 μM GDP, 10% (v/v) glycerol) were shock-frozen and stored at −80°C.
Biotinylation of the Gαi1 Protein for Library Screening.
NHS-biotin (2.5 eq.) in DMSO was added to the Gαi1 protein (1 eq.) and carefully stirred at room temperature for 30 min. Then, Tris buffer (2 M, pH 8.15) was added and the solution was stirred for 10 min. The biotinylated G protein was purified using a PD-10 column (SephadexTM G-25 M with 0.1% KathonTM CG, GE Healthcare), and the protein concentration was determined by Bradford assay according to the manufacturer’s instruction (ROTI® Nanoquant, Carl Roth GmbH + Co. KG).45 The Gαi1 protein solution was shock-frozen with 10% (v/v) glycerol and stored at −80 °C until usage.
One-Bead-One-Compound Library Screening.
The screening of the peptide library (X4[C/H/Y]0X420, where X is any of the proteinogenic amino acids and Nle, excluding Cys and Met), the hit selection, and identification was performed as earlier described.46,47
Peptide Synthesis and Purification.
Solid-phase peptide synthesis according to the Fmoc strategy was performed on a Rink Amide MBHA resin (0.53 mmol g−1) or a 2-chlorotrityl resin (KB-752: 0.40 mmol g-1, GPM-1b: 0.70 mmol g−1). Hits from the library screening (GPM-1, 2–13), Peptide 14, GPM-1 Y5A and KB-752 were synthesized with an automated ResPep SL peptide synthesizer from Intavis Bioanalytical Instruments GmbH using HBTU (4 eq.) as coupling reagent and N-methylmorpholine (NMM, 9 eq.) as base. The optimized hits were synthesized manually with HBTU (4 eq.) and HOBt (4 eq., GPM-1b, GPM-1c, 15) or HATU (4 eq., GPM-1d) as coupling reagents and DIPEA (8 eq.) or NMM (8 eq.) as base. The cleavage of the side-chain protecting groups together with the peptide from the resin was performed with reagent K as described previously.48 For GPM-1b, the linear sequence of GPM-1 was synthesized on the 2-chlorotrityl resin and, subsequently, Boc-Lys(Fmoc)-OH was coupled to the N-terminus. After cleavage of the linear precursor from the resin, the peptide was cyclized in solution with PyBOP (6 eq.) and DIPEA (12 eq.). The subsequent Fmoc group removal was achieved using diethylamine (20 eq.) in DMF. For GPM-1d, first Fmoc-Lys(Boc)-OH and the CPP sequence (F(2-Nal)RRRR) by using Fmoc-Phe-OH, Fmoc-(2-Nal)-OH and Fmoc-Arg(Pbf)-OH, were coupled to the resin and then, Fmoc-Dap(Alloc)-OH was coupled according to Lian et al.49 for later cyclization. Thereafter, the peptide was extended by the GPM-1 sequence (RWLRYLRYP) by using standard Fmoc-strategy. Subsequently, isophthalic acid was coupled and the Alloc group was cleaved by Pd(PPh3)4 (0.5 eq.) and phenylsilane (10 eq.) in DCM. After cyclization with PyBOP (10 eq.), HOBt (10 eq.) and NMM (20 eq.) on the resin, the peptide was cleaved from the resin with reagent K as described previously.48 The crude peptides were purified by semi-preparative reversed-phase HPLC using a Shimadzu LC-8A instrument equipped with a Knauer Eurospher column (C18, 250 × 32 mm, 5 μm particle size, 100 Å pore size) for amounts between 20–80 mg or a Vydac 218TP1022 column (C18, 250 × 22 mm, 5 μm particle size, 100 Å pore size) for amounts up to 20 mg with a gradient of 0.1% (v/v) TFA in water (eluent A) and 0.1% TFA in acetonitrile/water (90:10, eluent B). For each peptide, a gradient was selected according to the elution behavior in the analytical RP-HPLC by increasing 50% eluent B at a flow rate of 10 ml min−1 in 120 min. The peaks were detected at 220 nm. The purity of the peptides (>98%) was confirmed by analytical RP-HPLC from a Shimadzu LC-20AD system equipped with a Vydac 218TP column (C18, 250 × 4.6 mm, 5 μm particle size, 300 Å pore size) with a gradient system of 0.1% TFA in water (eluent A) and 0.1% (v/v) TFA in acetonitrile (eluent B) at a flow rate of 1 ml min−1 and 220 nm detection. The collected fractions were combined, freeze-dried, and stored at −20 °C. Detailed information on the individual peptides can be found in Supporting Information (Table S3).
Fluorescence Labeling of Peptides.
All linear peptides were N-terminally labeled on resin with 5(6)-carboxyfluorescein (Cf, 2 eq.) in DMF using PyBOP (2 eq.) as coupling reagent and DIPEA (3 eq.) as base. All cyclized peptides were selectively labeled with fluorescein isothiocyanate (FITC) in solution according to Trinh et al.34 on the side chain of the lysine previously inserted into the sequence. Therefore, after dissolving the purified peptide (1.5 mg) in DMSO (34 μl) and 100 mM NaHCO3 pH 8.5 (34 μl), FITC (15 μl, 10 mg ml−1 in DMSO) was added and the solution was incubated at 25 °C for 40 min in the dark. Then, 50% (v/v) TFA/water (7.5 μl) was added, and the labeled peptide was purified by reversed-phase HPLC using a Shimadzu LC-10AT system equipped with a Vydac 218TP column (C18, 250 × 4.6 mm, 5 μm particle size, 300 Å pore size), and a mobile phase system consisting of 0.1% (v/v) TFA in water (eluent A) and 0.1% (v/v) TFA in acetonitrile (eluent B). The purity of the peptides (>98%) was confirmed by analytical RP-HPLC and the identity of the peptides was validated with mass spectrometry. Detailed information on the fluorescence-labeled peptides can be found in Table S4.
Synthesis of Biotinylated Peptides.
For the peptides GPM-1 and 15, biotinylated analogs, Btn-GPM-1 and Btn-15, were synthesized on a Rink amide MBHA resin (0.53 mmol g−1) by solid phase peptide synthesis according to Fmoc strategy using an automated ResPep SL peptide synthesizer (Intavis Bioanalytical Instruments GmbH). First, Fmoc-Lys(Biotin)-OH and Fmoc-O2Oc-OH and then the corresponding sequence (GPM-1: RWLRYLRY; 15: F(2-Nal)RRRR) were coupled with HBTU (4 eq.) as coupling reagent and N-methylmorpholine (9 eq.) as base. The cleavage of the side chain protecting groups and the peptides from the resin took place as described before.48 GPM-1c and GPM-1d were biotinylated in solution with NHS-PEG4-Biotin (Thermo Fisher Scientific Inc.) at the lysine side chain. After the peptide (1–2 mg) was dissolved in DMSO (50 μl) and phosphate buffer (950 μl, 50 mM, pH 6.5), NHS-PEG4-Biotin (0.5 eq. for GPM-1c, 5 eq. for GPM-1d, 10 mM in DMSO) was added and the reaction was incubated for 90 min at 4 °C. The biotinylated peptides, Btn-GPM-1c and Btn-GPM-1d, were purified by reversed-phase HPLC using a Shimadzu LC-10AT instrument as described above. Subsequently, the correct biotinylation pattern of GPM-1c was determined by automated Edman degradation. The N-terminal sequence analysis was performed using a Shimdazu PPSQ-53A protein sequencer. Prior to analysis, the peptide was dried under vacuum, freshly dissolved in 0.5% (v/v) acetic acid, and 10 pmol of the peptide were applied to a polybrene treated Glass Fiber Disk. After drying under a stream of nitrogen for 10 min, two cycles of N-terminal sequence analysis were performed. The derivatized amino acids were separated isocratically by RP-HPLC on a Shimadzu LC-20AT with a Wakopak® Wakosil PTH-II column (C18, 4.6 mm × 250 mm, 5 μm particle size), detected at 269 nm, and identified by comparison with a PTH-standard mixture. The non- and double-biotinylated analogs were also examined in the same way. The purity of the peptides (>98%) was confirmed by analytical RP-HPLC and the identity of the peptides was validated with mass spectrometry. Detailed information about the biotinylated peptides can be found in Table S4.
Peptide Analysis.
The peptides were characterized by analytical RP-HPLC (see above) and mass spectrometry. The characterization of the peptides by mass spectrometry was performed as described previously.50 GPM-1 and peptides 2–13 as well as Cf-GPM-1 and peptides Cf-2–13 were additionally analyzed by amino acid analysis as reported earlier.50 In addition, the peptide content of all other peptides was determined with a GPM-1 or Cf-GPM-1 calibration curve by analytical RP-HPLC using the GPM-1 or Cf-GPM-1 peptide content from amino acid analysis as a basis. Detailed information on the individual peptides can be found in Tables S3, S4.
Supplementary Material
ACKNOWLEDGMENT
We thank E. Kostenis, M. Geyer and M. Famulok (University of Bonn) for access to cells, assays, and instruments, S. Linden, F. Steinbock, U. Rick (all University of Bonn), G. Schlipf (LMU Munich), and V. Uzunova (Cytiva Europe GmbH) for technical support, K. Hampel (Biaffin GmbH & Co KG) for support with SPR analysis, E. A. Galinski and M. Hecker (University of Bonn) for the collaboration on Gα protein expression, and Solvay GmbH for the friendly supply of chemicals.
Funding Sources
This work was supported by the University of Bonn, the Deutsche Forschungsgemeinschaft (DFG) within FOR 2372 and IM 97/14–1 (to D.I.), and the Bonner Universitätsstiftung (to B.N.).
ABBREVIATIONS
- AC
adenylyl cyclase
- cAMP
cyclic adenosine monophosphate
- CPP
cell-penetrating peptide
- CTX
cholera toxin
- GDI
guanine-nucleotide dissociation inhibitor
- GEF
guanine-nucleotide exchange factor
- GEM
guanine-nucleotide exchange modulator
- GPCR
G protein-coupled receptors
- MST
microscale thermophoresis
- PTX
pertussis toxin
- SPR
surface plasmon resonance
Footnotes
The authors declare no competing financial interests.
ASSOCIATED CONTENT
Supporting Information Available.
The Supporting Information is available free of charge at http://pubs.acs.org. Experimental procedures and instrumentation used, methods for binding and activity studies including characterization data (Figures S1–S18), computational analysis of protein-peptide interactions and corresponding data (Table S1–S12 and Video S1–S3) are supporting this article.
REFERENCES
- (1).Simon MI, Strathmann, and M. P., Gautam N (1991) Diversity of G proteins in signal transduction. Science 252, 802–808. [DOI] [PubMed] [Google Scholar]
- (2).Li J, Ge Y, Huang J-X, Stromgaard K, Zhang X, and Xiong X-F (2020) Heterotrimeric G proteins as therapeutic targets in drug discovery. J. Med. Chem. 63, 5013–5030. [DOI] [PubMed] [Google Scholar]
- (3).Nubbemeyer B, Pepanian A, Paul George AA, and Imhof D (2021) Strategies towards targeting Gαi/s proteins: scanning of protein-protein interaction sites to overcome inaccessibility. ChemMedChem. 16, 1697–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI, et al. (2017) A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).O’Hayre M, Vázquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, and Gutkind JS (2013) The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 13, 412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Campbell AP, and Smrcka AV (2018) Targeting G protein-coupled receptor signalling by blocking G proteins. Nat. Rev. Drug Discov. 17, 789–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Annala S, Feng X, Shridhar N, Eryilmaz F, Patt J, Yang J, Pfeil EM, Cervantes-Villagrana RD, Inoue A, Häberlein F, et al. (2019) Direct targeting of Gαq and Gα11 oncoproteins in cancer cells. Sci. Signal. 12, eaau548. [DOI] [PubMed] [Google Scholar]
- (8).Schrage R, Schmitz A-L, Gaffal E, Annala S, Kehraus S, Wenzel D, Büllesbach KM, Bald T, Inoue A, Shinjo Y, et al. (2015) The experimental power of FR900359 to study Gq-regulated biological processes. Nat. Commun. 6, 10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Reher R, Kühl T, Annala S, Benkel T, Kaufmann D, Nubbemeyer B, Odhiambo JB, Heimer P, Bäuml CA, Kehraus S, et al. (2018) Deciphering specificity determinants for FR900359-derived Gαq inhibitors based on computational and structure-activity studies. ChemMedChem 13, 1634–1643. [DOI] [PubMed] [Google Scholar]
- (10).Mangmool S, and Kurose H (2011) Gi/o-protein dependent and –independent actions of pertussis toxin (PTX). Toxins 3, 884–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Aktories K (2011) Bacterial protein toxins that modify host regulatory GTPases. Nat. Rev. Microbiol. 9, 487–498. [DOI] [PubMed] [Google Scholar]
- (12).Higashijima T, Uzu S, Nakajima T, and Ross EM (1988) Mastoparan, a peptide toxin from wasp venom, mimics receptors by activating GTP-binding regulatory proteins (G proteins) J. Biol. Chem. 263, 6491–6494. [PubMed] [Google Scholar]
- (13).Moreno M and Giralt E (2015) Three valuable peptides from bee and wasp venoms for therapeutic and biotechnological use: melittin, apamin and mastoparan. Toxins 7, 1126–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ja WW, and Roberts RW (2004) In vitro selection of state-specific peptide modulators of G protein signaling using mRNA display. Biochemistry 43, 9265–9275. [DOI] [PubMed] [Google Scholar]
- (15).Ja WW, Adhikari A, Austin RJ, Sprang SR, and Roberts RW (2005) A peptide core motif for binding to heterotrimeric G protein α subunits. J. Biol. Chem. 280, 32057–32060. [DOI] [PubMed] [Google Scholar]
- (16).Ja WW, Wiser O, Austin RJ, Jan LY, and Roberts RW (2006) Turning G proteins on and off using peptide ligands. ACS Chem. Biol. 1, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Austin RJ, Ja WW, and Roberts RW (2008) Evolution of class-specific peptides targeting a hot spot of the Gαs subunit. J. Mol. Biol. 377, 1406–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Millward SW, Fiacco S, Austin RJ, and Roberts RW (2007) Design of cyclic peptides that bind protein surfaces with antibody-like affinity. ACS Chem. Biol. 2, 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Howell SM, Fiacco SV, Takahashi TT, Jalali-Yazdi F, Millward SW, Hu B, Wang P, and Roberts RW (2014) Serum stable natural peptides designed by mRNA display. Sci. Rep. 4, 6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Fiacco SV, Kelderhouse LE, Hardy A, Peleg Y, Hu B, Ornelas A, Yang P, Gammon ST, Howell SM, Wang P, et al. (2016) Directed evolution of scanning unnatural-protease-resistant (SUPR) peptides for in vivo applucations. Chembiochem. 17, 1643–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Johnston CA, Willard FS, Jezyk MR, Fredericks Z, Bodor ET, Jones MB, Blaesius R, Watts VJ, Harden TK, Sondek J, et al. (2005) Structure of Galpha(i1) bound to GDP-selective peptide provides insight into guanine nucleotide exchange. Structure 13, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Johnston CA, Ramer JK, Blaesius R, Fredericks Z, Watts VJ, and Siderovski DP (2005) A bifunctional Galphai/Galphas modulatory peptide that attenuates adenylyl cyclase activity. FEBS Lett. 579, 5746–5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Johnston CA, Willard FS, Ramer JK, Blaesius R, Roques CN, and Siderovski DP (2008) State-selective binding peptides for heterotrimeric G-protein subunits: novel tools for investigating G-protein signaling dynamics. Comb. Chem. High Throughput Screen. 11, 370–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Aasland R, Abrams C, Ampe C, Ball LJ, Bedford MT, Cesareni G, Gimona M, Hurley JH, Jarchau T, Lehto VP, et al. (2002) Normalization of nomenclature for peptide motifs as ligands of modular protein domains. FEBS Lett. 513, 141–144. [DOI] [PubMed] [Google Scholar]
- (25).DiGiacomo V, Marivin A, and Garcia-Marcos M (2018) When heterotrimeric G proteins are not activated by GPCRs: structural insights and evolutionary conservation. Biochemistry 57, 255–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).de Opakua AI, Parag-Sharma K, DiGiacomo V, Merino N, Leyme A, Marivin A, Villate M, Nguyen LT, de la Cruz-Morcillo MA, Blanco-Canosa JB, et al. (2017) Molecular mechanism of Gαi activation by non-GPCR proteins with a Gα-binding and activating motif. Nat. Commun. 8, 15163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ghosh P, Rangamani P, and Kufareva I (2017) The GAPs, GEFs, GDIs and…now, GEMs: New kids on the heterotrimeric G protein signaling block. Cell Cycle 16, 607–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kalogriopoulos NA, Rees SD, Ngo T, Kopcho NJ, Ilatovskiy AV, Sun N, Komives EA, Chang G, Ghosh P, and Kufareva I (2019) Structural basis for GPCR-independent activation of heterotrimeric Gi proteins. Proc. Natl. Acad. Sci. U.S.A. 116, 16394–16403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kühl T, Sahoo N, Nikolajski M, Schlott B, Heinemann SH, and Imhof D (2011) Determination of hemin-binding characteristics of proteins by a combinatorial peptide library approach. Chembiochem. 12, 2846–2855. [DOI] [PubMed] [Google Scholar]
- (30).Sweeney MC, Wavreille A-S, Park J, Butchar JP, Tridandapani S, and Pei D (2005) Decoding protein-protein interactions through combinatorial chemistry: sequence specificity of SHP-1, SHP-2, and SHIP SH2 domains. Biochemistry 44, 14932–14947. [DOI] [PubMed] [Google Scholar]
- (31).Sweeney MC, and Pei D (2003) An improved method for rapid sequencing of support-bound peptides by partial edman degradation and mass spectrometry. J. Comb. Chem. 5, 218–222. [DOI] [PubMed] [Google Scholar]
- (32).Willard FS, and Siderovski DP (2006) The R6A-1 peptide binds to switch II of Galphai1 but is not a GDP-dissociation inhibitor. Biochem. Biophy. Res. Commun. 339, 1107–1112. [DOI] [PubMed] [Google Scholar]
- (33).Garcia-Marcos M, Ghosh P, and Farquhar MG (2009) GIV is a nonreceptor GEF for Gαi with a unique motif that regulates Akt signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 3178–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Trinh TB, Upadhyaya P, Qian Z, and Pei D (2016) Discovery of a Direct Ras inhibitor by screening a combinatorial library of cell-permeable bicyclic peptides. ACS Comb. Sci. 18, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Seidel SAI, Dijkman PM, Lea WA, van den Bogaart G, Jerabek-Willemsen M, Lazic A, Joseph JS, Srinivasan P, Baaske P, Simeonov A, et al. (2013) Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 59, 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Horton JK, Martin RC, Kalinka S, Cushing A, Kitcher JP, O’Sullivan MJ, and Baxendale PM (1992) Enzyme immunoassays for the estimation of adenosine 3’,5’ cyclic monophosphate and guanosine 3’,5’ cyclic monophosphate in biological fluids J. Immunol. Methods 155, 31–40. [DOI] [PubMed] [Google Scholar]
- (37).DiGiacomo V, de Opakua AI, Papakonstantinou MP, Nguywn LT, Merino N, Blanco-Canosa JB, Blanco FJ, and Garcia-Marcos M (2017) The Gαi-GIV binding interface is a druggable protein–protein interaction. Sci. Rep. 7, 8575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Dang CV, Reddy EP, Shokat KM, and Soucek L (2017) Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 17, 502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Suzuki T, Moriya K, Nagatoshi K, Ota Y, Ezure T, Ando E, Tsunasawa S, Utsumi T (2010) Strategy for comprehensive identification of human N-myristoylated proteins using an insect cell-free protein synthesis system. Proteomics 10, 1780–1793. [DOI] [PubMed] [Google Scholar]
- (40).McCusker E, Robinson AS (2008) Refolding of G protein α subunits from inclusion bodies expressed in Escherichia coli. Protein Expr. Purif. 58, 342–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Chen Z, Singer WD, Sternweis PC, Sprang SR (2005) Structure of the p115RhoGEF rgRGS domain–Gα13/i1 chimera complex suggests convergent evolution of a GTPase activator. Nat. Struct. Mol. Biol. 12, 191–197. [DOI] [PubMed] [Google Scholar]
- (42).Tesmer VM, Kawano T, Shankaranarayanan A, Kozasa T, Tesmer JJG (2005) Snapshot of activated G proteins at the membrane: the Galphaq-GRK2-Gbetagamma complex. Science. 310, 1686–1690. [DOI] [PubMed] [Google Scholar]
- (43).Lee E, Linder ME, Gilman AG (1994) Expression of G-protein α subunits in Escherichia coli. Meth. Enzymol. 237, 146–164. [DOI] [PubMed] [Google Scholar]
- (44).Burgess RR (2009) Refolding Solubilized Inclusion Body Proteins. Meth. Enzymol. 463, 259–282. [DOI] [PubMed] [Google Scholar]
- (45).Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. [DOI] [PubMed] [Google Scholar]
- (46).Sweeney MC, Wavreille A-S, Park J, Butchar JP, Tridandapani S, Pei D (2005) Decoding protein− protein interactions through combinatorial chemistry: sequence specificity of SHP-1, SHP-2, and SHIP SH2 domains. Biochemistry. 44, 14932–14947. [DOI] [PubMed] [Google Scholar]
- (47).Sweeney MC, Pei D (2003) An Improved Method for Rapid Sequencing of Support-Bound Peptides by Partial Edman Degradation and Mass Spectrometry. J. Comb. Chem. 5, 218–222. [DOI] [PubMed] [Google Scholar]
- (48).Kühl T, Sahoo N, Nikolaiski M, Schlott B, Heinemann SH, Imhof D (2011) Determination of hemin-binding characteristics of proteins by a combinatorial peptide library approach. Chembiochem. 12, 2846–2855. [DOI] [PubMed] [Google Scholar]
- (49).Lian W, Jiang B, Qian Z, Pei D (2016) Discovery of a direct Ras inhibitor by screening a combinatorial library of cell-permeable bicyclic peptides. ACS Comb. Sci. 18, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Bäuml CA, Paul George AA, Schmitz T, Sommerfeld P, Pietsch M, Podsiadlowski L, Steinmetzer T, Biswas A, Imhof D (2020) Distinct 3-disulfide-bonded isomers of tridegin differentially inhibit coagulation factor XIIIa: The influence of structural stability on bioactivity. Eur. J. Med. Chem. 201, 112474. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.