

Abstract
The RNA-guided endonuclease Cas9 has applied as an efficient gene-editing method in malaria parasite Plasmodium. However, the size (4.2 kb) of the commonly used Cas9 from Streptococcus pyogenes (SpCas9) limits its utility for genome editing in the parasites only introduced with cas9 plasmid. To establish the endogenous and constitutive expression of Cas9 protein in the rodent malaria parasite P. yoelii, we replaced the coding region of an endogenous gene Pysera1 with the intact SpCas9 coding sequence using the CRISPR/Cas9-mediated genome editing method, generating the cas9-knockin parasite (PyCas9ki) of the rodent malaria parasite P. yoelii. The resulted PyCas9ki parasite displays normal progression during the whole life cycle and possesses the Cas9 protein expression exclusively in asexual blood stage. By introducing the plasmid (pYCs) containing only sgRNA and homologous template elements, we successfully achieved both deletion and tagging modifications for different endogenous genes in the genome of PyCas9ki parasite. This cas9-knockin PyCas9ki parasite provides a new platform facilitating gene functions study in the rodent malaria parasite P. yoelii.
Keywords: Rodent malaria, Plasmodium yoelii, Genome modifications, Cas9, knockin
1. Introduction
Malaria remains the most serious infectious disease worldwide. To uncover the molecular targets for the future development of better anti-malaria medicine and vaccine, new technologies and methods are still urgently needed for studying the biology of malaria parasites and mechanism of malaria disease and pathology [1, 2]. CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats /CRISPR-associated protein 9), a powerful genome editing technology, has been recently successfully adapted for genome modification of malaria parasites, including Plasmodium falciparum and Plasmodium yoelii [3–6]. The CRISPR/Cas9 system was originated from a prokaryotic RNA programmable nuclease that can introduce a double-strand break (DSB) at a specific site on a chromosome through heterologous expression of two components: Cas9 endonuclease and a targeting single guide RNA (sgRNA) [7]. The DNA DSBs are repaired dominantly by homologous recombination (HR) pathways over the error-prone nonhomologous endjoining (NHEJ) pathway in Plasmodium [8, 9], therefore the donor templates were needed for HR repair in all the CRISPR/Cas9-mediated genome editing practices. For editing the genome of P. falciparum, the Cas9/sgRNA cassette and donor template were delivered into parasites in two separate vectors with each plasmid carrying a different drug resistant gene as selection marker [3, 5, 10–12]. For the rodent malaria parasite P. yoelii, a single vector system was applied because limited independent selection markers were available for the parasites [4, 13]. In the one-vector plasmid (pYC) design, all components, including the genes encoding the Cas9 protein, the human dihydrofolate reductase (hDHFR) for positive selection with pyrimethamine (Pyr), the sgRNA, and the donor template DNA were included in the plasmid vector. So far, the Streptococcus pyogenes-derived Cas9 (SpCas9) is the only endonuclease used for CRISPR/Cas9-mediated gene editing in malaria parasites [3–5], although the SpCas9 coding sequence is about 4200 bp in size [7]. Recently, a constitutive Cas9 expression was established to maximize the CRISPR/Cas9-mediated gene disruption and achieve genome-wide gene screening in another apicomplexan Toxoplasma.gondii [14]. We thus reasoned that endogenous and constitutive expressing Cas9 derived from the Cas9-knockin malaria parasite could increase the gene editing efficiency and facilitate the application for different goals, in which the genome editing would only require introduction of sgRNA and homologous template components, benefiting the plasmid construction procedures and plasmid delivering into the parasite.
In this study, we replaced the coding region of an endogenous gene Pysera1 with the intact SpCas9 coding sequence using the CRISPR/Cas9-mediated genome editing method, generating the cas9-knockin parasite (PyCas9ki) of the rodent malaria parasite P. yoelii. The resulted PyCas9ki parasite displays normal progression during the whole life cycle and possesses the Cas9 protein expression exclusively in asexual blood stage. By introducing the plasmid (pYCs) containing only sgRNA and homologous template elements, we successfully achieved both deletion and tagging modifications for different endogenous genes in the genome of PyCas9ki parasite. This cas9-knockin PyCas9ki parasite provides a new platform facilitating gene functions study in the rodent malaria parasite P. yoelii.
2. Materials and Methods
2.1. Plasmid construction
The procedure for generating constructs for gene deletion, tagging, and replacement was as described previously [4]. To construct pYCm vector for deleting the coding region of Pysera1 gene (PY17X_0305400), we amplified 526bp 5’ untranslated region (UTR) upstream of translation start codon as the left homologous arm and a 500bp 3’UTR region following translation stop codon as right homologous arms using PCR primers listed in Table S1. One sgRNA was designed to target the site in the coding region to be deleted. All the fragments were sequentially ligated into the pYCm vector by T4 connection. To construct pYCm vector for replacing the coding region of Pysera1 gene with the SpCas9 coding sequence, we amplified the full-length SpCas9 coding region from the plasmid pYC, tagged with a quadruple Myc epitope (4Myc) C-terminally, and inserted between the left and right homologous arms of the pYCm vector used for deleting Pysera1 construct. For gene editing purpose in the PyCas9ki parasite, we engineered the pYCm vector and removed the SpCas9 coding region via mutagenesis, resulting in a new vector, which we designed pYCs. To construct pYCs vectors for deleting the Pycdpk3 gene (PY17X_0410700) and Pyctrp gene (PY17X_0415800), and for tagging the Pysep1 (PY17X_0526200) and Pydhhc10 genes (PY17X_0946500), the procedures are similarly followed. All the primers and oligonucleotides used are listed in Table S1.
2.2. Malaria parasite and parasite transfection
The parasite was propagated in ICR mice (female, 5–6 weeks old) purchased from the Animal Care Center, Xiamen University. All mouse experiments were performed in accordance with approved protocols (XMULAC20140004) by the Committee for the Care and Use of Laboratory Animals at the School of Life Sciences, Xiamen University. All transfections were performed on the P. yoelii 17XNL parasite or the PyCas9ki (P.yoelii 17XNL Cas9 knockin) strain. The procedures for parasite transfection, Pyr selection, and cloning were as described previously [4]. Briefly, parasites were electroporated with purified plasmid DNA. Transfected parasites were immediately intravenously injected into a naive mouse. Pyr (6ug/ml) supplied in drinking water was provided to mice for drug selection from day 2 post-transfection. A small amount of blood sample was taken daily through tail clip and Giemsa-stained for infected red blood cells (iRBCs). Pyr resistant parasites usually appear 5–6 days after drug selection.
2.3. Negative selection with 5-Fluorocytosine
To remove the plasmid inside the parasite for next-round genome modification, the parasites were applied for negative selection with 5-Fluorocytosine (5-FC) as described previously [13]. Briefly, 5-FC (Sigma, USA) was prepared in water at a final concentration of 2.0 mg/ml and was provided to the animals in a dark drinking bottle. A naïve mouse receiving parasites containing residual plasmids after Pyr selection was subjected to 5-FC pressure for 8 days, with a change of new drug at day 4. The complete removal of plasmids in parasites was confirmed by PCR genotyping.
2.4. DNA preparation and detection of genetic modifications
Blood samples from infected mice were collected from the orbital sinus, and RBCs were lysed using 1% saponin in PBS. Parasite genomic DNAs were isolated using DNeasy Blood kits (Qiagen) after washing off hemoglobin and were used in PCR amplifications. For gene deletion and gene tagging, targeted modification was confirmed by PCR using two pairs of primer to detect 5’ and 3’ integrations respectively. To confirm the successful deletion of targeting region, another independent primer pairs were designed to amplify the region to be deleted. All the primers used are listed in Table S1.
2.5. In vitro ookinete differentiation
Ookinetes were prepared according to the procedure described previously [15]. Briefly, infected blood was injected intraperitoneously into mice that were made anemic by phenylhydrazine treatment (80 μg drug per mouse body weight) over a three-day period. Three days after infection, 200 μl of infected blood containing gametocytes was obtained from the orbital sinus and mixed immediately with 1 ml ookinete culture medium. The mixture was incubated at 22 °C for 20–24 h to allow gametogenesis, fertilization, and ookinete differentiation. Ookinetes formation was monitored by Giemsa staining of smears of the cultured cells.
2.6. In vitro ookinete gliding motility assay
Ookinete gliding motility was evaluated as previously described [15, 16]. All procedures were performed in a temperature (22°C)-controlled room. 20 μl of the ookinete cultures were suspended and mixed with an equal volume of Matrigel (BD, USA) on ice. The mixtures were transferred on a slide, sealed with nail varnish after adding a coverslip, and put at 22°C for 30 min before the microscopy analysis. After finding an eye field containing ookinetes under microscopy, time-lapse videos (1 frame every 20 s, for 20 min) were taken to monitor ookinete movement using a 40× objective lens on a Nikon ECLIPSE E100 microscope fitted with an ISH500 digital camera controlled by the ISCapture v3.6.9.3_N software (Tucsen, Fuzhou, Fujian, P. R. CHINA). Time-lapse movies were analyzed with Fiji and the Manual Tracking plugin. Ookinete gliding speed was calculated by dividing the distance that each individual ookinete moved by the tracking time. The experiments were performed in three times independently.
2.7. Mosquito infection and observation of parasites in mosquitoes
For mosquito infection, 40 female Anopheles stephensi mosquitoes were allowed to feed to one anaesthetized infected mice that carried comparable numbers of gametocytes as determined by Giemsa staining for 20 min. Twenty mosquitos were dissected day 7 post-infection, and oocysts in the midguts were counted. Salivary glands were isolated from 20–25 dissected mosquitos 14 days post-infection, and sporozoites were collected and counted using a hemocytometer under the microscopy.
2.8. Immunofluorescence assay
Parasite samples are harvested by centrifuging for 3 min at 2,000 rpm, washed twice with PBS, and resuspended in 4% freshly prepared paraformaldehyde on a poly-L-lysine coated glass slide for 15 min. Cells were washed twice with PBS, and permeabilized with 0.1% Triton X-100 for 8 min at room temperature. The sample was incubated with primary antibodies in 5% BSA in PBS for 12 h at 4°C, washed with PBS three times, and incubated with fluorescence conjugated secondary antibodies for 1 h. Subsequently, the cells were washed three times with PBS, and stained with Hoechst33342 for 8 min, mounted in 90% glycerol solution, and sealed with nail polish. All images were captured using identical settings in the Zeiss LSM 780 laser scanning confocal microscopy with a 100× oil objective. Results were obtained from three independent experiments.
2.9. Western blotting
Parasite protein extraction was performed using RIPA lysis buffer (50mM Tris (pH 7.4), 150mM NaCl, 1%Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 2mM sodium pyrophosphate, 25mM β-glycerophosphate, 1mM EDTA, 1mM Na3VO4, 0.5 μg/ml leupeptin) plus 1× complete protease inhibitor cocktail and 1mM PMSF. After ultrasonic, the sample was incubated on ice for 30 min, and centrifuged at 12,000 g for 10 min at 4°C. The supernatant was collected, mixed with 5× protein loading buffer. To completely denature the proteins, the samples were treated at 95°C for 5 min and store at −20°C for next analysis. About 40 μg total protein was loaded and separated in 4%−9% SDS-PAGE. Proteins were transferred to a 0.22 μm PVDF membrane by wet transfer blotting for 1 h, blocked with 5% skim milk for 1 h, and probed with the specific antibodies. The proteins were detected using an enhanced chemiluminescent substrate kit (ECL) and visualized by the film at dark room.
2.8. Software
To search a potent sequence targeting by Cas9/sgRNA in the genone, we used the online program, Eukaryotic Pathogen CRISPR guide RNA/DNA Design Tool (http://grna.ctegd.uga.edu). To quantify the fluorescent signal in the fluorescence microscopy images, we used the Image J (https://imagej.nih.gov/ij). To record the ookinete’s gliding route and calculate the motility speed, we used the Image J for the imaging processing and computing. For statistical analysis, we used the GraphPad Prism5 (https://www.graphpad.com).
3. Results
3.1. Generation of Cas9 knockin transgenic parasite
Because the Streptococcus pyogenes Cas9 (Spcas9) gene is a 4200 bp coding sequence in size, it is a challenging to incorporate Cas9 expression cassettes with necessary genetic elements, including promoters and polyadenlyation sequences, into the genome of malaria parasite. Alternatively, we attempt to replace the coding region of specific endogenous gene with cas9 coding sequence in the genome, and achieve expression of Cas9 protein driven by the regulatory sequence (promoter) of endogenous gene (Fig 1A). The parasite gene encoding a serine protease (P. yoelii sera1, Pysera1) has been shown to be nonessential for the parasite growth in the blood stage [4, 17]. To test the feasibility that Pysera1 serves as a safe harbor for cas9 integration and transcription in the transgenic parasite, we disrupted the Pysera1 gene in the P. yoelii 17XNL parasite using CRISPR/Cas9 methods described previously [13], and obtained two parasite knockout clones (ΔPysera1c1 and ΔPysera1c2) with deletion of the whole coding region (Fig S1A,S1B). Both ΔPysera1c1 and ΔPysera1c2 parasite clones display normal progression comparable with wildtype parasite in the life cycle, including asexual and sexual gametocytes stages in mice, mosquito stages, and mouse infectivity (Fig S1C to G), suggesting functional redundancy of Pysera1 gene in the life cycle of P. yoelii.
Fig. 1.
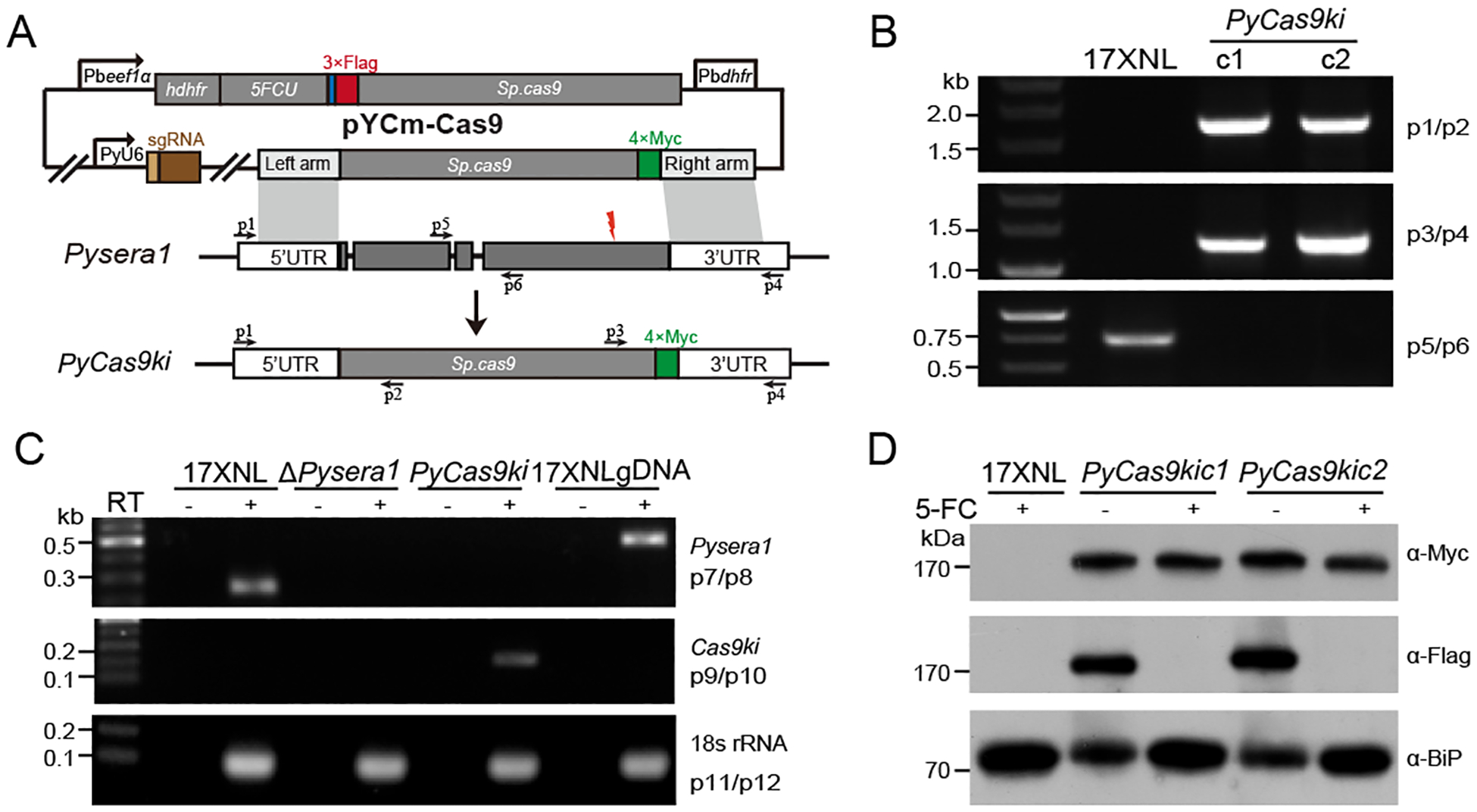
Generation of Cas9 knockin parasite of Plasmodium yoelii.
A. Schematic diagram of CRISPR/cas9 plasmid pYCm construct for replacing the endogenous sera1 gene coding region with cas9 gene in Plasmodium yoelii. The site for designed sgRNA recognition was indicated as red thunderbolt. Cas9 protein tagged with quadruple Myc epitope (4Myc, green) is for transgenic, and Cas9 protein tagged with triple Flag epitope (3Flag, Red) is for CRISPR/cas9 modification respectively.
B. Diagnostic PCR of two transgenic parasite clones (PyCas9kic1 and PyCas9kic1) with targeted modification in Pysera1 locus. Primers (p) used are shown in (A) and listed in Table S1.
C. RT-PCR detection of both sera1 and cas9 mRNAs expression in the asexual blood stage of 17XNL, ΔPysera1, and PyCas9ki parasites. PCR amplifications of the cDNA from the total RNA with (+) or without (−) reverse transcription are indicated. 18s rRNA mRNA serves as the internal control.
D. Western blotting analysis of Cas9 protein expression in wildtype 17XNL and PyCas9ki parasite clones. Anti-Flag and anti-Myc antibodies were used for detecting the expression of the 3Flag::Cas9 and Cas9::4Myc indicated in (A), respectively. BiP protein serves as an internal control. Parasite clones were treated with (+) or without (−) 5-FC to remove the pYCm plasmid episome.
Next, we constructed a plasmid pYCm-Cas9 containing an intact Cas9 encoding sequence tagged with quadruple Myc epitope (4Myc) C-terminally and flanked by two homologous regions of Pysera1 (0.5 kb of the 5’-flanking region and 0.5 kb of the 3’-flanking region) (Fig. 1A). Two parasite clones (PyCas9kic1 and PyCas9kic1) with targeted modification in Pysera1 locus were obtained and genotyped using diagnostic PCR (Fig. 1B). Corrected replacing Pysera1 with cas9 was further confirmed by RT-PCR analysis as the mRNA transcript of cas9, but not endogenous Pysera1, was specifically detected in the red blood stage of PyCas9ki parasites (Fig. 1C). Furthermore, we detected the expression of both Cas9::4Myc protein from integrated locus and 3Flag::Cas9 protein derived from the episomal pYCm plasmids using western blotting analysis (Fig 1D). To remove the episome plasmid in the parasites for next-round genetic modification, we applied the negative selection to the PyCas9ki parasite by treating with 5-FC [13] and confirmed the successful removal of episomal plasmid as no expression of 3Flag::Cas9 protein was detected in the parasites after 5-FC treatment (Fig 1D).
To evaluate the effect of endogenous Cas9 expression on the parasite development and differentiation, we performed detailed analysis to compare the progression in the life cycle between wildtype and PyCas9ki parasites. PyCas9ki parasites displayed similar asexual proliferation and gametocytes formation in mouse blood stage (Fig S2A,S2B), ookinete differentiation in vitro (Fig S2C), day 7 oocysts per mosquito (Fig S2D), day 14 salivary gland sporozoites (Fig S2E), and infection of mice with sporozoites compared with the results for WT, suggesting normal progression of PyCas9ki parasites during life cycle.
3.2. Specific expression of Cas9 protein in asexual blood stage
The expression of 4Myc-tagged Cas9 protein was detected within the nucleus of different asexual blood stages of PyCas9ki parasites, including rings, trophozoite, and schizont using anti-Myc antibody by immunofluorescence assay (IFA) (Fig 2A), which is similar with previous reports [4, 18]. In addition, no expression of Cas9 protein was detected in gametocytes, ookinetes, midgut oocysts, and salivary gland sporozoites (Fig 2B). These results indicated that the promoter of endogenous Pysera1 gene possesses the transcription activity specifically in the asexual blood stage of P. yoelii parasite. Interestingly, we observed significantly higher percentage of parasite cells expressing Cas9 protein in the asexual stage of PyCas9ki parasite over the parasite with episomal pYCm-Cas9 plasmids (Fig 2C).
Fig. 2.
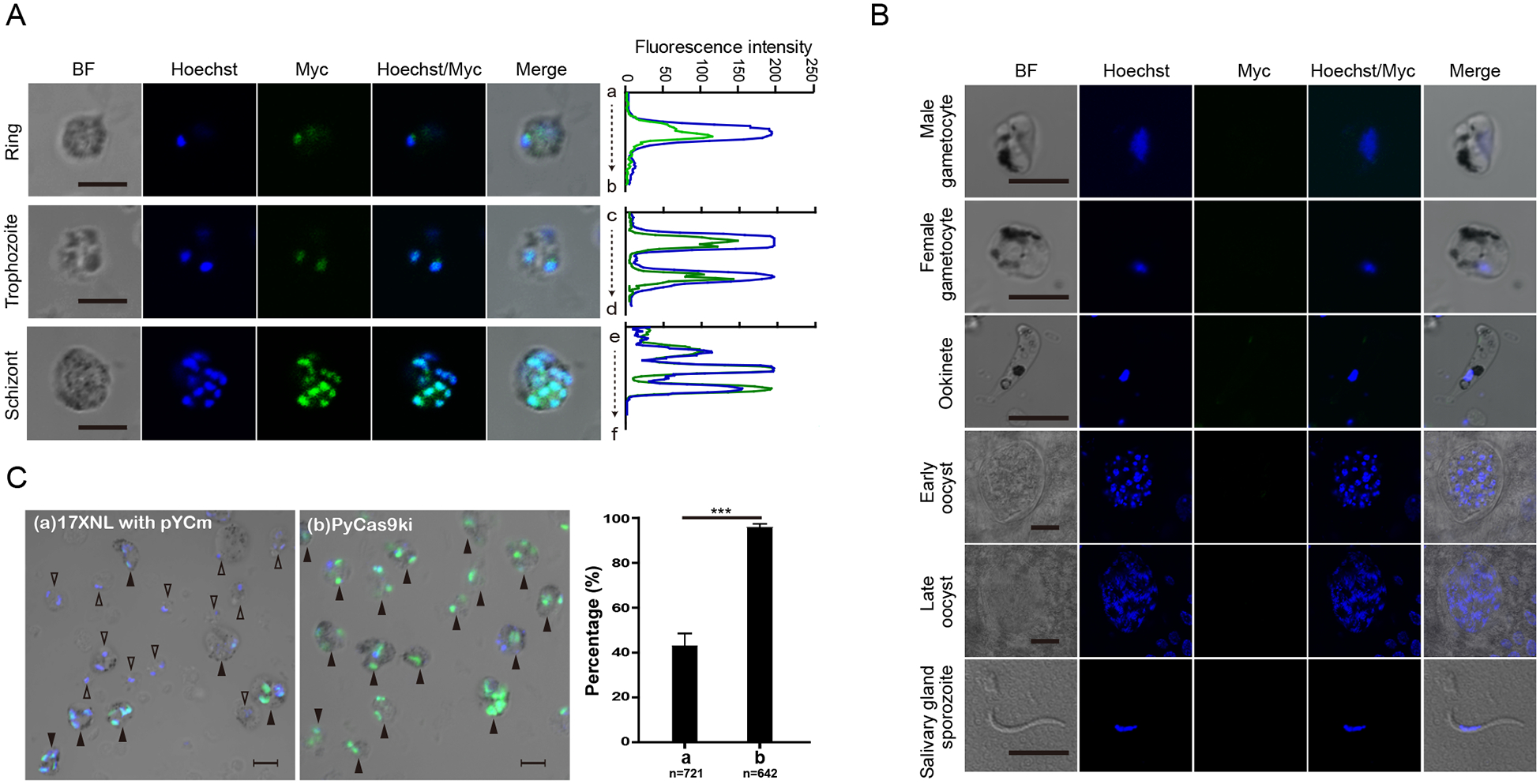
Expression and localization of Cas9 protein in the PyCas9ki parasite.
A. Immunofluorescence assay (IFA) of Cas9 protein expression in the asexual blood stage of PyCas9ki parasite, including ring, trophozoite, and scizhont. Nuclei were stained with Hoechst33342 (blue). Bar = 5 μm. Results are representative of three independent experiments. Co-localization analysis between Cas9 protein and nucleus was shown at the right pannel.
B. IFA analysis of Cas9 protein expression in sexual and mosquito stages of PyCas9ki parasite, including male and female gametocytes, ookinetes, oocysts in day 7 and 14, and salivary gland sporozites. Bar = 5 μm.
C. IFA analysis of Cas9 protein expression in the asexual blood stage of 17XNL parasite transfected with episomal pYCm and the PyCas9ki parasite. Parasites with Cas9 protein expressing and co-localizing with nuclei were indicated by solid triangle ▲, the parasites without Cas9 protein expression were indicated by empty triangle △. quantification results were shown in the right panel. Bar = 5 μm.
3.3. Endogenous gene deletion in the PyCas9ki parasite
Because only sgRNA cassette and homologous DNA template are needed to be introduced exogenously into the cell for CRISPR/cas9-mediated genome editing in the PyCas9ki parasite constitutively expressing Cas9 protein, we engineered the pYCm vector and removed the SpCas9 coding region via mutagenesis, resulting in a new plasmid vector with a smaller size, which we designed pYCs (Fig. S3). To test whether the Cas9 protein expressed endogenously could be applied for CRISPR/Cas9-medifated gene modification, we attempted to deleted two genes (Pyctrp and Pycdpk3) in the genome of the PyCas9ki parasites, separately. The ctrp and cdpk3 genes in both Plasmodium berghei and P. yoelii parasites were previously disrupted, leading to complete loss or severe defect in ookinete gliding motility, respectively, and absence of oocysts in the mosquito after infection [13, 19–22]. We constructed a plasmid pYCs-cdpk3 containing a 46-bp tag DNA (for PCR primers) flanked by two homologous regions of Pycdpk3 (0.49 kb of the 5’-flanking region and 0.53 kb of the 3’-flanking region) (Fig. 3A). Two sgRNAs targeting the exon1 and exon 2 of the Pycdpk3, respectively, were designed and inserted into the pYCs-cdpk3 vector, generating plasmids pYCs-cdpk3-sgRNA1 and pYCs-cdpk3-sgRNA2. One day after electroporation of the plasmids into the PyCas9ki strain, parasites were selected with Pyr supplied in drinking water. Pyr-resistant parasites were observed microscopically 5 to 6 days after electroporation. Expression of sgRNA1 and sgRNA2 transcripts was detected using RT-PCR in the transfected parasites (Fig. 3B). PCR analysis of genomic DNA from parental strain PyCas9ki and plasmid-transfected parasites indicated successful integration of left and right homologous arms at specific sites directed by both sgRNA1 and sgRNA2, but not by control sgRNA targeting irrelevant sequences (Fig. 3C). After limiting dilution cloning, two cloned parasites with disrupted Pycdpk3 gene were obtained and confirmed by PCR genotyping (Fig. 3D). Using the same method, we successfully disrupted the Pycdpk3 gene in the PyCas9ki parasites (Fig. 3E) and obtained two resulting mutant clones with targeted gene deletion (Fig. 3F). Both PyCas9ki parasite derived mutant clones (ΔPycdpk3 and ΔPyctrp) displayed normal asexual growth and gametocyte formation in the mouse (data not shown), and comparable conversion rate to mature ookinetes (Fig. 3G), but severe defect in ookinete gliding motility in vitro (Fig. 3F), which is consistent with the phenotype of cdpk3 KO in P. yoelii 17XNL parasite [13]. These results confirmed that successful gene deletion could be achieved via CRISPR/Cas9 in the PyCas9ki parasite in conjugation with pYCs plasmid system.
Fig. 3.
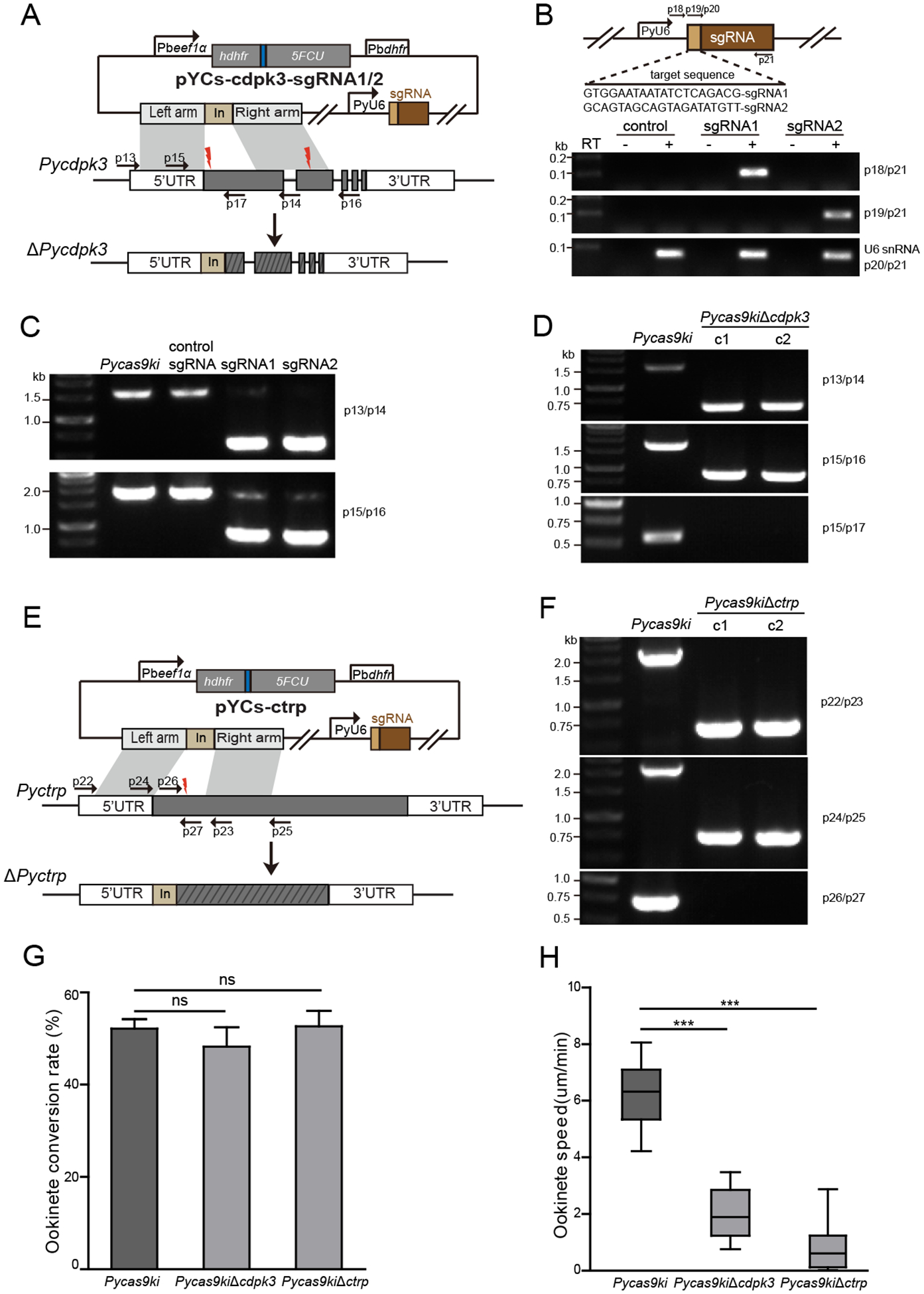
Deletion and mutant phenotype of endogenous cdpk3 gene in the PyCas9ki parasite
A. Schematic diagram of pYCs construct for deleting the endogenous cdpk3 gene. The site for designed sgRNA recognition was indicated as red thunderbolt.
B. Schematic of sgRNA expressing cassettes driven by P. yoelii U6 snRNA promoter in the pYCs plasmid. The protospacer sequences of sgRNA1 and sgRNA2 are indicated as light orange box. sgRNA transcripts were detected by RT-PCR using specific primer pair (p19/p21 and p20/p21) listed in Table S1. Endogenous U6 snRNA transcript serves as an internal control.
C. PCR analysis of genomic DNA from the 17XNL, PyCas9ki, and PyCas9ki parasites transfected with different sgRNA plasmids indicted. Successful integration of left and right homologous arms was detected at specific sites in the parasite directed by both sgRNA1 and sgRNA2, but not control sgRNA targeting irrelevant sequences.
D. PCR analysis of two clones with cdpk3 deletion in the PyCas9ki parasite.
E. In vitro ookinete differentiation of the parental PyCas9ki and PyCas9kiΔcdpk3 parasites.
F. Ookinete gliding motility of the parental PyCas9ki and PyCas9kiΔcdpk3 parasite using Matrigel motility assay. At least 20 cultured ookinetes were measured over a 20 min period. Results are representative of two independent experiments.
3.4. Tagging endogenous gene with epitope in the PyCas9ki parasite
Tagging endogenous genes with fluorescent proteins or epitope tags is widely used for studies of protein localization and protein interaction. To test this application in the PyCas9ki parasite, we built a construct (pYCs-Pysep1::6HA) containing a 498 bp C-terminal region of the Pysep1 gene followed by a sextuple HA tag (6HA) and a 512-bp 3’-flanking region (3’-UTR) of the Pysep1 gene (PY17X_0526200) (Fig 4A). The Pysep1 gene encodes an early transcribed membrane protein locating at the parasitophorous vacuole membrane (PVM) [23]. We detected the integration of both donor templates into the 3’ end of Pysep1 gene in the parasite 6 days after transfection and obtained two clones after limiting dilution cloning (Fig 4B). The expression of recombinant Sep1::6HA protein was detected using western blotting in the asexual blood stage of both parasite clones (Fig 4C). In asexual blood stages, the Sep1::6HA protein is localized at the PVM (Fig 4D), which is consistent with the protein localization reported previously [23]. In addition, we attempted to knock in a 4Myc epitope tag in another genes Pydhhc10 (PY17X_0946500) (Fig 4E), the orthologue of which encodes a S-acyl-transferase expressing only in gametocytes and ookinetes of P. berghei [24]. Targeted knockin of the 4Myc tag in the 3’ end of Pydhhc10 coding region was detected in the two parasite clones using genotypic PCR (Fig 4F). Furthermore, the expression of recombinant DHHC10::4Myc protein was detected in ookinetes of the two parasite clones in the analysis of western blotting (Fig 4G). In ookinetes, the DHHC10::4Myc protein is expressed in distinct cytoplasmic foci, the ookinete specific organelle crystalloid body (Fig 4H), consisting with previous observing [24]. Together, these results indicate that tagging endogenous genes could be achieved via CRISPR/Cas9 in the PyCas9ki parasite.
Fig. 4.
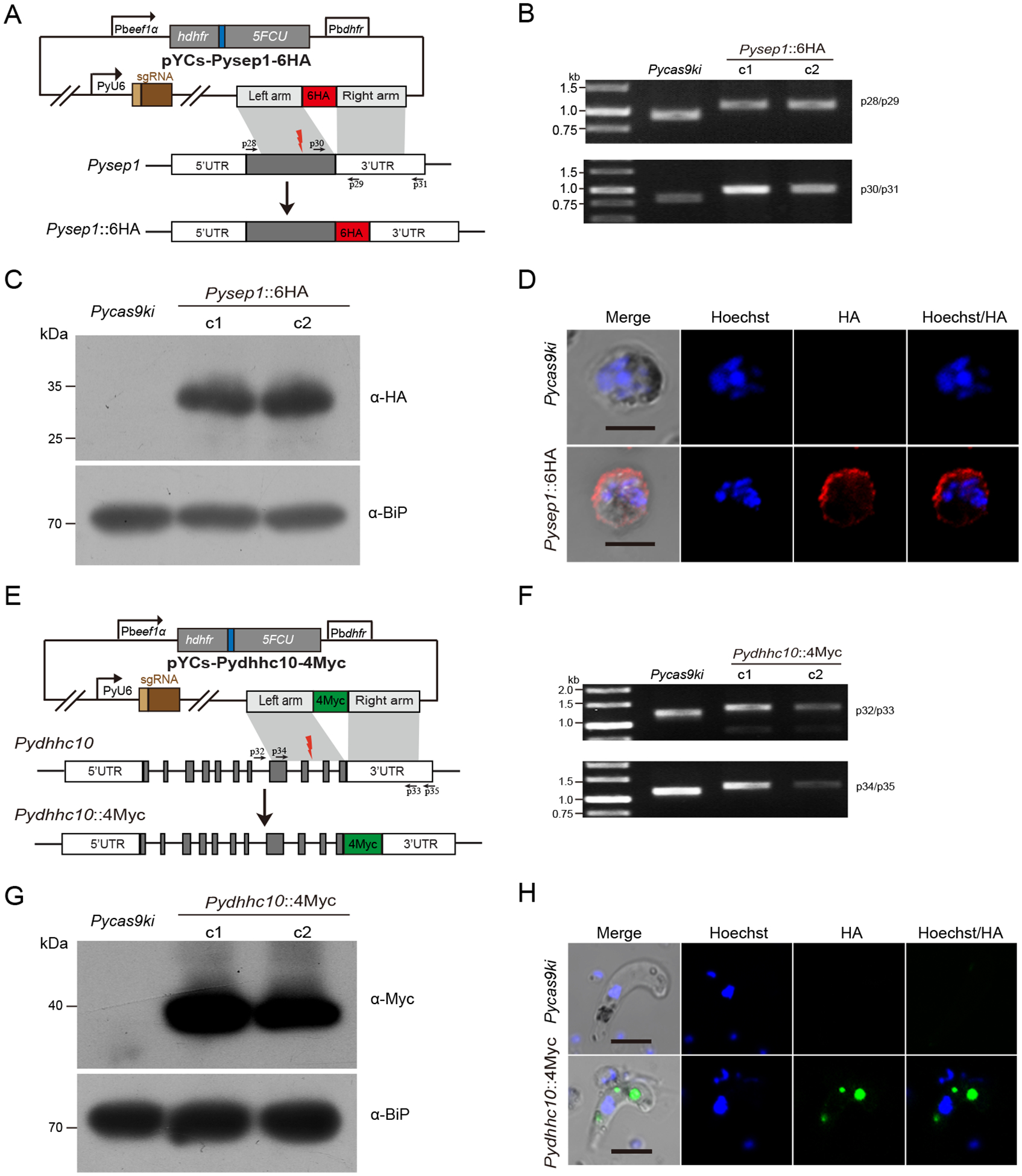
Tagging and protein expression of endogenous sep1 and dhhc10 genes in the PyCas9ki parasite
A. Schematic diagram of pYCs construct for tagging the sep1 gene with 6HA. The site for designed sgRNA recognition was indicated as red thunderbolt.
B. PCR detection of the PyCas9ki parasite and two PyCas9ki derived clones with sep1 gene tagging with 6HA C-terminally.
C. Western blotting analysis of 6HA tagged Sep1 protein expression in the asexual blood stage of the PyCas9ki parasite and two PyCas9ki derived clones. Anti-HA antibody was used for the detection. BiP protein serves as an internal control.
D. IFA analysis of 6HA tagged Sep1 protein expression in the asexual blood stage of the PyCas9ki parasite and one PyCas9ki derived clones. Nuclei were stained with Hoechst33342 (blue). Bar = 5 μm.
E. Schematic diagram of pYCs construct for tagging the dhhc10 gene with 4Myc. The site for designed sgRNA recognition was indicated as red thunderbolt.
F. PCR detection of the PyCas9ki parasite and two PyCas9ki derived clones with dhhc10 gene tagging with 4Myc C-terminally.
G. Western blotting analysis of the 4Myc tagged DHHC10 protein expression in the ookinetes of the PyCas9ki parasite and two PyCas9ki derived clones. Anti-Myc antibody was used for the detection. BiP protein serves as an internal control.
H. IFA analysis of the 4Myc tagged DHHC10 protein expression in the ookinetes of the PyCas9ki parasite and one PyCas9ki derived clones. Nuclei were stained with Hoechst33342 (blue). Bar = 5 μm.
4. Discussions
In this study, we present the generation of a transgenic rodent malaria parasite P. yoelii 17XNL strain PyCas9ki, where the CRPSPR associated protein Cas9 encoding gene was knocked in the endogenous Pysera1 gene locus and its expression is driven by the endogenous promoter of Pysera1 gene. The resulted PyCas9ki parasite displays normal progression during the whole life cycle and possesses the Cas9 protein expression exclusively in asexual blood stage. Furthermore, we demonstrate that both gene disruption and gene tagging, the most commonly used gene editing for gene function study, could be efficiently achieved in this PyCas9ki parasite conjugation with the pYCs vector.
So far, several CRISPR/Cas9-based applications have been described for the genome modification in P. falciparum and P. yoelii [3–5, 10–13]. In these Cas9 vector-based practice, the transcription and expression of exogenous Cas9 in the parasite was driven by the promoters, such as P. falciparum hsp86 promoter and P. yoelii eef1aa promoters, which possess the transcription activity for the whole parasite life cycle. Currently, the asexual blood stage is the only stage window for genetically manipulation in malaria parasites [2]. An idea design for exogenous Cas9 expression would be the asexual blood stage specific expression of the Cas9 because of the existence of the potent activity of caused by off-target effect of the endonuclease Cas9 [25, 26]. Fortunately, the endogenous Cas9 protein was expressed exclusively in the asexual blood stage of the resulted PyCas9ki parasite, including the ring, troph, and schizont (Fig 2A), excluding the possibility of the Cas9 derived off-target effect in the other stages of life cycle.
In our previous studies, we developed a CRISPR/Cas9-based single vector system pYC/pYCm to successfully modify P. yoelii genome, including gene deletion, gene tagging, and nucleotide replacement [4, 13]. In this one-vector plasmid pYCm design (Fig S3), all components, including the Cas9 and sgRNA expression cassettes, the multiple cloning site for insertion of donor template DNA, and the fused selectable markers (hdhfr and yfcu for sequential positive and negative selection) were included. We engineered an updated plasmid pYCs derived from the original pYCm, where the Cas9 encoding region was removed, and the sgRNA cassettes, the multiple cloning site, and the hdhfr/yfcu selection marker were sustained (Fig S3). Compared with the size (10.0 kb) of pYCm plasmid, this new “Cas9-free” plasmid pYCs (5.7 kb) thus can permit an introduction of larger donor DNA sequences, which could be required in some certain types of genome editing in the future.
In addition to gene deletion and gene tagging for the single endogenous gene, a particularly exciting category of future application will be the gene editing in multiple genes or locus simultaneously in the genome of this PyCas9ki parasite. The use of PyCas9ki parasite in conjunction with the pYCs vector containing multiplex sgRNA cassesste and their responding donor templetes for homologous recombination repair, technically make it feasible to easily introduce multiple genetic lesions in the same genome. As multigene interactions play an important role in many biological processes of eukaryotic organisms, multiplex genetic perturbation using the PyCas9ki parasite will enables the interrogation of complex effects for the multiple-members gene family in the life cycle development and differentiation of malaria parasite.
Supplementary Material
Fig. S2. Evaluation of developmental stages of Plasmodium yoelii 17XNL wildtype and PyCas9ki parasites in life cycle.
A. Parasitemia of 17XNL and PyCas9ki parasites. Naïve ICR mice (n = 3) were injected intravenously with 1,000 17XNL or 1,000 PyCas9ki parasite infected erythrocytes. Parasitemias were monitored daily by examining Giemsa-stained blood smears.
B. Gametocytemia of 17XNL and PyCas9ki parasites on day 7 post-infection. Gametocytes were monitored by Giemsa staining of blood smear and counted.
C. In vitro ookinetes differentiation of 17XNL and PyCas9ki parasites.
D. Midgut oocyst counts from dissected mosquito 7 day post-mosquito feeding with parasites indicated. Two independent experiments were performed.
E. Formation and infectivity to mouse of salivary gland sporozoites from parasites as indicated. Mosquitoes were dissected 14 days post blood feeding, and salivary gland sporozoites per mosquito were counted.
Fig. S1. Generation and evaluation of developmental stages of Plasmodium yoelii Δsera1 parasite.
A. Schematic diagram of pYCm construct for deleting the endogenous sera1 gene. One sgRNAs was designed and indicated as red thunderbolt.
B. Diagnostic PCR of two parasite clones with targeted deletion in Pysera1 locus. Primers (p) used are shown in (A) and listed in Table S1.
C. Parasitemia of 17XNL parasite and two mutant clones Δsera1c1 and Δsera1c1. Naïve ICR mice (n = 3) were injected intravenously with 1,000 indicated parasites infected erythrocytes. Parasitemias were monitored daily by examining Giemsa-stained blood smears.
D. Gametocytemia of 17XNL parasite and two mutant clones in the mice. Gametocytes were monitored by Giemsa staining of blood smear and counted.
E. In vitro ookinetes differentiation of 17XNL parasite and two mutant clones.
F. Midgut oocyst counts from dissected mosquito 7 day post-mosquito feeding with parasites indicated. Two independent experiments were performed.
G. Formation and infectivity to mouse of salivary gland sporozoites from parasites as indicated. Mosquitoes were dissected 14 days post blood feeding, and salivary gland sporozoites per mosquito were counted.
Fig. S3. Schematic of the pYCm and pYCs plasmids used in this study.
In the plasmid pYCm, all components, including Cas9 cassette, the sgRNA cassette, the selector marker human dihydrofolate reductase (hDHFR) for positive selection with pyrimethamine, and the selector marker yeast cytosine deaminase/uridyl phosphoribosyl transferase (yFCU) for negative selection with 5-FC, and the donor template DNA were included. The Cas9 encoding sequence region was removed from the pYCm, resulting the pYCs plasmid, which could be used for genome editing in the parasite with Cas9 protein expression endogenously.
Table S1. List of primers and sgRNA targeting sequences used in this study.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81522027 and 31772443, J.Y., and 31501912, H.C.), the China Thousand Youth Talents Plan, the Fundamental Research Funds for the Central Universities of China-Xiamen University (20720160069, 20720150165, and 2013121033), the “111” Project of the Ministration of Education of China (B06016), and the Division of Intramural Research, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH, X-z.S.). The authors thank Cindy Clark, NIH Library Writing Center, for manuscript editing assistance.
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at
References
- 1.Volkman SK, Neafsey DE, Schaffner SF, Park DJ, Wirth DF: Harnessing genomics and genome biology to understand malaria biology. Nature reviews Genetics 2012, 13(5):315–328. [DOI] [PubMed] [Google Scholar]
- 2.de Koning-Ward TF, Gilson PR, Crabb BS: Advances in molecular genetic systems in malaria. Nature reviews Microbiology 2015, 13(6):373–387. [DOI] [PubMed] [Google Scholar]
- 3.Ghorbal M, Gorman M, Macpherson CR, Martins RM, Scherf A, Lopez-Rubio JJ: Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nature biotechnology 2014, 32(8):819–821. [DOI] [PubMed] [Google Scholar]
- 4.Zhang C, Xiao B, Jiang Y, Zhao Y, Li Z, Gao H, Ling Y, Wei J, Li S, Lu M et al. : Efficient editing of malaria parasite genome using the CRISPR/Cas9 system. mBio 2014, 5(4):e01414–01414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wagner JC, Platt RJ, Goldfless SJ, Zhang F, Niles JC: Efficient CRISPR-Cas9-mediated genome editing in Plasmodium falciparum. Nature methods 2014, 11(9):915–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crawford ED, Quan J, Horst JA, Ebert D, Wu W, DeRisi JL: Plasmid-free CRISPR/Cas9 genome editing in Plasmodium falciparum confirms mutations conferring resistance to the dihydroisoquinolone clinical candidate SJ733. PloS one 2017, 12(5):e0178163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E: A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337(6096):816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Straimer J, Lee MC, Lee AH, Zeitler B, Williams AE, Pearl JR, Zhang L, Rebar EJ, Gregory PD, Llinas M et al. : Site-specific genome editing in Plasmodium falciparum using engineered zinc-finger nucleases. Nature methods 2012, 9(10):993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirkman LA, Lawrence EA, Deitsch KW: Malaria parasites utilize both homologous recombination and alternative end joining pathways to maintain genome integrity. Nucleic acids research 2014, 42(1):370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ng CL, Siciliano G, Lee MC, de Almeida MJ, Corey VC, Bopp SE, Bertuccini L, Wittlin S, Kasdin RG, Le Bihan A et al. : CRISPR-Cas9-modified pfmdr1 protects Plasmodium falciparum asexual blood stages and gametocytes against a class of piperazine-containing compounds but potentiates artemisinin-based combination therapy partner drugs. Molecular microbiology 2016, 101(3):381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knuepfer E, Napiorkowska M, van Ooij C, Holder AA: Generating conditional gene knockouts in Plasmodium - a toolkit to produce stable DiCre recombinase-expressing parasite lines using CRISPR/Cas9. Scientific reports 2017, 7(1):3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu J, Tong Y, Pan J, Yang Y, Liu Q, Tan X, Zhao S, Qin L, Chen X: A redesigned CRISPR/Cas9 system for marker-free genome editing in Plasmodium falciparum. Parasites & vectors 2016, 9:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang C, Gao H, Yang Z, Jiang Y, Li Z, Wang X, Xiao B, Su XZ, Cui H, Yuan J: CRISPR/Cas9 mediated sequential editing of genes critical for ookinete motility in Plasmodium yoelii. Molecular and biochemical parasitology 2017, 212:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sidik SM, Huet D, Ganesan SM, Huynh MH, Wang T, Nasamu AS, Thiru P, Saeij JPJ, Carruthers VB, Niles JC et al. : A Genome-wide CRISPR Screen in Toxoplasma Identifies Essential Apicomplexan Genes. Cell 2016, 166(6):1423–1435 e1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moon RW, Taylor CJ, Bex C, Schepers R, Goulding D, Janse CJ, Waters AP, Baker DA, Billker O: A cyclic GMP signalling module that regulates gliding motility in a malaria parasite. PLoS pathogens 2009, 5(9):e1000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brochet M, Collins MO, Smith TK, Thompson E, Sebastian S, Volkmann K, Schwach F, Chappell L, Gomes AR, Berriman M et al. : Phosphoinositide metabolism links cGMP-dependent protein kinase G to essential Ca(2)(+) signals at key decision points in the life cycle of malaria parasites. PLoS biology 2014, 12(3):e1001806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Putrianti ED, Schmidt-Christensen A, Arnold I, Heussler VT, Matuschewski K, Silvie O: The Plasmodium serine-type SERA proteases display distinct expression patterns and non-essential in vivo roles during life cycle progression of the malaria parasite. Cellular microbiology 2010, 12(6):725–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA et al. : Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339(6121):819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuda M, Sakaida H, Chinzei Y: Targeted disruption of the plasmodium berghei CTRP gene reveals its essential role in malaria infection of the vector mosquito. The Journal of experimental medicine 1999, 190(11):1711–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dessens JT, Beetsma AL, Dimopoulos G, Wengelnik K, Crisanti A, Kafatos FC, Sinden RE: CTRP is essential for mosquito infection by malaria ookinetes. The EMBO journal 1999, 18(22):6221–6227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishino T, Orito Y, Chinzei Y, Yuda M: A calcium-dependent protein kinase regulates Plasmodium ookinete access to the midgut epithelial cell. Molecular microbiology 2006, 59(4):1175–1184. [DOI] [PubMed] [Google Scholar]
- 22.Siden-Kiamos I, Ecker A, Nyback S, Louis C, Sinden RE, Billker O: Plasmodium berghei calcium-dependent protein kinase 3 is required for ookinete gliding motility and mosquito midgut invasion. Molecular microbiology 2006, 60(6):1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Birago C, Albanesi V, Silvestrini F, Picci L, Pizzi E, Alano P, Pace T, Ponzi M: A gene-family encoding small exported proteins is conserved across Plasmodium genus. Molecular and biochemical parasitology 2003, 126(2):209–218. [DOI] [PubMed] [Google Scholar]
- 24.Santos JM, Duarte N, Kehrer J, Ramesar J, Avramut MC, Koster AJ, Dessens JT, Frischknecht F, Chevalley-Maurel S, Janse CJ et al. : Maternally supplied S-acyl-transferase is required for crystalloid organelle formation and transmission of the malaria parasite. Proceedings of the National Academy of Sciences of the United States of America 2016, 113(26):7183–7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD: High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature biotechnology 2013, 31(9):822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O et al. : DNA targeting specificity of RNA-guided Cas9 nucleases. Nature biotechnology 2013, 31(9):827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S2. Evaluation of developmental stages of Plasmodium yoelii 17XNL wildtype and PyCas9ki parasites in life cycle.
A. Parasitemia of 17XNL and PyCas9ki parasites. Naïve ICR mice (n = 3) were injected intravenously with 1,000 17XNL or 1,000 PyCas9ki parasite infected erythrocytes. Parasitemias were monitored daily by examining Giemsa-stained blood smears.
B. Gametocytemia of 17XNL and PyCas9ki parasites on day 7 post-infection. Gametocytes were monitored by Giemsa staining of blood smear and counted.
C. In vitro ookinetes differentiation of 17XNL and PyCas9ki parasites.
D. Midgut oocyst counts from dissected mosquito 7 day post-mosquito feeding with parasites indicated. Two independent experiments were performed.
E. Formation and infectivity to mouse of salivary gland sporozoites from parasites as indicated. Mosquitoes were dissected 14 days post blood feeding, and salivary gland sporozoites per mosquito were counted.
Fig. S1. Generation and evaluation of developmental stages of Plasmodium yoelii Δsera1 parasite.
A. Schematic diagram of pYCm construct for deleting the endogenous sera1 gene. One sgRNAs was designed and indicated as red thunderbolt.
B. Diagnostic PCR of two parasite clones with targeted deletion in Pysera1 locus. Primers (p) used are shown in (A) and listed in Table S1.
C. Parasitemia of 17XNL parasite and two mutant clones Δsera1c1 and Δsera1c1. Naïve ICR mice (n = 3) were injected intravenously with 1,000 indicated parasites infected erythrocytes. Parasitemias were monitored daily by examining Giemsa-stained blood smears.
D. Gametocytemia of 17XNL parasite and two mutant clones in the mice. Gametocytes were monitored by Giemsa staining of blood smear and counted.
E. In vitro ookinetes differentiation of 17XNL parasite and two mutant clones.
F. Midgut oocyst counts from dissected mosquito 7 day post-mosquito feeding with parasites indicated. Two independent experiments were performed.
G. Formation and infectivity to mouse of salivary gland sporozoites from parasites as indicated. Mosquitoes were dissected 14 days post blood feeding, and salivary gland sporozoites per mosquito were counted.
Fig. S3. Schematic of the pYCm and pYCs plasmids used in this study.
In the plasmid pYCm, all components, including Cas9 cassette, the sgRNA cassette, the selector marker human dihydrofolate reductase (hDHFR) for positive selection with pyrimethamine, and the selector marker yeast cytosine deaminase/uridyl phosphoribosyl transferase (yFCU) for negative selection with 5-FC, and the donor template DNA were included. The Cas9 encoding sequence region was removed from the pYCm, resulting the pYCs plasmid, which could be used for genome editing in the parasite with Cas9 protein expression endogenously.
Table S1. List of primers and sgRNA targeting sequences used in this study.