

Key Points
Question
What is the safety and tolerability of zerlasiran and its effect on lipoprotein(a) serum concentrations?
Findings
The trial enrolled 32 healthy participants and 36 patients with cardiovascular disease and lipoprotein(a) concentrations 150 nmol/L or greater. No serious adverse event occurred. The median change from baseline in lipoprotein(a) concentration 365 days after single doses for placebo, 300 mg, and 600 mg were 14%, −30%, and −29%. The maximal median change from baseline after 2 doses of placebo, 200 mg, 300 mg, and 450 mg were 7%, −97%, −98%, and −99%.
Meaning
Zerlasiran was well tolerated and produced sustained reductions in lipoprotein(a) concentrations, supporting further development.
Abstract
Importance
Lipoprotein(a) is a causal risk factor for atherosclerotic cardiovascular disease (ASCVD) and calcific aortic stenosis, with no pharmacological treatments approved by regulatory authorities.
Objectives
To assess the safety and tolerability of zerlasiran, a short interfering RNA targeting hepatic synthesis of apolipoprotein(a), and effects on serum concentrations of lipoprotein(a).
Design, Setting, and Participants
Single- and multiple-dose study in healthy participants and patients with stable ASCVD, respectively, with lipoprotein(a) serum concentrations greater than 150 nmol/L, conducted at 7 research sites in the US, the Netherlands, UK, and Australia between November 18, 2020, and February 8, 2023, with last follow-up on August 23, 2023.
Interventions
Participants were randomized to receive (1) a single subcutaneous dose of placebo (n = 8), zerlasiran 300 mg (n = 6) or 600 mg (n = 6); or (2) 2 doses of placebo (n = 9), zerlasiran 200 mg (n = 9) at a 4-week interval or 300 mg (n = 9) or 450 mg (n = 9) at an 8-week interval.
Main Outcomes Measures
The primary outcome was safety and tolerability. Secondary outcomes included serum levels of zerlasiran and effects on lipoprotein(a) serum concentrations.
Results
Among 37 patients in the multiple-dose group (mean age, 56 [SD, 10.4] years; 15 [42%] women), 36 completed the trial. Among 14 participants with extended follow-up after single doses, 13 completed the trial. There were no serious adverse events. Median baseline lipoprotein(a) concentrations in the multiple-dose group were 288 (IQR, 199-352) nmol/L. Median changes in lipoprotein(a) concentration at 365 days after single doses were 14% (IQR, 13% to 15%) for the placebo group, −30% (IQR, −51% to −18%) for the 300 mg of zerlasiran group, and −29% (IQR, −39% to −7%) for the 600-mg dose group. After 2 doses, maximal median changes in lipoprotein(a) concentration were 19 (IQR, −17 to 28) nmol/L for the placebo group, −258 (IQR, −289 to −188) nmol/L for the 200 mg of zerlasiran group, −310 (IQR, −368 to −274) nmol/L for the 300-mg dose group, and −242 (IQR, −343 to −182) nmol/L for the 450-mg dose group, with maximal median percent change of 7% (IQR, −4% to 21%), −97% (IQR, −98% to −95%), −98% (IQR, −99% to −97%), and −99% (IQR, −99% to −98%), respectively, attenuating to 0.3% (IQR, −2% to 21%), −60% (IQR, −71% to −40%), −90% (IQR, −91% to −74%), and −89% (IQR, −91% to −76%) 201 days after administration.
Conclusions
Zerlasiran was well tolerated and reduced lipoprotein(a) concentrations with infrequent administration.
Trial Registration
ClinicalTrials.gov Identifier: NCT04606602
This phase 1 randomized study assesses the safety and tolerability of zerlasiran, a short interfering RNA targeting hepatic synthesis of apolipoprotein(a), and its effects on serum concentrations of lipoprotein(a), in healthy participants and patients with stable atherosclerotic cardiovascular disease.
Introduction
Following the initial description of lipoprotein(a) in 1963,1 accumulating evidence from observational and mendelian randomization studies has linked this lipid particle to a markedly increased risk of developing atherosclerotic cardiovascular disease (ASCVD) and calcific aortic valve stenosis.2 Lipoprotein(a) is formed via a covalent disulfide bond between apolipoprotein(a) and apolipoprotein B100. Lipoprotein(a) concentrations are largely genetically determined, with the rate limiting step in hepatic synthesis controlled by expression of the apolipoprotein(a) gene, LPA.3 Recently, advances in nucleic acid-based therapeutics have enabled substantial reductions in lipoprotein(a) serum concentrations by using RNA interference to degrade the mRNA that encodes for apolipoprotein(a).4,5,6,7 An oral drug, muvalaplin, that disrupts association between apolipoprotein(a) and apolipoprotein B100, has also achieved substantial reduction in lipoprotein(a) concentrations.8 However, limited data have characterized the safety and pharmacokinetic and pharmacodynamic effects of these treatments.
In a phase 1 trial of healthy volunteers with lipoprotein(a) concentrations 150 nmol/L or greater, we previously reported on the initial first-in-human safety and efficacy of single doses of zerlasiran (formerly SLN360), a short interfering RNA targeting lipoprotein(a).6 Although the protocol specified 150 days of follow-up, while the trial was ongoing the independent safety review committee (SRC) recommended 1 year follow-up for participants who received the 2 highest doses (300 mg or 600 mg). As prespecified in the protocol, this phase 1 trial subsequently enrolled additional cohorts of patients with established ASCVD administered 2 doses of zerlasiran or placebo. The current study reports on safety and efficacy during the extended 365 days of follow-up in healthy participants administered single doses and 201 days of follow-up for the patients with ASCVD administered 2 doses. The trial was designed to further characterize the safety and tolerability of zerlasiran and inform the optimal dosing regimen for the phase 2 and 3 development programs.
Methods
Study Organization and Oversight
The trial was managed by the sponsor (Silence Therapeutics, London, UK), the Cleveland Clinic Coordinating Center for Clinical Research (C5Research, Cleveland, Ohio), and a contract research organization (Medpace, Cincinnati, Ohio). At each trial site, prior to enrollment, the study protocol was approved by ethics committees or institutional review boards and written informed consent was obtained for all participants. The SRC monitored the trial and reviewed participant and patient data with support from an independent data analysis center at Medpace. After database lock, Medpace transferred a copy of the database to C5Research for statistical analysis.
Study Population
This randomized, double-blind, placebo-controlled trial consisted of 2 phases, an initial single-dose phase with a planned 150-day follow-up and a subsequent multiple-dose phase. The single-dose phase prespecified enrollment of 32 adults with no history of ASCVD and the multidose phase in 36 patients with stable ASCVD, both groups with lipoprotein(a) concentrations of 150 nmol/L or greater. Eligible participants were aged 18 to 70 years and excluded for moderate or severe hepatic cirrhosis. Stable doses for at least 8 weeks prior to enrollment were required for medications that could influence lipoprotein(a) levels including proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, prescription dose niacin, fibrates, statins, or ezetimibe. Full inclusion and exclusion criteria are described in the trial protocol (Supplement 1). The statistical analysis plan is available in Supplement 2.
Study Procedures
This study was conducted in accordance with the Declaration of Helsinki and international ethical guidelines. Both the single-dose and multiple-dose portions of the trial used an ascending-dose design shown schematically in eFigure 1 in Supplement 3. Of the 32 participants in the single-dose phase of the study, 14 continued beyond the prespecified 150-day follow-up as recommended by the SRC, with assessments extending to 365 days. This extended follow-up cohort included 5 healthy participants in the 300-mg dose group, 6 in the 600-mg dose group, and 3 in the placebo group. The multiple-dose group consisted of 36 patients with ASCVD in 3 cohorts of 12 patients with 3 in each dose group receiving placebo and 9 receiving zerlasiran. Two doses were administered to patients in each multiple-dose group, either 200 mg at a 4-week interval or 300 mg or 450 mg at an 8-week interval. Study drugs were administered by subcutaneous injection in the abdomen, including placebo (sodium chloride [0.9%]). Participants were monitored in a clinical research unit for 24 hours following injection. For each dose level, the SRC assessed safety and efficacy data and recommended dose escalation if deemed appropriate. The investigators, study participants, and sponsor were blinded to study drug assignment, but delegated site staff (eg, the site pharmacist) were unblinded.
Participants in the single-dose, extended follow-up cohort had additional scheduled visits beyond the originally prespecified 150-day follow-up at 210 days, 270 days, 330 days, and 365 days following study drug administration. The multiple-dose group had serial visits at up to 201 days following administration. The presence and extent of any adverse events, including injection site reactions, were assessed at each visit. Blood samples were obtained for lipoprotein(a) concentrations and safety laboratory studies, and a focused physical examination was performed if indicated by symptoms. Pharmacokinetic analyses were performed at frequent time points up to 36 hours and at visits up to 30 days after study drug administration. Lipoprotein(a) concentration was measured with a particle-enhanced turbidimetric assay on an autoanalyzer (c502; Roche).
Study End Points
The primary end point for this phase 1 human study was the safety and tolerability of zerlasiran. Treatment-emergent adverse events and adverse events of special interest were assessed. Injection site events were graded using both the National Cancer Institute Common Terminology Criteria for Adverse Events9 version 5 and the US Food and Drug Administration (FDA) vaccine product injection site reaction scale.
The efficacy outcome was change in serum lipoprotein(a) concentrations from baseline through 365 days for the healthy participants who received single doses of 300 mg or 600 mg and from baseline through 210 days for the patients with ASCVD who received 2 doses. Secondary outcomes included changes in other lipid parameters (low-density lipoprotein cholesterol [LDL-C], high-density lipoprotein cholesterol, total cholesterol, and triglycerides) and the pharmacokinetics of zerlasiran following administration. Exploratory objectives include changes in apolipoprotein B100, oxidized LDL-C, and C-reactive protein levels.
Sample Size
No formal sample size calculations were performed for this phase 1 study. The sample size of 32 healthy participants in the single-dose cohort and 36 patients with ASCVD in the multiple-dose cohort were deemed sufficient for initial assessment of the safety, pharmacokinetics, and efficacy of zerlasiran.
Statistical Analysis
Participants or patients assigned to placebo from the single-dose and multidose portions of the study were separately pooled to create placebo treatment groups. Descriptive statistics are used to summarize safety, efficacy, and pharmacokinetic end points for each treatment group. For categorical variables, summary tabulations of frequency and percentage of participants within each category were assessed. For continuous variables, the number of participants, mean and standard deviation, and median and interquartile range are presented by treatment group. Formal statistical testing of pharmacokinetics and efficacy were not conducted; therefore, no P values are presented. The statistical analysis plan in Supplement 2 provides full details of the analyses, which were performed using SAS version 9.4 (SAS Institute).
Results
Population and Disposition
The study was conducted between November 18, 2020, and February 8, 2023, with last follow-up on August 23, 2023, at 7 research sites in the US, the Netherlands, UK, and Australia. Enrollment included 68 participants, of which 50 are included in the current report, 14 in the single-dose phase with extended follow-up, and 36 in the multiple-dose phase. Figure 1 shows the flow of participants through the trial.
Figure 1. Disposition of Healthy Participants in the Single-Dose Cohort and Patients With Atherosclerotic Cardiovascular Disease in the Multiple-Dose Cohort.
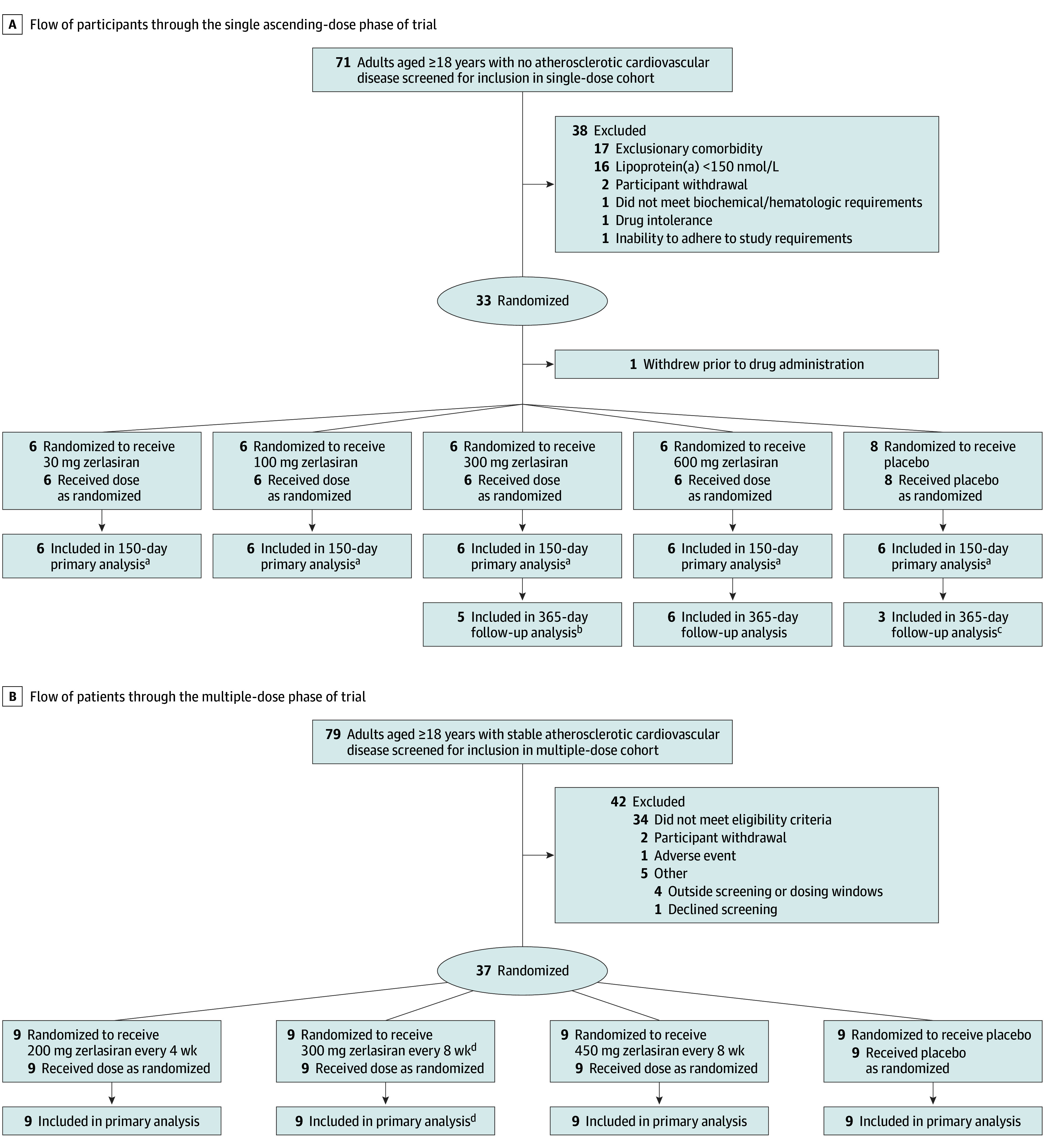
aPrimary analysis reported previously.
bOne participant in the 300-mg dose group only had follow-up to day 180 of the planned 365 days and then declined further participation.
cOne participant in the single-dose pooled placebo group did not consent to 365 days of follow-up.
dOne participant did not complete day-60 dosing and was excluded from the efficacy analyses but included in the safety assessments.
The baseline characteristics of the full single-dose cohort have been reported.6 Table 1 reports the baseline characteristics of participants for the 14 participants in the extended follow-up group and the multiple-dose participants, which included 15 female and 21 male patients (mean age, 56 [SD, 10.4] years) with a median baseline lipoprotein(a) concentration of 288 (IQR, 199-352) nmol/L. Baseline lipoprotein(a) concentrations for the multiple-dose patients are shown graphically in eFigure 2 in Supplement 3. Race was self-identified by patients and collected because lipoprotein(a) levels are known to vary among different racial groups. Most characteristics were similar across treatment groups except for lipid levels and cardiovascular risk factors, which differed between the healthy participants in the single-dose group and the patients with ASCVD in the multiple-dose group. The median baseline LDL-C level was 91 (IQR, 69-113) mg/dL (2.36 [1.79-2.93] mmol/L) in the healthy participants in the single-dose group and 61 (IQR, 47-84) mg/dL (1.58 [1.22-2.18] mmol/L) in the multiple-dose ASCVD group.
Table 1. Baseline Characteristics of Participants.
| Characteristic | Multiple-dose group | Single-dose group | |||||
|---|---|---|---|---|---|---|---|
| 200 mg Every 4 wk ×2 (n = 9) | 300 mg Every 8 wk ×2 (n = 9) | 450 mg Every 8 wk ×2 (n = 9) | Placebo (n = 9) | 300 mg (n = 5) | 600 mg (n = 6) | Placebo (n = 3) | |
| Demographics | |||||||
| Age, mean (SD), y | 59.8 (8.6) | 61.3 (6.8) | 56.9 (12.2) | 47.0 (8.2) | 57.8 (14.6) | 43.7 (17.5) | 54.3 (17.0) |
| Sex, No. (%) | |||||||
| Female | 2 (22.2) | 5 (55.6) | 4 (44.4) | 4 (44.4) | 3 (60.0) | 3 (50.0) | 3 (100.0) |
| Male | 7 (77.8) | 4 (44.4) | 5 (55.6) | 5 (55.6) | 2 (40.0) | 3 (50.0)0 | 0 |
| Individuals of childbearing potential, No. (%) | 0 | 1 (11.1) | 0 | 2 (22.2) | 0 | 0 | 1 (33.3) |
| Body mass index, mean (SD)a | 29.3 (5.4) | 29.9 (3.4) | 28.3 (4.7) | 29.6 (4.0) | 27.7 (3.3) | 26.8 (4.0) | 28.0 (4.1) |
| Race No. (%)b | |||||||
| Asian | 1 (11.1) | 0 | 0 | 0 | 0 | 0 | 0 |
| Black or African American | 0 | 1 (11.1) | 0 | 1 (11.1) | 2 (40.0) | 1 (16.7) | 0 |
| White | 8 (88.9) | 8 (88.9) | 9 (100.0) | 7 (77.8) | 3 (60.0) | 5 (83.3) | 3 (100.0) |
| Multiracial | 0 | 0 | 0 | 1 (11.1) | 0 | 0 | 0 |
| Past medical history, No. (%)c | |||||||
| Hypertension | 3 (33.3) | 6 (66.7) | 4 (44.4) | 4 (44.4) | 2 (40.0) | 1 (16.7) | 0 |
| Diabetes | 0 | 1 (11.1) | 0 | 3 (33.3) | 0 | 0 | 0 |
| Medications, No. (%) | |||||||
| Statin use | 9 (100) | 8 (88.9) | 8 (88.9) | 9 (100) | 3 (60.0) | 3 (50.0) | 1 (33.3) |
| PCSK9i use | 1 (11.1) | 3 (33.3) | 2 (22.2) | 0 | 0 | 0 | 0 |
| Laboratory findingsd | |||||||
| Lipoprotein(a), median (IQR), nmol/L | 266 (194-295) | 316 (276-377) | 247 (183-350) | 292 (217-389) | 313 (257-338) | 231 (179-276) | 211 (181-259) |
| Total cholesterol, mean (SD), mg/dL | 145 (53) | 137 (28) | 150 (50) | 137 (35) | 165 (18) | 180 (51) | 192 (90) |
| LDL-C, mean (SD), mg/dL | 69 (38.0) | 56 (19.5) | 77 (39.7) | 67 (23.9) | 96 (25) | 108 (54) | 111 (75) |
| HDL-C, mean (SD), mg/dL | 46 (14.8) | 59 (21.4) | 55 (13.0) | 50 (10.4) | 44 (9) | 55 (9) | 58 (16) |
| Triglycerides, median (IQR), mg/dL | 93 (77-129) | 85 (60-132) | 93 (74-106) | 73 (58-128) | 93 (76-179) | 75 (42-115) | 123 (79-139) |
| Apolipoprotein B100, mean (SD), md/dL | 71 (34) | 59 (14) | 69 (23) | 69 (23) | 89 (7) | 81 (25) | 87 (46) |
| Oxidized low-density lipoprotein, mean (SD) U/L | 48 (25.1) | 42 (12.5) | 57 (40.7) | 49 (16.5) | 55 (8) | 50 (15) | 63 (35) |
| C-reactive protein, median (IQR), mg/L | 0.5 (0.3-0.7) | 1.2 (1.0-1.4) | 0.4 (0.3-0.5) | 0.4 (0.3-1.0) | 1.7 (1.6-12.2) | 1.0 (0.4-2.1) | 0.4 (0.4-0.4) |
Abbreviations: PCSK9i, proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol.
SI conversion factors: To convert total cholesterol, LDL-C, and HDL-C values to mmol/L, multiply by 0.0259; triglyceride values to mmol/L, multiply by 0.0113.
Calculated as weight in kilograms divided by square of height in meters.
Race was self-identified by patients and collected because lipoprotein(a) levels are known to vary among racial groups. “Multiracial” indicates self-reported as multiracial Black, White, and Asian.
Participants reported medical history, which was verified by either primary care physician or provided directly by specialist.
Reference ranges: lipoprotein(a), less than 75 nmol/L; total cholesterol, 100 to 200 mg/dL; LDL-C, 50 to 130 mg/dL; HDL-C, 35 to 60 mg/dL; triglycerides, 50 to 150 mg/dL; apolipoprotein B100, 55 to 105 mg/dL; oxidized LDL-C, 24.6 to 97.0 U/L; C-reactive protein, 0 to 3 mg/L.
Primary Safety Outcome
As customary in phase 1 studies, safety assessments were designated as the primary outcome of interest. Table 2 summarizes treatment-emergent adverse events occurring in 3 or more participants in any of the active zerlasiran treatment groups. The most common adverse effects were headache in 9 (33%) and COVID-19 infection in 9 (33%), with none leading to withdrawal from the trial. Elevations in C-reactive protein level were present at 24 hours, but levels were within the normal range by day 7 and thereafter. Injection site adverse events were common, but mild to moderate, graded using the Common Terminology Criteria for Adverse Events scale. A total of 29 patients (81%) in the multiple-dose groups reported any injection site adverse event, 24 of which were reported as grade 1 and 5 as grade 2, with none reported as grade 3. Injection site reactions using the FDA vaccine product injection site reaction scale are reported in eTable 1 in Supplement 3. There were no serious adverse events or drug-induced liver injury.
Table 2. Adverse Events Reported by Investigators.
| Characteristic | No. (%) | ||||||
|---|---|---|---|---|---|---|---|
| Multidose cohort during 201-d follow-up (n = 36) | Single-dose cohort from 150- to 365-d follow-up (n = 14) | ||||||
| 200 mg Every 4 wk ×2 (n = 9) | 300 mg Every 8 wk ×2 (n = 9) | 450 mg Every 8 wk ×2 (n = 9) | Placebo (n = 9) | 300 mg (n = 5) | 600 mg (n = 6) | Placebo (n = 3) | |
| No. of participants with any treatment-emergent adverse eventa | 9 (100) | 9 (100) | 9 (100) | 8 (88.9) | 1(20) | 4 (66.6) | 2(66.6) |
| Treatment-emergent adverse events occurring in ≥3 participants | |||||||
| Headache | 3 (33.3) | 3 (33.3) | 3 (33.3) | 4 (44.4) | 0 | 1 (16.7) | 0 |
| Fatigue | 0 (0) | 1 (11.1) | 0 | 3 (33.3) | 0 | 0 | 0 |
| COVID-19 infection | 3 (33.3) | 5 (55.6) | 1 (11.1) | 3 (33.3) | 0 | 1 (16.7) | 0 |
| Upper respiratory tract infection | 1 (11.1) | 1 (11.1) | 1 (11.1) | 2 (22.2) | 0 | 0 | 0 |
| Treatment-emergent adverse events leading to withdrawal | 0 (0) | 0 | 0 | 0 | 0 | 0 | 0 |
| Injection site adverse eventsb | 9 (100) | 8 (89) | 9 (100) | 3 (33) | NA | NA | NA |
| Grade 1 | 6 (67) | 7 (78) | 9 (100) | 2 (22) | NA | NA | NA |
| Grade 2 | 3 (33) | 1 (11) | 0 | 1 (11) | NA | NA | NA |
| Grade 3 | 0 | 0 | 0 | 0 | NA | NA | NA |
| Serious adverse eventsc | 0 | 0 | 0 | 0 | 0 | 1 (16.7)d | 0 |
| Liver eventse | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| QTcF prolongationf | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| C-reactive protein (safety), median (IQR), mg/L | |||||||
| At 24 h | 2.3 (1.3-3.0) | 3.9 (2.1-6.2) | 4.5 (2.3-21.6) | 0.3 (0.2-1.1) | NA | NA | NA |
| Change from baseline at 24 h | 1.4 (0.7-1.9) | 2.2 (1.2-5.1) | 4.1 (2.0-19.3) | −0.1 (−0.2 to 0.01) | NA | NA | NA |
| At day 7 | 0.7 (0.5-1.0) | 1.0 (0.9-1.8) | 1.3 (0.8-4.1) | 0.5 (0.2-0.7) | NA | NA | NA |
| Change from baseline at day 7 | 0.1 (0.1-0.2) | 0.2 (−0.3 to 0.5) | 0.9 (0.5-1.8) | 0 (−0.1 to 0.03) | NA | NA | NA |
Abbreviation: NA, not available (previously reported for the entire single-dose cohort).
Defined as an event that emerged during treatment, having been absent pretreatment, or worsened relative to the pretreatment state.
Graded using the Common Terminology Criteria for Adverse Events. Grade 1 indicates asymptomatic or mild symptoms; grade 2 indicates moderate, minimal, local, or noninvasive intervention indicated; grade 3 indicates severe or medically significant (eg, ulceration or necrosis) but not immediately life-threatening.
Serious adverse events defined as those requiring hospitalization.
One participant experienced 2 unrelated serious adverse events: hospital admission for cholelithiasis and an acinic cell carcinoma of the right parotid gland requiring surgical resection.
Liver events include any alanine aminotransferase value greater than 3 times upper limit of normal, aspartate aminotransferase value greater than 3 times upper limit of normal, alkaline phosphatase value greater than 2 times upper limit of normal, or total bilirubin value greater than 2 times upper limit of normal.
A confirmed QT interval corrected (using the Fridericia formula) for heart rate (QTcF) greater than 500 ms and/or an increase of greater than 60 msec from baseline on repeated electrocardiograms performed at least 2 minutes apart.
Pharmacokinetics and Efficacy Outcomes
The pharmacokinetics of zerlasiran in the multiple-dose cohort from baseline through 36 hours is shown for each injection in eFigure 3, panels A and B, in Supplement 3. Plasma concentrations rose rapidly after the each of the 2 injections, reaching peak levels within 6 hours after administration. Zerlasiran rapidly disappeared from serum and was undetectable by 36 hours after dosing.
The efficacy of zerlasiran in reducing lipoprotein(a) concentrations after single doses of 300 mg or 600 mg with 365-day follow-up is shown in Figure 2A and in eTable 2 in Supplement 3, with the previously reported baseline to 150-day results included for reference purposes. The maximal median change was −10% (IQR, −16% to 1%) for the placebo group, −96% (IQR, −98% to −82%) for the 300-mg dose group, and −98% (IQR, −98 to −97) for the 600-mg dose group. For the 300-mg and 600-mg dose groups, levels gradually increased after 150 days but were still 50% and 45% below baseline after 210 days and 30% and 29% below baseline after 365 days, respectively. The absolute values for the single-dose cohort are displayed graphically in eFigure 4 in Supplement 3.
Figure 2. Percent Change in Lipoprotein(a) Serum Concentrations Following Zerlasiran Administration.

A, Dashed lines indicate previously reported data for change in lipoprotein(a) concentrations during the first 150 days following administration. Observed lipoprotein(a) values and absolute changes are reported in eFigure 4 and eTable 2, respectively, in Supplement 3. B, Observed lipoprotein(a) values and absolute changes are reported in eFigure 5 and eTable 3, respectively, in Supplement 3.
The multiple-dose group showed a maximal median change of 7% (IQR, −4% to 21%) for the placebo group, with a nadir after 30 days; −97% (IQR, −98% to −97%) for the 200-mg dose group, with a nadir at 60 days; −98% (IQR, −99% to −95%) for the 300-mg dose group, with a nadir at 90 days; and −99% (IQR, −99% to −98%) for the 450-mg dose group, with a nadir at 90 days (Figure 2B; eTable 3 in Supplement 3). After 201 days, the median change was 0% (IQR, −1.5% to 21%) for the placebo group, −60% (IQR, −71% to −40%) for the 200-mg dose group, −90% (IQR, −91% to −74%) for the 300-mg dose group, and −89% (IQR, −91% to −76%) for the 450-mg dose group. The observed values for lipoprotein(a) during follow-up in the multiple-dose group are shown in eFigure 5 in Supplement 3. Waterfall plots show the interpatient variability in efficacy at days 150 (Figure 3A) and 201 (Figure 3B) for the multiple-dose group and at day 365 (Figure 3C) for the single-dose group.
Figure 3. Waterfall Plot Showing Distribution of Lipoprotein(a) Responses.

aPatient did not complete day-60 dosing.
The effect of zerlasiran on LDL-C and apolipoprotein B100 levels after multiple doses was measured from baseline to 201 days after drug administration (eTables 4 and 5 and eFigures 6 and 7 in Supplement 3). For LDL-C level, maximal median change was 17% (IQR, −2% to 29%) observed at 150 days after drug administration for the placebo group, −35% (IQR, −45% to −26%) at 60 days for the 200-mg dose group, −47% (IQR, −64% to −12%) at 30 days for the 300-mg dose group, and −28% (IQR, −38% to −26%) at 90 days for the 450-mg dose group. For apolipoprotein B100, maximal median change was 12% (IQR, 2% to 17%) at 150 days after drug administration for the placebo group, −26% (IQR, −35% to −8%) at 43 days for the 200-mg dose group, −28% (IQR, −37% to −21%) at 90 days for the 300-mg dose group, and −23% (IQR, −34% to −22%) at 90 days for the 450-mg dose group. Effects on oxidized LDL-C are reported in eTable 6 in Supplement 3, showing dose-dependent reductions with a mean maximal change of −26% (SD, 23%).
Discussion
In this initial phase 1 study, zerlasiran appeared well-tolerated with no apparent dose-limiting toxicity, no serious adverse effects, and transient hepatic enzyme level elevations in 1 healthy participant after a single dose and in no patients with ASCVD after 2 doses. Mild to moderate injection site adverse events were observed in patients, primarily occurring during the first 24 hours after drug administration. Elevations in C-reactive protein level were observed at 24 hours but were no longer present at 7 days, the next measured time point. Reductions in lipoprotein(a) concentrations showed maximal effects after 2 doses of 300 mg, with little differentiation between the 300-mg and 450-mg doses. The initial phase 1 safety and efficacy findings have enabled initiation of a phase 2 trial, currently underway (NCT05537571).
The current report provides several insights beyond the initial first-in-human zerlasiran trial, including useful information on the potential dose and dosing regimens for future trials. Maximal reductions in lipoprotein(a) concentration were greater than 96% after single 300-mg and 600-mg doses, with efficacy attenuating after 150 days to 30% and 29% reductions, respectively, by 365 days. Two doses of 200 mg administered at a 30-day interval, or 300 mg or 450 mg given at a 60-day interval, produced greater than 97% maximal reductions in lipoprotein(a) concentration, gradually attenuated to 60%, 90%, and 89% reductions, respectively, by 201 days. Two doses, 300 mg or 450 mg, reduced lipoprotein(a) concentration by 90% and 92%, respectively, 120 days after the second dose. Waterfall plots show limited interpatient variability in efficacy at the higher doses (Figure 3, panels A and B). Taken together, these findings demonstrate that zerlasiran has the potential to sustainably reduce lipoprotein(a) concentration with relatively infrequent administration. Data from the current trial combined with findings from the ongoing phase 2 study will further inform the regimens necessary for the targeted lipoprotein(a) reduction and durability in later-phase development.
Zerlasiran is conjugated with N-acetyl galactosamine, an amino derivative of galactose, with high affinity for asialoglycoprotein receptors in hepatocytes, which facilitates rapid clearance from serum and concentration in hepatocytes.10 N-acetyl galactosamine conjugation is now commonly used in nucleic acid–based therapies to enhance hepatic update, thereby reducing the dose necessary to achieve the desired effect and limiting systemic exposure with the potential to reduce the likelihood of off-target effects. Using this strategy, serum concentrations of zerlasiran rose rapidly after subcutaneous injection, with rapid clearance from the circulation resulting in undetectable levels within 36 hours after administration.
The interest in developing these therapies to reduce lipoprotein(a) concentrations stems from strong genetic and observational evidence suggesting that lipoprotein(a) concentration represents an important and potentially modifiable cardiovascular risk factor.2,11,12,13,14,15,16 Lipoprotein(a) concentration is now identified in the current global lipid guidelines as a risk-enhancing factor for ASCVD, but until recently, no pharmacologic therapies have lowered lipoprotein(a) concentration with large enough reductions to potentially affect cardiovascular outcomes. Niacin and PCSK9 inhibitors modestly reduce lipoprotein(a) concentrations, but no randomized clinical trials have demonstrated that lowering lipoprotein(a) concentration with these therapies reduces ASCVD events.17,18
Zerlasiran is one of several nucleic acid–based therapies currently under development to substantially reduce serum concentration of lipoprotein(a). These include pelacarsen and olpasiran, currently in phase 3 trials, and lepodisiran, currently in phase 2 and 3 trials.4,5,7 The length of the current study, 365 days, represents the longest follow-up data available after a single dose for nucleic acid–based therapies designed to lower lipoprotein(a) concentrations. The phase 1 trial of olpasiran reported 225 days of follow-up and the pelacarsen trial 27 weeks of follow-up. The phase 1 lepodisiran trial reported data following single doses for 48 weeks, nearly as long as the current zerlasiran trial.
None of the drugs currently under development has demonstrated that reductions in serum lipoprotein(a) concentrations reduce major adverse cardiovascular events. Nonrandomized observational studies of apheresis have shown some evidence of clinical benefit, and this treatment has been approved by the FDA, but is not practical for most patients with an elevated lipoprotein(a) concentration.19,20 The current trial also showed significant reductions in LDL-C and apolipoprotein B100 levels after administration of zerlasiran. This finding is not unexpected, since lipoprotein(a) particles contain both LDL-C and apolipoprotein B100. Accordingly, reduction in lipoprotein(a) concentration should also reduce the measured mass of LDL-C and apolipoprotein B100.21 The magnitude of reductions in LDL-C and apolipoprotein B100 mass with zerlasiran was similar to the effects reported for olpasiran and pelacarsen.4,5 It is uncertain whether these reductions in LDL-C and apolipoprotein B100 mass might contribute to clinical benefits of lipoprotein(a)-lowering therapies.
How much reduction in lipoprotein(a) concentrations may be required to achieve a beneficial effect on cardiovascular outcomes has been debated.2,22,23 Post hoc analyses of outcomes trials of drugs that more moderately lower lipoprotein(a) concentration have yielded encouraging results. Both monoclonal antibody inhibitors of PCSK9, alirocumab and evolucumab, showed moderate reductions in lipoprotein(a) concentration, in the range of 20% to 25% in large outcome trials. For both drugs, hypothesis-generating post hoc analyses suggest that these reductions independently associate with the observed benefit on major adverse cardiovascular outcomes.24,25 However, other analyses have suggested that much greater reductions are required for likely cardiovascular benefits.22,23
Limitations
The current trial has limitations. First, the study was small, with only 52 mostly White participants exposed to active drug in the single-dose and multiple-dose portions of the trial and only 14 followed up for 365 days. Second, comprehensive evaluation of safety will require larger phase 2 and phase 3 trials. The larger phase 2 trial currently underway should provide additional insight into both safety and efficacy.
Conclusions
Zerlasiran was safe and well tolerated after single doses up to 600 mg and 2 doses of 300 mg or 450 mg. This therapy reduced lipoprotein(a) concentration as much as 99% at the nadir for the single 600-mg dose. Two doses of 300 mg or 450 mg given at a 60-day interval produced nearly identical 98% and 99% reductions at the nadir, with 90% and 89% reductions present at 201 days of follow-up. These findings support further development of zerlasiran in phase 2 and 3 clinical trials.
Trial Protocol
Statistical Analysis Plan
Acknowledgments
eTable 1. Injection Site Assessment 2 Days After Injection Using the FDA Vaccine Product Injection Site Reaction Grading Scale
eTable 2. Lipoprotein(a) Concentration Over Time During Extended Follow-Up in the Single-Dose Cohort
eTable 3. Lipoprotein (a) Concentration Over Time in Multiple-Dose Cohort
eTable 4. Change in LDL-C During Multidose Phase of the Trial
eTable 5. Change in Apo B During Multiple-Dose Phase of the Trial
eTable 6. Absolute and Percent Change in Oxidized LDL-C During Multiple-Dose Phase of the Trial
eFigure 1. Schematic of the Phase 1 Trial Showing the Ascending-Dose Approach for Single and Multiple Doses
eFigure 2. Distribution of Baseline Lipoprotein(a) Levels
eFigure 3. Plasma Concentrations of Zerlasiran After the First and Second Administered Doses
eFigure 4. Observed Lipoprotein(a) Concentrations During 365 Days Follow-Up After a Single Dose
eFigure 5. Observed Lipoprotein(a) Concentrations During 201 Days Follow-Up After Multiple Doses
eFigure 6. Percent Change in LDL-C During 201 Days Follow-up After Multiple Doses
eFigure 7. Percent Change in Apo B During 201 Days Follow-Up After Multiple Doses
Data Sharing Statement
References
- 1.Berg K. A new serum type system in man—the Lp system. Acta Pathol Microbiol Scand. 1963;59:369-382. doi: 10.1111/j.1699-0463.1963.tb01808.x [DOI] [PubMed] [Google Scholar]
- 2.Burgess S, Ference BA, Staley JR, et al. ; European Prospective Investigation Into Cancer and Nutrition–Cardiovascular Disease (EPIC-CVD) Consortium . Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a mendelian randomization analysis. JAMA Cardiol. 2018;3(7):619-627. doi: 10.1001/jamacardio.2018.1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boerwinkle E, Leffert CC, Lin J, Lackner C, Chiesa G, Hobbs HH. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J Clin Invest. 1992;90(1):52-60. doi: 10.1172/JCI115855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, et al. ; AKCEA-APO(a)-LRx Study Investigators . Lipoprotein(a) reduction in persons with cardiovascular disease. N Engl J Med. 2020;382(3):244-255. doi: 10.1056/NEJMoa1905239 [DOI] [PubMed] [Google Scholar]
- 5.Koren MJ, Moriarty PM, Baum SJ, et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein(a). Nat Med. 2022;28(1):96-103. doi: 10.1038/s41591-021-01634-w [DOI] [PubMed] [Google Scholar]
- 6.Nissen SE, Wolski K, Balog C, et al. Single ascending dose study of a short interfering RNA targeting lipoprotein(a) production in individuals with elevated plasma lipoprotein(a) levels. JAMA. 2022;327(17):1679-1687. doi: 10.1001/jama.2022.5050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nissen SE, Linnebjerg H, Shen X, et al. Lepodisiran, an extended-duration short interfering RNA targeting lipoprotein(a): a randomized dose-ascending clinical trial. JAMA. 2023;330(21):2075-2083. doi: 10.1001/jama.2023.21835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicholls SJ, Nissen SE, Fleming C, et al. Muvalaplin, an oral small molecule inhibitor of lipoprotein(a) formation: a randomized clinical trial. JAMA. 2023;330(11):1042-1053. doi: 10.1001/jama.2023.16503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. Published November 27, 2017. Accessed February 27, 2024. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf
- 10.Springer AD, Dowdy SF. GalNAc-siRNA conjugates: leading the way for delivery of RNAi therapeutics. Nucleic Acid Ther. 2018;28(3):109-118. Published online May 24, 2018. doi: 10.1089/nat.2018.0736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117:176-184. doi: 10.1161/CIRCULATIONAHA.107.715698 [DOI] [PubMed] [Google Scholar]
- 12.Cybulska B, Kłosiewicz-Latoszek L, Penson PE, Banach M. What do we know about the role of lipoprotein(a) in atherogenesis 57 years after its discovery? Prog Cardiovasc Dis. 2020;63(3):219-227. Published online April 8, 2020. doi: 10.1016/j.pcad.2020.04.004 [DOI] [PubMed] [Google Scholar]
- 13.Kamstrup PR, Tybjærg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63(5):470-477. doi: 10.1016/j.jacc.2013.09.038 [DOI] [PubMed] [Google Scholar]
- 14.Danesh J, Collins R, Peto R. Lipoprotein(a) and coronary heart disease: meta-analysis of prospective studies. Circulation. 2000;102(10):1082-1085. doi: 10.1161/01.CIR.102.10.1082 [DOI] [PubMed] [Google Scholar]
- 15.Kamstrup PR, Tybjærg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63(5):470-477. doi: 10.1016/j.jacc.2013.09.038 [DOI] [PubMed] [Google Scholar]
- 16.Tsimikas S. A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69(6):692-711. doi: 10.1016/j.jacc.2016.11.042 [DOI] [PubMed] [Google Scholar]
- 17.Safarova MS, Trukhacheva EP, Ezhov MV, et al. Pleiotropic effects of nicotinic acid therapy in men with coronary heart disease and elevated lipoprotein(a) levels. Article in Russian. Kardiologiia. 2011;51(5):9-16. [PubMed] [Google Scholar]
- 18.Cao YX, Liu HH, Li S, Li JJ. A meta-analysis of the effect of PCSK9-monoclonal antibodies on circulating lipoprotein (a) levels. Am J Cardiovasc Drugs. 2019;19(1):87-97. doi: 10.1007/s40256-018-0303-2 [DOI] [PubMed] [Google Scholar]
- 19.Roeseler E, Julius U, Heigl F, et al. ; Pro(a)LiFe-Study Group . Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of follow-up and apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol. 2016;36(9):2019-2027. doi: 10.1161/ATVBAHA.116.307983 [DOI] [PubMed] [Google Scholar]
- 20.Moriarty PM, Gray JV, Gorby LK. Lipoprotein apheresis for lipoprotein(a) and cardiovascular disease. J Clin Lipidol. 2019;13(6):894-900. doi: 10.1016/j.jacl.2019.09.010 [DOI] [PubMed] [Google Scholar]
- 21.Yeang C, Witztum JL, Tsimikas S. Novel method for quantification of lipoprotein (a)-cholesterol: implications for improving accuracy of LDL-C measurements. J Lipid Res. 2021:62:100053. doi: 10.1016/j.jlr.2021.100053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madsen CM, Kamstrup PR, Langsted A, Varbo A, Nordestgaard BG. Lipoprotein(a)-lowering by 50 mg/dL (105 nmol/L) may be needed to reduce cardiovascular disease 20% in secondary prevention: a population-based study. Arterioscler Thromb Vasc Biol. 2020;40(1):255-266. doi: 10.1161/ATVBAHA.119.312951 [DOI] [PubMed] [Google Scholar]
- 23.Lamina C, Kronenberg F; Lp(a)-GWAS-Consortium . Estimation of the required lipoprotein(a)-lowering therapeutic effect size for reduction in coronary heart disease outcomes: a mendelian randomization analysis. JAMA Cardiol. 2019;4(6):575-579. doi: 10.1001/jamacardio.2019.1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bittner VA, Szarek M, Aylward PE, et al. ; ODYSSEY OUTCOMES Committees and Investigators . Effect of alirocumab on lipoprotein(a) and cardiovascular risk after acute coronary syndrome. J Am Coll Cardiol. 2020;75(2):133-144. doi: 10.1016/j.jacc.2019.10.057 [DOI] [PubMed] [Google Scholar]
- 25.O’Donoghue ML, Fazio S, Giugliano RP, et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation. 2019;139(12):1483-1492. doi: 10.1161/CIRCULATIONAHA.118.037184 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial Protocol
Statistical Analysis Plan
Acknowledgments
eTable 1. Injection Site Assessment 2 Days After Injection Using the FDA Vaccine Product Injection Site Reaction Grading Scale
eTable 2. Lipoprotein(a) Concentration Over Time During Extended Follow-Up in the Single-Dose Cohort
eTable 3. Lipoprotein (a) Concentration Over Time in Multiple-Dose Cohort
eTable 4. Change in LDL-C During Multidose Phase of the Trial
eTable 5. Change in Apo B During Multiple-Dose Phase of the Trial
eTable 6. Absolute and Percent Change in Oxidized LDL-C During Multiple-Dose Phase of the Trial
eFigure 1. Schematic of the Phase 1 Trial Showing the Ascending-Dose Approach for Single and Multiple Doses
eFigure 2. Distribution of Baseline Lipoprotein(a) Levels
eFigure 3. Plasma Concentrations of Zerlasiran After the First and Second Administered Doses
eFigure 4. Observed Lipoprotein(a) Concentrations During 365 Days Follow-Up After a Single Dose
eFigure 5. Observed Lipoprotein(a) Concentrations During 201 Days Follow-Up After Multiple Doses
eFigure 6. Percent Change in LDL-C During 201 Days Follow-up After Multiple Doses
eFigure 7. Percent Change in Apo B During 201 Days Follow-Up After Multiple Doses
Data Sharing Statement