

Abstract
The NLRP3 inflammasome, an essential component of the innate immune system, has been emerging as a viable drug target and a potential biomarker for human diseases. In our efforts to develop novel small molecule NLRP3 inhibitors, a 1-(5-chloro-2-methoxybenzyl)-4-phenyl-1H-1,2,3-triazole scaffold was designed via a rational approach based on our previous leads. Structure-activity relationship studies and biophysical studies identified a new lead compound 8 as a potent (IC50: 0.55 ± 0.16 μM), selective and direct NLRP3 inhibitor. Positron emission tomography (PET) imaging studies of [11C]8 demonstrated its rapid and high brain uptake as well as fast washout in mice and rhesus macaque. Notably, plasma kinetic analysis of this radiotracer from the PET/MRI studies in rhesus macaque suggested radiometabolic stability. Collectively, our data not only encourage further studies of this lead compound, but also warrant further optimization to generate additional novel NLRP3 inhibitors and suitable CNS PET radioligands with translational promise.
Introduction
As a key regulator of innate immunity, the NOD-like receptor (NLR) family pyrin domain containing 3 (NLRP3) inflammasome is responsible for the maturation and release of pro-inflammatory cytokines interleukin (IL)-1β and IL-18.1 In the central nervous system (CNS), the NLRP3 inflammasome is predominantly expressed in glial cells, however, expression of NLRP3 is also detected in neurons.2,3 Architecturally, the NLRP3 inflammasome contains a sensor component---NLRP3, an adaptor component---the apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and an effector component---pro-caspase-1. Upon assembly and activation of the inflammasome complex, pro-caspase-1 will be cleaved to provide active caspase-1, which subsequently cleaves and releases IL-1β and IL-18, ultimately leading to inflammatory responses and pyroptosis.4,5 Compared to other members of the NLR family, the NLRP3 inflammasome can be activated by a plethora of stimuli, e.g., extracellular ATP, β-amyloid (Aβ), nigericin, alum, calcium pyrophosphate dihydrate, monosodium urate (MSU), silica, and asbestos, via mechanisms that are not yet understood.6 Therefore, it is not surprising that growing evidence has revealed the involvement of the NLRP3 inflammasome in various inflammatory and neurodegenerative disorders including Alzheimer’s disease (AD),7,8 Parkinson’s disease (PD),9,10 Huntington’s disease (HD),11 multiple sclerosis (MS),12,13 brain injury,14 stroke,15 and epilepsy.16 Notably, the NLRP3 inflammasome has been indicated important roles as a linkage from the commonly observed pathologies to neuroinflammation, and ultimately functional deficit in neurodegenerative disorders. For example, both Aβ oligomers and tau aggregates can activate the NLRP3 inflammasome in microglia and astrocytes, leading to deficits of synaptic plasticity in AD.17 NLRP3 inflammasome is also linked to the aggravation of brain tissue injury upon stimulation by thioredoxin interacting protein (TXNIP).18 Collectively, the results highlight the NLRP3 inflammasome as a promising drug target to achieve disease interventions.
Besides its therapeutic potential, the NLRP3 inflammasome has recently been exploited as a potential biomarker for inflammatory conditions to assist diagnosis and clinical evaluation of new therapeutics, given its association with the innate immune responses and development of inflammation. The level of NLRP3 in cerebrospinal fluid (CSF) has been studied to predict the severity and discriminate between community-acquired bacterial meningitis (CABM) and viral meningitis (VM) in patients.19 The changes of the CSF level of NLRP3 have also been tested as a potential biomarker for septic patients. Serum protein levels of ASC, caspase-1, IL-1β and IL-18 have been investigated as biomarkers for traumatic brain injury (TBI), MS and stroke patients.20 Researchers also investigated NLRP3 as a predictive biomarker for glioma patients21 and cardiometabolic syndrome.22 Notably, a most recent study suggested that activation of the NLRP3 inflammasome could serve as a predictor for COVID-19 patients.23
In the past decade, a number of small molecule NLRP3 inflammasome inhibitors have been reported to interfere with the inflammasome pathway via different mechanisms of actions (MOAs). Among them, some of the direct binders of the NLRP3 protein showed promising effects in preclinical animal models.24 Structurally, most of them are either derived from a sulfonylurea chemical scaffold or containing an electrophilic moiety, i.e., Michael reaction acceptor, exemplified by MCC950,25 CY-09,26 Tranilast,27 and Oridonin28 (Fig. 1A). Interestingly, only MCC950 was attempted as a positron emission tomography (PET) radiotracer to image the NLRP3 inflammasome activity. Unfortunately, the radiotracer, [11C]MCC950 (Fig. 1C), showed poor brain uptake, thus limiting its further use as a radioligand for CNS applications.29 Therefore, CNS penetrant NLRP3 inhibitors and PET radiotracers are urgently needed, given the important roles of this inflammasome in neurodegenerative disorders, to explore their therapeutic potential and as tools to validate the NLRP3 inflammasome as a potential biomarker for neuroinflammation.
Figure 1.
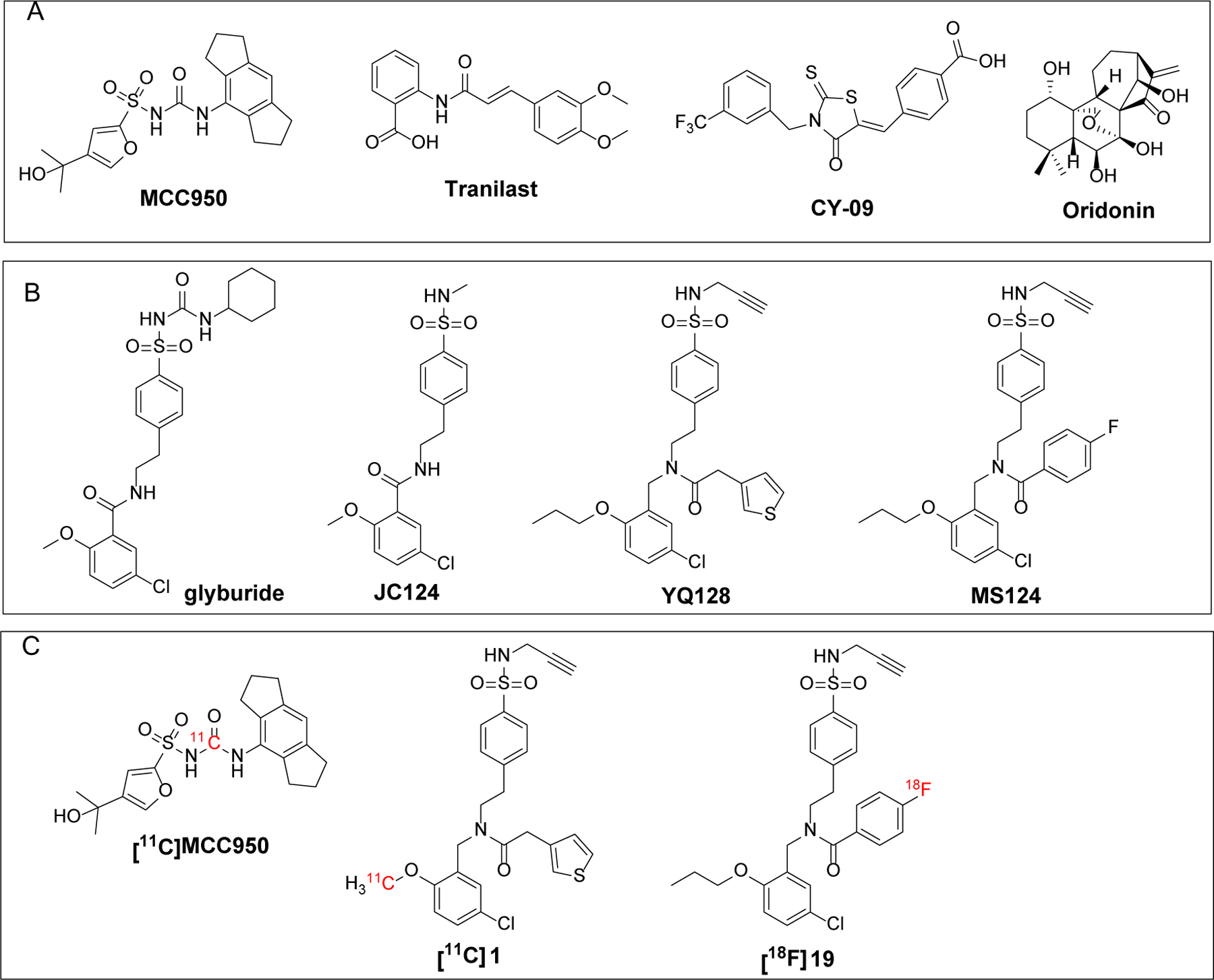
(A) Structures of the reported direct NLRP3 inhibitors and (B) Structures of JC124 and its analogues YQ128 and MS124. (C) Reported PET radiotracers of NLRP3.
Recently, our laboratory designed and developed a series of NLRP3 inhibitors based on a sulfonamide containing scaffold, derived from the structure of glyburide (Fig. 1B) that showed anti-inflammatory effects by inhibiting the NLRP3 inflammasome pathway.30 The change to a sulfonamide moiety was to eliminate the concerns for hypoglycemic effects caused by the sulfonylurea moiety.31 Our 1st-generation lead compound, JC124 (Fig. 1B), showed inhibition on the NLRP3 inflammasome with an IC50 of 3.25 ± 1.34 μM,32 and demonstrated brain penetration and in vivo efficacy in AD animal models.33 Recent medicinal chemistry campaign led to new lead compounds YQ128 and MS124 with significantly improved potency (IC50 = 0.30 ± 0.01 and 0.12 ± 0.01 μM, respectively) (Fig. 1B).34,35 Since both compounds show good binding affinity to human recombinant NLRP3 protein with a Kd of 117 and 84 nM, respectively, we also attempted development of PET radiotracers based on these two lead compounds. Notably, both radiotracers, [11C]1 and [18F]19 (Fig. 1C), can rapidly cross the blood brain barrier (BBB) and exhibit moderate brain uptake (maximum ~ 1.7% and 2.5% ID/cc, respectively).35,36 Autoradiography studies using brain tissues from wild type and APP/PS1 AD mice also demonstrated in vitro binding specificity of [18F]19 and higher labeling by this radiotracer in the brain tissues of APP/PS1 mice. However, YQ128 and MS124 suffer from rapid metabolism in mouse liver microsomes, suggesting limitations for further development. Herein, we report the design and discovery of a new lead compound 8 from a triazole containing scaffold as a potent, selective, and CNS penetrant NLRP3 inhibitor. Furthermore, a carbon-11 labeled 8 demonstrated promising properties as a CNS radiotracer in mice and non-human primates (NHPs).
Results and Discussion
Design of a 1-(5-chloro-2-methoxybenzyl)-4-phenyl-1H-1,2,3-triazole scaffold to develop potent and selective NLRP3 inhibitors.
Given that JC124 exhibited relatively better metabolic stability in mouse liver microsome compared to other lead NLRP3 inhibitors, and a triazole moiety typically favors interactions with the ATP binding site and brain penetration,37–39 we designed a 1,2,3-triazole-containing scaffold to replace the amide moiety of JC124, as exemplified by compound 1 (Fig. 2). The calculated physicochemical properties of 1, e.g., molecular weight (M.Wt) of 327.8, ClogP of 5.1, tPSA of 37.19, and CNS multiparameter optimization (MPO) score of 3.9, also predict its suitability as a CNS agent.40 The design of such triazole scaffold will also allow quick derivatization and SAR exploration by the well-established “click chemistry”.41
Figure 2.
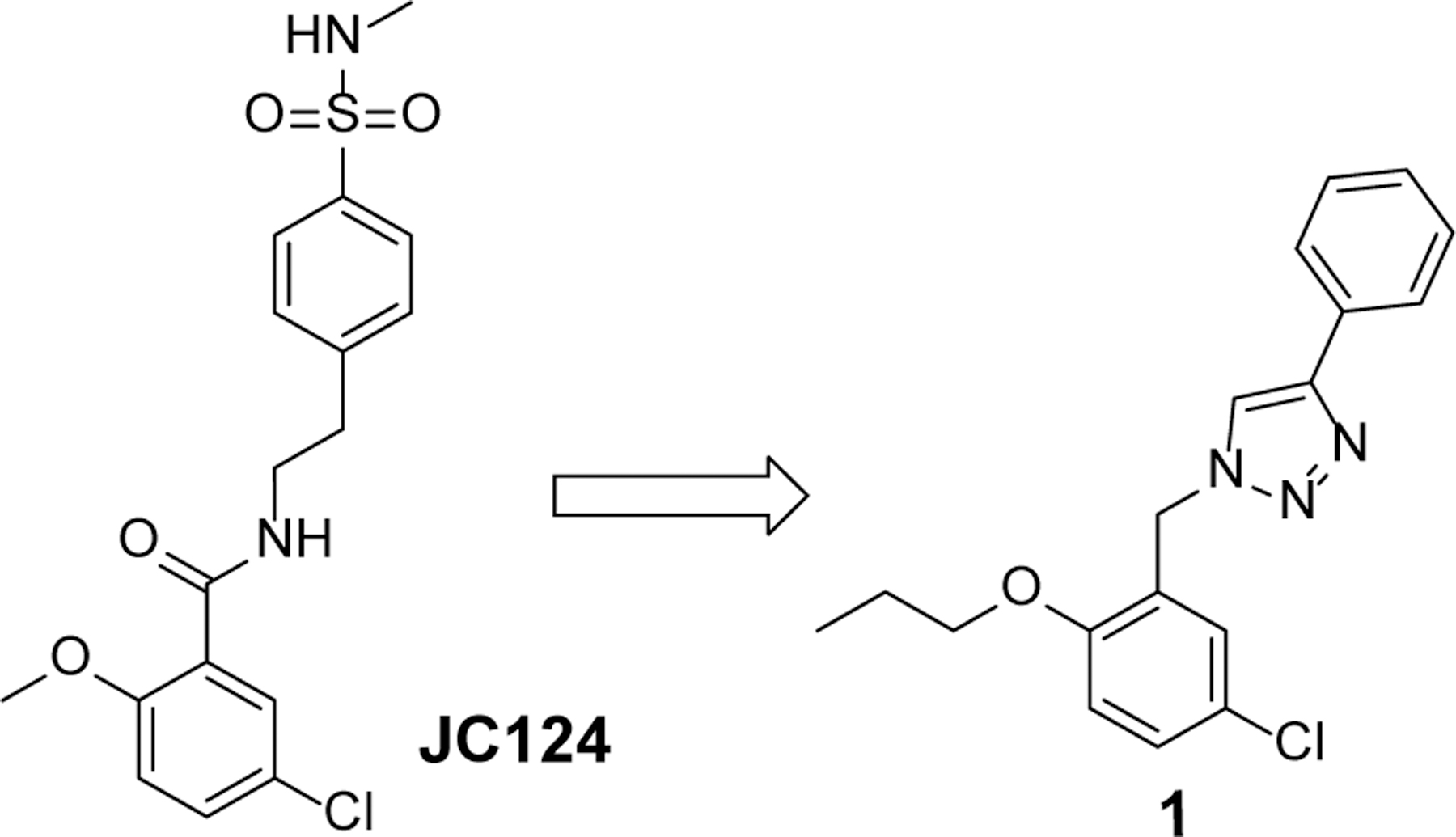
Rational design of 1 from JC124.
Initial biological characterization demonstrated that compound 1 dose-dependently inhibited the release of IL-1β upon activation of the NLRP3 inflammasome by LPS/ATP treatment with an IC50 of 2.09 ± 0.26 μM (Fig. 3A). Notably, the chemical transformation to replace the amide with a 1,2,3-triazole moiety led to ~2-fold increase in potency compared to JC124. Inhibition was only observed for the NLRC4 and AIM2 inflammasomes at higher concentrations (3 and 10 μM, Fig. 3B. C), suggesting reasonable selectivity to the NLRP3 inflammasome. Compound 1 also did not show interference with the priming by LPS in the same cellular model at concentrations ≤ 3 μM (Fig. 3D), further suggesting its specific inhibition on the NLRP3 inflammasome. Binding studies of compound 1 using a microscale thermophoresis (MST) assay and the full length recombinant NLRP3 proteins revealed a Kd of 55 nM (Fig. 3E). The binding interaction of compound 1 with NLRP3 was further confirmed by a thermal shift assay (TSA) using cell lysates from Expi293F cells overexpressing NLRP3 (Fig. 3F). Taken together, the results from the initial characterizations suggest that further structural modifications of this triazole-containing chemical scaffold may hold promise to deliver candidate compounds as potent and selective NLRP3 inhibitors.
Figure 3.
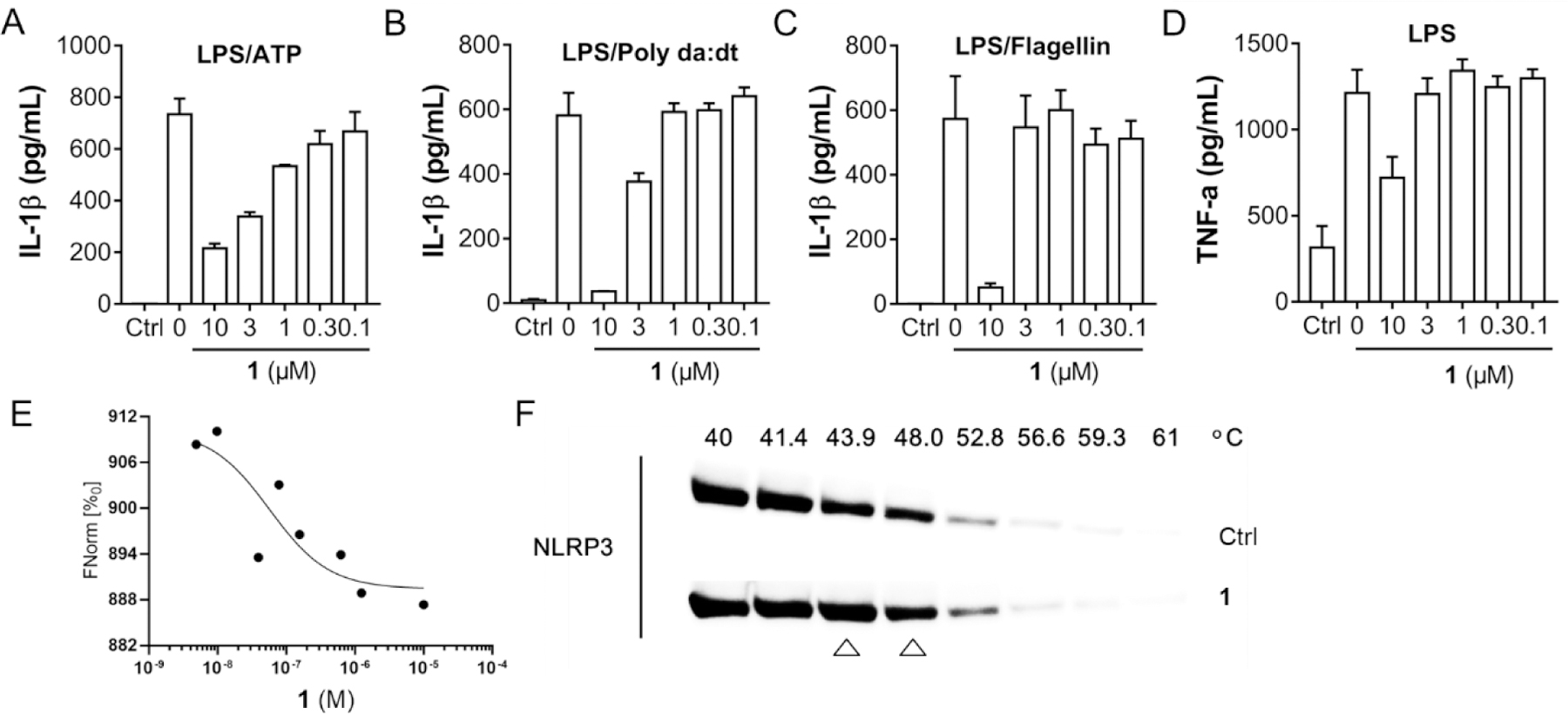
Compound 1 inhibits NLRP3 inflammasome and interacts with recombinant NLRP3. (A) J774A.1 cells were primed with LPS (1 μg/mL) for 4.5 h and then treated with various concentrations of compound 1, followed by stimulation with ATP (5 mM) for 30 min. IL-1β in the culture media was measured by ELISA. (B) J774A.1 cells were treated with LPS (1 μg/mL) and 1 (10 μM) for 1 h, followed by stimulation with (poly(dA:dT)) (4 μg/mL) for 4 h or (C) Flagellin (1 μg/mL) for 6 h. The supernatants were collected and levels of IL-1β were measured by ELISA. (D) J774A.1 cells were incubated with or without various concentrations of 1, followed by treatment with LPS (1 μg/mL) for 4.5 h, TNF-α in the culture media was measured by ELISA. (E) Binding affinity of compound 1 with human recombinant NLRP3 were tested by MST. (F) Cell lysate of Expi293F cells overexpressing NLRP3 was incubated with or without 1 and heated at temperature gradients, in which NLRP3 levels were visualized by Western blot.
Structure-activity relationship (SAR) studies of compound 1.
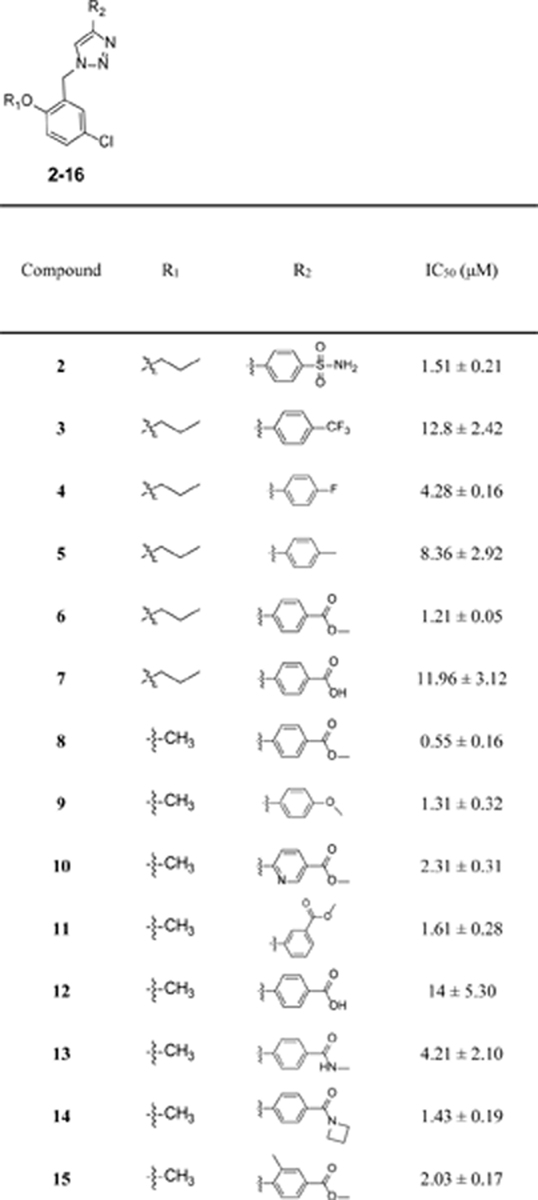
With the confirmation of compound 1 as a relatively selective NLRP3 inhibitor, we then conducted SAR studies to understand and explore the chemical space around this hit structure. The design of congeners of compound 1 was initially centered on the benzyl moiety and the phenyl ring connected to the triazole ring. As shown in Fig. 4, in compounds 2–7, different substituents were introduced to the para-position of the phenyl ring and to evaluate whether commonly encountered functional groups in drug molecules will provide analogs with improved potency. The incorporation of a sulfonamide substituent in 2 was to compare directly with JC124 whether this triazole-containing scaffold shows any advantage as NLRP3 inhibitors. The introduction of an ester or carboxylic acid moiety in compounds 6 and 7 will provide a handle for further structural expansion and modifications, provided they will retain the activity as NLRP3 inhibitors. In compounds 8–16, a methoxy moiety was used to replace the propoxyl moiety of compound 1 while on the para-position of the phenyl ring, an ester or amide was incorporated. Again, the design of such analogs was to compare the activity with that of JC124 as well as the parent compound 1. The design of such analogs also took future SAR expansions into considerations in case promising activities were observed.
Figure 4.
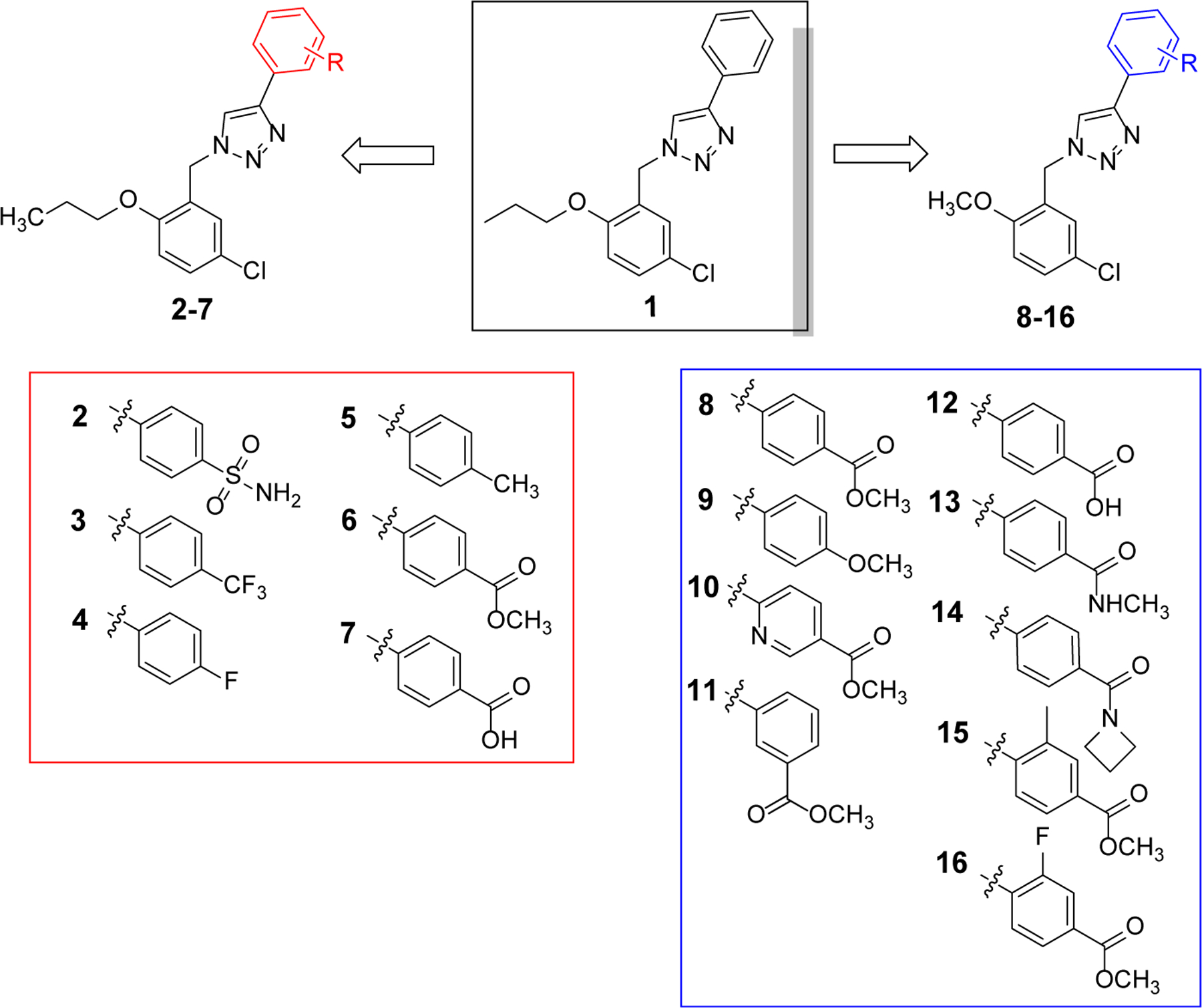
Chemical structures of the designed analogues of hit 1.
The chemical syntheses for the designed compounds are outlined in Scheme 1. Briefly, the alkyne precursors 19a-c were prepared via Sonogashira coupling reactions using commercially available 17a-c with trimethylsilylacetylene followed by the deprotection of trimethylsilyl (TMS) group with TBAF. Alkylation of 20 with 1-bromo-propane or iodomethane followed by the aldehyde reduction gave alcohol intermediates 22a-b. Bromination of 22a-b using phosphorus tribromide provided bromide precursors 23a-b. Cu(I)-catalyzed one pot click reaction (CuAAC) of 23a-b with NaN3 and various alkynes 19 yielded the target compounds 1–6, 8–11 and 15–16. Hydrolysis of the ester group from compounds 6 and 8 with LiOH in methanol gave compounds 7 and 12, respectively. Amides 13 and 14 were prepared from 12 with typical coupling reactions in the presence of HATU.
Scheme 1.
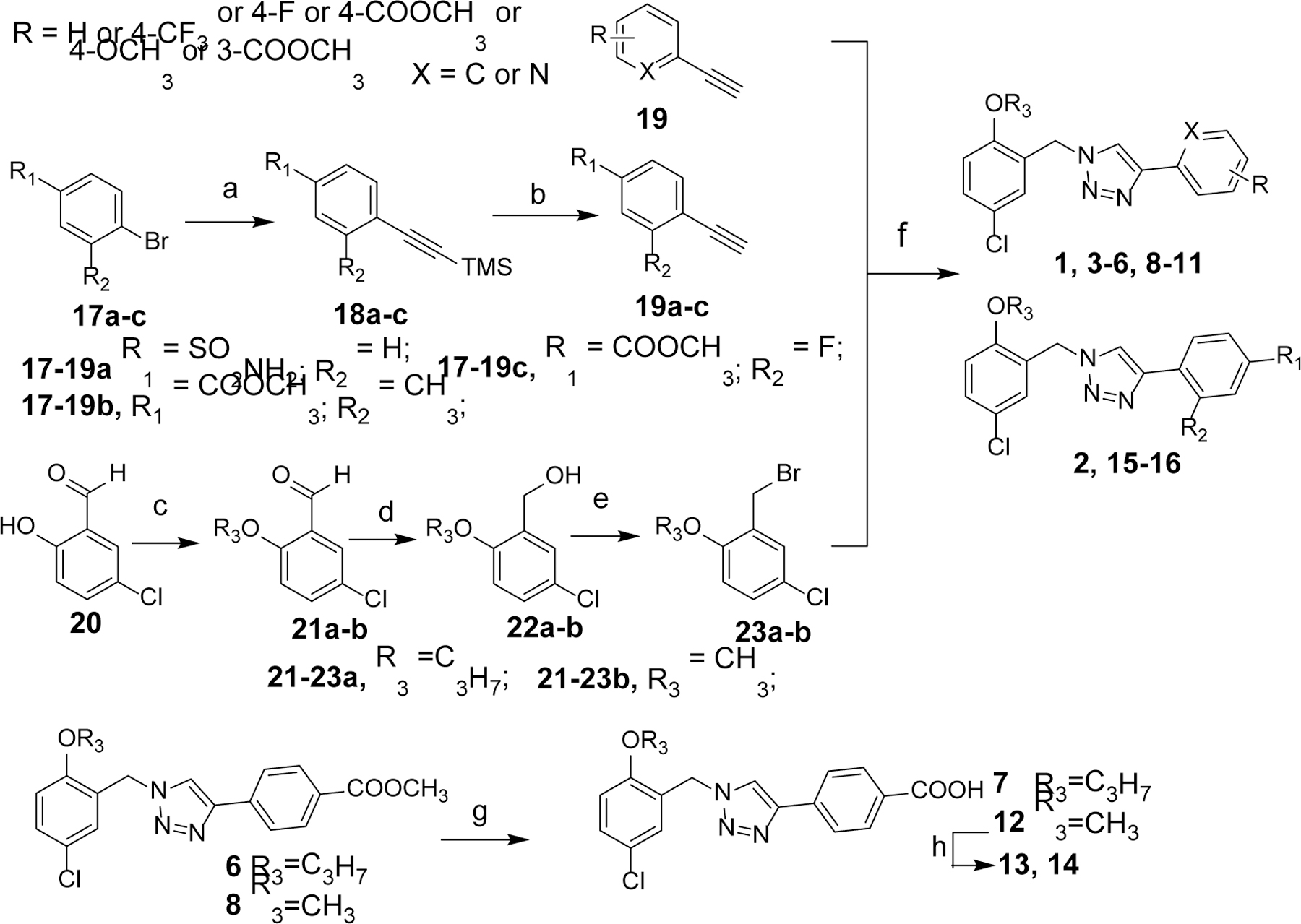
Scheme 1a
aReagents and conditions: (a) trimethylsilylacetylene, CuI, Pd(pph3)2Cl2, TEA, THF, 60 °C, overnight; (b) 1M TBAF, r.t., 1 h; (c) 1-Bromopropane or iodomethane, K2CO3, DMF, 40 °C, 4 h; (d) NaBH4, methanol, r.t., 40 min; (e) PBr3, DCM, 0 °C, 30 min; (f) various alkynes, NaN3, CuSO4, sodium ascorbate, H2O/EtOH, 50 °C, 2~3 h; (g) LiOH, MeOH, 50 °C, overnight; (h) amines, HATU, Et3N, r.t., 2~4 h.
Inhibition of IL-1β release in J774A.1 cells upon stimulation with LPS/ATP.
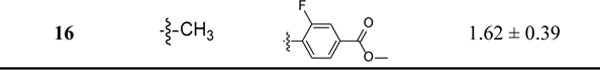
After chemical synthesis, we first tested the compounds for their inhibitory potency on the release of IL-1β in J774A.1 cells upon stimulation with LPS/ATP, a well-established cellular model to evaluate the activity of the NLRP3 inflammasome. As shown in Table 1, introduction of a sulfonamide moiety to the para-position on the phenyl ring of 1 led to a compound with comparable potency as evidenced by 2 with an IC50 of 1.51 μM. However, introduction of a -CF3, -F or -CH3 to the same position led to compounds (3–5) with significantly decreased inhibitory potency. This is consistent with our previous SAR studies of JC12432 and confirms that substituents at this specific position are essential as NLRP3 inhibitors. Compared to 1, compound 6 with an ester moiety at the para-position exhibited increased potency (∼2-fold). Notably, the potency of the carboxylic acid congener (7) is ~10 fold lower compared to that of 6. The results may suggest the importance of H-bond interactions and hydrophobicity on this position. This could also be due to cell permeability difference between the ester and carboxylic acid compounds.42,43 Surprisingly, a simple conversion of the propoxyl group in 6 to a methoxy moiety led to an analog 8 with significantly increased potency (IC50 of 0.55 μM), suggesting possible steric interactions with the target protein on this position. Again, the results here echo our previous SAR studies of JC124.32 The results from compounds 9-12 further suggested the importance of the phenyl ring and the para-ester moiety on the inhibitory potency as NLRP3 inhibitors. Similar to compound 7, compound 12 is significantly less potent than compound 8. Moving the ester moiety to a meta-position as demonstrated by compound 11 also led to a decrease of inhibitory potency. Bioisosteric replacement of the ester of 8 with an amide (13-14) resulted in decreased inhibitory potency. With an intention to increase the dihedral angle between the triazole and the phenyl ring, a -CH3 or -F was introduced in compounds 15 and 16, respectively. Unfortunately, such modifications led to a 3–4 fold decrease in inhibitory potencies compared to that of 8.
Table 1.
Inhibitory potency of compounds 2–16 on the release of IL-1β in J774A.1 cells upon stimulation with LPS/ATP.
|
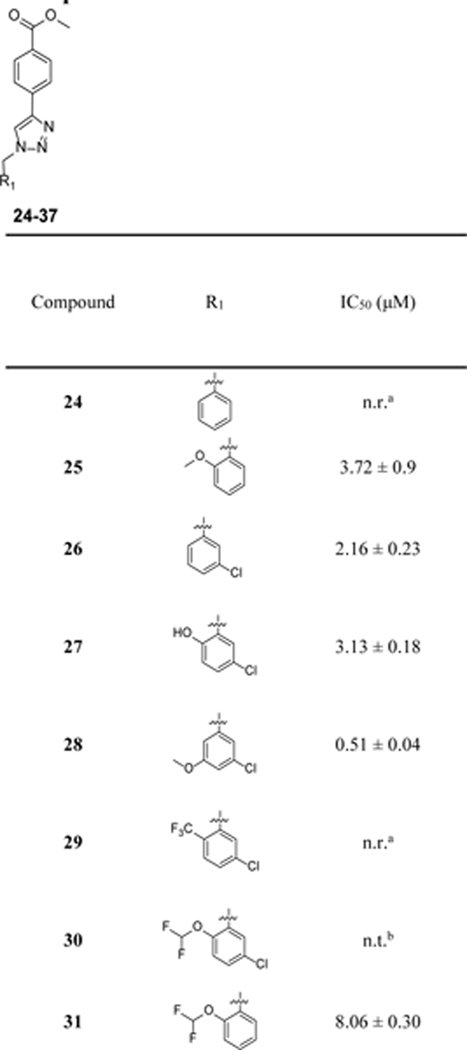
Given the promising potency demonstrated by compound 8, further SAR studies were conducted to explore the modifications on the benzyl moiety (Table 2). Compounds 24–27 were designed to evaluate the importance of the CH3O and Cl on the observed inhibitory potency. Compound 28 was designed to evaluate how moving the CH3O to the meta-position will impact the potency. Compounds 29–34 were designed to shed light on whether these two substituents on the benzyl moiety can be modified to improve potency. In compounds 35–37, di-F substituents were introduced. The chemical syntheses for the designed compound were accomplished by following the CuAAC reaction conditions described in Scheme 1 using various halides, NaN3 and methyl 4-ethynylbenzoate. Compound 27 was obtained by demethylation of 8 with BBr3.
Table 2.
Inhibitory potency of compounds 24–37 on the release of IL-1β in J774A.1 cells upon stimulation with LPS/ATP.
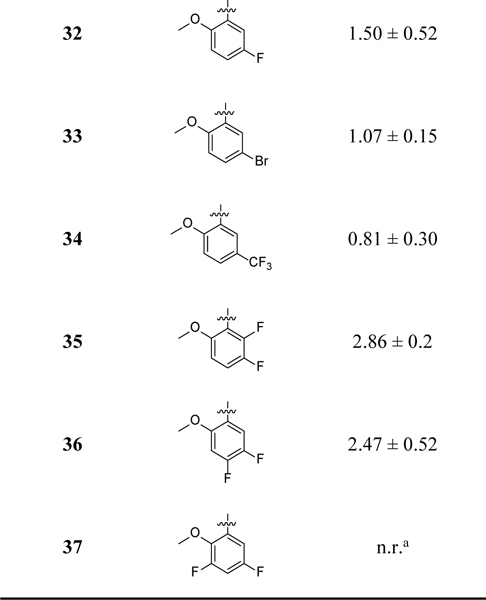
|
n.r.: no reduction.
n.t.: no trend.
The inhibitory potency for the compounds on IL-1β release under LPS/ATP stimulation in J774A.1 cells was obtained and listed in Table 2. Removal of the 5-Cl (24) or 2-OCH3 (25) or both (26) led to ~3–7-fold decrease or complete loss of potency. Changing the CH3O- of compound 8 to a OH- led to ~5-fold decrease in potency as evidenced by analog 27. These results strongly suggest the importance of -Cl and -OCH3 substituents on the observed biological activities of 8. Notably, moving the ortho-CH3O to a meta-position (28) led to a compound with comparable potency as that of 8. The results of 29–31 further supported the importance of CH3O and Cl on the observed inhibitory potency. Replacing the 5-Cl with a 5-F (32) or 5-Br (33) led to ~2–3-fold decrease in potency, while with 5-CF3 (34) retained comparable potency as compound 8. Difluorinated analogs 35-37 showed reduced or loss of potency. Collectively, the SAR studies provide valuable information on the structural features of this chemical scaffold to develop NLRP3 inhibitors.
Further characterization of compound 8 as a selective NLRP3 inhibitor.
Considering its potency and structural similarity to our previous lead JC124, compound 8 was selected for further characterization. In addition, the calculated physicochemical properties of 8 (M.Wt = 357.7, ClogP = 4.1, tPSA = 63.49, MPO score of 4.5)40 also suggested better CNS penetration than hit compound 1. As shown in Fig. 5A, compound 8 consistently suppressed the production of IL-1β under both LPS/ATP and LPS/nigericin stimulation conditions. However, this compound did not show inhibitory effects on the NLRC4 and AIM2 inflammasomes (Fig. 5B). Furthermore, no inhibitory effect was observed for the LPS priming step as demonstrated in Fig. 5C. Biophysical studies of compound 8 using MST and fluorescence spectroscopy (FS) assays demonstrated strong binding interactions of this compound with recombinant full length NLRP3 proteins with a Kd value of 18 and 25 nM, respectively (Fig. 5D & 5E). Taken together, the results strongly suggest that compound 8 is a potent and direct inhibitor of the NLRP3 protein.
Figure 5.
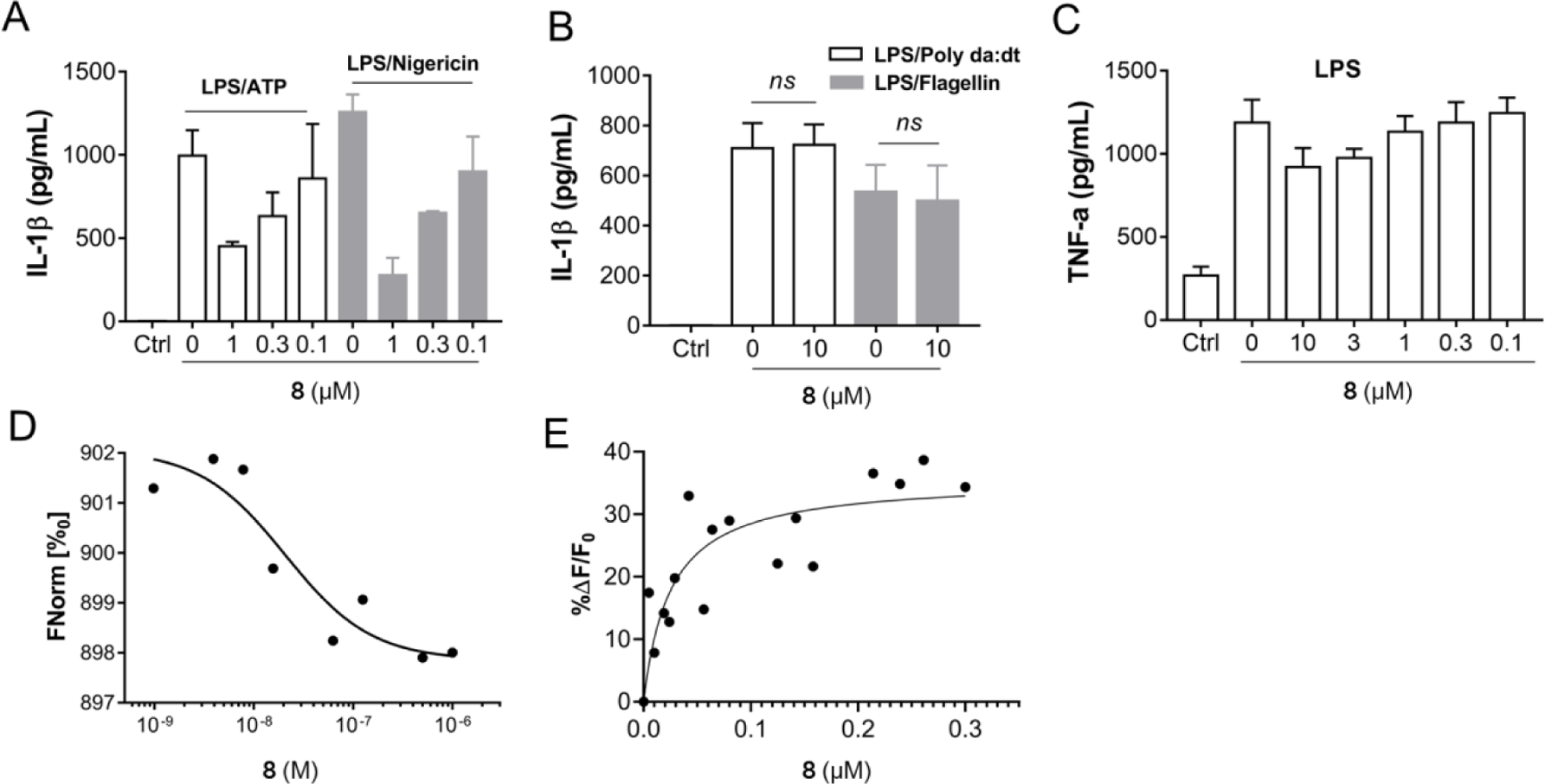
Compound 8 selectively inhibits NLRP3 inflammasome and interacts with recombinant NLRP3. (A) J774A.1 cells were primed with LPS (1 μg/mL) for 4.5 h and then treated with various concentrations of 8, followed by stimulation with ATP (5 mM) or nigericin (5 μM) for 30 min. IL-1β in the culture media was measured by ELISA. (B) J774A.1 cells were treated with LPS (1 μg/mL) and 8 (10 μM) for 1 h, followed by stimulation with Flagellin (1 μg/mL) for 6 h or (poly(dA:dT)) (4 μg/mL) for 4 h. The supernatants were collected and levels of IL-1β were measured by ELISA. (C) J774A.1 cells were incubated with or without various concentrations of 8, followed by treatment with LPS (1 μg/mL) for 4.5 h, TNF-α in the culture media was measured by ELISA. (D) Binding affinity of 8 with human recombinant NLRP3 tested by MST. (E) Binding affinity of 8 with recombinant NLRP3 tested by FS.
Development of [11C]8 as a CNS PET radiotracer.
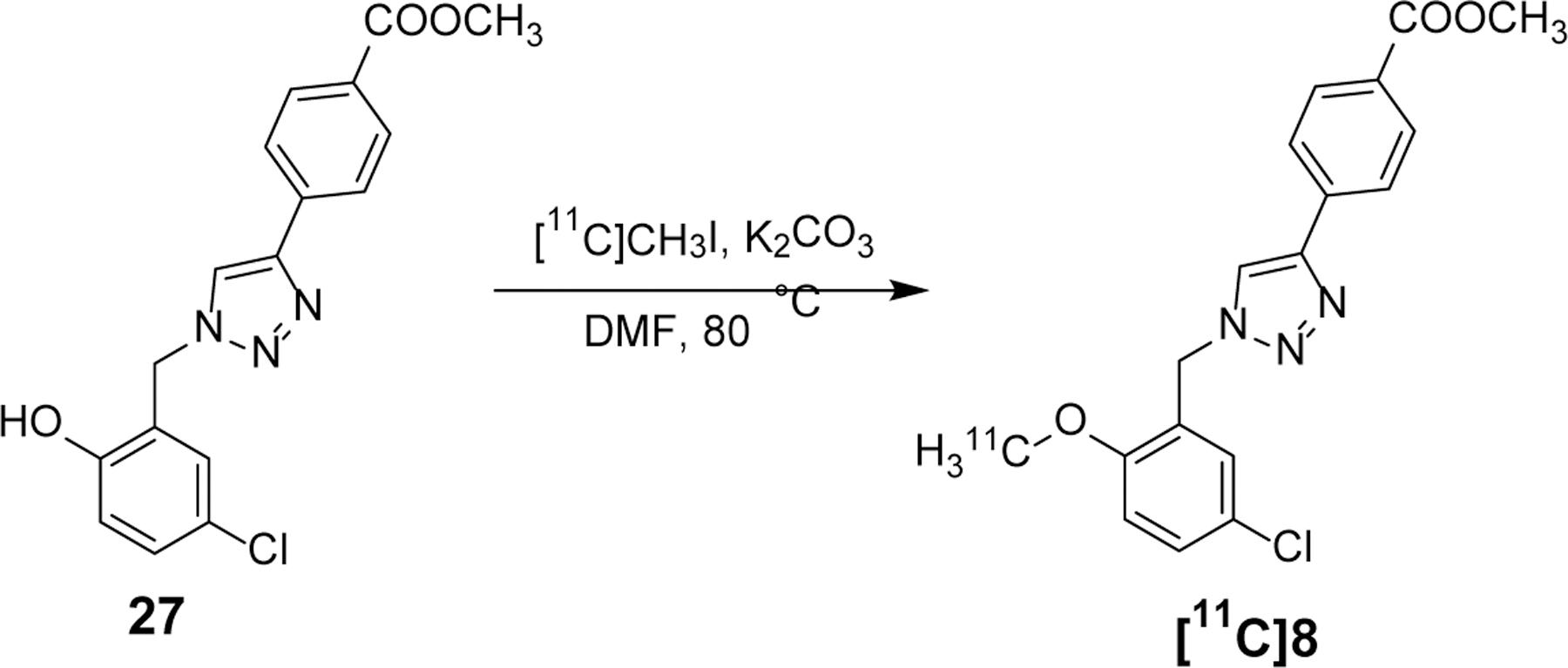
Although the presence of an ester moiety in the structure of 8 poses metabolic liability, a trait that disfavors drug discovery, we decided to convert this compound to a radiotracer to explore its utility as a PET imaging ligand, given that (1) it shows strong binding affinity to the NLRP3 protein and (2) development of CNS PET radiotracers has different structural requirements than traditional drug development.44 As shown in Scheme 2, radiosynthesis of [11C]8 ([11C]YM8) was accomplished by employing our previous reported procedure: methylation of compound 27 with [11C]CH3I at 80 °C to afford [11C]8 in 38 ± 5% (n = 6, decay corrected) yield. Total synthesis time from the end of cyclotron bombardment was 35 ± 5 min. The radiochemical purity was > 99 %, and the molar radioactivity averaged 732 GBq/μmol at the time of injection. The resultant [11C]8 was reconstituted in sterile saline containing < 10 % (v/v) ethanol for in vivo studies.
Scheme 2.
Determination of the Bmax and expression levels of NLRP3 in human brains.
We first attempted to measure the Bmax of NLRP3 in human brain tissues in order to guide the selection and optimization of future NLRP3 PET radiotracers, given that a suitable CNS PET radiotracer typically has a Bmax/Kd ratio > 5.45,46 To this end, we used western blotting analysis to quantify the levels of NLRP3 from healthy human brain tissues. The calculated Bmax of human brain NLRP3 is around 202 nM (Fig. S1 A–C). Therefore, [11C]8 already demonstrates to be a suitable PET tracer with a Bmax/Kd ratio > 8. As our ultimate goal is to develop suitable CNS NLRP3 PET radiotracers to validate the NLRP3 inflammasome as a potential biomarker for neuroinflammation in AD patients, we next compared the difference of NLRP3 expression in the brain tissues of healthy controls and AD patients. The expression of NLRP3 is significantly elevated in AD patients compared to the healthy controls (Fig. S1 D&F), consistent with the literatures47,48 and suggesting the potential of NLRP3 as an AD biomarker.
[11C]8 specifically recognizes and labels NLRP3.
Next, we performed autoradiography studies using wild type (WT) and nlrp3−/− mouse brain tissues to establish the binding specificity of [11C]8. As shown in Fig. 6A and 6B, the results demonstrated strong labeling of the brain tissues from WT C57BL/6 mice by [11C]8. The results also suggest the presence of NLRP3 in the brain tissues of WT mice. The binding of [11C]8 was significantly blocked by both 8 and MCC950, thus suggesting its binding specificity to the NLRP3 protein and the potential use of [11C]8 as a general tool to develop PET tracers as well as NLRP3 inhibitors. Notably, the labeling differences observed from the brain tissues of nlrp3−/− mice and WT mice (Fig. 6C and 6D) clearly confirmed the specific labeling of NLRP3 by this radiotracer. Given the elevated NLRP3 proteins in AD patients and mice47,48 and demonstrated binding specificity of this radiotracer, we expect that the binding signal of [11C]8 would be significantly higher in AD mice than that in healthy control mice.
Figure 6.
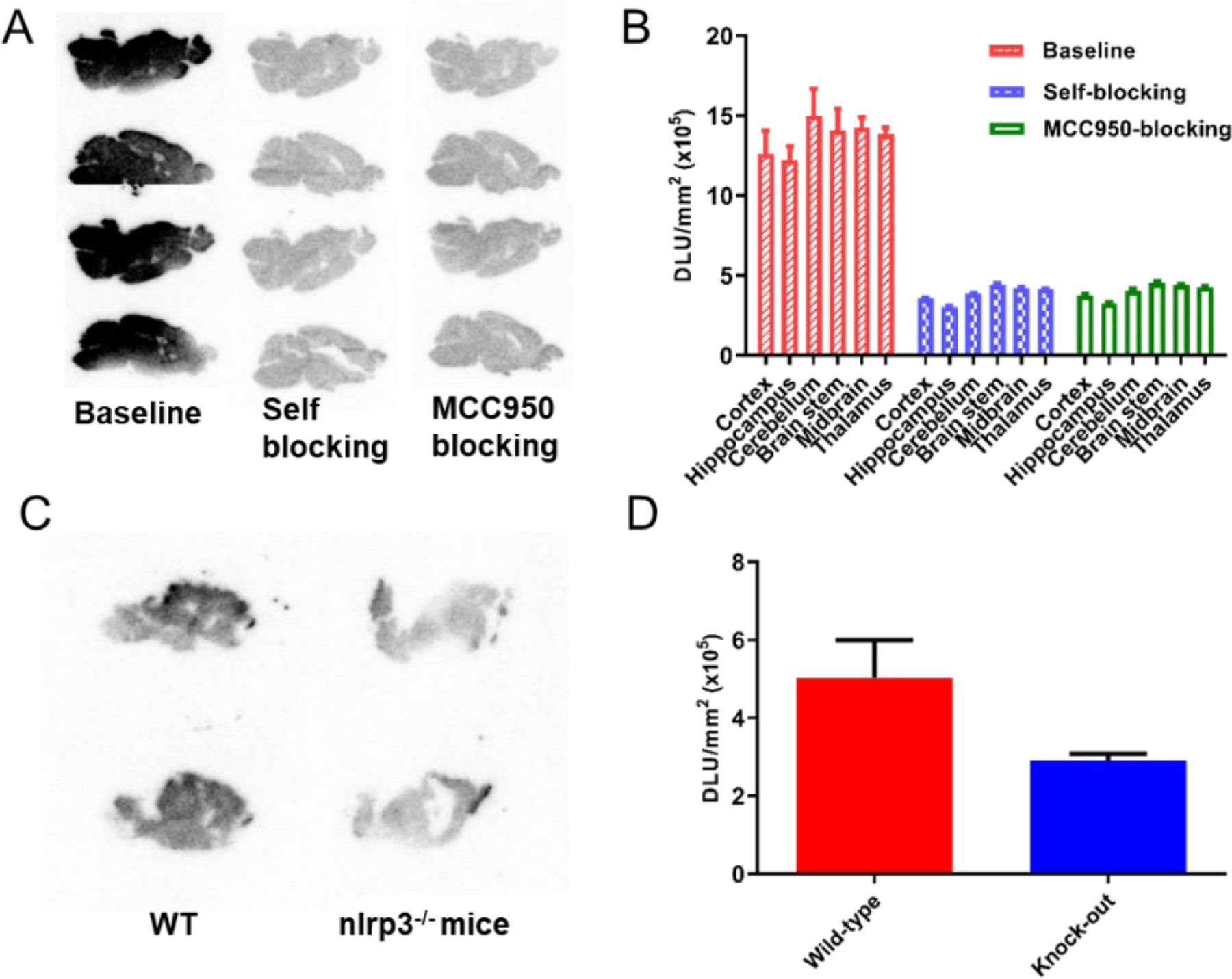
In vitro autoradiography of [11C]8 binding in mouse brain tissues. (A) Representative autoradiographic images of mice brains (sagittal). The baseline mice brain sections were treated with [11C]8 only, and the blocking mice brain sections were co-treated with [11C]8 and unlabeled 8 (10 μM) and MCC950 (10 μM). (B) Relative radioactive uptake under baseline and blocking conditions. (C) Representative autoradiographic images of mice brains (sagittal) from WT and nlrp3−/− mice. (D) Relative radioactive uptake of [11C]8.
PET/CT Studies of [11C]8 in mice.
With the confirmation of its specific labeling for the NLRP3 protein in mouse brain tissues, we next conducted PET/CT studies using [11C]8 in male C57BL/6 mice (25–30 g, n = 3). Since the primary objectives of the PET studies in mice are to assess the in vivo attributes of the tracer, e.g., its distribution, brain penetration, binding characteristics, specificity, etc., differentiating between the sexes of the mice was not deemed necessary at this stage. Therefore, only male mice were used in the experiments. After administration via tail vein injection in male C57BL/6 mice, PET scanning was performed to evaluate the biodistribution of [11C]8 at four time points (2, 10, 30, and 60 min). The radioactivity accumulation is presented as the percentage of injected dose per cubic centimeter (% ID/cc). As shown in Fig. 7, several organs, including brain, blood, liver and kidney, exhibited quick initial uptakes (2.58, 4.28, 3.95 and 3.56% ID/cc at 2 min post injection, respectively). The levels of radioactivity in brain and blood showed a relatively slow clearance (0.69% ID/cc and 2.07% ID/cc at 60 min post injection, respectively), while the liver and kidney uptakes increased gradually within the scanning timeframe (5.65% ID/cc and 6.19% ID/cc at 60 min post injection, respectively). This might suggest hepatobiliary and urinary elimination of [11C]8. The uptake in heart and lung peaked at 2 min (2.32 and 1.39% ID/cc, respectively) and was washed out slowly over time. The lowest uptake was observed in muscle (0.51% ID/cc at 2 min and 0.66% ID/cc at 60 min post injection).
Figure 7.
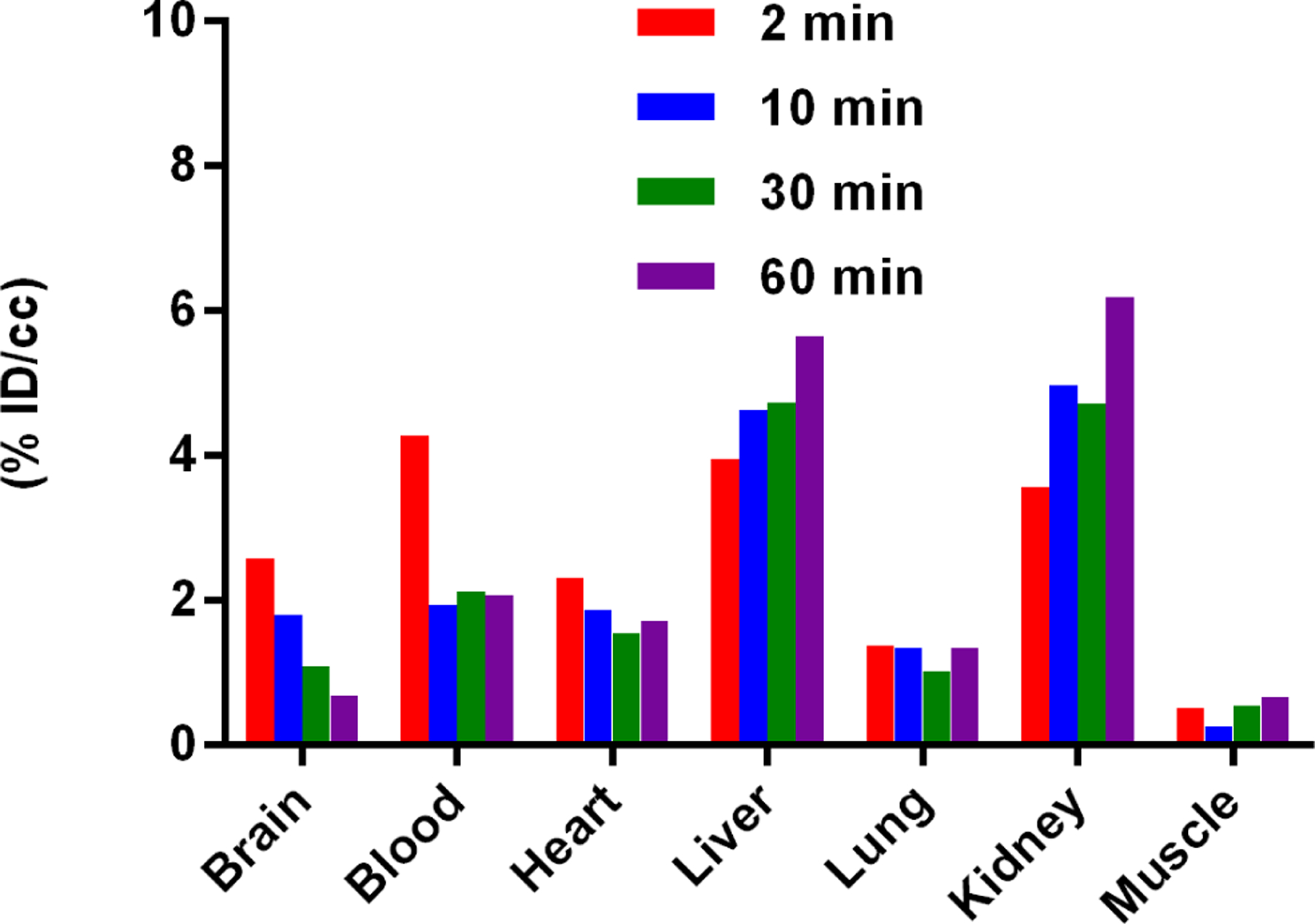
Biodistribution in C57BL/6 mice at four different time points (2, 10, 30, and 60 min) post injection of [11C]8 (n=3). All data are the mean. Data are expressed as the percentage of injected dose per cubic centimeter (% ID/cc).
Next, the brain uptake of [11C]8 was investigated as a potential CNS PET radiotracer. The representative PET/CT images (sagittal, summed images 30–60 min) and time-activity curves (TACs) are shown in Fig. 8A and 8B. The baseline studies in mice demonstrated that [11C]8 rapidly crossed the BBB, reached the brain homogeneously 5 minutes post-injection, and the signal declined gradually within the scanning timeframe. Notably, [11C]8 exhibited high brain uptake with a maximum of 2.7% ID/cc, significantly higher than our previously reported NLRP3 PET radiotracers ([11C]1 and [18F]19: maximum ~ 1.7% and 1.8% ID/cc, respectively).35,36 Furthermore, the brain labeling by [11C]8 can be significantly suppressed (22%) by the unlabeled parent compound 8, similar to our previously reported radiotracer [11C]1 (29% reduction),36 thus suggesting the specific binding of this radiotracer in the mouse brains (Fig. 8C).
Figure 8.
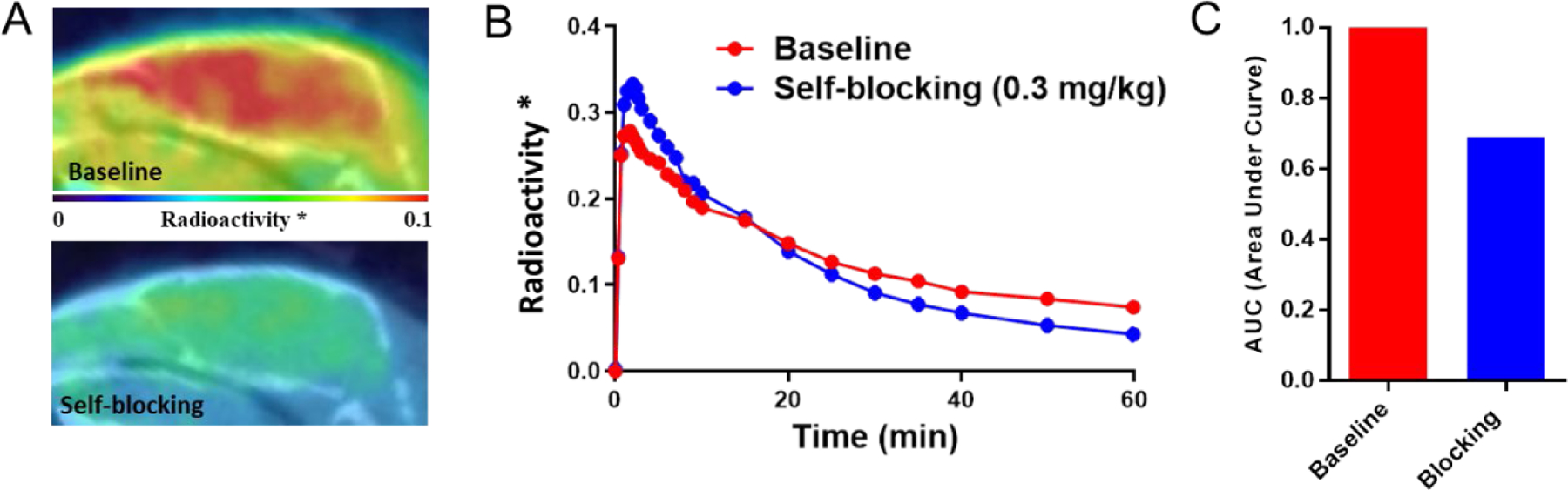
Baseline and blocking analysis of [11C]8 in C57BL/6 Mouse brains (n=3). (A) Sagittal PET/CT images of [11C]8 in baseline and blocking (1 mg/kg, a structurally unrelated sulfonamide type and unlabeled compound) experiments (summed 30–60 mins). (B) TACs of whole brain uptake of [11C]8. “*” means whole brain radioactivity normalized by maximal blood radioactivity. (C) AUC for TACs of baseline and blocking (30–60 mins, ∼22% reduction in AUC).
PET/MR studies of [11C]8 in NHP.
To assess its translational potential, a 90-min dynamic PET/MR scan was carried out in a male rhesus macaque after intravenous injection of [11C]8. As shown in Fig. 9A and 9B, [11C]8 demonstrated excellent brain uptake with a peak SUV > 3 in the whole brain. Regional analysis showed the highest uptake in the cerebellum while comparable distribution was observed in other brain regions (Fig. 9B). During the PET/MR scan, blood plasma samples were acquired for analysis and metabolite correction was used for kinetic modeling analysis. Notably, the parent fraction of [11C]8 in plasma (Fig. 9C, red line) showed the presence of 70% of the parent tracer at 90 min even though the presence of an ester moiety in the radioligand, indicating radiometabolic stability of this radiotracer. The overall plasma activity (Fig. 9C, blue line) indicates a fast washout of this radiotracer from blood. The results clearly support the consensus that different structural features are needed in PET radiotracer development compared to the traditional drug discovery.
Figure 9.
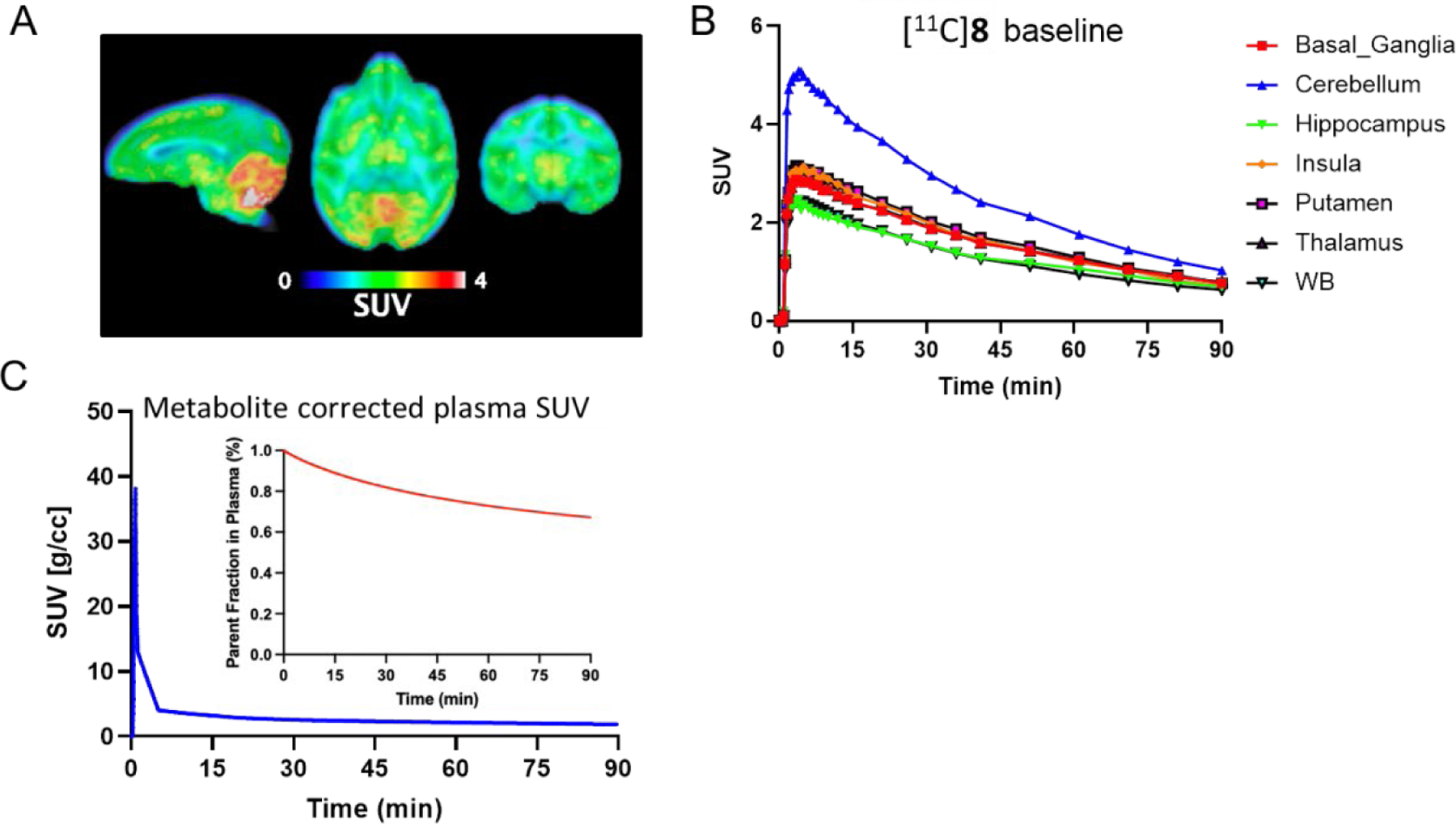
PET/MR imaging of [11C]8 (i.v.) in NHP (n=1). (A) Representative PET/MR image of [11C]8 in rhesus macaque brain (summed 0–90 min); (B) TACs of [11C]8 in the whole brain and brain regions; (C) Plasma TACs of [11C]8 and parent tracer fraction of the metabolites in plasma.
Kinetic analysis of NHP blood distribution volumes.
Logan plot, One- and Two-tissue compartmental models (1TCM and 2TCM) were then employed for the estimation of distribution volumes (VT) and the results are shown in Table 3 in descending order. The highest VT was observed in the cerebellum, consistent with the observations from the SUV images (Fig. 9A and 9B). As can be seen in the table, analysis by 1TCM and 2TCM delivered similar VT values, each of which has stable fits and comparable VT values to those derived from Logan plots (equilibrium time, t*, of 40 min). To identify which fit best describes [11C]8, we compared the Akaike Information Criterion (AIC) and Model Selection Criterion (MSC), wherein lower AIC and higher MSC values are indicative of a better fit. As shown in Table 3, the 2TCM kinetic fits apparently seem to slightly better than the 1TCM ones. Since this pilot kinetic studies were conducted in only one rhesus macaque, future work needs to extend the scope of this preliminary analysis by increasing animal numbers to achieve higher power for statistical comparisons between kinetic models in the NHP and to evaluating the test-retest variability of [11C]8 to further its translatability.
Table 3.
Kinetic analysis of NHP blood distribution volumes (Vt). Formatting for 1TCM and 2TCM are as VT (AIC; MSC)
| Vt [mL/cm3] | |||
|---|---|---|---|
| ROI | Logan Plot (t*=40 min) | 1TCM | 2TCM |
| Cerebellum | 0.827 | 0.943 (65.066; 3.044) | 0.981 (51.550; 3.485) |
| Midbrain | 0.625 | 0.697 (66.685; 2.971) | 0.719 (59.148; 3.231) |
| NAc | 0.625 | 0.684 (51.732; 3.327) | 0.699 (46.671; 3.511) |
| Putamen | 0.574 | 0.634 (56.864; 3.219) | 0.640 (53.451; 3.354) |
| Hypothalamus | 0.563 | 0.588 (54.284; 3.238) | 0.612 (31.348; 3.964) |
| Amygdala | 0.552 | 0.559 (48.973; 3.333) | 0.565 (44.267; 3.507) |
| Insula | 0.548 | 0.516 (60.197; 3.139) | 0.619 (58.833; 3.212) |
| Thalamus | 0.543 | 0.587 (58.195; 3.163) | 0.594 (53.592; 3.334) |
| Basal Ganglia | 0.540 | 0.585 (57.559; 3.173) | 0.590 (53.058; 3.340) |
| Hippocampus | 0.455 | 0.461 (52.123; 3.282) | 0.476 (33.629; 3.873) |
| White Matter | 0.451 | 0.496 (69.540; 2.865) | 0.495 (65.179; 3.028) |
| WB | 0.434 | 0.470 (64.429; 2.996) | 0.471 (59.502; 3.176) |
Conclusion
In our continuing efforts to develop novel small molecule inhibitors of the NLRP3 inflammasome, we have successfully designed a 4-(1-(5-chloro-2-methoxybenzyl)-1H-1,2,3-triazol-4-yl)benzoate scaffold as NLRP3 inhibitors. Initial characterization established compound 1 as a selective hit NLRP3 inhibitor. To explore the chemical space of compound 1 and to identify structure features that are important for biological activities of this chemical scaffold, the following SAR studies of the hit compound 1 were focused on the modifications of the phenyl ring and the propoxyl substituent on the benzyl moiety. The SAR results revealed that the CH3O and Cl substituents on the benzyl moiety are essential for the observed activity as NLRP3 inhibitors, while the CH3O substituent can be located to different positions. Modifications on the phenyl ring of this scaffold demonstrated that a para-ester substituent provides the optimal activity as NLRP3 inhibitors. The SAR studies identified compound 8 as a potent and selective NLRP3 inhibitor. Biophysical studies using recombinant NLRP3 proteins also suggested compound 8 as a direct binder of NLRP3 proteins. Given the growing interest in identifying novel biomarkers for neuroinflammation and the promising binding affinity to NLRP3 by 8, we attempted to convert compound 8 into a carbon-11 labeled PET radiotracer. Autoradiography studies of [11C]8 employing brain tissues from WT and nlrp3−/− mice and blocking studies demonstrated specific labeling of NLRP3 by this radiotracer. Following PET/CT studies of [11C]8 in mice demonstrated rapid and high uptake of this tracer into brain, suggesting its potential as a CNS PET radiotracer. Self-blocking study results in mice also supported specific labeling of radiotracer in vivo. Notably, PET/MR studies of [11C]8 in rhesus macaque demonstrated excellent brain uptake with a peak SUV>3, consistent with the PET/CT studies in mice. Kinetic analysis of the plasma samples suggested promising radiometabolic stability of this radiotracer despite the presence of an ester functional group in the structure. Taken together, the results from this study strongly encourage further development and optimization of the 4-(1-(5-chloro-2-methoxybenzyl)-1H-1,2,3-triazol-4-yl)benzoate scaffold to provide novel NLRP3 inhibitors and CNS PET radiotracers as potential therapeutic and diagnostic agents.
Experimental Details
Chemistry.
All chemical reagents and solvents were obtained from commercial suppliers, including VWR International, Sigma-Aldrich, and used as received. Silica gel flash chromatography (200–300 mesh) was purchased from Fisher Scientific. The 1H and 13C nuclear magnetic resonance (NMR) spectra of compounds were collected using a Bruker ARX 400 spectrometer with CDCl3 or DMSO-d6 as a solvent. All chemical shifts (δ) are assigned as parts per million (ppm) using tetramethylsilane (TMS) as the internal standard. Signals are described as s (singlet), d (doublet), t (triplet), q (quartet), or m (multiplet). Coupling constants (J) are quoted in hertz. The purity of target compounds was determined to be ≥ 95% by HPLC using a Varian 100–5 C18 250 × 4.6 mm column with UV detection (288 nm) (60% acetonitrile/40% H2O with 0.1% TFA). High-resolution mass spectrometry (HRMS) was performed on an Applied BioSystems 3200 Q trap with a turbo V source for Turbolon Spray.
Method A.
1-(5-chloro-2-propoxybenzyl)-4-phenyl-1H-1,2,3-triazole (1).
Compounds 23a (225 mg, 0.86 mmol), NaN3 (56 mg, 0.86 mmol), phenylacetylene (95 μL, 0.86 mmol), CuSO4·5H2O (11 mg, 0.043 mmol) and sodium ascorbate (17 mg, 0.086 mmol) were placed in flask in H2O/EtOH (6 mL, 1:2, v/v). The mixture was stirred at 50 °C for 2–3 h. H2O (10 mL) was added to the reactants followed by extraction with DCM (3 × 5 mL). The combined organic layers were washed with brine (2 × 10 mL), dried over anhydrous Na2SO4 and concentrated under vacuum. The crude product was purified by crystallization (DCM/EtOH) to afford 1 (151 mg, 53%) as a white solid. 1H NMR (400 MHz, Chloroform-d) δ 7.76 (d, J = 7.5 Hz, 3H), 7.34 (t, J = 7.0 Hz, 2H), 7.30 – 7.19 (m, 2H), 7.12 (d, J = 2.6 Hz, 1H), 6.77 (d, J = 8.7 Hz, 1H), 5.49 (s, 2H), 3.90 (t, J = 6.4 Hz, 2H), 1.77 (m, 2H), 0.97 (t, J = 7.4 Hz, 3H). HRMS (AP-ESI) m/z: calcd for C18H18ClN3O [M + H]+, 328.1211; found, 328.1201.
4-(1-(5-chloro-2-propoxybenzyl)-1H-1,2,3-triazol-4-yl)benzenesulfonamide (2).
The procedure for the synthesis of 2 followed Method A using 23a and 19a as starting materials in 65% yield as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.63 (s, 1H), 8.04 (d, J = 8.2 Hz, 2H), 7.88 (d, J = 8.2 Hz, 2H), 7.47 – 7.28 (m, 4H), 7.08 (d, J = 8.8 Hz, 1H), 5.60 (s, 2H), 3.96 (t, J = 6.3 Hz, 2H), 1.69 (m, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 155.92, 145.38, 143.56, 134.31, 130.32, 130.14, 126.81, 125.81, 125.63, 124.33, 123.26, 114.25, 70.20, 48.87, 22.32, 10.79. HRMS (AP-ESI) m/z: calcd for C18H19ClN4O3S [M + Na]+, 429.0759; found, 429.0741.
1-(5-chloro-2-propoxybenzyl)-4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazole (3).
The procedure for the synthesis of 3 followed Method A using 23a and 1-ethynyl-4-(trifluoromethyl)benzene as starting materials in 65% yield as a white solid. 1H NMR (400 MHz, Chloroform-d) δ 8.03 – 7.45 (m, 5H), 7.29 – 7.10 (m, 2H), 6.77 (d, J = 8.8 Hz, 1H), 5.49 (s, 2H), 3.89 (t, J = 6.4 Hz, 2H), 1.76 (m, 2H), 0.96 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 155.92, 145.28, 135.16, 130.26, 130.13, 128.39 (q, JC-F = 31.9 Hz), 126.32 (q, JC-F = 3.8 Hz), 126.15, 125.67, 124.73 (q, JC-F = 272.7 Hz), 124.36, 123.41, 114.27, 70.23, 48.87, 22.33, 10.77. 19F NMR (376 MHz, DMSO) δ −60.98. HRMS (AP-ESI) m/z: calcd for C19H17ClF3N3O [M + Na]+, 418.0904; found, 418.0918.
1-(5-chloro-2-propoxybenzyl)-4-(4-fluorophenyl)-1H-1,2,3-triazole (4).
The procedure for the synthesis of 4 followed Method A using 23a and 1-ethynyl-4-fluorobenzene as starting materials in 67% yield as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.49 (s, 1H), 7.97 – 7.77 (m, 2H), 7.39 (m, 1H), 7.35 – 7.16 (m, 3H), 7.07 (d, J = 8.8 Hz, 1H), 5.57 (s, 2H), 3.96 (t, J = 6.3 Hz, 2H), 1.70 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 162.21 (d, JC-F = 245.2 Hz), 155.83, 145.79, 130.05 (d, JC-F = 5.5 Hz), 127.73 (d, JC-F = 3.2 Hz), 127.63 (d, JC-F = 8.1 Hz), 125.85, 124.33, 122.04, 116.35, 116.13, 114.22, 70.20, 48.69, 22.34, 10.80. 19F NMR (376 MHz, DMSO) δ −114.20. HRMS (AP-ESI) m/z: calcd for C18H17ClFN3O [M + Na]+, 368.0936; found, 368.0950.
1-(5-chloro-2-propoxybenzyl)-4-(p-tolyl)-1H-1,2,3-triazole (5).
The procedure for the synthesis of 5 followed Method A using 23a and 4-methylphenylacetylene as starting materials and purified by column using Hexane/EA=4:1 as elution to give white solid in 63% yield. 1H NMR (400 MHz, Chloroform-d) δ 7.71 (d, J = 7.8 Hz, 3H), 7.33 – 7.29 (m, 1H), 7.26 – 7.17 (m, 3H), 6.86 (d, J = 8.8 Hz, 1H), 5.57 (s, 2H), 3.99 (t, J = 6.5 Hz, 2H), 2.39 (s, 3H), 1.86 (m, 2H), 1.06 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 155.23, 147.94, 137.92, 129.92, 129.89, 129.46, 127.84, 125.64, 125.60, 124.77, 119.38, 112.75, 70.18, 48.82, 22.51, 21.26, 10.56. HRMS (AP-ESI) m/z: calcd for C19H20ClN3O [M + Na]+, 364.1187; found, 364.1188.
Methyl 4-(1-(5-chloro-2-propoxybenzyl)-1H-1,2,3-triazol-4-yl)benzoate (6).
The procedure for the synthesis of 6 followed Method A using 23a and methyl 4-ethynylbenzoate as starting materials in 67% yield as a yellowish solid. 1H NMR (400 MHz, DMSO-d6) δ 8.65 (s, 1H), 8.01 (s, 4H), 7.56 – 7.20 (m, 2H), 7.07 (d, J = 8.8 Hz, 1H), 5.59 (s, 2H), 3.91 (m, 5H), 1.87 – 1.48 (m, 2H), 0.90 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.38, 155.88, 145.58, 135.67, 130.32, 130.22, 130.10, 129.09, 125.67, 124.33, 123.40, 114.23, 70.20, 52.59, 48.83, 22.33, 10.78. HRMS (AP-ESI) m/z: calcd for C20H20ClN3O3 [M + Na]+, 408.1085; found, 408.1080.
Method B.
4-(1-(5-chloro-2-propoxybenzyl)-1H-1,2,3-triazol-4-yl)benzoic acid (7).
To a solution of compound 6 (60 mg, 0.156 mmol) in MeOH (2 mL), LiOH (37.2 mg, 1.56 mmol) was added. The reaction mixture was heated to 50 °C and stirred for 2–3 h (monitored by TLC). MeOH was evaporated and the residue was partitioned between H2O and DCM (10 mL, 1:1, v/v). The layers were separated, and the aqueous layer was acidified to pH 3–5 with 1 N HCl. The aqueous solution was extracted with DCM (2 × 5 mL). The combined organic layers were washed with brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was purified by crystallization (DCM) to afford 7 (42 mg, 72% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.63 (s, 1H), 8.07 – 7.90 (m, 4H), 7.40 (m, 1H), 7.34 (d, J = 2.7 Hz, 1H), 7.08 (d, J = 8.8 Hz, 1H), 5.60 (s, 2H), 3.96 (t, J = 6.3 Hz, 2H), 1.69 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.46, 155.87, 145.74, 135.23, 130.44, 130.38, 130.21, 130.09, 125.70, 125.52, 124.33, 123.25, 114.23, 70.20, 48.81, 22.33, 10.79. HRMS (AP-ESI) m/z: calcd for C19H18ClN3O3 [M-H]−, 370.0964; found, 370.0962.
Methyl 4-(1-(5-chloro-2-methoxybenzyl)-1H-1,2,3-triazol-4-yl)benzoate (8).
The procedure for the synthesis of 8 followed Method A using 23b and methyl 4-ethynylbenzoate as starting materials in 58% yield as a yellowish solid. 1H NMR (400 MHz, Chloroform-d) δ 8.00 (d, J = 7.7 Hz, 2H), 7.92 – 7.60 (m, 3H), 7.23 (m, 1H), 7.14 (d, J = 2.6 Hz, 1H), 6.80 (d, J = 8.8 Hz, 1H), 5.48 (s, 2H), 3.85 (s, 3H), 3.81 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.38, 156.40, 145.69, 135.64, 130.32, 130.13, 130.01, 129.10, 125.73, 125.67, 124.55, 123.49, 113.61, 56.53, 52.62, 48.56. HRMS (AP-ESI) m/z: calcd for C18H16ClN3O3 [M + Na]+, 380.0772; found, 380.0761.
1-(5-chloro-2-methoxybenzyl)-4-(4-methoxyphenyl)-1H-1,2,3-triazole (9).
The procedure for the synthesis of 9 followed Method A using 23b and 4-ethynylanisole as starting materials. Purification of 9 was achieved by crystallization using DCM/Hexane to get a white solid in 68% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (s, 1H), 7.84 – 7.70 (m, 2H), 7.42 (m, 1H), 7.23 (d, J = 2.7 Hz, 1H), 7.11 (d, J = 8.8 Hz, 1H), 7.04 – 6.95 (m, 2H), 5.56 (s, 2H), 3.84 (s, 3H), 3.79 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 159.48, 156.30, 146.73, 129.96, 129.70, 127.02, 126.05, 124.57, 123.73, 121.26, 114.74, 113.61, 56.53, 55.62, 48.31. HRMS (AP-ESI) m/z: calcd for C17H16ClN3O2 [M + Na]+, 352.0823; found, 352.0832.
Methyl 6-(1-(5-chloro-2-methoxybenzyl)-1H-1,2,3-triazol-4-yl)nicotinate (10).
The procedure for the synthesis of 10 followed Method A using 23b and methyl 6-ethynylnicotinate as starting materials. Purification was achieved by column using Hexane/EA=2:1 as elution to give a white solid in 54% yield. 1H NMR (400 MHz, DMSO-d6) δ 9.10 (s, 1H), 8.71 (s, 1H), 8.40 (d, J = 8.2 Hz, 1H), 8.18 (d, J = 8.4 Hz, 1H), 7.43 (d, J = 8.8 Hz, 1H), 7.31 (s, 1H), 7.12 (d, J = 8.9 Hz, 1H), 5.64 (s, 2H), 3.92 (s, 3H), 3.84 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.45, 156.34, 153.91, 150.81, 146.71, 138.53, 130.11, 129.88, 125.77, 125.35, 124.80, 124.60, 119.72, 113.67, 56.59, 52.87, 48.57. HRMS (AP-ESI) m/z: calcd for C17H15ClN4O3 [M + Na]+, 381.0725; found, 381.0720.
Methyl 3-(1-(5-chloro-2-methoxybenzyl)-1H-1,2,3-triazol-4-yl)benzoate (11).
The procedure for the synthesis of 11 followed Method A using 23b and methyl 3-ethynylbenzoate as starting materials. Purification was achieved by column using Hexane/EA=2:1 as elution to give a yellow oil in 52% yield. 1H NMR (400 MHz, Chloroform-d) δ 8.30 (s, 1H), 8.10 (s, 1H), 7.92 (d, J = 7.6 Hz, 2H), 7.54 – 7.34 (m, 1H), 7.23 (m, 1H), 7.14 (d, J = 2.6 Hz, 1H), 6.80 (d, J = 8.8 Hz, 1H), 5.49 (s, 2H), 3.86 (s, 3H), 3.81 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.50, 156.37, 145.77, 131.68, 130.76, 130.33, 130.08, 129.93, 128.90, 126.00, 125.75, 124.54, 122.89, 113.60, 56.53, 52.76, 48.50. HRMS (AP-ESI) m/z: calcd for C18H16ClN3O3 [M + Na]+, 380.0772; found, 380.0765.
4-(1-(5-chloro-2-methoxybenzyl)-1H-1,2,3-triazol-4-yl)benzoic acid (12).
The procedure for the synthesis of 12 followed Method B using 8 as a starting material. The crude product was purified by crystallization (DCM) to afford 12 in 75% yield as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.67 (s, 1H), 7.99 (s, 4H), 7.43 (m, 1H), 7.29 (d, J = 2.7 Hz, 1H), 7.11 (d, J = 8.8 Hz, 1H), 5.59 (s, 2H), 3.83 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.46, 156.40, 145.86, 135.19, 130.43, 130.11, 129.97, 125.72, 125.58, 124.56, 123.32, 113.63, 56.54, 48.54. HRMS (AP-ESI) m/z: calcd for C17H14ClN3O3 [M-H]−, 342.0651; found, 342.0636.
Method C.
4-(1-(5-chloro-2-methoxybenzyl)-1H-1,2,3-triazol-4-yl)-N-methylbenzamide (13).
To a solution of compound 12 (30 mg, 0.087 mmol), methyl amine hydrochloride (7.1 mg, 0.105 mmol), HATU (40 mg, 0.105 mmol) in anhydrous DMF (3 mL), Et3N (34 uL, 0.192 mmol) was added. The mixture was stirred at r.t. for 2–4 h. After the reaction was completed, DCM (5 mL) was added to dilute the reactant. The organic layer was washed with 1 N HCl, sat. NaHCO3 aqueous, brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The crude product was purified by crystallization (EtOH) to afford 13 (15 mg, 48% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.68 (s, 1H), 8.51 (d, J = 4.7 Hz, 1H), 8.00 (d, J = 8.5 Hz, 2H), 7.95 (d, J = 8.5 Hz, 2H), 7.48 (m, 1H), 7.34 (d, J = 2.7 Hz, 1H), 7.16 (d, J = 8.8 Hz, 1H), 5.64 (s, 2H), 3.89 (s, 3H), 2.85 (d, J = 4.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.61, 156.38, 146.04, 134.07, 133.54, 130.09, 129.93, 128.15, 125.76, 125.37, 124.56, 122.94, 113.63, 56.54, 48.50, 26.71. HRMS (AP-ESI) m/z: calcd for C18H17ClN4O2 [M + Na]+, 379.0932; found, 379.0936.
Azetidin-1-yl(4-(1-(5-chloro-2-methoxybenzyl)-1H-1,2,3-triazol-4-yl)phenyl)methanone (14).
The procedure for the synthesis of 14 followed Method C using 12 as a starting material. The crude product was purified by crystallization (DCM/Hexane) to afford 14 in 30% yield as a white solid. 1H NMR (400 MHz, Chloroform-d) δ 7.83 – 7.73 (m, 2H), 7.73 – 7.64 (m, 1H), 7.61 (m, 2H), 7.23 (d, J = 9.0 Hz, 1H), 7.13 (s, 1H), 6.88 – 6.70 (m, 1H), 5.48 (d, J = 2.5 Hz, 2H), 4.20 (m, 4H), 3.88 – 3.72 (m, 3H), 2.28 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.83, 156.35, 145.98, 133.35, 132.77, 130.07, 129.88, 128.84, 125.79, 125.45, 124.54, 123.03, 113.60, 56.52, 53.46, 49.01, 48.51, 16.00. HRMS (AP-ESI) m/z: calcd for C20H19ClN4O2 [M + Na]+, 405.1089; found, 405.1068.
Methyl 4-(1-(5-chloro-2-methoxybenzyl)-1H-1,2,3-triazol-4-yl)-3-methylbenzoate (15).
The procedure for the synthesis of 15 followed Method A using 23b and 19b as starting materials. Purification was achieved by column using Hexane/Acetone=5:1 as elution to give a white solid in 38% yield. 1H NMR (400 MHz, Chloroform-d) δ 7.96 – 7.91 (m, 1H), 7.91 – 7.85 (m, 2H), 7.72 (s, 1H), 7.31 (m, 1H), 7.22 (d, J = 2.6 Hz, 1H), 6.87 (d, J = 8.8 Hz, 1H), 5.58 (s, 2H), 3.92 (s, 3H), 3.88 (s, 3H), 2.51 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.95, 155.71, 146.32, 135.46, 134.38, 132.09, 130.07, 130.02, 129.41, 128.74, 127.21, 126.02, 124.63, 122.55, 112.08, 55.89, 52.06, 48.76, 21.47. HRMS (AP-ESI) m/z: calcd for C19H18ClN3O3 [M + Na]+, 394.0929; found, 394.0943.
Methyl 4-(1-(5-chloro-2-methoxybenzyl)-1H-1,2,3-triazol-4-yl)-3-fluorobenzoate (16).
The procedure for the synthesis of 16 followed Method A using 23b and 19c as starting materials. Purification was achieved by column using Hexane/Acetone=5:1 as elution to give a white solid in 35% yield. 1H NMR (400 MHz, Chloroform-d) δ 8.40 (m, 1H), 8.00 (d, J = 3.9 Hz, 1H), 7.92 (m, 1H), 7.79 (m, 1H), 7.30 (m, 1H), 7.21 (d, J = 2.6 Hz, 1H), 6.86 (d, J = 8.8 Hz, 1H), 5.57 (s, 2H), 3.94 (s, 3H), 3.88 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 165.79, 158.66 (d, JC-F = 248.3 Hz), 155.75, 140.47, 130.96, 130.07, 127.68, 125.97, 125.77, 124.47, 123.92, 123.14, 117.00, 116.76, 112.10, 55.89, 52.39, 48.85. 19F NMR (376 MHz, DMSO-d6) δ −113.52. HRMS (AP-ESI) m/z: calcd for C18H15ClFN3O3 [M + Na]+, 398.0678; found, 398.0672.
Method D.
4-((trimethylsilyl)ethynyl)benzenesulfonamide (19a).
A mixture of 17a (1 g, 4.20 mmol), trimethylsilylacetylene (0.70 mL, 5.08 mmol), CuI (80 mg, 0.42 mmol), Pd(pph3)2Cl2 (297 mg, 0.42 mmol) and TEA (5.9 mL, 42.3 mmol) was dissolved in anhydrous 1,4-dioxane (10 mL) under N2. The mixture was stirred at 100 °C overnight. The solvent was removed and DCM (30 mL) was added. Then the organic layer was washed with H2O, brine, dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The crude 18a was dissolved in 1M tetrabutylammonium fluoride (TBAF) in THF (8 mL) and stirred at r.t. for 1 h, the reaction was quenched with saturated NH4Cl (30 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel (DCM/MeOH = 30:1) afford 19a (329 mg, 43%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.96 – 7.83 (m, 2H), 7.78 – 7.67 (m, 2H), 7.51 (s, 2H), 4.49 (s, 1H).
Methyl 4-ethynyl-3-methylbenzoate (19b).
The procedure for the synthesis of 18b followed Method D using 17b and trimethylsilylacetylene as starting materials. Then the crude 18b was treated with TBAF and resulting product 19b was purified by silica gel column chromatography (Hexane/EtOAc = 60:1) in 41% yield as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.89 – 7.78 (m, 1H), 7.73 (m, 1H), 7.44 (d, J = 8.0 Hz, 1H), 3.84 (d, J = 2.5 Hz, 3H), 3.35 (s, 1H), 2.42 (s, 3H).
Methyl 4-ethynyl-3-fluorobenzoate (19c).
The procedure for the synthesis of 18c followed Method D using 17c and trimethylsilylacetylene as starting materials. Then the crude 18c was treated with TBAF and resulting product 19c was purified by silica gel column chromatography (Hexane/EtOAc = 60:1) in 38% yield as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.76 – 7.61 (m, 2H), 7.47 (m, 1H), 3.86 (d, J = 1.9 Hz, 3H), 3.38 (s, 1H).
Method E.
5-chloro-2-propoxybenzaldehyde (21a).
To a solution of compound 20 (5 g, 32.05 mmol) and K2CO3 (6.64 g, 48.07 mmol) in DMF (50 mL), 1-Bromopropane (4.37 mL, 48.07 mmol) was added at 0 °C. Then the reactant was heated at 40 °C for 4 h. DMF was evaporated under reduced pressure, and the residue was dissolved in DCM (50 mL). The organic layer was washed with H2O, brine, dried over anhydrous Na2SO4, and concentrated to afford 21a (5.1 g, 80%) as a yellow solid without any further purification. 1H NMR (400 MHz, CDCl3) δ 10.46 (s, 1H), 7.79 (d, J = 2.8 Hz, 1H), 7.61 – 7.43 (m, 1H), 6.95 (d, J = 8.9 Hz, 1H), 4.05 (t, J = 6.4 Hz, 2H), 1.90 (m, 2H), 1.09 (t, J = 7.4 Hz, 3H).
(5-chloro-2-propoxyphenyl)methanol (22a).
To a solution of 21a (1 g, 5.05 mmol) in MeOH (20 mL), NaBH4 (382 mg, 10.1 mmol) was added portionwise. After stirring at r.t. for 40 min, the solvent was evaporated and the residue was taken up to DCM (20 mL). The organic layer was washed with H2O, brine, dried over anhydrous Na2SO4, concentrated to afford 22a (0.75 g, 74 %) as a colorless oil without further purification. 1H NMR (400 MHz, CDCl3) δ 7.20 (d, J = 2.7 Hz, 1H), 7.12 (m, 1H), 6.70 (d, J = 8.7 Hz, 1H), 4.58 (s, 2H), 3.88 (t, J = 6.5 Hz, 2H), 1.75 (m, 2H), 0.97 (t, J = 7.5 Hz, 3H).
2-(bromomethyl)-4-chloro-1-propoxybenzene (23a).
To a solution of 22a (2 g, 10 mmol) in anhydrous DCM (30 mL), PBr3 (1 mL, 11 mmol) was added dropwise at 0 °C. The mixture was stirred at the same temperature for 30 min. Then, the reactant was washed with brine, dried over Na2SO4 and concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel (Hexane/EtOAc= 30:1) to afford 23a (1.9 g, 72%) as a colorless oil. 1H NMR (400 MHz, Chloroform-d) δ 7.23 (d, J = 2.6 Hz, 1H), 7.14 (m, 1H), 6.71 (d, J = 8.8 Hz, 1H), 4.42 (s, 2H), 3.90 (t, J = 6.4 Hz, 2H), 1.79 (m, 2H), 1.01 (t, J = 7.4 Hz, 3H).
2-(bromomethyl)-4-chloro-1-methoxybenzene (23b).
The procedure for the synthesis of 23b followed Method E using 20 as a starting material. 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J = 2.6 Hz, 1H), 7.29 – 7.24 (m, 1H), 6.83 (d, J = 8.8 Hz, 1H), 4.51 (s, 2H), 3.90 (s, 3H).
Methyl 4-(1-benzyl-1H-1,2,3-triazol-4-yl)benzoate (24).
The procedure for the synthesis of 24 followed Method A using methyl 4-ethynylbenzoate and benzyl bromide as starting materials. The crude product was purified by crystallization (DCM/EtOH) to afford 24 in 36% yield as a yellowish solid. 1H NMR (400 MHz, DMSO-d6) δ 8.80 (s, 1H), 8.10 – 7.97 (m, 4H), 7.46 – 7.28 (m, 5H), 5.68 (s, 2H), 3.87 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.37, 146.07, 136.28, 135.63, 130.34, 129.29, 129.16, 128.68, 128.41, 125.70, 123.28, 53.61, 52.59. HRMS (AP-ESI) m/z: calcd for C17H15N3O2 [M + Na]+, 316.1056; found, 316.1044.
Methyl 4-(1-(2-methoxybenzyl)-1H-1,2,3-triazol-4-yl)benzoate (25).
The procedure for the synthesis of 25 followed Method A using methyl 4-ethynylbenzoate and 1-(bromomethyl)-2-methoxybenzene as starting materials. The crude product was purified by column using Hexane/Acetone=4:1 as elution to give a white solid in 61% yield. 1H NMR (400 MHz, Chloroform-d) δ 8.08 (d, J = 7.8 Hz, 2H), 7.89 (d, J = 8.0 Hz, 2H), 7.81 (s, 1H), 7.38 (t, J = 7.9 Hz, 1H), 7.28 (s, 1H), 7.09 – 6.88 (m, 2H), 5.62 (s, 2H), 3.94 (s, 3H), 3.92 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.80, 157.22, 146.68, 135.16, 130.52, 130.43, 130.13, 129.41, 125.42, 122.76, 121.05, 120.60, 110.88, 55.57, 52.09, 49.31. HRMS (AP-ESI) m/z: calcd for C18H17N3O3 [M + Na]+, 346.1162; found, 346.1145.
Methyl 4-(1-(3-chlorobenzyl)-1H-1,2,3-triazol-4-yl)benzoate (26).
The procedure for the synthesis of 26 followed Method A using methyl 4-ethynylbenzoate and 3-chlorobenzyl bromide as starting materials. The crude product was purified by crystallization (DCM/EtOH) to afford 26 in 56% yield as a white solid. 1H NMR (400 MHz, Chloroform-d) δ 8.18 – 8.02 (m, 2H), 7.96 – 7.82 (m, 2H), 7.78 (s, 1H), 7.42 – 7.28 (m, 3H), 7.20 (m, 1H), 5.57 (s, 2H), 3.93 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.73, 147.44, 136.38, 135.18, 134.67, 130.54, 130.22, 129.76, 129.17, 128.18, 126.14, 125.53, 120.35, 53.66, 52.15. HRMS (AP-ESI) m/z: calcd for C17H14ClN3O2 [M + Na]+, 350.0667; found, 350.0678.
Methyl 4-(1-(5-chloro-2-hydroxybenzyl)-1H-1,2,3-triazol-4-yl)benzoate (27).
To a stirred solution of 8 (52 mg, 0.145 mmol) in anhydrous DCM (2 mL), cooled at −78 °C, a solution of BBr3 (69 uL, 0.727 mmol) in DCM (1 mL) was added dropwise. Stirring was continued for 1 h, and then the solution was treated with MeOH (5 mL) with cooling and stirred at r.t. for 1–2 h. Saturated NaHCO3 (5 mL) was added and the reactant was extracted with DCM (2 × 5 mL). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The crude product was purified by crystallization (DCM) to afford 27 (42 mg, 84%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.25 (s, 1H), 8.68 (s, 1H), 8.02 (s, 4H), 7.24 (d, J = 7.7 Hz, 2H), 6.89 (d, J = 8.4 Hz, 1H), 5.56 (s, 2H), 3.87 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.39, 154.83, 145.64, 135.67, 130.31, 130.06, 129.89, 129.08, 125.70, 124.06, 123.40, 122.87, 117.50, 52.59, 48.73. HRMS (AP-ESI) m/z: calcd for C17H14ClN3O3 [M + Na]+, 366.0616; found, 366.0599.
Methyl 4-(1-(3-chloro-5-methoxybenzyl)-1H-1,2,3-triazol-4-yl)benzoate (28).
The procedure for the synthesis of 28 followed Method A using methyl 4-ethynylbenzoate and 1-(bromomethyl)-3-chloro-5-methoxybenzene as starting materials. The crude product was purified by crystallization (EA/Hexane) to afford 28 in 31% yield as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.81 (s, 1H), 8.11 – 7.95 (m, 4H), 7.02 (m, 2H), 6.94 (m, 1H), 5.65 (s, 2H), 3.87 (s, 3H), 3.78 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.37, 160.86, 146.10, 139.31, 135.54, 134.57, 130.35, 129.21, 125.75, 123.43, 120.46, 114.07, 113.71, 56.19, 52.85, 52.60. HRMS (AP-ESI) m/z: calcd for C18H16ClN3O3 [M + Na]+, 380.0772; found, 380.0783.
Methyl 4-(1-(5-chloro-2-(trifluoromethyl)benzyl)-1H-1,2,3-triazol-4-yl)benzoate (29).
The procedure for the synthesis of 29 followed Method A using methyl 4-ethynylbenzoate and 5-chloro-2-(trifluoromethyl)benzyl bromide as starting materials. The crude product was purified by crystallization (DCM/EtOH) to afford 29 in 65% yield as a white solid. 1H NMR (400 MHz, Chloroform-d) δ 8.13 – 8.07 (m, 2H), 7.94 – 7.87 (m, 2H), 7.82 (s, 1H), 7.69 (d, J = 8.4 Hz, 1H), 7.48 – 7.43 (m, 1H), 7.28 (t, J = 1.3 Hz, 1H), 5.77 (s, 2H), 3.93 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.71, 147.63, 139.39, 134.98, 134.46, 130.37, 130.27, 129.91, 129.21, 127.80 (q, JC-F = 5.8 Hz), 126.43, 125.60, 123.81 (q, JC-F = 274.6 Hz), 120.74, 52.17, 49.82. 19F NMR (376 MHz, DMSO-d6) δ −58.20. HRMS (AP-ESI) m/z: calcd for C18H13ClF3N3O2 [M + Na]+, 418.0541; found, 418.0553.
Methyl 4-(1-(5-chloro-2-(difluoromethoxy)benzyl)-1H-1,2,3-triazol-4-yl)benzoate (30).
The procedure for the synthesis of 30 followed Method A using methyl 4-ethynylbenzoate and 2-(bromomethyl)-4-chloro-1-(difluoromethoxy)benzene as starting materials. The crude product was purified by crystallization (DCM/EtOH) to afford 30 in 40% yield as a white solid. 1H NMR (400 MHz, Chloroform-d) δ 8.21 – 8.03 (m, 2H), 7.92 (d, J = 8.2 Hz, 2H), 7.88 (s, 1H), 7.39 (m, 1H), 7.33 (d, J = 2.6 Hz, 1H), 7.19 (d, J = 8.7 Hz, 1H), 6.61 (t, J = 72.7 Hz, 1H), 5.64 (s, 2H), 3.95 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.39, 148.32, 145.85, 135.55, 130.95, 130.56, 130.35, 129.86, 129.22, 128.84, 125.76, 123.56, 120.63, 116.76 (t, JC-F = 260.8 Hz), 52.60, 48.26. 19F NMR (376 MHz, DMSO-d6) δ −82.18 (d, J = 73.1 Hz). HRMS (AP-ESI) m/z: calcd for C18H14ClF2N3O3 [M + Na]+, 416.0584; found, 416.0569.
Methyl 4-(1-(2-(difluoromethoxy)benzyl)-1H-1,2,3-triazol-4-yl)benzoate (31).
The procedure for the synthesis of 31 followed Method A using methyl 4-ethynylbenzoate and 1-(bromomethyl)-2-(difluoromethoxy)benzene as starting materials. The crude product was purified by crystallization (DCM/EtOH) to afford 31 in 48% yield as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.71 (s, 1H), 8.08 – 7.98 (m, 4H), 7.50 – 7.43 (m, 1H), 7.38 – 7.32 (m, 1H), 7.32 – 7.24 (m, 3H), 5.70 (s, 2H), 3.87 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.37, 149.36 (t, JC-F = 2.8 Hz), 145.82, 135.61, 130.97, 130.73, 130.34, 129.17, 126.77, 126.05, 125.72, 123.48, 118.71, 116.86 (t, JC-F = 259.7 Hz), 52.60, 48.63. 19F NMR (376 MHz, DMSO-d6) δ −81.62 (d, J = 73.6 Hz). HRMS (AP-ESI) m/z: calcd for C18H15F2N3O3 [M + Na]+, 382.0974; found, 382.0982.
Methyl 4-(1-(5-fluoro-2-methoxybenzyl)-1H-1,2,3-triazol-4-yl)benzoate (32).
The procedure for the synthesis of 32 followed Method A using methyl 4-ethynylbenzoate and 2-(bromomethyl)-4-fluoro-1-methoxybenzene as starting materials. The crude product was purified by crystallization (DCM/EtOH) to afford 32 in 67% yield as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.69 (s, 1H), 8.03 (m, 4H), 7.21 (m, 1H), 7.10 (m, 2H), 5.61 (s, 2H), 3.87 (s, 3H), 3.82 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.38, 156.47 (d, JC-F = 236.6 Hz), 153.83 (d, JC-F = 2.0 Hz), 145.72, 135.66, 130.30, 129.11, 125.73, 125.26 (d, JC-F = 7.6 Hz), 123.45, 117.06 (d, JC-F = 24.2 Hz), 116.41 (d, JC-F = 22.5 Hz), 113.12 (d, JC-F = 8.2 Hz), 56.66, 52.59, 48.65. 19F NMR (376 MHz, DMSO-d6) δ −123.59. HRMS (AP-ESI) m/z: calcd for C18H16FN3O3 [M + Na]+, 364.1068; found, 364.1080.
Methyl 4-(1-(5-bromo-2-methoxybenzyl)-1H-1,2,3-triazol-4-yl)benzoate (33).
The procedure for the synthesis of 33 followed Method A using methyl 4-ethynylbenzoate and 4-bromo-2-(bromomethyl)-1-methoxybenzene as starting materials. The crude product was purified by column using Hexane/EA=3:1 as elution to give a white solid in 37% yield. 1H NMR (400 MHz, Chloroform-d) δ 8.07 (d, J = 7.8 Hz, 2H), 7.89 (d, J = 8.1 Hz, 2H), 7.80 (s, 1H), 7.45 (d, J = 8.8 Hz, 1H), 7.36 (s, 1H), 6.82 (d, J = 8.8 Hz, 1H), 5.55 (s, 2H), 3.92 (s, 3H), 3.88 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.75, 156.29, 146.88, 134.97, 133.15, 132.93, 130.15, 129.56, 125.47, 124.90, 120.55, 113.13, 112.66, 55.90, 52.07, 48.69. HRMS (AP-ESI) m/z: calcd for C18H16BrN3O3 [M + Na]+, 424.0267; found, 424.0276.
Methyl 4-(1-(2-methoxy-5-(trifluoromethyl)benzyl)-1H-1,2,3-triazol-4-yl)benzoate (34).
The procedure for the synthesis of 34 followed Method A using methyl 4-ethynylbenzoate and 2-(bromomethyl)-1-methoxy-4-(trifluoromethyl)benzene as starting materials. The crude product was purified by column using Hexane/EA=3:1 as elution to give a white solid in 71% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.70 (s, 1H), 8.02 (s, 4H), 7.77 (m, 1H), 7.66 (d, J = 2.3 Hz, 1H), 7.28 (d, J = 8.7 Hz, 1H), 5.68 (s, 2H), 3.91 (s, 3H), 3.87 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.40, 160.51, 145.69, 135.65, 130.32, 129.16, 128.16 (q, JC-F = 3.8 Hz), 127.56 (q, JC-F = 3.6 Hz), 126.16, 125.76, 124.81 (q, JC-F = 272.0 Hz), 124.64, 123.50, 121.50 (q, JC-F = 32.4 Hz), 112.41, 56.71, 52.59, 48.73. 19F NMR (376 MHz, DMSO-d6) δ −59.83. HRMS (AP-ESI) m/z: calcd for C19H16F3N3O3 [M + Na]+, 414.1036; found, 414.1041.
Methyl 4-(1-(2,3-difluoro-6-methoxybenzyl)-1H-1,2,3-triazol-4-yl)benzoate (35).
The procedure for the synthesis of 35 followed Method A using methyl 4-ethynylbenzoate and 2,3-difluoro-6-methoxybenzyl bromide as starting materials. The crude product was purified by crystallization (DCM/EtOH) to afford 35 in 65% yield as a white solid. 1H NMR (400 MHz, Chloroform-d) δ 8.10 – 8.02 (m, 2H), 7.91 – 7.85 (m, 2H), 7.82 (s, 1H), 7.18 (m, 1H), 6.66 (m, 1H), 5.67 (d, J = 1.8 Hz, 2H), 3.92 (s, 3H), 3.88 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.79, 154.08 (dd, JC-F = 5.2, 2.3 Hz), 149.49 (dd, JC-F = 250.3, 14.3 Hz), 146.75, 145.06 (dd, JC-F = 242.6, 13.1 Hz), 134.99, 130.15, 129.53, 125.48, 120.47, 117.79 (dd, JC-F = 18.1, 2.1 Hz), 112.37 (d, JC-F = 13.9 Hz), 106.16 (dd, JC-F = 6.0, 3.7 Hz), 56.50, 52.12, 42.12. 19F NMR (376 MHz, DMSO-d6) δ −139.75 (d, J = 23.1 Hz), −147.97 (d, J = 22.9 Hz). HRMS (AP-ESI) m/z: calcd for C18H15F2N3O3 [M + Na]+, 382.0974; found, 382.0987.
Methyl 4-(1-(4,5-difluoro-2-methoxybenzyl)-1H-1,2,3-triazol-4-yl)benzoate (36).
The procedure for the synthesis of 36 followed Method A using methyl 4-ethynylbenzoate and 4,5-difluoro-2-methoxybenzyl bromide as starting materials. The crude product was purified by crystallization (DCM/EtOH) to afford 36 in 66% yield as a white solid. 1H NMR (400 MHz, Chloroform-d) δ 8.08 (d, J = 8.1 Hz, 2H), 7.88 (d, J = 8.0 Hz, 2H), 7.80 (s, 1H), 7.10 (t, J = 9.4 Hz, 1H), 6.77 (m, 1H), 5.53 (s, 2H), 3.93 (s, 3H), 3.87 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.74, 153.41 (dd, JC-F = 7.7, 2.4 Hz), 150.85 (dd, JC-F = 250.2, 13.6 Hz), 146.96, 144.28 (dd, JC-F = 242.6, 12.7 Hz), 134.86, 130.17, 129.61, 125.46, 120.51, 118.80 (d, JC-F = 2.1 Hz), 118.61 (d, JC-F = 2.0 Hz), 101.21 (d, JC-F = 21.5 Hz), 56.40, 52.12, 48.26. 19F NMR (376 MHz, DMSO-d6) δ −135.30 (d, J = 23.1 Hz), −149.03 (d, J = 23.1 Hz). HRMS (AP-ESI) m/z: calcd for C18H15F2N3O3 [M + Na]+, 382.0974; found, 382.0974.
Methyl 4-(1-(3,5-difluoro-2-methoxybenzyl)-1H-1,2,3-triazol-4-yl)benzoate (37).
The procedure for the synthesis of 37 followed Method A using methyl 4-ethynylbenzoate and 1-(bromomethyl)-3,5-difluoro-2-methoxybenzene as starting materials. The crude product was purified by crystallization (DCM/EtOH) to afford 37 in 61% yield as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.74 (s, 1H), 8.07 – 7.96 (m, 4H), 7.39 (m, 2H), 5.67 (s, 2H), 3.87 (s, 3H), 3.86 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.36, 154.10 (dd, JC-F = 222.2, 10.8 Hz), 151.66 (dd, JC-F = 228.2, 11.1 Hz), 145.94, 144.40 (dd, JC-F = 10.6, 3.0 Hz), 135.54, 130.31, 129.18, 125.74, 123.12, 118.40 (dd, J = 16.1, 4.2 Hz), 116.27, 105.61 (dd, J = 27.2, 22.6 Hz), 57.06, 52.60, 47.50. 19F NMR (376 MHz, DMSO-d6) δ −124.01 (d, J = 3.5 Hz), −130.07 (d, J = 3.6 Hz). HRMS (AP-ESI) m/z: calcd for C18H15F2N3O3 [M + Na]+, 382.0974; found, 382.0958.
Radiosynthesis of [11C]8.
[11C]CO2 was obtained via the 14N (p, α) 11C reaction on nitrogen with 2.5% oxygen, with 11 MeV protons (Siemens Eclipse cyclotron), and trapped on molecular sieves in a TRACERlab FX-MeI synthesizer (GE Healthcare). [11C]CH4 was obtained by the reduction of [11C]CO2 in the presence of Ni/hydrogen at 350 °C. The resulting [11C]CH4 passed through an oven containing I2 to produce [11C]CH3I via a radical reaction.
The prepared [11C]CH3I was trapped in anhydrous DMF (300 μL) containing precursor 27 (1.0 mg) and K2CO3 (5.0 mg). The reaction vessel was heated at 80 °C and kept there for 3 min. The radioactive mixture containing [11C]8 was quenched by addition of an HPLC mobile phase (0.7 mL) and then applied to a reverse phase semipreparative HPLC (Phenomenex Gemini-NX 5u C18 110A, 250 × 10 mm, 5.0 mL/min, a gradient of 10–90 % CH3CN in H2O). A radioactive fraction having a retention time of 10.2 min was collected in a flask, and diluted in water (30 mL). The final product was reformulated by loading onto a solid-phase exchange (SPE) C-18 cartridge (Waters WAT020515 Sep-Pak Plus Short C18), rinsing with water (4 × 5 mL), eluting with EtOH (0.3 mL), and diluting with saline (2.7 mL). The chemical and radiochemical purity of the final product was tested by analytical HPLC (VARIAN Puruit XRs 5 C18, 150 × 4.6 mm), eluting with a gradient of 10–90 % CH3CN in H2O of 0.1% TFA, at a flow rate of 2 mL/min. Confirmation of the identity of [11C]8 was achieved by co-injection with 8 as reference standard. For the determination of molar radioactivity, mass (μmol) of [11C]8 with a known radioactivity was determined by HPLC comparison of UV absorbance at 254 nm with those of known concentrations of non-radioactive 8.
Animals.
All the animal studies were approved and carried out following the guidelines of the Subcommittee on Research Animals of the Massachusetts General Hospital and Harvard Medical School. These guidelines are in accordance with the Guide of NIH for the Care and Use of Laboratory Animals.
Human brain postmortem tissues.
All human tissues are requested from NIH NeuroBioBank.
Inhibition of the NLRP3 Inflammasome Assays.
J774A.1 cells were plated into 96-well plates (1 × 105 cells/well) in growth medium overnight. Cells were primed with Escherichia coli 0111:B4 LPS (Sigma-Aldrich) (final concentration: 1 μg/mL) for 4.5 h. Test compounds were then added and incubated for 30 min. After incubation, ATP (5 mM) was added to activate NLRP3 inflammasome for 30 min. Supernatants were then collected and IL-1β release was measured with a mouse IL-1β ELISA kit (DuoSet ELISA, R&D Systems) following the manufacturer’s instructions.
Inhibition of NLRC4 and AIM2 Inflammasome Assays.
J774A.1 cells were plated into 96-well plates (1 × 105 cells/well) in growth medium overnight. Next day, cells were treated with LPS (1 μg/mL) and various concentrations of compounds for 1 h. Flagellin (1 μg/mL) (Enzo Life Sciences, Farmingdale, NY) or poly-deoxyadenylic-deoxy-thymidylic acid sodium salt (poly(dA:dT)) (4 μg/mL) (InvivoGen, San Diego, CA) in DMEM was added to activate NLRC4 or AIM2 inflammasome, respectively. Flagellin cell-transfection was accomplished utilizing the Polplus transfection kit (PULSin, New York, NY) and allowed to incubate for 6 h. For AIM2 activation, cells were incubated with poly(dA:dT) delivered by Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific) for 4 h. The supernatants were collected and IL-1β release was measured with a mouse IL-1β ELISA kit following the manufacturer’s instructions.
NLRP3 protein expression.
Expi293F cells were transfected with either 10His-Flag-hNLRP3_pcDNA3.1 (+) or pcDNA3-FLAG-mNLRP3 (Addgene #75127) using the ExpiFectamine™ 293 Transfection Kit (Thermo Fisher Scientific). Cells were harvested 48 h after initial transfection, washed with cold PBS and pelleted. Cell pellets were resuspended in lysis buffer (50 mM HEPES, 150 mM NaCl, 1 mM TCEP, 10 mM MgCl2, 1x protease inhibitors, 10% glycerol, and 1% DDM, pH 7.5). Cell lysates were centrifuged at 50,000×g at 4 °C for 1 h. Supernatant was incubated with Mouse IgG agarose beads (Sigma Aldrich) at 4 °C, followed by incubation with M2 Anti-FLAG beads (Sigma Aldrich) at 4 °C for 2–3 h. The beads were then washed with lysis buffer and eluted with elution buffer (50 mM HEPES, 500 mM NaCl, 10% glycerol, 0.1% DDM, and 100 µg/mL FLAG peptide (Thermo Fisher Scientific), pH 7.5). The eluted fractions were pooled and concentrated by centrifugation via ultrafiltration device to remove proteins <100 kDa.
MST assay.
Monolith NT. Automated instrument was used to measure the Kd value. Recombinant human NLRP3 protein (500 nM) was incubated with 50 nM RED-tris-NTA dye (SKU: NT-L118) for 30 min at room temperature. Then, the samples were centrifuged for 10 min at 4 °C and 15,000 g. The supernatant was transferred to a 384-wells plate, following the addition of a range of concentrations of compounds and incubated for 30 min in PBS-T assay buffer (1 × PBS with 0.05% Tween 20). The samples were loaded into Monolith NT.Automated Premium capillary chips (SKU: MO-AK005) and ligand-induced change in thermophoresis curves were analyzed using an excitation power of 80% and an MST power of high. Kd values were calculated using the mass action equation with NanoTemper software from duplicate reads of an experiment.
FS assay.
The Cary Eclipse Fluorescence Spectrophotometer (Agilent Technologies) was used to measure the intrinsic fluorescent intensity (ex: 280 nm; em: 340 nm) of the recombinant mouse NLRP3 protein (500 nM). The protein was added into a fluorescence micro cell cuvette (SKU: 6610021600) and measured for initial fluorescence, followed by compound titration, measuring the fluorescent intensity at each titration point. The percent change in fluorescence was calculated for each point and the titration was carried out until a plateau in the curve was achieved. Data was fitted using a hyperbolic nonlinear curve regression in the Prism software to determine the Kd value.
TSA assay.
The supernatant for NLRP3 protein purification was collected and incubated with the compound 1 (50 μM) or DMSO control at ambient temperature for 1 h. The lysate was equally divided into eight PCR tubes and heated at the temperatures of 40, 41.4, 43.9, 48, 52.8, 56.6, 59.3, and 61 °C for 3 min separately. The NLRP3 levels of samples were determined by Western blotting analysis.
Autoradiography studies in mouse brain tissues.
Mouse brain tissue was cut and stored at −80 °C until they were used for experiment. The tissue sections were preincubated with Tris·HCl buffer (pH 7.4, 50 mM) solution for 30 min at ambient temperature, followed by incubation with [11C]8 (5 mCi/L, ~ 1 nM) for 20 min at ambient temperature. For blocking studies, unlabeled 8 (10 μM) or MCC950 (10 μM) (n = 4) was added to incubation solution in advance to determine the specificity of radioligand binding. After incubation, tissue sections were washed with Tris·HCl buffer (3 × 5 min). The tissue sections were dried with cold air, then placed on imaging plates for 120 min. Autoradiograms were obtained and ROIs were carefully drawn with the reference of naked-eye observation. Data were analyzed using OptiQuant software (PerkinElmer) and expressed as the density light units per square millimeter (DLU/mm2).
NLRP3 measurements in human brain tissues.
Postmortem hippocampus tissues from both healthy controls and AD patients (n = 8) were utilized and applied by Western blotting (WB) analysis, following previously reported methods49. Sample lysates were obtained by homogenization in RIPA buffer (89900, Thermo Fisher) containing Protease/Phosphatase Inhibitor Cocktail (5872, Cell Signaling Technology) with a PRO200 homogenizer (PK-01200P, Pro Scientific). Supernatants were collected, and total protein concentrations were measured using a Pierce TM BCA Protein Assay Kit (23227, Thermo Scientific). Equal amounts of sample proteins (25 μg) were used for electrophoresis on 4–20% Criterion TGX stain-free precast gels (5678095, Bio-Rad) and electrophoretically transferred onto PVDF membranes. The membranes were probed overnight at 4 ℃ with antibodies against NLRP3 (1:1000, Abcam, ab263899) and anti-GAPDH (1:10000, Abcam, ab8245) in TBST containing 3% BSA (w/v). The blots were then incubated for 1 hour with an anti-rabbit secondary antibody (1:5000, Cell Signaling Technology, 7074) at room temperature. The immune complex was detected by using an ECL detection kit (GE, RPN2235V). The luminescence was visualized on the Bio-Rad Imaging System (Bio-Rad, ChemiDoc MP). Image J (National Institutes of Health, Bethesda, MD) analyzed the densitometry of protein bands. Subsequently, normalized band intensities were divided by the control group's average to determine normalized fold change vs. the control group.
PET/CT imaging studies.
All animal studies were carried out at Massachusetts General Hospital (PHS Assurance of Compliance No. A3596–01). The Subcommittee on Research Animal Care (SRAC) serves as the Institutional Animal Care and Use Committee (IACUC) for the Massachusetts General Hospital. SRAC reviewed and approved all procedures detailed in this paper.
PET/CT imaging was performed in anesthetized (isoflurane) male mice (n = 3) to minimize discomfort. Highly trained animal technicians monitored animal safety throughout all procedures, and veterinary staff were responsible for daily care. All mice were socially housed in cages appropriate for the physical and behavioral health of the individual animal and were given unlimited access to food and water, with additional nutritional supplements provided as prescribed by the attending veterinary staff. All animals were anesthetized with inhalational isoflurane at 3% in a carrier of 2 L/min medical oxygen, and maintained at 2% isoflurane for the duration of the scan. The mice were fixed on the bed of a Triumph Trimodality PET/CT scanner (Gamma Medica, Northridge, CA) in the prone position, and injected with [11C]8 (100 μL, ~100 μCi) via a lateral tail vein catheterization at the start of PET acquisition. Dynamic PET acquisition lasted for 60 min and was followed by CT for anatomic coregistration. PET data were reconstructed using a 3D-MLEM method resulting in a full width at half-maximum resolution of 1 mm. Reconstructed images were exported from the scanner in DICOM format along with an anatomic CT. These files were imported to PMOD software (PMOD Technologies LLC, version 4.0). Volumes of interests, including the whole brain, striatum, cortex, thalamus, midbrain, cerebellum, brain stem, and hippocampus were placed referencing the MRI template software. TACs were exported in terms of decay corrected activity at specified time points with gradually increasing intervals. TACs were expressed as the percentage of injected dose per cubic centimeter (%ID/cc).
PET/MRI of Resus Macaque.
A male rhesus macaque (weight = 11.25 kg) was deprived of food for 12 hours prior to the simultaneous PET/MR scanning. Anesthesia was induced with intramuscular xylazine (0.5–2.0 mg/kg) and ketamine (10 mg/kg). After endotracheal intubation, V-line and A-line was inserted and anesthetic state was maintained using isoflurane. The macaque was antecubital catheterized for radiotracer injection and a radial arterial line was placed for metabolite analysis. PET/MR images of the brain were acquired on a 3T Siemens TIM-Trio scanner with a BrainPET insert (Siemens, Munich, Germany), using an in-house built 8-channel head coil. Dynamic PET image acquisition was initiated followed by radioligand administration (Dose: 5.20 mCi). A MEMPRAGE MRI sequence was acquired 30 minutes after the start of the scan to enable anatomic co-registration. Dynamic data from the PET scan was recorded in list mode and corrected for attenuation prior to reconstruction. PET data was reconstructed using a 3D-OSEM method and binned into progressively longer time frames. Reconstructed images were converted into standard uptake values (SUV) by ratio of the PET recorded radioactivity and injected dose multiplied by the weight of the NHP. PET data was motion-corrected followed by registration to the INIA19 Template and NeuroMaps Atlas for brain imaging analysis. Image registration was carried out on high-resolution MPRAGE T1 image using a twelve degree-of-freedom linear algorithm and a nonlinear algorithm to the atlas brain. This transformation was subsequently applied to the simultaneously collected dynamic PET data. Regions of interest (VOIs) were segmented according to the brain atlas. Representative SUV images were generated by averaging the dynamic PET data from 0–90 minutes following tracer injection and overlaying on the INIA19 brain template. Kinetic modeling was performed in PMOD (PMOD3.9; PMOD Technologies). VOIs were exported from brain regions for kinetic analysis. Estimations of regional VT (mL/cm3) were performed by 1TCM, 2TCM, and Logan Plot modeling with metabolite-corrected plasma TAC (see below). Goodness of fit was assessed for 1TCM and 2TCM via the Akaike Information Criterion (AIC) and Model Selection Criterion (MSC). Lower AIC and higher MSC values were indicative of a better fit.
Plasma and metabolite HPLC analysis.
Arterial blood samples were collecting while the macaque underwent PET imaging. Metabolite extraction and High Performance Liquid Chromatography (HPLC) were based on previously reported methods from our institution to generate radioHPLC chromatograms for each blood sample. In brief, blood samples were centrifuged to isolate plasma for 4 minutes at 4000 RPM and 4 °C. Protein precipitation was attained via addition and mixing of plasma (1 mL) to acetonitrile (1 mL). This was then centrifuged again at for 4 minutes at 4000 RPM and 4 °C to obtain protein-free plasma. The protein-free supernatant (1 mL) was diluted in deionized water (4 mL) and underwent HPLC to distinguish radiometabolites from the parent radiotracer. HPLC setup included a column-switching valve for sample concentration (online solid phase extraction; Agilent Bond Elut Online SPE, PLRP-S, 4.6 × 12.5 mm) and subsequent separation (Agilent Eclipse Plus C18, 4.6 × 100 mm, 3.5 μm). Radiometabolite HPLC was conducted as follows: Plasma samples were placed onto the SPE concentrator column, 1% ACN and 99% H2O at 2 mL/min. After 3 min, the sample was flowed from the SPE column to the separation column under gradient conditions (Mobile Phase A: water + 0.1% formic acid; Mobile Phase B: acetonitrile + 0.1% formic acid; separation method = 95/5 – 50/50 A/B from 3 – 8 min linear gradient; 50/50 – 5/95 A/B from 8–10 min linear gradient; 5/95 A/B from 10–11 min isocratic; flow rate 2 mL/min). Analytes were then monitoring for ~10 min after sample injection via dual opposing bismuth germanium oxide detectors for coincidence detection (Eckert and Ziegler). Once generated, chromatograms were corrected for radioactive decay and subsequently integrated to quantify area under the curve for each respective metabolite and comparison to the original parent tracer. Each plasma sample’s parent fraction was fit, and then applied to the plasma input curve, allowing derivation of the radiometabolite-corrected plasma input function used for tissue compartment modeling discussed previously.
Supplementary Material
Acknowledgements
The work was supported in part by the NIA of the NIH under award number U01AG076481 and R01AG076912 (SZ) and the Alzheimer’s Drug Discovery Foundation (ADDF) RC-201804-2015064 (SZ).
Abbreviations
- AD
Alzheimer’s disease
- AIM2
absent in melanoma 2
- ASC
apoptosis-associated speck-like protein containing a caspase recruitment domain
- ATP
adenosine triphosphate
- Aβ
β-amyloid
- AIC
Akaike Information Criterion
- BBB
blood brain barrier
- CNS
central nervous system
- CSF
cerebrospinal fluid
- CABM
community-acquired bacterial meningitis
- CuAAC
copper-catalyzed alkyne-azide-cycloaddition
- DMEM
Dulbecco’s modified Eagle medium
- DMF
N,N-dimethylformamide
- DCM
dichloromethane
- ELISA
enzyme-linked immunosorbent assay
- FS
fluorescence spectrophotometer
- FBS
fetal bovine serum
- HPLC
high performance liquid chromatography
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uronium
- HD
Huntington’s disease
- IL-1β
interleukin-1β
- IL-18
interleukin-18
- IACUC
Institutional Animal Care and Use Committee
- %ID/cc
percentage of injected dose per cubic centimeter
- LPS
lipopolysaccharide
- MST
microscale thermophoresis
- MSU
monosodium urate
- MS
multiple sclerosis
- MRI
magnetic resonance imaging
- MOAs
mechanisms of actions
- MSC
Model Selection Criterion
- M.Wt
molecular weight
- MPO
Multiparameter Optimization
- NOD
nucleotide-binding oligomerization domain
- NLRP3
NOD-like receptor family pyrin-domain-containing 3
- NMR
nuclear magnetic resonance
- NLRC4
NLR Family CARD Domain Containing 4
- NHPs
non-human primates
- PD
parkinson’s diseases
- PET
positron emission tomography
- Poly(dA:dT)
poly-deoxyadenylic–deoxythymidylic acid sodium salt
- PBS
phosphate-buffered saline
- SAR
structure-activity relationship
- SPE
solid-phase exchange
- SRAC
Subcommittee on Research Animal Care
- TXNIP
thioredoxin interacting protein
- TEA
triethylamine
- TNF-α
tumor necrosis factor-α
- TCM
tissue compartmental models
- TACs
time-activity curves
- TSA
thermal shift assay
- TLC
thin-layer chromatography
- THF
tetrahydrofuran
- TBS
tert-butyldimethylsilyl
- TBAF
tetra-n-butylammonium fluoride
- TBI
traumatic brain injury
- VM
viral meningitis
- WT
wild type
Footnotes
Supporting Information
The Supporting Information is available free of charge.
Additional experimental details and results, including supplementary figures, NMR spectra, HPLC spectra, and HRMS spectra (PDF)
Molecular formula string (CSV)
Conflicts of interest
The authors declare no conflict of interest.
References:
- (1).Lamkanfi M; Dixit VM Mechanisms and functions of inflammasomes. Cell 2014, 157 (5), 1013–1022. [DOI] [PubMed] [Google Scholar]
- (2).Maturana CJ; Aguirre A; Sáez JC High glucocorticoid levels during gestation activate the inflammasome in hippocampal oligodendrocytes of the offspring. Dev. Neurobiol 2017, 77, 625–642. [DOI] [PubMed] [Google Scholar]
- (3).Johann S; Heitzer M; Kanagaratnam M; Goswami A; Rizo T; Weis J; Troost D; Beyer C NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 2015, 63, 2260–2273. [DOI] [PubMed] [Google Scholar]
- (4).Lu A; Wu H Structural mechanisms of inflammasome assembly. FEBS J 2015, 282 (3), 435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Schroder K; Tschopp J The inflammasomes. Cell 2010, 140 (6), 821–32. [DOI] [PubMed] [Google Scholar]
- (6).Swanson KV; Deng M; Ting JP The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol 2019, 19, 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sita G; Graziosi A; Hrelia P; Morroni F NLRP3 and Infections: β-Amyloid in Inflammasome beyond Neurodegeneration. Int. J. Mol. Sci 2021, 22, 6984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Milner MT; Maddugoda M; Götz J; Burgener SS; Schroder K The NLRP3 inflammasome triggers sterile neuroinflammation and Alzheimer's disease. Curr. Opin. Immunol 2021, 68, 116–124. [DOI] [PubMed] [Google Scholar]
- (9).Pike AF; Szabo I; Veerhuis R; Bubacco L The potential convergence of NLRP3 inflammasome, potassium, and dopamine mechanisms in Parkinson's disease. NPJ Parkinsons Dis 2022, 8, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Nguyen LTN; Nguyen HD; Kim YJ; Nguyen TT; Lai TT; Lee YK; Ma H; Kim YE Role of NLRP3 Inflammasome in Parkinson’s Disease and Therapeutic Considerations. J. Parkinson's Dis 2022, 12, 2117–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Paldino E; Fusco FR Emerging Role of NLRP3 Inflammasome/Pyroptosis in Huntington’s Disease. Int. J. Mol. Sci 2022, 23, 8363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gris D; Ye Z; Iocca HA; Wen H; Craven R,R; Gris P; Huang M; Schneider M; Miller SD; Ting JP. NLRP3 Plays a Critical Role in the Development of Experimental Autoimmune Encephalomyelitis by Mediating Th1 and Th17 Responses. J. Immunol 2010, 185 (2), 974–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Olcum M; Tastan B; Kiser C; Genc S; Genc K Microglial NLRP3 inflammasome activation in multiple sclerosis. Adv. Protein Chem. Struct. Biol 2020,119, 247–308. [DOI] [PubMed] [Google Scholar]
- (14).Irrera N; Russo M; Pallio G; Bitto A; Mannino F; Minutoli L; Altavilla D; Squadrito F The Role of NLRP3 Inflammasome in the Pathogenesis of Traumatic Brain Injury. Int. J. Mol. Sci 2020, 21, 6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Alishahi M; Farzaneh M; Ghaedrahmati F; Nejabatdoust A; Sarkaki A; Khoshnam SE NLRP3 inflammasome in ischemic stroke: As possible therapeutic target. Int. J. Stroke 2019, 14, 574–591. [DOI] [PubMed] [Google Scholar]
- (16).Mohseni-Moghaddam P; Roghani M; Khaleghzadeh-Ahangar H; Sadr SS; Sala C A literature overview on epilepsy and inflammasome activation. Brain Res. Bull 2021, 172, 229–235. [DOI] [PubMed] [Google Scholar]
- (17).He X; Xu J; Li G; Li M; Li L; Pei Z; Zhang L; Hu X NLRP3-dependent microglial training impaired the clearance of amyloid-beta and aggravated the cognitive decline in Alzheimer’s disease. Cell Death Dis 2020, 11 (10), 849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nasoohi S; Ismael S; Ishrat T Thioredoxin-Interacting Protein (TXNIP) in Cerebrovascular and Neurodegenerative Diseases: Regulation and Implication. Mol. Neurobiol 2018, 55, 7900–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Gong Z; Zhang C; Li Y; Jing L; Duan R; Yao Y; Teng J; Jia Y NLRP3 in the Cerebrospinal Fluid as a Potential Biomarker for the Diagnosis and Prognosis of Community-Acquired Bacterial Meningitis in Adults. Front. Cell. Infect. Microbiol 2022, 11, 803186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).O'Brien WT; Pham L; Symons GF; Monif M; Shultz SR; McDonald SJ The NLRP3 inflammasome in traumatic brain injury: potential as a biomarker and therapeutic target. J. Neuroinflamm 2020, 17, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Li L; Liu Y Aging-related gene signature regulated by Nlrp3 predicts glioma progression. Am. J. Cancer Res 2015, 5, 442–449. [PMC free article] [PubMed] [Google Scholar]
- (22).Suceveanu A; Mazilu L; Katsiki N; Parepa I; Voinea F; Pantea-Stoian A; Rizzo M; Botea F; Herlea V; Serban D et al. NLRP3 Inflammasome Biomarker-Could Be the New Tool for Improved Cardiometabolic Syndrome Outcome. Metabolites 2020, 10, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Courjon J; Dufies O; Robert A; Bailly L; Torre C; Chirio D; Contenti J; Vitale S; Loubatier C; Doye A et al. Heterogeneous NLRP3 inflammasome signature in circulating myeloid cells as a biomarker of COVID-19 severity. Blood Adv 2021, 5, 1523–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Xu Y; Biby S; Kaur B; Zhang S A patent review of NLRP3 inhibitors to treat autoimmune diseases. Expert Opin. Ther. Pat 2023, 33, 455–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Coll RC; Robertson AAB; Chae JJ; Higgins SC; Muñoz-Planillo R; Inserra MC; Vetter I; Dungan LS; Monks BG; Stutz A et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med 2015, 21, 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jiang H; He H; Chen Y; Huang W; Cheng J; Ye J; Wang A; Tao J; Wang C; Liu Q et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med 2017, 214, 3219–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Huang Y; Jiang H; Chen Y; Wang X; Yang Y; Tao J; Deng X; Liang G; Zhang H; Jiang W et al. Tranilast directly targets NLRP3 to treat inflammasome‐ driven diseases. EMBO Mol. Med 2018, 10 (4), e8689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).He H; Jiang H; Chen Y; Ye J; Wang A; Wang C; Liu Q; Liang G; Deng X; Jiang W et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun 2018, 9 (1), 2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hill JR; Shao X; Massey NL; Stauff J; Sherman PS; Robertson AAB; Scott PJH Synthesis and evaluation of NLRP3-inhibitory sulfonylurea [11C]MCC950 in healthy animals. Bioorg. Med. Chem. Lett 2020, 30, 127186. [DOI] [PubMed] [Google Scholar]
- (30).Lamkanfi M; Mueller JL; Vitari AC; Misaghi S; Fedorova A; Deshayes K; Lee WP; Hoffman HM; Dixit VM Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol 2009, 187, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kemmer FW; Tacken M; Berger M Mechanism of exercise-induced hypoglycemia during sulfonylurea treatment. Diabetes 1987, 36, 1178–1182. [DOI] [PubMed] [Google Scholar]
- (32).Fulp J; He L; Toldo S; Jiang Y; Boice A; Guo C; Li X; Rolfe A; Sun D; Abbate A et al. Structural Insights of Benzenesulfonamide Analogues as NLRP3 Inflammasome Inhibitors: Design, Synthesis, and Biological Characterization. J. Med. Chem 2017, 61, 5412–5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Yin J; Zhao F; Chojnacki JE; Fulp J; Klein WL; Zhang S; Zhu X NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol 2018, 55, 1977–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Jiang Y; He L; Green J; Blevins H; Guo C; Patel SH; Halquist MS; McRae M; Venitz J; Wang X et al. Discovery of Second-Generation NLRP3 Inflammasome Inhibitors: Design, Synthesis, and Biological Characterization. J. Med. Chem 2019, 62, 9718–9731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Xu Y; Xu Y; Blevins H; Guo C; Biby S; Wang X; Wang C; Zhang S Development of sulfonamide-based NLRP3 inhibitors: Further modifications and optimization through structure-activity relationship studies. Eur. J. Med. Chem 2022, 238, 114468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Xu Y; Xu Y; Blevins H; Lan Y; Liu Y; Yuan G; Striar R; Zagaroli JS; Tocci DR; Langan AG et al. Discovery of carbon-11 labeled sulfonamide derivative: A PET tracer for imaging brain NLRP3 inflammasome. Bioorg. Med. Chem. Lett 2021, 34, 127777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Okumura Y; Maya Y; Onishi T; Shoyama Y; Izawa A; Nakamura D; Tanifuji S; Tanaka A; Arano Y; Matsumoto H Design, Synthesis, and Preliminary Evaluation of SPECT Probes for Imaging β-Amyloid in Alzheimer's Disease Affected Brain. ACS Chem. Neurosci 2018, 9, 1503–1514. [DOI] [PubMed] [Google Scholar]
- (38).Taddei M; Ferrini S; Giannotti L; Corsi M; Manetti F; Giannini G; Vesci L; Milazzo FM; Alloatti D; Guglielmi MB et al. Synthesis and Evaluation of New Hsp90 Inhibitors Based on a 1,4,5-Trisubstituted 1,2,3-Triazole Scaffold. J. Med. Chem 2014, 57, 2258–2274. [DOI] [PubMed] [Google Scholar]
- (39).Willis NJ; Mahy W; Sipthorp J; Zhao Y; Woodward HL; Atkinson BN; Bayle ED; Svensson F; Frew S; Jeganathan F et al. Design of a Potent, Selective, and Brain-Penetrant Inhibitor of Wnt-Deactivating Enzyme Notum by Optimization of a Crystallographic Fragment Hit. J. Med. Chem 2022, 65, 7212–7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wager TT; Hou X; Verhoest PR; Villalobos A Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci 2016, 7, 767–775. [DOI] [PubMed] [Google Scholar]
- (41).Kolb HC; Finn MG; Sharpless KB Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- (42).Ecker AK; Levorse DA; Victor DA; Mitcheltree MJ Bioisostere Effects on the EPSA of Common Permeability-Limiting Groups. ACS Med. Chem. Lett 2022, 13, 964–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Lassalas P; Gay B; Lasfargeas C; James MJ; Tran V; Vijayendran KG; Brunden KR; Kozlowski MC; Thomas CJ; Smith AB et al. Structure Property Relationships of Carboxylic Acid Isosteres. J. Med. Chem 2016, 59, 3183–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Pike VW PET radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol. Sci 2009, 30, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Patel S; Gibson R In vivo site-directed radiotracers: a mini-review. Nucl. Med. Biol 2008, 35, 805–815. [DOI] [PubMed] [Google Scholar]
- (46).Van de Bittner GC; Ricq EL; Hooker JM A philosophy for CNS radiotracer design. Acc. Chem. Res 2014, 47, 3127–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Heneka MT; Kummer MP; Stutz A; Delekate A; Schwartz S; Vieira-Saecker A; Griep A; Axt D; Remus A; Tzeng T et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Saresella M; La Rosa F; Piancone F; Zoppis M; Marventano I; Calabrese E; Rainone V; Nemni R; Mancuso R; Clerici M The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer's disease. Mol. Neurodegener 2016, 11, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Bai P; Mondal P; Bagdasarian FA; Rani N; Liu Y; Gomm A; Tocci DR; Choi SH; Wey HY; Tanzi RE; Zhang C. Development of a potential PET probe for HDAC6 imaging in Alzheimer's disease. Acta Pharm. Sin. B 2022, 12, 3891–3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.