

ABSTRACT
The inflammasome is a pivotal component of the innate immune system, acting as a multiprotein complex that plays an essential role in detecting and responding to microbial infections. Salmonella Enteritidis have evolved multiple mechanisms to regulate inflammasome activation and evade host immune system clearance. Through screening S. Enteritidis C50336ΔfliC transposon mutant library, we found that the insertion mutant of dinJ increased inflammasome activation. In this study, we demonstrated the genetic connection between the antitoxin DinJ and the toxin YafQ in S. Enteritidis, confirming their co-transcription. The deletion mutant ΔfliCΔdinJ increased cell death and IL-1β secretion in J774A.1 cells. Western blotting analysis further showed elevated cleaved Caspase-1 product (p10 subunits) and IL-1β secretion in cells infected with ΔfliCΔdinJ compared to cells infected with ΔfliC. DinJ was found to inhibit canonical inflammasome activation using primary bone marrow-derived macrophages (BMDMs) from Casp-/- C57BL/6 mice. Furthermore, DinJ specifically inhibited NLRP3 inflammasome activation, as demonstrated in BMDMs from Nlrp3-/- and Nlrc4-/- mice. Fluorescence resonance energy transfer (FRET) experiments confirmed the translocation of DinJ into host cells during infection. Finally, we revealed that DinJ could inhibit the secretion of IL-1β and IL-18 in vivo, contributing to S. Enteritidis evading host immune clearance. In summary, our findings provide insights into the role of DinJ in modulating the inflammasome response during S. Enteritidis infection, highlighting its impact on inhibiting inflammasome activation and immune evasion.
KEYWORDS: Salmonella Enteritidis, DinJ, NLRP3 inflammasome, immune evasion
INTRODUCTION
The inflammasome is a multiprotein complex that plays a pivotal role in the innate immune system against bacterial infections. It operates as a molecular platform for sensing and responding to danger signals such as pathogenic microbes, cellular damage, or metabolic stress (1–4). Assembly of the inflammasome leads to the activation of Caspase-1, triggering the maturation and secretion of pro-inflammatory cytokines like IL-1β and IL-18. These cytokines facilitate neutrophil migration to infected tissues and against pathogens (5). Additionally, activation of Caspase-1 leads to gasdermin D cleavage, resulting in cell rupture via pyroptosis, and inflammatory cell death mediated through the inflammasome (6). Various pathogens could activate the inflammasome, leading to the secretion of inflammatory cytokines (7–10). Moderate inflammation aids the host in pathogen clearance, while excessive inflammation could cause tissue damage (11). Pathogens have evolved multiple mechanisms to maintain the homeostasis of the inflammasome activation, balancing an effective immune response with the potential tissue damage caused by excessive inflammation.
Salmonella, a facultative intracellular pathogen, has the potential to cause severe illness in both humans and animals. The invasion of host cells by Salmonella is facilitated by a type III secretion system (T3SS) encoded by Salmonella pathogenicity island I (SPI-1) (12). Subsequently, Salmonella replicates within the host cell that is established by the second T3SS encoded by SPI-2 (13). Salmonella invasion into macrophages induces a form of cell death known as pyroptosis, and the T3SS could directly secrete the effectors into host cells to modulate the activation of the inflammasome (14, 15). This inflammatory form of cell death acts as a host defense mechanism, contributing to the elimination of Salmonella-infected cells and enhancing the immune response against the invading pathogen. Salmonella SopB effector could inhibit the NLRC4 inflammasome activation by specifically preventing apoptosis-associated speck-like protein containing a CARD (ASC) oligomerization (16). Salmonella AcnB could inhibit the NLPR3 inflammasome activation by mitochondrial reactive oxygen species (mtROS), contributing to Salmonella persistence infection in vivo (3). Furthermore, the Salmonella SiiD protein could inhibit NLRP3 inflammasome activation through mtROS-ASC signaling and plays an essential role in evading host immune clearance (17). Therefore, Salmonella have evolved mechanisms to inhibit or evade pyroptosis, aiding in their persistence within the host and establishment of infection.
The toxin and antitoxin (TA) systems are genetic modules commonly found in bacteria, playing a pivotal role in regulating cellular processes and responding to various stresses (18, 19). The TA systems consist of a pair of closely linked genes, one encodes a toxin protein and the other encodes an antitoxin to neutralize the toxin effects. Investigation into the Salmonella genome has revealed the presence of 24 TA loci, including five type I and 19 type II TA systems (20). DinJ-YafQ belongs to the type II TA system, is classified as a ribosome-dependent mRNA interferase, and is extensively studied in Escherichia coli (21, 22). The dinJ-yafQ genes form an operon with the antitoxin gene (dinJ) located upstream of the toxin gene (yafQ). The expression of this operon is directly repressed by the antitoxin DinJ or the DinJ-YafQ complex (23). Moreover, the DinJ-YafQ TA system has been reported to regulate persister formation by controlling indole production in E. coli (24). Similarly, the Salmonella Typhimurium TA system plays a role in mediating the phenotypic switch of persisters, contributing to persistent infections (25). However, the functions of Salmonella DinJ-YafQ TA system in the context of persistent infection and immune response remain poorly researched. Further exploration is needed to unravel its specific roles in Salmonella pathogenicity.
In our previous study, we identified that the insertion mutant of dinJ could cause an increase in the activation of inflammasome (26). However, the mechanisms by which DinJ inhibits inflammasome activation have not been thoroughly investigated. Therefore, we constructed dinJ deletion mutant and complemented strains, assessing inflammasome activation in both J774A.1 cells and bone marrow-derived macrophages (BMDMs). Significantly, the deletion of dinJ induced NLRP3-dependent inflammasome activation in Salmonella-infected macrophages. Furthermore, the deletion of dinJ also could increase the activation of inflammasome in the mice, contributing to the clearance of Salmonella by host immune systems. Collectively, our results suggest that the mediating activation of the NLRP3 inflammasome is beneficial for Salmonella infection.
RESULTS
Salmonella Enteritidis DinJ inhibits the activation of inflammasome in J774A.1 cells
Inhibition of the inflammasome activation could facilitate Salmonella to evade the clearance of the host immune system. In our previous studies, we used a transposon mutant library to screen the genes involved in inhibiting inflammasome activation and we found that DinJ insertion mutant strain increased the activation of inflammasome (26). In S. Enteritidis, we identified that the antitoxin dinJ and toxin yafQ were connected and showed 83% and 78% identity to the E. coli dinJ and yafQ genes, respectively (Fig. 1). RT-PCR yielded a product of the expected size with primers spanning the regions from dinJ to yafQ. No product was obtained using RNA as the template, while a product of the same size was observed when genomic DNA (gDNA) was used as the template (Fig. 1). This indicates that the dinJ and yafQ genes are co-transcribed in S. Enteritidis.
Fig 1.
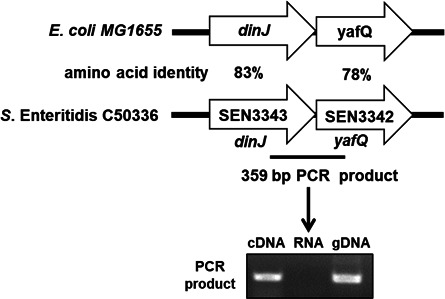
Co-transcription of dinJ and yafQ. A schematic representation of the predicted dinJ-yafQ genes in S. Enteritidis. Numbers represent the amino acid identity of the S. Enteritidis C50336 compared with E. coli MG1655. RT-PCR was used to determine the co-transcription of dinJ and yafQ.
Therefore, we constructed the dinJ deletion mutant and complemented strains to investigate cell death and the associated release of IL-1β. As shown in Figure 2, cells infected with ΔfliCΔdinJ released significantly increased levels of cell death (Fig. 2A) and IL-1β secretion (Fig. 2B) compared to ΔfliC-infected cells, while the complemented strain was restored to the parental strain. The release of inflammasome-independent cytokine IL-6 showed no significant difference in J774A.1 cells infected with ΔfliC and its derived strains (Fig. 2C), indicating that DinJ specifically inhibited the release of inflammasome-dependent cytokines. Western blotting analysis also revealed a significant increase in the cleaved Caspase-1 product p10 subunits and IL-1β in cells infected with ΔfliCΔdinJ compared to ΔfliC-infected cells (Fig. 2D). Furthermore, growth curves showed no significant difference among ΔfliC, ΔfliCΔdinJ, complemented, and empty vector-complemented strains (Fig. S1A). The deletion of dinJ did not affect the adhesion (Fig. S1B) and invasion (Fig. S1C) ability of S. Enteritidis, indicating that the inhibition of DinJ to inflammasome activation is unrelated to the growth, adhesion, and invasion ability of Salmonella. Taken together, these results indicated that DinJ inhibits the activation of inflammasome during S. Enteritidis infection.
Fig 2.
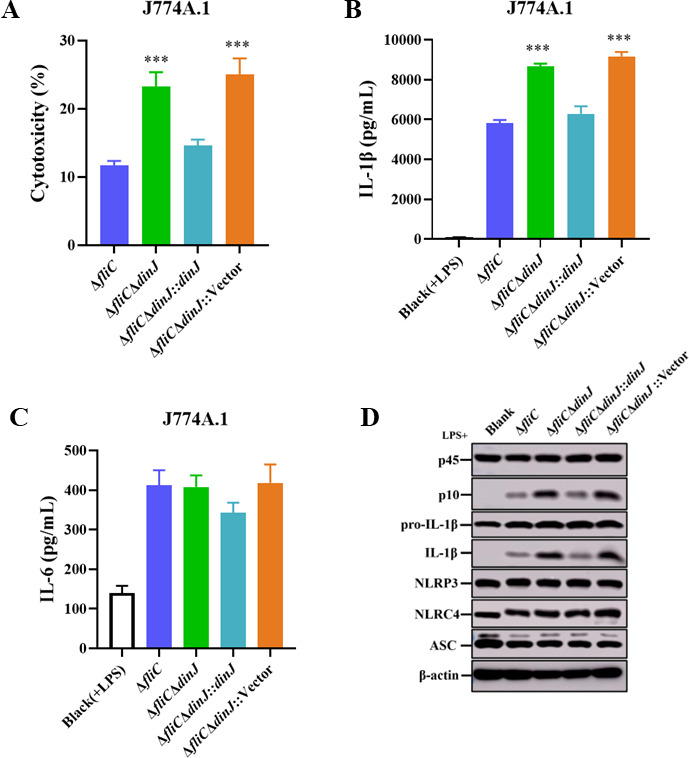
DinJ inhibits the activation of inflammasome in J774A.1 cells. (A) Evaluation of cytotoxicity in J774A.1 cells infected with ΔfliC, ΔfliCΔdinJ, ΔfliCΔdinJ::dinJ, or empty vector-complemented strain ΔfliCΔdinJ::pMMB207 at an MOI of 100 for 4 h. (B and C) Enzyme-linked immunosorbent assay (ELISA) analysis assessing the secretion of IL-1β (B) and IL-6 (C) in the infected cells. (D) Western blotting analysis of the cleavage of Caspase-1, secretion of IL-1β, and the expression levels of NLRP3, NLRC4, and ASC in J774A.1 cells. β-actin was used as the loading control. Statistical significance was determined at P values of <0.001 (***) using the Student’s t-test.
Both NLRP3 and NLRC4 have been reported as the main inflammasomes involved in the host immune system against Salmonella infection (27, 28). Therefore, we measured the expression levels of NLRP3, NLRC4, and ASC to assess whether the inhibition of DinJ to inflammasome activation depended on the expression of these proteins. The expression levels of NLRP3, NLRC4, and ASC showed no significant differences among the cells infected with ΔfliC, ΔfliCΔdinJ, complemented, and empty vector-complemented strains (Fig. 2D), suggesting that DinJ inhibits the activation of inflammasome independently of the expression levels of NLRP3, NLRC4, and ASC proteins.
DinJ inhibits the canonical inflammasome activation through Caspase-1
Primary BMDMs were obtained from wild-type (WT) C57BL/6 mice to confirm the inhibitory effect of DinJ on inflammasome activation. The cytotoxicity of BMDMs infected with ΔfliCΔdinJ significantly increased compared to ΔfliC-infected BMDMs (Fig. 3A). Similar results were observed in IL-1β secretion (Fig. 3B) and Caspase-1 activation (Fig. 3E). Furthermore, the expression levels of NLRP3 and NLRC4 showed no significant difference in the BMDMs infected with the indicated strains (Fig. 3E). These results further confirm that the S. Enteritidis DinJ could inhibit the activation of inflammasome in primary macrophages from mice.
Fig 3.
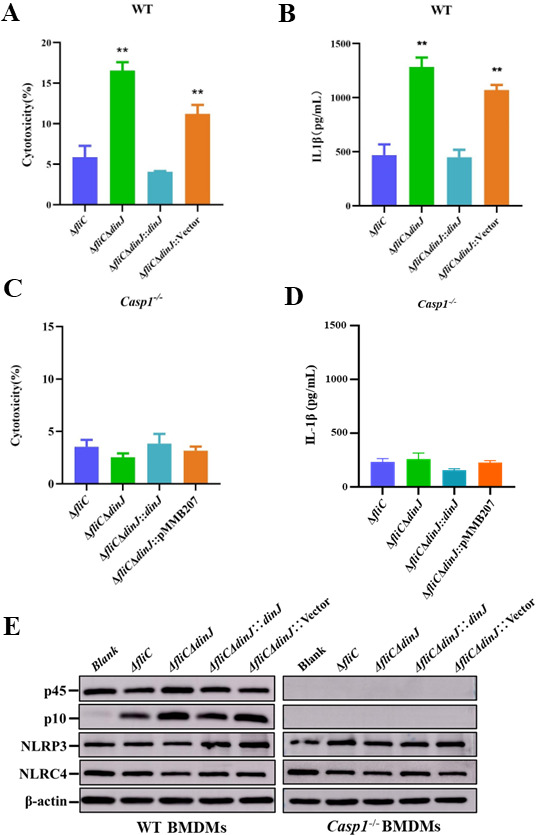
DinJ inhibits the canonical inflammasome activation through Caspase-1. (A and C) Cytotoxicity of the BMDMs obtained from WT (A) and Casp1-/- (C) C57BL/6 mouse. (B and D) ELISA analysis measuring the secretion of IL-1β in BMDMs obtained from WT (B) and Casp1-/- (D) C57BL/6 mouse. (E) Western blotting analysis evaluating the cleavage of Caspase-1 and the expression levels of NLRP3 and NLRC4. β-actin served as the loading control. Statistical significance was determined at P values of < 0.01 (**) using the Student’s t-test.
Caspase-1 is a key factor for the canonical inflammasome and essential for inflammasome-mediated inflammatory secretion. Therefore, the BMDMs obtained from Casp-1 C57BL/6 mice were utilized to confirm whether DinJ inhibits inflammasome activation through Caspase-1. Cell death (Fig. 3C) and IL-1β secretion (Fig. 3D) showed no significant differences among cells infected by the indicated strains. Western blotting failed to detect the expression of Caspase-1 and its cleaved subunit (Fig. 3E), further confirming the knockout of casp1 gene. These results indicate that S. Enteritidis DinJ inhibits the canonical inflammasome activation.
DinJ specifically inhibits NLRP3-dependent inflammasome activation
To further confirm the role of DinJ in regulating inflammasome activation, we obtained the BMDMs from Nlrp3-/- and Nlrc4-/- C57BL/6 mice and infected them with ΔfliC, ΔfliCΔdinJ, complemented, and empty vector-complemented strains. Nlrc4-/-BMDMs infected with ΔfliCΔdinJ and ΔfliCΔdinJ::Vector strains lead to significantly increased cytotoxicity compared to the cells infected with ΔfliC (Fig. 4A). In contrast, Nlrp3-/- BMDMs showed lower levels of cytotoxicity in response to infection by ΔfliCΔdinJ and ΔfliCΔdinJ::Vector strains, suggesting that the absence of dinJ induces activation of NLRP3 inflammasome. Furthermore, ΔfliCΔdinJ and ΔfliCΔdinJ::Vector strains induced significantly enhanced secretion of IL-1β in Nlrc4-/- BMDMs, while this phenotype was not observed in the Nlrp3-/- BMDMs (Fig. 4D and E), indicating that NLRP3 inflammasome activation was essential for the increased secretion of IL-1β. The secretion of inflammasome-independent cytokine IL-6 was not affected by DinJ in any of the BMDMs (Fig. 4C and F). Western blotting analysis also showed that the cleaved Caspase-1 subunit P10 and mature IL-1β were increased in the ΔfliCΔdinJ-infected WT and Nlrc4-/- BMDMs compared to those infected by ΔfliC, but not in the Nlrp3-/- BMDMs (Fig. 4G and H). All these observed phenotypes in BMDMs infected with the complemented strain were restored to ΔfliC-infected BMDMs. Taken together, these results indicate that S. Enteritidis DinJ protein specifically inhibits NLPR3-dependent inflammasome activation.
Fig 4.
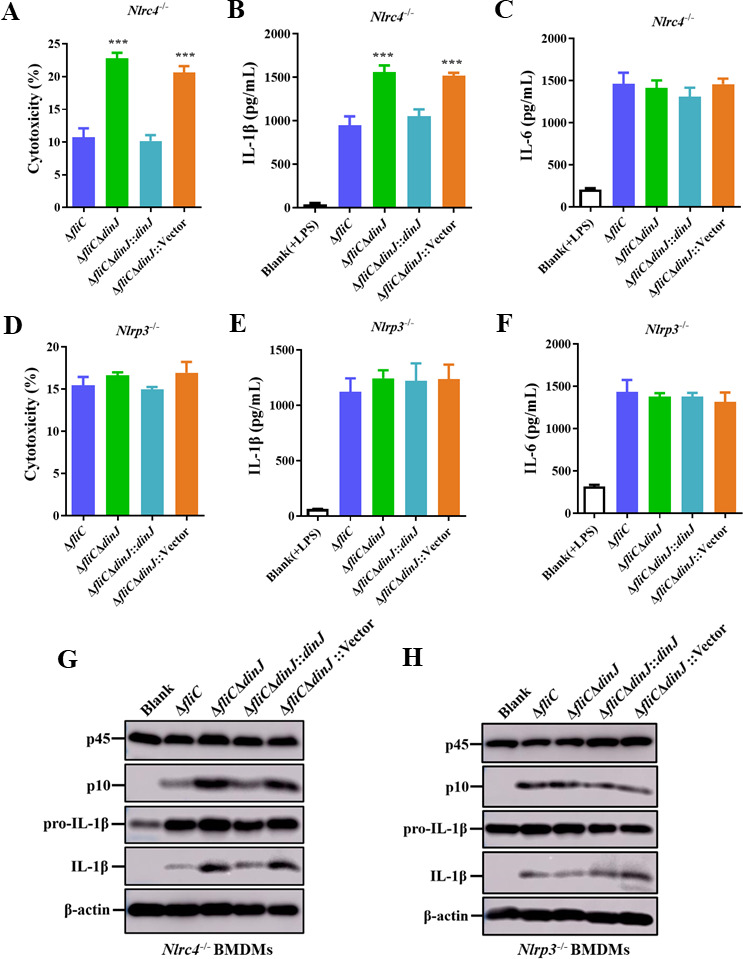
Deletion of S. Enteritidis dinJ induces NLRP3 inflammasome activation. (A–C) Evaluation of cytotoxicity (A), IL-1β (B), and IL-6 (C) levels in the supernatants of Nlrc4-/- BMDMs infected with the indicated strains. (D–F). Evaluation s of cytotoxicity (D), IL-1β (E), and IL-6 (F) levels in the supernatants of Nlrp3-/- BMDMs infected with the indicated strains. (G and H) Western blotting analysis of Caspase-1 activation and IL-1β secretion in Nlrc4-/- BMDMs (G) and Nlrp3-/- BMDMs (H) infected with the indicated strains. Statistical significance was determined at P values of < 0.01 (**) using the Student’s t-test.
DinJ can be translocated into host cells
Salmonella secretion systems can deliver effectors into host cells to modulate inflammasome activation. Therefore, we employed fluorescence resonance energy transfer (FRET) to verify the translocation of DinJ protein. The DinJ protein was fused with the β-lactamase TEM-1, and the fused protein was confirmed by Western blotting (Fig. 5A). As shown in Fig. 5B, blue fluorescent cells were observed in the HeLa cells infected with ΔfliC-pCX340-dinJ, while no blue fluorescent cells were observed in the blank control and HeLa cells infected with ΔfliC-pCX340. Moreover, we randomly selected more than 600 cells to calculate the percentage of the cells exhibiting blue fluorescent, and the transfer efficiency was approximately 3.9% (Fig. 5C). These results demonstrate that DinJ can be translocated into host cells during infection.
Fig 5.
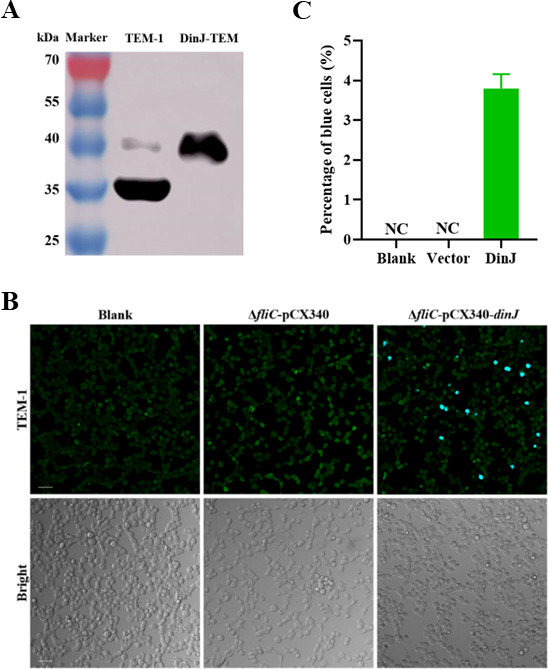
DinJ can be translocated into HeLa cells. (A) Western blotting analysis confirming the presence of DinJ-TEM-1 fusion protein. (B) HeLa cells were infected with S. Enteritidis ΔfliC strains carrying either pCX340 or pCX340-dinJ. Translocation of the DinJ-TEM-1 fusion protein into HeLa cells results in the cleavage of CCF2-AM, leading to the emission of blue fluorescence, while the uncleaved CCF2-AM emitted green fluorescence. Scale bar = 50 µm. (C) The percentage of the blue fluorescent cells. Three images were captured per cell well, and approximately 600 cells were counted to calculate the percentage of the blue fluorescent cells.
DinJ enables S. Enteritidis evasion of the NLPR3 inflammasome in vivo
Several bacterial and viral pathogens can inhibit the activation of NLRP3 inflammasome to evade clearance by the host immune system. To confirm the function of DinJ in the virulence of S. Enteritidis, we investigated whether DinJ could inhibit inflammasome activation and antibacterial defense in vivo. The mice were pretreated with streptomycin before being orally infected with ΔfliC and ΔfliCΔdinJ. The secretion of inflammasome-dependent cytokines IL-1β and IL-18, as well as the inflammasome-independent cytokine IL-6, in the serum was assessed. Compared to ΔfliC-infected mice, IL-1β (Fig. 6A) and IL-18 (Fig. 6B) were significantly increased in mice infected with ΔfliCΔdinJ, while the secretion of IL-6 showed no significant difference in the infected mice (Fig. 6C). Furthermore, bacterial burdens in the liver and spleen of the infected mice were also evaluated. The results showed that the bacterial CFUs of the liver (Fig. 6D) and spleen (Fig. 6E) in ΔfliCΔdinJ-infected mice were significantly lower than those in ΔfliC-infected mice. These results indicate that the S. Enteritidis DinJ protein inhibits NLRP3-dependent inflammasome activation and contributes to Salmonella evading the clearance of the host immune system in vivo.
Fig 6.
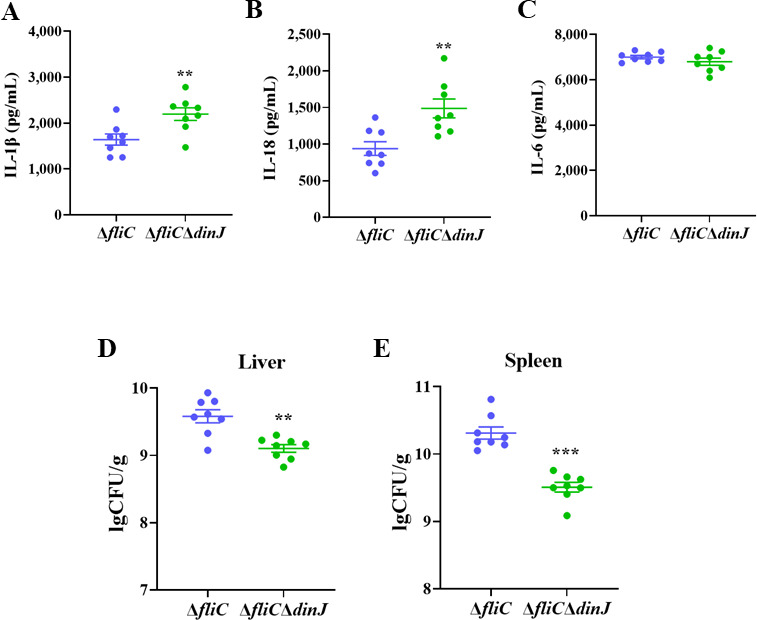
DinJ inhibits inflammasome activation to evade clearance of host immune system. (A–C) ELISA analysis of IL-1β (A), IL-18 (B), and IL-6 (C) in the serum of WT mouse infected with ΔfliC or ΔfliCΔdinJ. (D and E) Assessment of bacterial loading in the liver (D) and spleen (E) of WT mouse infected with ΔfliC or ΔfliCΔdinJ. Statistical significance was determined at P values of <0.01 (**) or <0.001 (***) using the Student’s t-test.
DISCUSSION
During infection, the host immune system recognizes Salmonella and activates both the NLRC4 and NLRP3 inflammasome, contributing to the host defense against Salmonella infection (29, 30). The NLRC4 inflammasome recognizes Salmonella flagellin, T3SS needle, and basal rod proteins, while the NLRP3 inflammasome responds to various structurally and chemically diverse signals (4, 31, 32). Deletion or downregulation of the flagellin gene impairs the ability to induce NLRC4 inflammasome activation in Salmonella-infected macrophages (33, 34). Furthermore, purified flagellin could activate Caspase-1 in an NLRC4-dependent manner (32, 34). Another study demonstrated that Salmonella flagellin induces NLRC4 inflammasome activation in human macrophages (35). In the late logarithmic growth phase, flagellin genes are highly expressed, inducing robust inflammasome activation within 1 h in vitro (30). Therefore, in our previous studies, we used the fliC deletion mutant strain as the parental strain to construct the insertion mutant library and to screen for genes of S. Enteritidis involved in inhibiting inflammasome activation (17, 26). We have demonstrated that S. Enteritidis SiiD and GalE protein could inhibit the activation of inflammasome in an NLRP3-dependent way (17, 26). In this study, we report for the first time that S. Enteritidis DinJ can inhibit NLRP3-dependent inflammasome activation. Notably, all of the S. Enteritidis SiiD, GalE, and DinJ proteins inhibit NLRP3-dependent canonical inflammasome activation, which is likely due to the deletion of the fliC gene, which could avoid the NLRC4 inflammasome activation. Therefore, we only screened for the proteins that inhibit NLRP3-dependent inflammasome activation.
The YafQ-DinJ system is classified as a type II toxin-antitoxin system and is recognized for its role in regulating persister formation. YafQ is an endoribonuclease-type toxin, exerting strict control on the expression of tnaA. Conversely, DinJ is an antitoxin protein, strongly inhibiting the effects of toxin (24). Under a stress condition, the antitoxin protein DinJ may undergo degradation by the protease, allowing the toxin protein YafQ to degrade the transcripts of target genes. However, there are fewer works to explore the contribution of YafQ-DinJ TA systems in S. Enteritidis infection. Our previous study found that the insertion mutant of SEN3342 in S. Enteritidis caused the increased activation of the inflammasome, sharing 83% identity with the dinJ gene in E. coli (Fig. 1). Generally, the deletion of the antitoxin gene would increase the level of free toxin, leading to growth arrest or cell death (36). However, in our current study, the deletion mutant strain of antitoxin gene dinJ exhibited no significant difference in growth curves compared to the parent strain ΔfliC in Luria-Bertani broth (LB) medium (Fig. S1). Similar results were observed in other type II TA systems Hha-TomB of S. Typhimurium and E. coli, where the deletion of hha or tomB did not affect growth in the minimal or complex medium compared to the WT (37, 38). However, it’s noteworthy that Hha was found to induce cell death under biofilm or acid stress conditions (39, 40). Previous findings also showed that DinJ-YafQ could be induced under a high temperature (24). Therefore, it is plausible that ΔfliCΔdinJ may exhibit a distinct growth curve under stress conditions compared to ΔfliC.
Toxin-antitoxin systems and their impact on Salmonella infection have been studied extensively. S. Typhimurium type II TA systems Hha-TomB have emerged as a significant regulator of virulence and host immune response (41). In our study, the deletion of the antitoxin dinJ resulted in higher cytotoxicity in J774A.1 cells compared to the parent strain ΔfliC-infected cells; whereas, no attenuation was observed in mice infected with ΔfliCΔdinJ. Moreover, the deletion of the toxin gene yafQ did not affect the cytotoxicity of the infected cells (Fig. S2), suggesting that the elevated cytotoxicity caused by ΔdinJ was not attributed to an increase in the expression of yafQ. Furthermore, our investigation revealed that the deletion of the dinJ gene did not influence the adhesion and invasion capabilities of S. Enteritidis. This observation contrasts with the impact of deleting the antitoxin gene tomB, which did not affect the adhesion rate but impaired the invasion rate in ΔtomB compared to the WT (41). These findings highlight the complex and multifaceted nature of TA systems in the context of host-pathogen interactions, in which different components may contribute differently to virulence and immune evasion mechanisms. Taken together, the higher cytotoxicity observed in ΔfliCΔdinJ-infected cells suggests a potential role for DinJ in modulating inflammasome activation. Further exploration of these mechanisms will provide valuable insights into the nuanced dynamics of bacterial pathogenesis and immune evasion.
To further elucidate the mechanisms associated with inflammasome activation and immune escape attributes of DinJ, we conducted in vivo analyses. The results showed a higher production of inflammatory cytokines IL-1β (Fig. 6A) and IL-18 (Fig. 6B) in the serum of mice infected with ΔfliCΔdinJ as compared to ΔfliC, indicating an increased inflammasome activation in vivo. Additionally, we observed a significantly lower bacterial load in the liver and spleen of mice infected with ΔfliCΔdinJ relative to ΔfliC (Fig. 6D and E). These findings align with previous studies suggesting that enhanced inflammasome activation and secretion of IL-1β and IL-18 contribute to the clearance of Salmonella in vivo (17, 26). To promote host survival, bacteria have evolved various mechanisms to escape host inflammasome signaling and clearance. Previous studies have shown that Dam can increase inflammasome activation, while GalE and SiiD can inhibit NLRP3 inflammasome activation (17, 26, 42). Altogether, our results indicate that the deletion of dinJ induces the NLRP3 inflammasome activation and triggers the immune responses that facilitate the clearance of S. Enteritidis. What is the mechanism by which dinJ-deficient S. Enteritidis induces NLRP3 inflammasome activation? The expression levels of NLRP3 and NLRC4 showed no significant differences in the BMDMs infected with the ΔfliC or ΔfliCΔdinJ strains (Fig. 2D), indicating the inhibition of NLRP3 inflammasome activation by DinJ is independent of regulating its expression. S. Typhimurium AcnB and S. Enteritidis SiiD have been reported to inhibit NLRP3 inflammasome activation through mtROS (3, 17). Furthermore, the bacterial metabolites potentially serve as the signals that trigger inflammasome activation (3). Therefore, further studies will explore the mechanism of how DinJ inhibits NLRP3 inflammasome activation.
Our previous studies also showed that S. Enteritidis could translocate Dam, GalE, and SiiD proteins into the cells, and modulate the activation of inflammasome. In this study, we also found that DinJ could be translocated into the cells with a low translocation efficiency of 3.9%. TEM-1-based effector translocation assays identified nine candidate effectors of Edwardsiella piscicida, which also showed a low translocation efficiency of around 5% (43). The translocation efficiency of GalE was 4.3%, which also could inhibit the activation of inflammasome (26). TEM-1 assays rely on β-lactamase, which is a relatively large tag that may influence the translocation efficiency of the protein.
This study addressed a significant gap in previous research by investigating the role of toxin-antitoxin systems in NLRP3 inflammasome activation. Despite the well-documented influence of Salmonella flagellin, T3SS, and T4SS effector proteins, along with metabolic signaling pathways, on inflammasome activation (3, 4, 16, 17, 44), the exploration of toxin-antitoxin systems in inflammasome activation was limited. In summary, our studies revealed that the antitoxin protein DinJ, for the first time, can inhibit NLRP3-dependent inflammasome activation. Supported by in vivo experiments, the results uncovered the involvement of DinJ in assisting Salmonella to evade the host immune system by suppressing inflammasome activation. This newly established link between the inhibition of NLRP3 inflammasome activation and bacterial immune escape deepens our understanding of intricate host-pathogen interactions, confirming the significant roles of inflammasome activation in the persistence of Salmonella.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions
The bacterial strains and plasmids used in this study are listed in Table 1. The S. Enteritidis C50336 was obtained from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). S. Enteritidis C50336, E. coli, and derivatives were cultured at 37°C in LB medium with the appropriate antibiotics.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Source |
|---|---|---|
| Escherichia coli strains | ||
| X7213λpir | Host for π requiring plasmids, conjugal donor | (17) |
| Salmonella Enteritidis strains | ||
| C50336 | Wild type | China Medical Culture Collection Center, National Institute for the Control of Pharmaceutical and Biological |
| C50336ΔfliC | C50336, in-frame deletion in fliC (parent strain) | (17) |
| ΔfliCΔdinJ | ΔfliC, in-frame deletion in dinJ | This study |
| ΔfliCΔdinJ::dinJ | ΔfliCΔdinJ with pMMB207 expressing the dinJ gene, Cmr | This study |
| ΔfliCΔdinJ::Vector | ΔfliCΔdinJ with pMMB207, Cmr | This study |
| ΔfliC-pCX340 | ΔfliC with pCX340, Tetr | This study |
| ΔfliC-pCX340-dinJ | ΔfliC with pCX340 expressing the dinJ gene, Tetr | This study |
| Plasmids | ||
| pDM4 | Suicide vector, pir dependent, R6K, SacBR, Cmr | (45, 46) |
| pMMB207 | Expression vector, Cmr | (47) |
| pMMB207-dinJ | pMMB207 derivative containing dinJ, Cmr | This study |
| pCX340 | pBR322 derivative, cloning vector used to fuse effectors to TEM-1-β-lactamase, Tetr | (33, 48) |
| pCX340-dinJ | pCX340 derivative containing dinJ, Cmr | This study |
Construction of dinJ mutant and complemented strains
The deletion mutant strain C50336ΔfliCΔdinJ was constructed by double exchange of homologous recombination with the plasmid pDM4 based on the parental strain C50336ΔfliC (17, 26). Briefly, the primers listed in Table 2 were used to amplify the upstream and downstream fragments of dinJ. The products were cloned into the suicide plasmid pDM4 by the ClonExpress MultiS One Step Cloning Kit (Vazyme, Nanjing, China). The recombined plasmid ΔdinJ::pDM4 was transferred into C50336ΔfliC by conjugation and was screened for the dinJ deletion mutant strain on LB plates with 15% sucrose. The complemented strain was constructed with the plasmid pMMB207. The PCR products of the dinJ gene were ligated into plasmid pCX340 with the restriction enzymes Nde I and Kpn I, and then the recombined plasmids were electroporated into ΔfliCΔdinJ and ΔdinJ for FRET.
TABLE 2.
Primers used in this study
| Primer name | Primer sequence (5′ to 3′) | Target |
|---|---|---|
| dinJ-yafQ-F | AGGGGAAAAGGGTGGTGA | Co-transcribed |
| dinJ-yafQ-R | CGACGAAACGCTGAAGGA | Co-transcribed |
| dinJ-up-F | GAGCGGATAACAATTTGTGGAATCCCGGGATGGAGGTTCACGGCGGGACGCATAA | Deletion mutant |
| dinJ-up-R | CCCATAGTCACATCGATTAGTCTCCTGTACTGTGT | Deletion mutant |
| dinJ-down-F | CTAATCGATGTGACTATGGGGCAAAGGGAAATTGA | Deletion mutant |
| dinJ-down-R | AGCGGAGTGTATATCAAGCTTATCGATACCGCAGGTGTTAGGGCTTATCTATTCC | Deletion mutant |
| dinJ-in-F | ATGTCCTGGCTGAAATGGGG | Deletion mutant |
| dinJ-in-R | ATCAACGCCAGCTTCGCTAT | Deletion mutant |
| dinJ-out-F | AGCCGAACGAAAGCGAAGAAAC | Deletion mutant |
| dinJ-out-R | CCGCAATCTGCTGTTACTCAATCA | Deletion mutant |
| pCX340-F | AGACAATCTGTGTGGGCACTCGACC | FRET |
| pCX340-R | TTCTGAGAATAGTGTATGCGGCGAC | FRET |
| pCX340-dinJ-F | AATAAGGAGGAATAACATATGATGGCTGCAAATGCGCTTGTTCGTG | FRET |
| pCX340-dinJ-R | CGAATTCTCCGCGGAGGTACCGATCCCTAACTGGTCAAACAAA | FRET |
Mice and cell culture conditions
Wild-type (WT) specific pathogen-free (SPF) female C57BL/6 mice were purchased from Vital River Laboratory Animal Technology Co., Ltd (Beijing, China). Casp1-/-, Nlrp3-/-, and Nlrc4-/-mice were bred based on the C57BL/6 strain mice and were purchased from Shanghai Model Organisms Center, Inc (Shanghai, China).
J774A.1 and HeLa cells were purchased from the American Tissue Culture Collection (ATCC, Manassas, VA, USA). The BMDMs were obtained from the femoral and tibial bone marrow of the mice, and their maturation was induced with 25 ng/mL macrophage colony-stimulating factor (M-CSF) (PeproTech, Rocky Hill, NJ, USA). All cells were grown in a cell incubator at 37°C with 5% CO2 and were cultured in complete Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin.
Salmonella infection
Overnight cultured S. Enteritidis and derivatives were diluted into fresh LB medium with a concentration of 1:100 and were cultured at 37°C for 3.5 h. The J774A.1 cells or BMDMs were seeded into 48-, 24-, or 12-well with a concentration of 1 × 105, 2 × 105, or 5 × 105 cells per well and were cultured at 37°C with 5% CO2 for 24 h. The J774A.1 cells or BMDMs were pre-activated for 5 h with lipopolysaccharide (LPS) diluted in Opti-MEM reduced serum medium (Gibco Thermo Fisher Scientific, Waltham, MA, USA). After they were washed twice with phosphate-buffered saline (PBS), the bacteria strains were added to J774A.1 cells and BMDMs at a multiplicity of infection (MOI) of 100:1 and were incubated at 37°C for 1.5 h. The cell supernatant was discarded, and the cells were incubated in Opti-MEM with 50 µg/mL gentamicin at 37°C for another 3 h.
Adhesion and invasion assays
After infecting the J774A.1 cells with the indicated strains for 30 min, the cells were washed twice with PBS and were subsequently incubated in PBS with 0.2% Triton X-100 at 37°C for 15 min. The cell lysate was collected and diluted with PBS for the determination of bacterial adhesion ratio. For assessment of bacterial invasion, the J774A.1 cells were infected with the indicated strains for 30 min, washed twice with PBS, and then incubated in DMEM with 100 µg/mL gentamicin at 37°C for 1 h. Subsequently, the cells were incubated in PBS with 0.2% Triton X-100 at 37°C for 15 min. The resulting cell lysate was collected and diluted with PBS for determination of bacterial invasion ratio.
Cytotoxicity analysis
After the J774A.1 cells or BMDMs were infected with bacteria as described above, the supernatants were harvested for cytotoxicity analysis. Quantification of cytotoxicity was performed using the LDH Cytotoxicity Assay Kit according to the manufacturer’s instructions (Beyotime, Haimen, China).
ELISA
After the J774A.1 cells or BMDMs were infected with bacteria as described above, the supernatants were harvested for quantification of cytokine. Expression of the cytokines IL-1β, IL-18, and IL-6 were quantified using Mouse IL-1 beta/IL-1F2 DuoSet ELISA, Mouse IL-18 DuoSet ELISA, and Mouse IL-6 DuoSet ELISA according to the manufacturer’s manual (R&D Systems, Minneapolis, MN, USA).
Western blotting analysis
Following the infection of J774A.1 cell or BMDMs with bacteria as described above and harvesting of supernatants, the cells were lysed with cell lysis buffer for Western blotting. The supernatants and lysates were mixed, supplemented with a protease inhibitor, and subjected to centrifugation at 2,000 rpm for 5 min to remove the bacteria and cell debris. The cleared supernatants were then mixed with an equal volume of methanol and a 0.25 volume of chloroform. Following centrifugation at 12,000 rpm for 5 min, the upper aqueous phase was removed, and an additional equal volume of methanol was added. The supernatants were discarded after centrifugation, the remaining protein pellets were dried at 55°C for 10 min, and then resuspended in 1 × SDS-PAGE loading buffer. Protein samples were boiled at 100°C for 10 min, separated by 12% SDS-PAGE, transferred onto digested nitrocellulose membranes, and blocked in PBS with 3% skim milk for 1.5 h. Membranes were then incubated with anti-Caspase-1 p10 antibody (AG-20B-0044-C100, AdipoGen, San Diego, CA, USA), anti-NLRP3 antibody (AG-20B-0014-C100, AdipoGen, San Diego, CA, USA), anti-NLRC4 antibody (ab201792, Abcam, Cambridge, UK), anti-IL-1β antibody (12242S, Cell Signaling Technology, Danvers, MA, USA), and anti-β-actin antibody (A5441, Sigma-Aldrich, St. Louis, MO, USA). Following incubation, the membranes were exposed using Amersham Imager 600 Imaging System with enhanced chemiluminescence (ECL) substrate exposure.
FRET
The overnight cultured S. Enteritidis strains ΔfliC-pCX340 and Δ fliC-pCX340-dinJ were diluted into fresh LB medium with a concentration of 1:100 and were cultured for 3.5 h. Bacteria were washed twice with DMEM and infected HeLa cells with an MOI of 100:1. The bacteria were removed after infection for 3 h, and the HeLa cells were washed four times with DMEM before being subjected to another 4-h infection in DMEM. Following a single wash with DMEM, CCF2/AM was added, and the cells were incubated for 2 h in the dark. Cell images were acquired using Leica confocal microscope with excitation at 405 nm and emission at 450–470 nm and 520–540 nm.
Mouse infection
Female C57BL/6 mice (6–8 wk) were randomly divided into three groups, each consisting of eight mice. After a 4-h period of dietary restriction, each group orally received 7.5 mg of streptomycin, followed by a 20-h restoration of the regular diet. After another 4-h dietary restriction, each group was orally infected with 300 µL of ΔfliC or ΔfliCΔdinJ at a dose of 5 × 106 CFU per mouse. Control mice received 300 µL PBS under the same conditions. After infection for 5 d, the mice were euthanized, and the serum, liver, and spleen samples were collected. The levels of IL-1β, IL-18, and IL-6 in the serum were quantified using ELISA. The liver and spleen tissues were homogenized and diluted in PBS to calculate the bacterial load in the organs.
Statistical analysis
All experiments were conducted a minimum of three times. All results were expressed as the mean ± SEM using GraphPad Prism 8.0 software. The statistical analysis used a Student’s t-test; *** represents a P value < 0.001, ** is a P value < 0.01, and * is a P value < 0.05.
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (32161143011, 31972685, and 31920103015).
Contributor Information
Xinan Jiao, Email: jiao@yzu.edu.cn.
Zhiming Pan, Email: zmpan@yzu.edu.cn.
Manuela Raffatellu, University of California San Diego School of Medicine, La Jolla, California, USA.
ETHICS APPROVAL
All animal experiments were approved by the Animal Welfare and Ethics Committees of Yangzhou University and complied with the guideline of the Institutional Administrative Committee and Ethics Committee of Laboratory Animals (IACU license number: YZUEWLL-201811-001). All animals were handled with care to minimize distress during the experiments.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/iai.00505-23.
Impact of DinJ on S. Enteritidis growth, adhesion, and invasion.
Cytotoxicity of S. Enteritidis yafQ deletion mutant strain.
Legends for Fig. S1 and S2.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Rathinam VAK, Fitzgerald KA. 2016. Inflammasome complexes: emerging mechanisms and effector functions. Cell 165:792–800. doi: 10.1016/j.cell.2016.03.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, Hardy LL, Garceau V, Sweet MJ, Ross IL, Hume DA, Stacey KJ. 2009. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323:1057–1060. doi: 10.1126/science.1169841 [DOI] [PubMed] [Google Scholar]
- 3. Wynosky-Dolfi MA, Snyder AG, Philip NH, Doonan PJ, Poffenberger MC, Avizonis D, Zwack EE, Riblett AM, Hu B, Strowig T, Flavell RA, Jones RG, Freedman BD, Brodsky IE. 2014. Oxidative metabolism enables Salmonella evasion of the NLRP3 inflammasome. J Exp Med 211:653–668. doi: 10.1084/jem.20130627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhao Y, Yang JL, Shi JJ, Gong YN, Lu QH, Xu H, Liu LP, Shao F. 2011. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477:596–600. doi: 10.1038/nature10510 [DOI] [PubMed] [Google Scholar]
- 5. Oliveira SHP, Canetti C, Ribeiro RA, Cunha FQ. 2008. Neutrophil migration induced by IL-1beta depends upon LTB4 released by macrophages and upon TNF-alpha and IL-1beta released by mast cells. Inflammation 31:36–46. doi: 10.1007/s10753-007-9047-x [DOI] [PubMed] [Google Scholar]
- 6. Shi JJ, Zhao Y, Wang K, Shi XY, Wang Y, Huang HW, Zhuang YH, Cai T, Wang FC, Shao F. 2015. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526:660–665. doi: 10.1038/nature15514 [DOI] [PubMed] [Google Scholar]
- 7. Higa N, Toma C, Koizumi Y, Nakasone N, Nohara T, Masumoto J, Kodama T, Iida T, Suzuki T. 2013. Vibrio parahaemolyticus effector proteins suppress inflammasome activation by interfering with host autophagy signaling. PLoS Pathog 9:e1003142. doi: 10.1371/journal.ppat.1003142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ratner D, Orning MPA, Proulx MK, Wang D, Gavrilin MA, Wewers MD, Alnemri ES, Johnson PF, Lee B, Mecsas J, Kayagaki N, Goguen JD, Lien E. 2016. The Yersinia pestis effector YopM inhibits pyrin inflammasome activation. PLoS Pathog 12:e1006035. doi: 10.1371/journal.ppat.1006035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jiang JT, Wang WW, Sun F, Zhang YX, Liu Q, Yang DH. 2021. Bacterial infection reinforces host metabolic flux from arginine to spermine for NLRP3 inflammasome evasion. Cell Rep 34:108832. doi: 10.1016/j.celrep.2021.108832 [DOI] [PubMed] [Google Scholar]
- 10. Bierschenk D, Boucher D, Schroder K. 2017. Salmonella-induced inflammasome activation in humans. Mol Immunol 86:38–43. doi: 10.1016/j.molimm.2016.11.009 [DOI] [PubMed] [Google Scholar]
- 11. Crowley SM, Knodler LA, Vallance BA. 2016. Salmonella and the inflammasome: battle for intracellular dominance. Curr Top Microbiol Immunol 397:43–67. doi: 10.1007/978-3-319-41171-2_3 [DOI] [PubMed] [Google Scholar]
- 12. Zhang K, Riba A, Nietschke M, Torow N, Repnik U, Pütz A, Fulde M, Dupont A, Hensel M, Hornef M. 2018. Minimal SPI1-T3SS effector requirement for Salmonella enterocyte invasion and intracellular proliferation in vivo. PLoS Pathog 14:e1006925. doi: 10.1371/journal.ppat.1006925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang L, Wang P, Song X, Zhang H, Ma S, Wang J, Li W, Lv R, Liu X, Ma S, Yan J, Zhou H, Huang D, Cheng Z, Yang C, Feng L, Wang L. 2021. Salmonella Typhimurium reprograms macrophage metabolism via T3SS effector SopE2 to promote intracellular replication and virulence. Nat Commun 12:879. doi: 10.1038/s41467-021-21186-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bierschenk D, Monteleone M, Moghaddas F, Baker PJ, Masters SL, Boucher D, Schroder K. 2019. The Salmonella pathogenicity island-2 subverts human NLRP3 and NLRC4 inflammasome responses. J Leukoc Biol 105:401–410. doi: 10.1002/JLB.MA0318-112RR [DOI] [PubMed] [Google Scholar]
- 15. Naseer N, Egan MS, Reyes Ruiz VM, Scott WP, Hunter EN, Demissie T, Rauch I, Brodsky IE, Shin S. 2022. Human NAIP/NLRC4 and NLRP3 inflammasomes detect Salmonella type III secretion system activities to restrict intracellular bacterial replication. PLoS Pathog 18:e1009718. doi: 10.1371/journal.ppat.1009718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hu GQ, Song PX, Chen W, Qi S, Yu SX, Du CT, Deng XM, Ouyang HS, Yang YJ. 2017. Cirtical role for Salmonella effector SopB in regulating inflammasome activation. Mol Immunol 90:280–286. doi: 10.1016/j.molimm.2017.07.011 [DOI] [PubMed] [Google Scholar]
- 17. Guo YX, Gu D, Huang TT, Li A, Zhou Y, Kang XL, Meng C, Xiong D, Song L, Jiao XA, Pan ZM. 2023. Salmonella Enteritidis T1SS protein SiiD inhibits NLRP3 inflammasome activation via repressing the mtROS-ASC dependent pathway. PLoS Pathog 19:e1011381. doi: 10.1371/journal.ppat.1011381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jurėnas D, Fraikin N, Goormaghtigh F, Van Melderen L. 2022. Biology and evolution of bacterial toxin-antitoxin systems. Nat Rev Microbiol 20:335–350. doi: 10.1038/s41579-021-00661-1 [DOI] [PubMed] [Google Scholar]
- 19. Hu Y, Benedik MJ, Wood TK. 2012. Antitoxin DinJ influences the general stress response through transcript stabilizer CspE. Environ Microbiol 14:669–679. doi: 10.1111/j.1462-2920.2011.02618.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Di Cesare A, Losasso C, Barco L, Eckert EM, Conficoni D, Sarasini G, Corno G, Ricci A. 2016. Diverse distribution of toxin-antitoxin II systems in Salmonella enterica serovars. Sci Rep 6:28759. doi: 10.1038/srep28759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Motiejūnaite R, Armalyte J, Markuckas A, Suziedeliene E. 2007. Escherichia coli dinJ-yafQ genes act as a toxin-antitoxin module. FEMS Microbiol Lett 268:112–119. doi: 10.1111/j.1574-6968.2006.00563.x [DOI] [PubMed] [Google Scholar]
- 22. Ruangprasert A, Maehigashi T, Miles SJ, Dunham CM. 2017. Importance of the E. coli DinJ antitoxin carboxy terminus for toxin suppression and regulated proteolysis. Mol Microbiol 104:65–77. doi: 10.1111/mmi.13641 [DOI] [PubMed] [Google Scholar]
- 23. Ruangprasert A, Maehigashi T, Miles SJ, Giridharan N, Liu JX, Dunham CM. 2014. Mechanisms of toxin inhibition and transcriptional repression by Escherichia coli DinJ-YafQ. J Biol Chem 289:20559–20569. doi: 10.1074/jbc.M114.573006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Masuda Y, Sakamoto E, Honjoh KI, Miyamoto T. 2020. Role of toxin-antitoxin-regulated persister population and indole in bacterial heat tolerance. Appl Environ Microbiol 86:e00935-20. doi: 10.1128/AEM.00935-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wiradiputra MRD, Khuntayaporn P, Thirapanmethee K, Chomnawang MT. 2022. Toxin-antitoxin systems: a key role on persister formation in Salmonella enterica serovar Typhimurium. Infect Drug Resist 15:5813–5829. doi: 10.2147/IDR.S378157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang T, Gu D, Guo Y, Li A, Kang X, Jiao X, Pan Z. 2022. Salmonella Enteritidis GalE protein inhibits LPS-induced NLRP3 inflammasome activation. Microorganisms 10:911. doi: 10.3390/microorganisms10050911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pandeya A, Zhang Y, Cui J, Yang L, Li J, Zhang G, Wu C, Li Z, Wei Y. 2023. Inflammasome activation and pyroptosis mediate coagulopathy and inflammation in Salmonella systemic infection. Microbiol Res 275:127460. doi: 10.1016/j.micres.2023.127460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. 2010. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med 207:1745–1755. doi: 10.1084/jem.20100257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Franchi L. 2011. Role of inflammasomes in Salmonella infection. Front Microbiol 2:8. doi: 10.3389/fmicb.2011.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miao EA, Rajan JV. 2011. Salmonella and Caspase-1: a complex interplay of detection and evasion. Front Microbiol 2:85. doi: 10.3389/fmicb.2011.00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang J, Zhao Y, Shi J, Shao F. 2013. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci U S A 110:14408–14413. doi: 10.1073/pnas.1306376110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A. 2006. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol 7:569–575. doi: 10.1038/ni1344 [DOI] [PubMed] [Google Scholar]
- 33. Winter SE, Winter MG, Atluri V, Poon V, Romão EL, Tsolis RM, Bäumler AJ. 2015. The flagellar regulator TviA reduces pyroptosis by Salmonella enterica serovar Typhi. Infect Immun 83:1546–1555. doi: 10.1128/IAI.02803-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozören N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, Grant EP, Núñez G. 2006. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in Salmonella-infected macrophages. Nat Immunol 7:576–582. doi: 10.1038/ni1346 [DOI] [PubMed] [Google Scholar]
- 35. Gram AM, Wright JA, Pickering RJ, Lam NL, Booty LM, Webster SJ, Bryant CE. 2021. Salmonella flagellin activates NAIP/NLRC4 and canonical NLRP3 inflammasomes in human macrophages. J Immunol 206:631–640. doi: 10.4049/jimmunol.2000382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sonika S, Singh S, Mishra S, Verma S. 2023. Toxin-antitoxin systems in bacterial pathogenesis. Heliyon 9:e14220. doi: 10.1016/j.heliyon.2023.e14220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jaiswal S, Paul P, Padhi C, Ray S, Ryan D, Dash S, Suar M. 2016. The Hha-TomB toxin-antitoxin system shows conditional toxicity and promotes persister cell formation by inhibiting apoptosis-like death in S. Typhimurium. Sci Rep 6:38204. doi: 10.1038/srep38204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barrios AFG, Zuo R, Ren D, Wood TK. 2006. Hha, YbaJ, and OmpA regulate Escherichia coli K12 biofilm formation and conjugation plasmids abolish motility. Biotechnol Bioeng 93:188–200. doi: 10.1002/bit.20681 [DOI] [PubMed] [Google Scholar]
- 39. García-Contreras R, Zhang XS, Kim Y, Wood TK. 2008. Protein translation and cell death: the role of rare tRNAs in biofilm formation and in activating dormant phage killer genes. PLoS One 3:e2394. doi: 10.1371/journal.pone.0002394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ryan D, Pati NB, Ojha UK, Padhi C, Ray S, Jaiswal S, Singh GP, Mannala GK, Schultze T, Chakraborty T, Suar M. 2015. Global transcriptome and mutagenic analyses of the acid tolerance response of Salmonella enterica serovar Typhimurium. Appl Environ Microbiol 81:8054–8065. doi: 10.1128/AEM.02172-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Paul P, Patel P, Verma SK, Mishra P, Sahu BR, Panda PK, Kushwaha GS, Senapati S, Misra N, Suar M. 2022. The Hha-TomB toxin-antitoxin module in Salmonella enterica serovar Typhimurium limits its intracellular survival profile and regulates host immune response. Cell Biol Toxicol 38:111–127. doi: 10.1007/s10565-021-09587-z [DOI] [PubMed] [Google Scholar]
- 42. Guo Y, Gu D, Huang T, Cao L, Zhu X, Zhou Y, Wang K, Kang X, Meng C, Jiao X, Pan Z. 2020. Essential role of Salmonella Enteritidis DNA adenine methylase in modulating inflammasome activation. BMC Microbiol 20:226. doi: 10.1186/s12866-020-01919-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu Y, Zhao L, Yang M, Yin K, Zhou X, Leung KY, Liu Q, Zhang Y, Wang Q. 2017. Transcriptomic dissection of the horizontally acquired response regulator EsrB reveals its global regulatory roles in the physiological adaptation and activation of T3SS and the cognate effector repertoire in Edwardsiella piscicida during infection toward turbot. Virulence 8:1355–1377. doi: 10.1080/21505594.2017.1323157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kofoed EM, Vance RE. 2011. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477:592–595. doi: 10.1038/nature10394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang SY, Lauritz J, Jass J, Milton DL. 2002. A ToxR homolog from Vibrio anguillarum serotype O1 regulates its own production, bile resistance, and biofilm formation. J Bacteriol 184:1630–1639. doi: 10.1128/JB.184.6.1630-1639.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Milton DL, O’Toole R, Horstedt P, Wolf-Watz H. 1996. Flagellin A is essential for the virulence of Vibrio anguillarum. J Bacteriol 178:1310–1319. doi: 10.1128/jb.178.5.1310-1319.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Morales VM, Bäckman A, Bagdasarian M. 1991. A series of wide-host-range low-copy-number vectors that allow direct screening for recombinants. Gene 97:39–47. doi: 10.1016/0378-1119(91)90007-x [DOI] [PubMed] [Google Scholar]
- 48. Charpentier X, Oswald E. 2004. Identification of the secretion and translocation domain of the enteropathogenic and enterohemorrhagic Escherichia coli effector Cif, using TEM-1 beta-lactamase as a new fluorescence-based reporter. J Bacteriol 186:5486–5495. doi: 10.1128/JB.186.16.5486-5495.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Impact of DinJ on S. Enteritidis growth, adhesion, and invasion.
Cytotoxicity of S. Enteritidis yafQ deletion mutant strain.
Legends for Fig. S1 and S2.