

Abstract
Necrotizing enterocolitis (NEC) is a severe and potentially fatal intestinal disease that has been difficult to study due to its complex pathogenesis, which remains incompletely understood. The pathophysiology of NEC includes disruption of intestinal tight junctions, increased gut barrier permeability, epithelial cell death, microbial dysbiosis, and dysregulated inflammation. Traditional tools to study NEC include animal models, cell lines, and human or mouse intestinal organoids. While studies using those model systems have improved the field’s understanding of disease pathophysiology, their ability to recapitulate the complexity of human NEC is limited. An improved in vitro model of NEC using microfluidic technology, named NEC-on-a-chip, has now been developed. The NEC-on-a-chip model consists of a microfluidic device seeded with intestinal enteroids derived from a preterm neonate, co-cultured with human endothelial cells and the microbiome from an infant with severe NEC. This model is a valuable tool for mechanistic studies into the pathophysiology of NEC and a new resource for drug discovery testing for neonatal intestinal diseases. In this manuscript, a detailed description of the NEC-on-a-chip model will be provided.
Introduction
Necrotizing enterocolitis (NEC) affects preterm infants, with an incidence of up to 10% in those born weighing < 1500 g1. The pathophysiology of NEC is complex and includes damage to the intestinal epithelium, disruption of intestinal tight junctions, increased gut barrier permeability, immune dysregulation, and epithelial cell death2, 3. Our understanding of the mechanisms involved in the pathogenesis of NEC remains incomplete, and despite decades of research, there are still no effective targeted therapies.
A significant barrier to advancing NEC research is the limited availability and small size of primary intestinal tissue isolated from human infants. Intestinal tissue resected from infants with NEC is often necrotic and severely damaged, which complicates studies into mechanisms that precede disease onset. For example, the small intestine of infants with NEC is inundated with immune cells, and a reduced number of intestinal stem cells, decreased epithelial cell proliferation, and increased epithelial cell apoptosis are also observed4, 5, 6, 7. This leads to difficulties in culturing intestinal epithelial cells from these samples and in isolating RNA and proteins, which can be degraded in this hostile inflammatory environment. Additionally, since the disease process is already advanced in infants with surgical NEC, mechanistic studies into factors that induce disease are unfeasible. These limitations have led to a reliance on animal models for mechanistic studies of NEC.
Animal models of NEC have been established for mice, rats, piglets, rabbits, and baboons5, 8, 9, 11. A strength of animal models is that NEC-like intestinal disease is induced by factors associated with NEC onset in humans, including a dysbiotic microbiome, repeated episodes of hypoxia, and the absence of breast milk feeds5, 8, 10, 11. In addition, the inflammatory response and pathologic changes observed during experimental NEC parallel human disease5, 9, 12. While these models mimic many of the features of human NEC, there are inherent differences between the pathophysiology of NEC in animals and humans. For example, the murine model of NEC is induced in mice born full-term, and although their intestinal development is incomplete, the pathophysiology of NEC is inherently different in this clinical context. Murine intestinal gene expression at birth is similar to a pre-viable human fetus and does not approximate that of a preterm neonate of 22–24 weeks gestation until day 14 (P14)13. This confounds the murine NEC model because intestinal injury cannot generally be induced in mice after P10. In addition, inbred strains of mice lack the immunologic14 and microbiologic diversity of human neonates15, which serves as another confounding factor. Thus, increased incorporation of primary human samples into NEC research improves the clinical relevance of studies in this field.
Studies into the mechanisms of NEC in vitro have traditionally utilized monotypic cell lines derived from adult intestinal cancer cells, such as colorectal adenocarcinoma (Caco2) and human colon adenocarcinoma (HT-29) cells16. These models are convenient but limited in physiologic relevance due to their growth from adult cancer cells, non-polarized architecture, and phenotypic changes related to repeated passages in culture. Intestinal enteroids improve upon those models since they can be grown from the crypts of intestinal tissue, differentiated into all intestinal epithelial subtypes, and form a three-dimensional (3D) villus-like structure17, 18, 19, 20. Recently, intestinal enteroids have been combined with microfluidic technology to develop a small intestine-on-a-chip model and provide a more physiologically relevant in vitro model system21.
The initial organ-on-a-chip microfluidic devices were introduced in the early 2000s22, 23, 24. The first organ-on-a-chip model was the human breathing lung-on-a-chip25. This was followed by numerous single-organ models such as intestine21, liver26, kidneys27, bone marrow28, blood-brain barrier29, and heart30. These organ-on-a-chip models have been used to study acute, chronic, and rare diseases, including acute radiation syndrome,31 chronic obstructive pulmonary disease,32 and neurodegenerative diseases33. The polarized nature of the cells on these chips and the presence of two cellular compartments separated by a porous membrane allows for the modeling of complex physiologic processes such as perfusion, chemical concentration gradients, and immune cell chemotaxis34, 35. These microfluidic systems thus provide a new tool for studying the pathophysiology and mechanisms of human disease.
The small intestine-on-a-chip model was described by Kasendra et al. in 2018, who utilized pediatric (ages 10–14 years old) small intestinal biopsy specimens differentiated into enteroids and cultured on a microfluidic device21. Vascular endothelial cells, continuous media flow, and stretch/relaxation were also incorporated into this model. They observed intestinal epithelial subtype differentiation, formation of 3D villus-like axes, mucus production, and small intestinal gene expression patterns21. This microfluidic model was applied to neonatal disease with the development of the NEC-on-a-chip system, which incorporates neonatal intestinal enteroids, endothelial cells, and the microbiome from a neonate with NEC36. NEC-on-a-chip recapitulates many of the critical features of human NEC, including inflammatory gene expression, loss of specialized epithelial cells, and reduced gut barrier function36. Thus, this model has numerous applications in the study of NEC, including mechanistic studies and drug discovery. In this manuscript, a detailed protocol for the performance of the NEC-on-a-chip model is provided.
Protocol
Enteroids were derived from small intestinal samples from premature infants (born at 22 to 36 weeks gestation) obtained at the time of surgery for NEC or other intestinal conditions with non-inflammatory etiologies. All specimen collection and processing was performed after informed consent and approval from the Institutional Review Boards at Washington University in St. Louis (IRB Protocol numbers 201706182 and 201804040) and the University of North Carolina at Chapel Hill (IRB protocol number 21-3134).
1. Isolation and plating of crypts from human neonatal small intestine to establish enteroids
- Media preparation
- Prepare all media required as described in Table 1. Ensure aseptic technique is used for the preparation of all media. Filter sterilize media using a 0.2 μm filter and store at 4 °C until use.
- Washing and mincing intestinal tissue
- Place the cell culture matrix hydrogel on ice to thaw. Obtain human intestinal tissue from the operating room. Immediately place the sample in 5 mL of ice-cold tissue washing media to inactivate endogenous proteases.
- Flush intestinal contents with ice-cold Dulbecco’s phosphate-buffered saline (D-PBS) using a 10 mL syringe fitted with a trimmed P1000 pipette tip. Transfer the intestinal tissue to a 35 mm dish containing 5 mL of ice-cold D-PBS.
- Cut the intestinal tissue longitudinally to expose the lumen, and cut it into small pieces using fine scissors. Rinse intestinal tissue by gently agitating in D-PBS in the tissue culture dish.
- Transfer tissue fragments to a clean 35 mm dish. Mince tissue with fine scissors. The optimal size is 0.5 cm2 per piece. The intestinal tissue should pass through a trimmed 1 mL pipette tip.
- Dissociating intestinal tissue
- Add 1 mL of prewarmed collagenase I to the tissue. Mix tissue with collagenase by pipetting 10–15 times, and transfer to a 15 mL conical tube.
- Secure the tube horizontally on a shaker set at 50 rpm. Shake for 20 min at room temperature.
- After the incubation, remove the tubes from the shaker, and resuspend the solution by pipetting 10–15 times. Optional: Remove a small aliquot to verify the presence of single crypts using phase contrast microscopy.
- Isolation of intestinal crypts
- Filter crypts through a 70 μm strainer into a 50 mL conical tube on ice. Wash the strainer with 9 mL of ice-cold tissue washing media. Transfer the filtrate to a 15 mL conical tube.
- Centrifuge at 300 x g for 10 min at 4 °C, and gently remove the supernatant. There will be approximately 200 μL of media remaining in the tube. Resuspend the pellet in 5 mL of the ice-cold tissue washing media.
- Centrifuge at 300 x g for 10 min at 4 °C. Resuspend the pellet in 1 mL of tissue washing media and transfer to a 1.5 mL microcentrifuge tube.
- Centrifuge at 300 x g for 5 min at 4 °C to pellet the crypts. Carefully and completely aspirate the supernatant as much as possible using a pipette. Place the tube on ice.
- Plating intestinal crypts
-
Resuspend the pellet in cell culture matrix hydrogel (20 μL/well of a 48-well tissue culture plate) using a pre-chilled pipette tip to make a homogenous suspension of crypts while the tube remains on ice. Avoid making bubbles when pipetting. Keep the tube on ice until ready for use.NOTE: The cell culture matrix hydrogel will polymerize at temperatures above 8 °C. Crypts isolated from a piece of intestinal tissue 0.5 cm-1 cm in length are generally plated in 10 wells of a 48-well tissue culture plate.
- Plate the crypt/cell culture matrix hydrogel suspension in a pre-warmed 48-well tissue culture plate. Warm the plate to aid in the solidification of the matrix. Place the suspension in the center of the well. Avoid bubbles when plating.
- Incubate the plates for 20 min at 37 °C to polymerize the matrix. It is essential for the cell culture matrix hydrogel to fully polymerize before adding media.
- After the matrix polymerizes, add 300 μL of prewarmed 50% L-WRN conditioned media to each well. Incubate at 37 °C, 5% CO2.
-
Change media every 2 to 3 days. Replace with warm 50% L-WRN conditioned media. Passage every 7 to 10 days. Passage enteroids when they are 50%−90% confluent and exhibit bud formation.NOTE: The media may need to be changed more frequently if it becomes yellow. The frequency of passage will depend on the enteroid density and the rate of growth of a particular patient’s cells. Enteroids will need to be passaged if their centers turn dark in appearance.
-
- Passaging the enteroids
- Gently aspirate media from the wells. Carefully wash wells with 500 μL of warm D-PBS. Be careful not to disturb the matrix.
- Add 250 μL of cold cell recovery solution to each well. Scratch the bottom of each well with a fresh pipette tip to disrupt the cell culture matrix hydrogel. Incubate plate on ice for 30 min.
- Dissociate enteroids by vigorous pipetting (~25–50 times with P200 pipette) and add 500 μL of tissue washing media.
- Transfer enteroid suspension into a 15 mL tube and add an additional 5 mL of cold tissue washing media. Pipette gently to form a homogenous solution.
-
Centrifuge at 400 x g for 5 min at 4 °C to pellet the enteroids. Place the tube on ice and aspirate the supernatant as completely as possible. There will be approximately 30–60 μL remaining in the tube.NOTE: If there are difficulties breaking up the enteroids with pipetting, 0.25% trypsin-EDTA or enzymatic dissociation reagent can be utilized to facilitate this process.
- Resuspend enteroids in the residual volume of wash media with a P200 pipette. Avoid bubble formation while pipetting.
- Add 20 μL of cell culture matrix hydrogel per new well of a 48 well plate to the enteroid suspension. Split the enteroids 1:3, 1:4, or 1:5, depending on their density.
- Add the suspension to a pre-warmed 48-well plate. Incubate the plate at 37 °C for 20 min to solidify the matrix.
- After polymerization, add 300 μL of pre-warmed 50% L-WRN conditioned media to each well. Incubate at 37 °C, 5% CO2.
Table 1. Media composition for crypt isolation and enteroid growth.
Refer to Van Dussen et al.40 and Miyoshi et al.41 for detailed methods describing the preparation of L-WRN conditioned media.
| Media | Reagent | Volume | Final Concentration |
|---|---|---|---|
| Tissue Washing Media | 500 mL | ||
| Advanced DMEM/F12 | 435 mL | ||
| 1 M HEPES | 5.0 mL | 10 mM | |
| 200 mM L-glutamine | 5.0 mL | 2 mM | |
| 10,000 U/mL penicillin/streptomycin | 5.0 mL | 100 U/mL | |
| Fetal bovine serum (Heat Inactivated) | 50 mL | 10% | |
| L-WRN primary culture media | 637.5 mL | ||
| Advanced DMEM/F12 | 500 mL | ||
| 200 mM L-glutamine | 6.25 mL | 2 mM | |
| 10,000 U/mL penicillin/streptomycin | 6.25 mL | 100 U/mL | |
| Fetal bovine serum (Heat Inactivated) | 125 mL | 20% | |
| 50% L-WRN conditioned media | 1000 mL | ||
| L-WRN conditioned media | 500 mL | 50% | |
| L-WRN primary culture media | 500 mL | 50% |
2. Neonatal intestine-on-a-chip model
NOTE: For detailed instructions on handling the microfluidic chips and using this equipment, please see the manufacturer’s duodenum intestine-chip culture protocol37.
- Media preparation
- Prepare all media required as described in Table 2. Ensure aseptic technique is used. Filter sterilize media using a 0.2 μm filter and store at 4 °C until use.
- Day (−3): Thawing and expanding human intestinal microvascular endothelial cells (HIMECs)
- Thaw a vial of HIMECs and plate per the manufacturer’s recommendations.
- Coat a 25 cm2 flask with gelatin-based coating solution for 2 min. Remove excess solution. Add HIMECs (approximately 0.5–1 × 106 cells) resuspended in 6 mL of HIMEC media to the flask. Incubate at 37 °C, 5% CO2.
- Change HIMEC media every 48 h. Add HIMECs to the endothelial (bottom) channel of the chip within 48–72 h of becoming 100% confluent.
- Day (−1): Activating and coating the chips prior to the addition of the enteroids
-
Reconstitute the chip activation reagent 1 (CR-1) powder to make CR-1 solution. Ensure CR-1 powder and chip activation reagent 2 (CR-2) solution are at room temperature for this step.NOTE: Keep the CR-1 powder and the CR-1 solution that is prepared in the following steps protected from light at all times. The light should be off in the hood for these steps.
- Place the chip and chip carrier into the chip cradle, then place them in a tissue culture dish in the cell culture hood. See the manufacturer’s protocol for the proper technique for chip placement in the carrier37.
- Add 1 mL of CR-2 solution to the vial of CR-1 powder and then directly transfer the solution from the CR-1 powder vial to a 15 mL tube wrapped in aluminum foil to protect it from light. Do not mix the solution with the pipette. The goal is to avoid bubble formation.
- Add another 1 mL of CR-2 solution to the CR-1 vial, and transfer to the 15 mL tube. Repeat for a total of 4 rinses and a total of 4 mL of CR-2 rinsed through the CR-1 vial. Add the cap to the vial and invert for the last rinse to remove any residual powder.
- Add an additional 6 mL of CR-2 to the 15 mL tube containing CR-1 for a final volume of 10 mL (0.5 mg/mL). Mix this solution with a pipette without introducing bubbles. Ensure that CR-1 is fully dissolved prior to proceeding to the next step.
-
Addition of CR-1 solution to chipNOTE: It is critical to avoid introducing bubbles to the channels while adding these solutions. CR-1 solution should remain protected from light for this step.
-
Using a 200 μL pipette tip, add 20 μL of CR-1 solution to the bottom channel inlet until the solution begins to exit the bottom channel outlet. Add 50 μL of CR-1 solution through the top channel inlet until the solution begins to exit the top channel outlet. Remove any excess CR-1 solution from the surface of the chip by gentle aspiration.NOTE: Solutions should always be added to the bottom channel before the top channel. If bubbles are noted, flush both channels with CR-1 solution and repeat 2.3.2.1.
- If the lid of the tissue culture dish containing the chips is in place, remove it for this step. Place chips under UV light for 15 min. Remove CR-1 from the top and bottom channels.
- Repeat steps 2.3.2.1 through 2.3.2.2 for a total of two UV activation steps. After this step, the chips are no longer light-sensitive.
- Remove CR-1 from the top and bottom channels. Wash both channels with 100 μL of CR-2 solution. Remove the CR-2 solution from the top and bottom channels.
- Wash both channels with 100 μL of sterile D-PBS. Repeat the washes 2x. Add 100 μL of D-PBS to both channels. Remove excess from the surface of the chips but leave the channels full.
-
-
Coat channels with the extracellular matrix.
- Thaw fibronectin, collagen IV, and cell culture matrix hydrogel on ice immediately prior to this step and keep these solutions on ice for the remainder of this section.
-
Prepare the following extracellular matrix (ECM) solutions for the top and bottom channels of the chip:Top Channel: 100 μL of 200 μg/mL of type IV collagen and 100 μg/mL of cell culture matrix hydrogel in D-PBS per chip.Bottom Channel: 100 μL of 200 μg/mL of type IV collagen and 30 μg/mL of fibronectin in D-PBS per chip.
- Completely remove D-PBS from the top and bottom channels. Add 50 μL of ECM to the bottom channel inlet until a drop appears on the outlet. Repeat for the upper channel. Make sure to use the appropriate ECM solutions for each channel and add the solution to the bottom channel first.
- Add D-PBS to the chip cradle reservoir and replace the lid on the tissue culture plate. Place the chips in a humidified 37 °C, 5% CO2 incubator overnight.
-
- Day (0): Seeding chips with HIMECs and enteroids
- Vacuum equilibration of media
- Add the required volume of HIMEC media and chip expansion media, to complete the experiment, to a sterile 50 mL conical tube. Equilibrate media to 37 °C in a water bath for at least 1 h.
- Connect the vacuum filtration device to 50 mL tubes containing warmed media. The tubes should be oriented with the prewarmed media at the bottom of the 50 mL conical tube. Vacuum at −70 kPa or higher for 10 s.
- Flip the tubes so that the media moves through the filter. Continue to vacuum for 5 min. Upon completion of vacuum filtration, remove the vacuum filtration device and place the 50 mL tubes containing media into the 37 °C incubator.
- Washing the chips
-
Wash the endothelial channel by flushing with 100 μL of prewarmed HIMEC media.Wash the epithelial channel by flushing with 100 μL of prewarmed chip expansion media. Remove any media that spread onto the surface of the chips.
- Repeat washing step. Media should remain in the channels as well as inlet and outlet ports after these flushes. Replace the lid of the culture dish containing the chips and return to the incubator until ready for use.
-
- Harvesting HIMECs
- Aspirate media from the 25 cm2 flask containing HIMECs. Wash the monolayer gently with D-PBS. Add 2 mL of prewarmed 0.05% trypsin-EDTA to the flask and rinse gently over the monolayer.
- Incubate the flask at 37 °C for 2–5 min. Neutralize trypsin with 10 mL of HIMEC media. Resuspend cells in media and transfer to a 15 mL tube.
- Spin at cells 150 x g for 5 min. at 4 °C. Resuspend cells in 200 μL of HIMEC media.
- Remove 10 μL of cells and place the tube on ice. Count cells using a hemocytometer or automated cell counter. The desired concentration of HIMECs is 6 × 106 to 8 × 106 cells/mL.
- Seeding HIMECs onto the chip
- Remove the chip from the incubator and bring it into the hood. Aspirate any media that may have leaked on the surface of the chip.
- Flush the epithelial (top) channel of the chip with 100 μL of prewarmed chip expansion media. Flush the endothelial (bottom) channel of the chip with 100 μL of prewarmed HIMEC media.
-
Gently resuspend HIMECs to obtain a homogenous distribution. Completely remove the media from the endothelial (bottom) channel. Add 10 μL of HIMECs (~36,000 cells/chip) to the endothelial (bottom) channel.NOTE: If it is difficult to fill the HIMEC channel without bubble formation when using 10 μL of HIMECs, increase the volume of cells added. It should be the same number of cells per chip.
- Add additional organoid growth media to the epithelial (top) channel if it is not full. Check the seeding density of the HIMECs. If they are not uniformly distributed and do not cover 80%−90% of the chip surface, flush the channel and repeat seeding.
- Invert the chip into the chip cradle in the cell culture dish. Add D-PBS to the reservoir in the chip cradle. Replace the lid to the cell culture dish and place it in the incubator.
- Leave the chip inverted at 37 °C for 2–3 h to allow HIMECs to attach to the chip membrane. After incubation is completed, return the chip/chip cradle to an upright position.
- Gently wash the endothelial (bottom) channel with 100 μL of warm HIMEC media. Leave media in the channel. Gently wash the epithelial (top) channel with 100 μL of organoid growth media. Leave media in the channel.
- Return chips to the 37 °C incubator while completing the next steps.
-
Harvesting enteroids and seeding on chipNOTE: Chips are seeded with enteroids that were split 10–14 days prior, are 50%−75% confluent, and at passage 7 and 20. Please see Figure 2 for the appearance of enteroids on Day 0. The time required for enteroids to be ready for seeding will depend on the rate of growth of a particular patient’s cells.
- Gently aspirate media from the wells. Carefully wash wells containing cell culture matrix hydrogel with 500 μL of warm D-PBS. Be careful not to disturb the cell culture matrix hydrogel.
- Add 250 μL of cold cell recovery solution to each well. Scratch the bottom of each well with a fresh pipette tip to disrupt cell culture matrix hydrogel. Add contents of wells to a cold 15 mL tube on ice.
- Incubate tube on ice for 45 min, and invert the tube every 3–5 min. During this step, prepare enteroid dissociation media. Place this media in the 37 °C water bath when 10 min are remaining in the incubation step.
- Centrifuge at 300 x g for 5 min at 4 °C to pellet the enteroids. Remove the supernatant.
- Add 2 mL of enteroid dissociation media per harvested plate to the pellet and place in a 37 °C water bath for 60 s to 120 s. Gently swirl tube during incubation. Incubate long enough to achieve fragmented enteroids but without dissociation into a single cell suspension.
- After incubation, add 10 mL of cold tissue washing media to each 15 mL tube. Centrifuge at 300 x g for 5 min at 4 °C to pellet the enteroids. Aspirate the supernatant completely.
- Resuspend the pellet in 200 μL of chip expansion media. Dissociate enteroids by vigorous pipetting (~25–50 times with P200 pipette).
- Combine cells into a sterile 1.5 mL microcentrifuge tube. Pipette gently to form a homogenous solution. The goal is to seed the chip with fragmented enteroids in small clusters of 10–30 cells.
-
Remove 10 μL of cell suspension for counting. Place the remaining cell suspension on ice. Adjust the volume of chip expansion media to achieve a concentration of 6 × 106 cells/mL.NOTE: It may be necessary to centrifuge the enteroids and resuspend them in a smaller volume of media to achieve the required density.
-
Bring the microfluidic chips into the hood and aspirate the media from the epithelial (top) channel of the chip. Load the top channel of the chip with 30 μL of resuspended cells (~180,000 cells/chip). Ensure complete and uniform coverage of the epithelial channel of the chip for the formation of a monolayer. If this is not achieved, rinse the enteroids through the chip and reseed.NOTE: If there are difficulties achieving a uniform suspension while loading multiple chips, the enteroids can be separated into 40 μL aliquots in individual 1.5 mL microcentrifuge tubes. This will minimize variability in the enteroid numbers and fragment size as loading proceeds.
- Return the chips to the chip cradle in the cell culture dish (if they have been removed). Add D-PBS to the reservoir in the chip cradle. Replace the lid to the cell culture dish, and place chips at 37 °C, 5% CO2 overnight.
- Day (1): Preparing pods and introducing flow to chips
- Bring the chips into the tissue culture hood. Gently flush the epithelial channel twice with 100 μL of chip expansion media. Gently flush the endothelial channel twice with 100 μL of HIMEC media. Remove excess media from chip surface.
- Prime pods and regulate per the manufacturer’s protocol37. Add 2 mL of appropriate media to the inlet reservoirs. Add 300 μL of the appropriate media to the outlet reservoirs. The flow rate will be 30 μL/h. Stretch will not be initiated yet.
- Day (2): Monitoring monolayer development and changing media
- Pause the culture module and bring the pods into the hood.
- Inspect chips and pods for bubbles. Examine the epithelial monolayer using a phase contrast microscope. Capture images in consistent locations across the chip to monitor progress. Image the chips while they remain in the pods.
- Remove the media from the upper channel inlet reservoirs and replace it with 2 mL of chip differentiation media. Add 1 mL of HIMEC media to the lower channel inlet reservoir. Leave enough media in the inlet reservoirs to ensure that the bottom of the reservoir remains covered with a thin layer of media.
- Return the pods to the culture module and restart flow at 30 μL/h.
- Day (3): Monitoring monolayer development, initiating stretch, and adding media
-
Repeat steps 2.6.1.1. and 2.6.1.2. Add 1 mL of chip differentiation media to the upper channel inlet reservoir and add 1 mL of HIMEC media to the lower channel inlet reservoir.NOTE: Remove media from the outlet reservoirs of both channels once they begin to reach 50–75% capacity.
- Return the pods to the culture module. Restart flow at 30 μL/h, and initiate stretch at 2% strain and a frequency of 0.2 Hz.
-
- Day (4): Monitoring monolayer development, increasing stretch, and adding media
-
Repeat steps 2.6.1.1. and 2.6.1.2. Add 1 mL of chip differentiation media to the upper channel inlet reservoir and add 1 mL of HIMEC media to the lower channel inlet reservoir.NOTE: Remove media from the outlet reservoirs of both channels once they begin to reach 50–75% capacity.
- Return the pods to the culture module. Restart flow at 30 μL/h and increase stretch to 10% strain and a frequency of 0.2 Hz.
-
- Day (5) through Day (7): Monitoring monolayer development and adding media
- Repeat steps 2.6.1.1. and 2.6.1.2. Add 1 mL of chip differentiation media to the upper channel inlet reservoir and add 1 mL of HIMEC media to the lower channel inlet reservoir.
- Return the pods to the culture module, and restart flow and stretch. Once villus-like axes are visualized, proceed to the NEC-on-a-chip model.
Table 2.
Media composition for the NEC-on-a-chip model.
| Media | Reagent | Volume | Final Concentration |
|---|---|---|---|
| Chip expansion media | 25 mL | ||
| 50% L-WRN condition media | 23.4 mL | ||
| 1 M HEPES | 250 µL | 10 mM | |
| 10 mM Y-27632 | 25 µL | 10 µM | |
| 5 mM CHIR99021 | 25 µL | 5 µM | |
| 10 mM SB 431542 | 25 µL | 10 µM | |
| 1 µM human [Leu15]-gastrin I | 250 µL | 10 nM | |
| 5 mM A83-01 | 2.5 µL | 500 nM | |
| 50x B-27 Supplement | 500 µL | 1x | |
| 100x N-2 Supplement | 250 µL | 1x | |
| 500 mM N-acetylcysteine | 50 µL | 1mM | |
| 50 µg/mL murine epidermal growth factor (mEGF) | 25 µL | 50 ng/mL | |
| 1M Nicotinamide | 250 µL | 10mM | |
| Chip differentiation media | 25 mL | ||
| 50% L-WRN condition media | 23.4 mL | ||
| 1 M HEPES | 250 µL | 10 mM | |
| 10 mM SB 431542 | 25 µL | 10 µM | |
| 1 µM human [Leu15]-gastrin I | 250 µL | 10 nM | |
| 5 mM A83-01 | 2.5 µL | 500 nM | |
| 50x B-27 Supplement | 500 µL | 1x | |
| 100x N-2 Supplement | 250 µL | 1x | |
| 500 mM N-acetylcysteine | 50 µL | 1mM | |
| 50 µg/mL murine epidermal growth factor (mEGF) | 25 µL | 50 ng/mL | |
| 1M Nicotinamide | 250 µL | 10mM | |
| HIMEC media | 500 mL | ||
| Complete human endothelial cell media with the kit | |||
| Fetal bovine serum (Heat Inactivated) | 25 mL | 5% |
Figure 2: Progression of intestinal epithelial cells grown using the neonatal intestine-on-a-chip model.
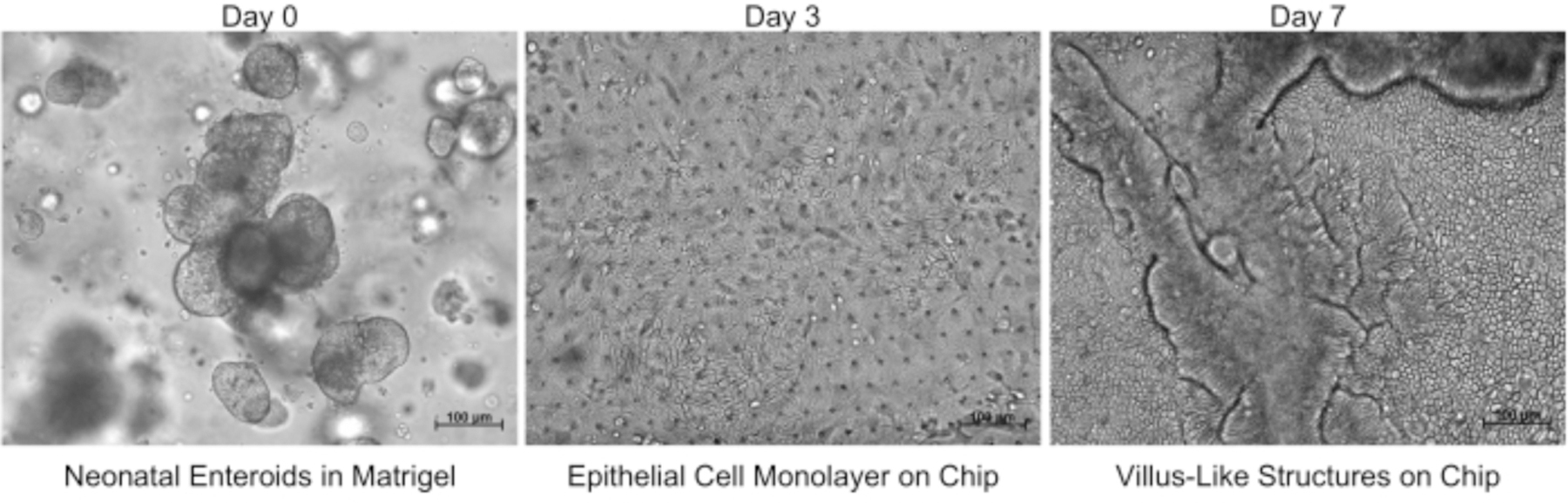
Images acquired using brightfield microscopy demonstrate neonatal enteroids on day 0, an epithelial cell monolayer in the chip on day 3, and visible villus-like structures on day 7. Scale bars = 100 μm.
3. NEC-on-a-chip model
-
Intestinal bacteria culture
NOTE: This step requires a pre-titrated frozen stock of enteric bacteria from a patient with NEC that is prepared in advance of initiating this protocol. For these experiments, a previously described polymicrobial enteric bacteria slurry from one patient with severe surgical NEC was used38.
Make a 50% glycerol stock of enteric bacteria and store in a −80 °C freezer until further use.
Thaw an aliquot of the intestinal bacteria and inoculate 10 μL into 3 mL of Luria-Bertani (LB) media. Incubate at 37 °C with shaking at 150 rpm overnight.
On the same day, gently wash channels with 100 μL of pre-warmed antibiotic-free chip differentiation media (top channel) or 100 μL of pre-warmed antibiotic free HIMEC media (bottom channel). Fully aspirate the medium and repeat flush for a total of 3 washes.
After aspirating the third wash, replace the media in the channels and leave in place.
Rinse top and bottom channel inlet reservoirs with sterile D-PBS and then fill with 3 mL of the appropriate antibiotic-free media. Prime pods and regulate as in 2.5.2.
Return the chip to the culture module, and re-initiate stretch and flow.
The following morning, take 1 mL of the overnight bacterial culture and inoculate it into 25 mL of fresh LB media.
Return this new culture to the 37 °C shaker until the OD600 reaches 0.6 ± 0.02 measured using a 1 cm cuvette. Dilute the bacterial culture to 7 × 108 colony forming units/mL in antibiotic-free chip expansion media.
Save an aliquot of this culture to confirm the concentration of the inoculum39. It may be necessary to adjust the concentration of the bacteria depending on the response of the epithelium.
Remove the media from the epithelial (top) channel. Add 30 μL of the bacterial culture diluted in antibiotic-free chip expansion media to the top channel of the chip. Be very careful to avoid cross-contamination of the HIMEC (endothelial) channel with bacteria. Remove any extra media from the surface of the chip.
Allow the chips to remain under static conditions for 30 min to facilitate bacterial adherence to the apical side of the neonatal epithelium. After the incubation, flush the top channel with 100 μL of antibiotic-free expansion media to remove unattached bacteria. Return chips to the culture module and re-initiate stretch and flow.
Every 24 h, remove the pods from the culture module, and slowly flush 100 μL of antibiotic-free chip expansion media through the epithelial channel. This will aid in preventing bacterial overgrowth.
4. Intestinal permeability assay
NOTE: This can be performed at any step during the protocol. If initiated when chips are seeded, the intestinal permeability assay can be used to serially assess monolayer confluence. If initiated upon the addition of intestinal bacteria, this assay can be used to determine the influence of intestinal bacteria on gut epithelial monolayer integrity.
Remove media from the inlet reservoir for the epithelial (top) channel. Add appropriate media containing permeability dye (50 μg/mL) to the inlet reservoir. Use antibiotic-free chip differentiation media for the NEC-on-a-chip model.
Collect the effluents from the epithelial and endothelial channels every 24 h. Quantify the concentration of permeability dye by using a microplate reader as per Lanik et al.36.
5. Immunohistochemistry
Gently wash the bottom channel and top channel of the chip with 200 μL of D-PBS. Completely aspirate D-PBS from the channels.
Fix the cells by flushing both channels with 4% paraformaldehyde, leaving solution in the channels. Aspirate excess from the chip surface. Incubate at room temperature for 30 min.
Wash both channels with 200 μL of D-PBS. Leave D-PBS in the bottom channel and remove completely from the top channel.
Permeabilize the epithelial layer by flushing the top channel with 100 μL of 0.1% detergent solution, leaving solution in the channels. Aspirate excess from the chip surface, and incubate at room temperature for 45 min.
Wash both channels with 200 μL of D-PBS. Leave D-PBS in the bottom channel and remove completely from the top channel.
Add 10% donkey serum (or an appropriate blocking agent) to the top channel for 1 h at room temperature. Wash both channels with 200 μL of D-PBS. Leave D-PBS in the bottom channel and remove completely from the top channel.
Dilute primary antibodies in 5% donkey serum and add to the top channel of the chip (antibodies and concentrations previously tested are available in Lanik et al.36 ). Incubate at 4 °C overnight.
Wash both channels 3x with 200 μL of D-PBS. Leave D-PBS in the bottom channel and remove completely from the top channel.
Dilute fluorescent-labeled secondary antibodies diluted 1:200 in 5% donkey serum in D-PBS and either Hoechst 33342 or DAPI (nuclear stains). Add secondary antibodies to the top channel of the chip. Incubate at room temperature for 1 h, protected from light.
-
Wash both channels 3x with 200 μL of D-PBS. Leave channels filled with D-PBS after the final wash. Place the chip in an imaging dish with D-PBS for image acquisition using confocal microscopy.
NOTE: Fixed chips, either stained or unstained, can be stored at 4 °C for up to 1 week in PBS. Ensure channels do not dry up during this period.
6. Isolation of RNA, preparation of cDNA, and quantitative real-time PCR
Gently wash the bottom channel and top channel of the chip with 200 μL of D-PBS. Completely aspirate D-PBS from the channels.
Extract total RNA from the cells using an RNA extraction reagent per the manufacturer’s instructions. To isolate RNA from the epithelium only, infuse through the top channel only.
Quantify the RNA concentration using a nano-volume spectrophotometer. Reverse-transcribe 1 μg of total RNA. Perform quantitative real-time PCR. Primers previously used in this model are available in Lanik et al. 36.
Representative Results
Enteroids were seeded onto the microfluidic device (Figure 1) and cultured as described above. Growth of the enteroids in cell culture matrix hydrogel prior to seeding and then the subsequent expansion of the intestinal epithelial cell monolayer after seeding the device was monitored via brightfield microscopy (Figure 2). A confluent intestinal epithelial cell monolayer formed and subsequently developed into a mature 3D villus-like structure (Figure 2). This microfluidic device seeded with an enteroid-derived neonatal intestinal epithelium and HIMECs is known as the neonatal intestine-on-a-chip model36.
Figure 1: Schematic representation of enteroid culture and the microfluidics platform.
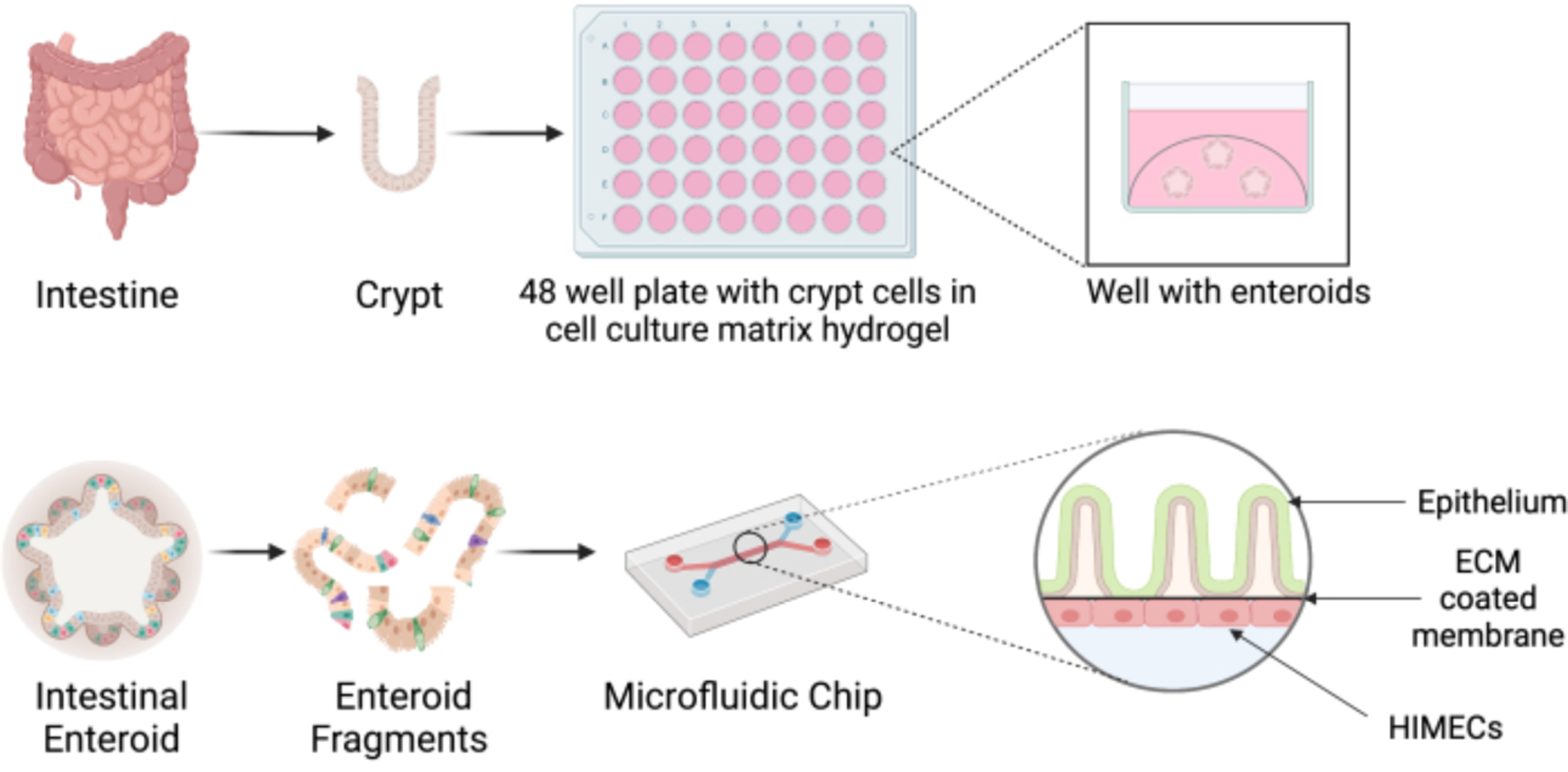
Crypts are isolated from a surgically resected piece of neonatal intestine, and they are seeded in cell culture matrix hydrogel and grown into intestinal enteroids. Enteroids are dissociated and seeded in the top channel of the microfluidics device coated with extracellular matrix (ECM). Endothelial cells (HIMECs) are then added to the bottom channel. Figure created with Biorender.com.
The NEC-on-a-chip model was developed to recapitulate the microbial dysbiosis present during neonatal NEC. This model includes the addition of a dysbiotic microbiome from a neonate with severe NEC to the neonatal intestine-on-a-chip model. With the addition of this dysbiotic microbiome, significantly increased expression of an array of proinflammatory cytokines, including tumor necrosis factor alpha (TNFα; Figure 3), interleukin (IL)-1 beta (IL-1β)36, and IL-8 was detected36. This increase in proinflammatory cytokines mirrors what is observed for human NEC36.
Figure 3. NEC-on-a-chip intestinal epithelial cell TNFα mRNA expression.
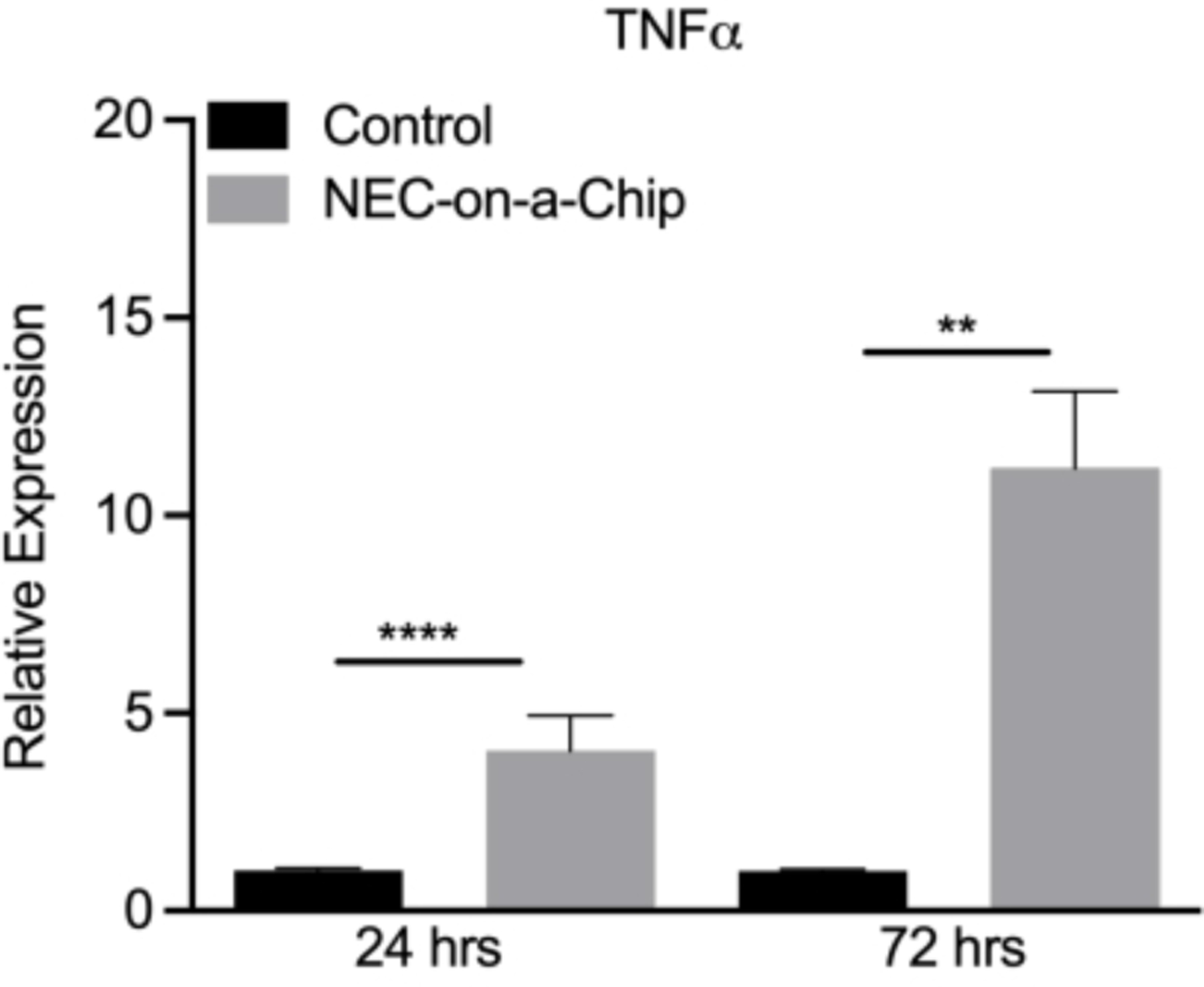
Comparison of TNFα mRNA levels upon incubation with media alone (control) or a dysbiotic microbiome from an infant with NEC (NEC-on-a-chip) for 24 h or 72 h. **** p < 0.0001 vs. control 24 h; ** p < 0.005 vs. control 72 h by Mann-Whitney U test. n=9 for control 24 h; n=10 for NEC-on-a-chip 24 h; n=6 for control 72 h; n=5 for NEC-on-a-chip 72 h. Data is mean ± SEM.
Discussion
This NEC-on-a-chip system is a powerful new tool that can be used to model the pathophysiology of NEC. This platform provides a complex microenvironment that more closely resembles the in vivo intestinal milieu than previous models by incorporating a co-culture system with continuous luminal flow and stretch. These conditions promote the development of 3D villus-like architecture lined by a highly polarized epithelium consisting of mature epithelial subtypes and tight junctions (Figure 2)36. Additionally, the apical epithelial surface is easily accessible for exposure to experimental stimuli, such as patient-derived microbiota or novel therapeutics. Finally, this model could also be used to study a variety of inflammatory and non-inflammatory intestinal diseases, as well as mechanisms of normal intestinal epithelial development, by utilizing enteroids derived from the relevant patient populations. This model allows for the maximization of data acquisition from a single patient sample, which is essential given the limited availability and size of neonatal intestinal samples.
Using this model, many of the physiologic changes in the intestinal epithelium that occur during NEC in infants were observed. The addition of the dysbiotic microbiome from a patient with severe NEC led to the upregulation of inflammatory cytokines (Figure 3), increased gut-on-a-chip barrier permeability, enhanced cell death, decreased proliferation, and the loss of mature epithelial sub-types36. Thus, future studies utilizing this model can focus on determining the mechanisms underlying the development of NEC and possible therapeutic interventions.
There are inherent limitations in implementing a complex model such as NEC-on-a-chip. This system requires specialized equipment, reagents, and training to utilize the microfluidics platform. In addition, the model requires close attention to detail with accurate preparation of the various media, proper seeding of the chip, and maintenance of sterile technique, being critical for success. Investigators must also have access to intestinal samples or enteroids from neonatal patients, which are generally only available at quaternary care centers with pediatric surgeons unless obtained through collaborators. Lastly, incorporation of additional important components in the pathophysiology of NEC, such as immune cells or hypoxia, further increases the difficulty of this model although increases the physiologic relevance.
In summary, NEC-on-a-chip is an important advance in the field of NEC research that will improve the quality of mechanistic studies and facilitate the testing of novel therapeutics for NEC. The ultimate goal of this research is to design and test targeted therapies that improve outcomes for infants with intestinal inflammation and NEC.
Acknowledgments
This manuscript was supported by R01DK118568 (MG), R01DK124614 (MG), and R01HD105301 (MG) from the National Institutes of Health, the Chan Zuckerberg Initiative Grant 2022-316749 (MG), a Thrasher Research Fund Early Career Award (LCF), a UNC Children’s Development Early Career Investigator Grant (LCF) through the generous support of donors to the University of North Carolina at Chapel Hill, and the Department of Pediatrics at the University of North Carolina at Chapel Hill.
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/65605.
Disclosures
The authors declare no conflicts of interest related to this manuscript.
References
- 1.Alsaied A, Islam N,Thalib L Global incidence of Necrotizing Enterocolitis: a systematic review and Meta-analysis. BMC Pediatrics. 20 (1), 344 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neu J,Walker WA Necrotizing enterocolitis. The New England Journal of Medicine. 364 (3), 255–264 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frazer LC,Good M Intestinal epithelium in early life. Mucosal Immunology. 15 (6), 1181–1187 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Good M et al. The human milk oligosaccharide 2’-fucosyllactose attenuates the severity of experimental necrotising enterocolitis by enhancing mesenteric perfusion in the neonatal intestine. The British Journal of Nutrition. 116 (7), 1175–1187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mihi B, Lanik WE, Gong Q,Good M A Mouse Model of Necrotizing Enterocolitis. Methods in Molecular Biology. 2321, 101–110 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Afrazi A et al. Toll-like receptor 4-mediated endoplasmic reticulum stress in intestinal crypts induces necrotizing enterocolitis. The Journal of Biological Chemistry. 289 (14), 9584–9599 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neal MD et al. Toll-like receptor 4 is expressed on intestinal stem cells and regulates their proliferation and apoptosis via the p53 up-regulated modulator of apoptosis. The Journal of Biological Chemistry. 287 (44), 37296–37308 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sodhi C, Richardson W, Gribar S,Hackam DJ The development of animal models for the study of necrotizing enterocolitis. Disease models & mechanisms. 1 (2–3), 94–98 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ares GJ, McElroy SJ,Hunter CJ The science and necessity of using animal models in the study of necrotizing enterocolitis. Seminars in pediatric surgery. 27 (1), 29–33 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu P et al. Animal models of gastrointestinal and liver diseases. Animal models of necrotizing enterocolitis: pathophysiology, translational relevance, and challenges. American journal of physiology. Gastrointestinal and liver physiology. 306 (11), G917–G928 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nolan LS, Gong Q, Hofmeister HN,Good M A protocol for the induction of experimental necrotizing enterocolitis in neonatal mice. STAR Protocol. 2 (4), 100951 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Egan CE et al. Toll-like receptor 4-mediated lymphocyte influx induces neonatal necrotizing enterocolitis. The Journal of Clinical Investigation. 126 (2), 495–508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stanford AH et al. A direct comparison of mouse and human intestinal development using epithelial gene expression patterns. Pediatric Research. 88 (1), 66–76 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noll KE, Ferris MT,Heise MT The Collaborative Cross: A Systems Genetics Resource for Studying Host-Pathogen Interactions. Cell Host Microbe. 25 (4), 484–498 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ericsson AC,Franklin CL The gut microbiome of laboratory mice: considerations and best practices for translational research. Mammalian Genome. 32 (4), 239–250 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Fazio L et al. Necrotizing Enterocolitis: Overview on In Vitro Models. International Journal of Molecular Sciences. 22 (13), 6761 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato T et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 459 (7244), 262–265 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Foulke-Abel J et al. Human enteroids as an ex-vivo model of host-pathogen interactions in the gastrointestinal tract. Experimental Biology and Medicine. 239 (9), 1124–1134 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato T,Clevers H Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science. 340 (6137), 1190–1194 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Sato T et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 141 (5), 1762–1772 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Kasendra M et al. Development of a primary human Small Intestine-on-a-Chip using biopsy-derived organoids. Scientific Reports. 8 (1), 2871 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Middendorp S et al. Adult stem cells in the small intestine are intrinsically programmed with their location-specific function. Stem Cells. 32 (5), 1083–1091 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Sung JH, Kam C, Shuler ML A microfluidic device for a pharmacokinetic-pharmacodynamic (PK-PD) model on a chip. Lab Chip. 10 (4), 446–455 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Sung JH,Shuler ML A micro cell culture analog (microCCA) with 3-D hydrogel culture of multiple cell lines to assess metabolism-dependent cytotoxicity of anti-cancer drugs. Lab Chip. 9 (10), 1385–1394 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Huh D et al. Reconstituting organ-level lung functions on a chip. Science. 328 (5986), 1662–1668 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jang KJ et al. Reproducing human and cross-species drug toxicities using a Liver-Chip. Science translational medicine,. 11 (517), eaax5516 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Musah S et al. Mature induced-pluripotent-stem-cell-derived human podocytes reconstitute kidney glomerular-capillary-wall function on a chip. Nature biomedical engineering. 1, 0069 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chou DB et al. On-chip recapitulation of clinical bone marrow toxicities and patient-specific pathophysiology. Nature biomedical engineering. 4 (4), 394–406 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park TE et al. Hypoxia-enhanced Blood-Brain Barrier Chip recapitulates human barrier function and shuttling of drugs and antibodies. Nature Communications. 10 (1), 2621 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Agarwal A, Goss JA, Cho A, McCain ML,Parker KK Microfluidic heart on a chip for higher throughput pharmacological studies. Lab Chip. 13 (18), 3599–3608 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jalili-Firoozinezhad S et al. Modeling radiation injury-induced cell death and countermeasure drug responses in a human Gut-on-a-Chip. Cell Death & Disease. 9 (2), 223 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benam KH et al. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nature Methods. 13 (2), 151–157 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Osaki T, Uzel SGM,Kamm RD On-chip 3D neuromuscular model for drug screening and precision medicine in neuromuscular disease. Nature Protocols. 15 (2), 421–449 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Chen YC et al. Single-cell Migration Chip for Chemotaxis-based Microfluidic Selection of Heterogeneous Cell Populations. Scientific Reports. 5, 9980 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiang Y et al. Gut-on-chip: Recreating human intestine in vitro. Journal of tissue engineering. 11 2041731420965318 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lanik WE et al. Microfluidic device facilitates in vitro modeling of human neonatal necrotizing enterocolitis-on-a-chip. JCI Insight. 8 (8), e146496, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emulate. Duodenum Intestine-Chip Protocol. https://emulatebio.wpenginepowered.com/wp-content/uploads/2022/10/EP203-Rev-A-Duodenum-Intestine-Chip-Protocol.pdf. (2022).
- 38.Good M et al. Lactobacillus rhamnosus HN001 decreases the severity of necrotizing enterocolitis in neonatal mice and preterm piglets: evidence in mice for a role of TLR9. American journal of physiology. Gastrointestinal and liver physiology. 306 (11), G1021–1032 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.JoVE Science Education Database. Serial Dilutions and Plating: Microbial Enumeration. JoVE. (2023). [Google Scholar]
- 40.VanDussen KL, Sonnek NM,Stappenbeck TS L-WRN conditioned medium for gastrointestinal epithelial stem cell culture shows replicable batch-to-batch activity levels across multiple research teams. Stem Cell Research. 37, 101430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miyoshi H,Stappenbeck TS In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nature Protocols. 8 (12), 2471–2482 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]