

Abstract
Background:
While transplant associated thrombotic microangiopathy (TA-TMA) commonly complicates pediatric hematopoietic cellular therapy (HCT), pulmonary manifestations and histology of TA-TMA (pTA-TMA) are rarely reported with scant data regarding timing, risk factors, pathogenesis, nor outcomes. Pulmonary hypertension (PH) and diffuse alveolar hemorrhage (DAH) are recognized manifestations of pTA-TMA. The objective of this study was to characterize the pathologic findings, outcomes, and co-incident diagnoses preceding biopsy-proven pTA-TMA.
Methods:
In IRB approved retrospective studies, available lung tissue was reviewed at two institutions between Jan 2016 and August 2021 to include those with pulmonary vascular pathology.
Results:
Histologic features of pulmonary TA-TMA (pTA-TMA) were present in 10 children with prior respiratory decline after an allogeneic (allo, n= 9) or autologous (n=1) HCT. Pathologic lesions included muscular medialization, microthrombi, and red cell fragments in addition to perivasculitis and intimal arteritis. Parenchymal findings included diffuse alveolar damage, organizing pneumonia and plasmocytic infiltrates. Six were clinically diagnosed with TA-TMA and all were treated with eculizumab a median of 2.5 days after clinical diagnosis (range 0–11 days). Four were identified post-mortem. Co-incident pulmonary infection was confirmed in 8/10 patients. Five (56%) of allo-HCT patients experienced graft versus host disease (4 acute, 1 chronic) prior to respiratory symptoms. Two patients had clinically recognized diffuse alveolar hemorrhage (DAH), though 9 (90%) had evidence of DAH on histology. While all patients had an echocardiogram (ECHO) at the time of symptoms and 9 serial ECHOs, only two patients had PH detected. Treatments varied but included sildenafil (n=3), steroids (n=1), and eculizumab (n=6). One patient is alive; the remaining 9 died a median of 52 days after respiratory symptoms (range 4–440), a median of 126 days post HCT (range 13–947).
Conclusion:
Pulmonary TA-TMA is a heterogeneous histologic disease characterized by arteriole inflammation, microthrombi, and often diffuse alveolar hemorrhage. pTA-TMA presented with respiratory decline with systemic TA-TMA in all patients. While DAH was clinically noted in 2 (20%), nine (90%) had DAH on histology. Clinicians should maintain a high degree of suspicion for DAH in patients with TA-TMA and pulmonary symptoms. Co-incident rates of GVHD and pulmonary infections were high while PH identified by echocardiogram occurred in few (20%). Outcomes were poor despite early use of eculizumab and other therapies. Our data merit consideration of pTA-TMA in patients with acute respiratory decline in the setting of systemic TA-TMA, GVHD, and infection. Investigation of additional therapies for pTA-TMA are needed.
Graphical Abstract

Background:
Transplant associated thrombotic microangiopathy (TA-TMA) is a common complication of both allogeneic and autologous pediatric hematopoietic cellular therapy (HCT) recipients and associated with significant morbidity and mortality1–4. Published risk factors include a underlying HCT indications (i.e, neuroblastoma, aplastic anemia, sickle cell disease) myeloablative preparative regimen, graft-versus-host disease, HLA mismatch, and infection2,5–8. TA-TMA is thought to be part of a spectrum of early endothelial disorders post HCT including veno-occlusive disease, engraftment syndrome, and diffuse alveolar hemorrhage9,10. The working hypothesis of TA-TMA pathogenesis includes injury to the microvascular endothelium in the setting of immune activation, leading to endothelial injury, complement activation, and microthrombi formation11. Although TA-TMA is often a multi-systemic disorder, it most commonly affects the kidneys. Little is known about the pulmonary disease process and whether the risk factors and treatments differ from the systemic non-pulmonary TA-TMA.
Biopsy proven pulmonary involvement of TA-TMA (pTA-TMA) is rarely reported12,13. Clinical manifestations attributed to pTA-TMA include pulmonary hypertension (PH), diffuse alveolar hemorrhage (DAH), and pleural effusions12–15. However, these findings are not specific to pTA-TMA and may be due to infections or other etiologies. Few reports have documented the histology findings of patients with pTA-TMA12,16. In these reports, pulmonary arterioles are described as having a thickened wall with separated endothelium, red cell extravasation and microthrombi12, similar to the vascular findings of TA-TMA in the kidney and intestines17–19. Because lung biopsy is morbid after HCT, pTA-TMA is likely underappreciated and little is known about incidence, pathologic findings, etiology, risk factors, nor outcomes.
Given the pathologic findings of pTA-TMA which can include obstructive remodeling of the pulmonary vascular bed, it is not surprising that the end organ effect of pulmonary hypertension (PH) has been linked to pTA-TMA.12,20 PH is defined as increased pulmonary arterial pressure and vascular resistance assessed by right heart catheterization. This invasive procedure is rarely performed in children post HCT, and instead echocardiogram (ECHO) is typically used to diagnose PH. Findings of elevated right ventricular pressure and/or right heart failure are supportive of PH21. Further, while PH may reflect pTA-TMA, PH can also be the downstream effect of another vascular pulmonary processes described after HCT including pulmonary veno-occlusive disease (pVOD)22. Both pulmonary arterial hypertension and pVOD disease are reported etiologies of PH in TA-TMA12,23.
We hypothesized that children with biopsy-proven pTA-TMA would have concurrent clinically diagnosed TA-TMA with similar risk factors, but may not be appreciated prior to autopsy, and that PH may be a late finding of pTA-TMA conferring poor outcomes. To answer this research question, we reviewed all biopsy and autopsy cases of children after HCT, identified tissue samples with pulmonary microvascular changes, and described the co-incident infections, inflammation, endothelial disorders, pulmonary histologic changes, and captured treatments and outcomes.
Methods:
In these IRB approved retrospective studies, we reviewed all consecutive available lung biopsies or autopsy tissue at Children’s Healthcare of Atlanta (CHOA) and Children’s National between January 2016 and July 2022. All patients with pulmonary vascular pathology identified on lung tissue and a clinical diagnosis of TA-TMA or TA-TMA in other organs were then re-reviewed with a pathologist to confirm findings and describe histologic features. There was 100% concordance between pathologists. Additional data extracted from the medical records included: patient and transplant characteristics, TA-TMA diagnosis, ECHO results (pre-and post pTA-TMA diagnosis), concurrent co-morbidities, associated treatments, and outcomes.
A clinical diagnosis of TA-TMA was made using Jodele et al criteria24. Patients met ≥4/7 of the following within a 14 day period: (1) lactate dehydrogenase (LDH) above normal for age; (2) schistocytes on blood smear; (3) de novo thrombocytopenia or increased transfusion requirements; (4) de novo anemia or increased transfusion requirements; (5) hypertension ≥ 99% for age (<18 years) or ≥140/90 (≥18 years of age); (6) proteinuria ≥30 mg/dL or random urine protein/creatinine ratio (rUPCR) ≥2 mg/mg; and (7) elevated soluble C5b-9 (≥ 244 ng/mL). Acute graft versus host disease was staged and graded using MAGIC criteria25. Chronic GVHD severity was defined using NIH Criteria26. Infections were diagnosed via bacterial culture, fungal culture, viral culture, or viral PCR. Response to eculizumab was defined as a complete response if patients attained transfusion independence and normalization of LDH. Unchanged or increased transfusion requirements and/or death with active TA-TMA was considered a nonresponse. All other responses were considered partial hematologic responses. Complete organ response was defined as recovery of organ function back to pre-HCT baseline, no response as death with TA-TMA or organ failure that was not reversed. All other responses were considered partial organ responses.
Results:
During the study period, there were 8 lung biopsies and 33 autopsies available for review. Among these, 10 children had pulmonary arterial endothelial or microvascular changes after an allogeneic (allo, n=9) or autologous (n=1) HCT. All samples were obtained for an indication of respiratory failure. Of these 10 with pTA-TMA, seven samples were obtained post-mortem, with just 3 children with tissue obtained while alive. Among patients who had a biopsy (n=3), the median time from respiratory symptoms to time of tissue obtained was 12 days (range 4 to 22). Of those with autopsy tissue (n=6), the median time from respiratory symptoms to time of tissue was 27 days (range 4 to 440 days). Two patients had tissue obtained before mechanical ventilation. Seven were intubated and ventilated a median of 12 days (range 4–25 days) before tissue obtained. Patient 10 was an outlier and had an autopsy which demonstrated pulmonary vascular changes nearly 440 days after clinical diagnosis in the setting of leukemia relapse and infection.
Sixty percent of patients (n=6/10) were clinically diagnosed with TA-TMA on a median of day + 73.5 (range 14 to 716 days); the remaining patients were diagnosed with TA-TMA postmortem. However, on retrospective review of the clinical data, all met clinical criteria for TA-TMA (prior to clinical screening programs for this disease). All patients had previously described high-risk features of TA-TMA at the time of diagnosis including one of the following: elevated sC5b-9, nephrotic range proteinuria, refractory hypertension, multiorgan dysfunction, or LDH ≥2x the upper limit of normal2,3. Of the five patients with other organs sampled, four had evidence of TA-TMA in the vessels from the kidney (n=2), intestines (n=1), heart (n=1), and pleura (n=1). The clinical diagnosis of TA-TMA preceded pulmonary symptoms in 3/6 (50%) patients by a median of 11 days (range 7 to 99 days). In three patients with concurrent pulmonary infections, respiratory symptoms preceded clinical TA-TMA diagnosis (Table 1).
Table 1:
Clinical characteristics and tissue description of children with pulmonary TA-TMA
| Pt | Age | Transplant Characteristics | Infections | GVHD | TA-TMA Features | Pulmonary Features | Pulm HTN* | Tissue diagnosis | Tx (Day post HCT treated) | Status |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 7 yr | 8/8 URD BMT for T-ALL | Culture negative sepsis | Stage 4, grade 4 lower gut acute GVHD | Clinical Dx D+ 197 sC5b9 308 UPC 4.22 mg/dl LDH 1,805 (ULN 237) Other organ involvement: pro-BNP 13,833 pg/mL (nl <1100), haptoglobin undetectable, bloody stools | RD, Hypoxia D+ 194, RF D +206 DAH | No, 5 ECHOs Pulm HTN protocol | Biopsy Day +216
|
Eculizumab (D+200) Sildenafil | Alive D+ 1273 |
| 2 | 3 yr | Autologous #2 for NB | CMV reactivation RSV | -- | Clinical Dx D+ 24 Recurrence D+ 108 sC5b9 522 ng/dL UPC 4.25 mg/mg LDH 627 U/L (ULN 321) | RD, Hypoxia, D +31, RF D +36 | Yes, 16 ECHOs, Pulm HTN protocol | Biopsy Day +35
|
Eculizumab (D+24) Sildenafil Steroids | Dead D +136 |
| 3 | 19 yr | 7/8 URD BMT for B-ALL | EBV viremia | Severe, extensive chronic GVHD | Clinical Dx D+ 716 sc5b-9 422 ng/dL UPC 0.7mg/mg | RD, hypoxia, D+ 580, RF D+733 | No, 5 ECHOs Pulm HTN protocol | Biopsy Day 592
|
Rituximab Eculizumab (D 718) | Dead D + 947 |
| 4 | 2 yr | 7/8 PBSC for MDS | Adenovirus pneumonitis | Stage 2, grade 3 lower gut acute GVHD | Clinical Dx D + 57 sC5b-9 298 ng/dL UPC 23.3 mg/mg LDH 680 U/L (ULN 321 U/L) Renal failure Refractory HTN | RD, Hypoxia D+ 31, RF D+ 57 | Yes, 6 ECHOs, Pulm HTN protocol | Autopsy (lung only)
|
Eculizumab (D+68) Sildenafil | Dead D+ 83 |
| 5 | 14 yr | 10/10 URD BMT for MDS | Candida Pneumonia | stage 2, grade 3 lower gut acute GVHD | Post Mortem Diagnosis LDH 1233 U/L (ULN 272 U/L) Renal failure | RD, Hypoxia, D+16, RF D+ 25 DAH | No, 3 ECHOs, Pulm HTN protocol | Autopsy
|
Steroids | Dead D+ 43 |
| 6 | 19 yr | 7/8 URD PBSC for HLH | CMV reactivation, Staphylococc us Epidermidis Bacteremia and Klebsiella Pneumoniae UTI | Stage 2 lower GI, Stage 1 upper GI, overall grade 3 acute GVHD | Clinical Dx D+ 14 sC5b-9 509 ng/dL UPC 2.94 mg/mg LDH 1680 u/L (ULN 246 u/L) Renal failure Refractory HTN | RD, hypoxia D+ 113 RF D+ 119 | No, 6 ECHOs, Pulm HTN protocol | Autopsy:
|
Eculizimab (D+ 15) | Dead D+ 134 |
| 7 | 13 yr | 10/10 MSD for AML | Rhinovirus, culture negative sepsis | None | Post mortem diagnosis LDH 516 Proteinuria Renal Failure | RD, hypoxia D+8 RF D+8 | No, 3 ECHOs Pulm HTN protocol | Autopsy:
|
None | Dead D+13 |
| 8 | 1 yr | 7/8 MUD for MDS/SAMD9L | Culture negative sepsis | None | Post mortem diagnosis LDH 345 U/L Proteinuria Renal failure | RD D+19 DAH | No, 1 ECHO, Pulm HTN protocol | Autopsy:
|
None | Dead D+23 |
| 9 | 18 yr | 7/8 MUD for mycosis fungoides | CMV viremia with pneumonitis | None | Post mortem diagnosis LDH 595 U/L Cytopenias, low hapto Renal failure Pericardial effusion | D +56 pericardial/pleur al effusions requiring drainage RD D +70 RF D+114 | No, 19 ECHOs, Pulm HTN protocol | Autopsy (lung only):
|
None | Dead D + 126 |
| 10 | 5 yr | 7/10 haplo related for AML | Aspergillus, stenotrophom onas, adenovirus, PJP, pseudomonas | Severe chronic lung (BO) and liver | Dx D+90 Schistocytes, proteinuria, sC5b-9 448 ng/dL, GI biopsy TMA and GvHD | RD and hypoxia D + 101 D+ 480 relapse | No. 5 ECHOs, Pulm HTN protocol | Autopsy:
|
Eculizumab (D+ 101) Steroids | Dead D+ 541 |
Transplant associated thrombotic microangiopathy (TA-TMA), , Pulmonary HTN detected on ECHO,
DAH not clinically diagnosed, but noted on tissue,
not clinically diagnosed, but retrospectively met criteria and autopsy of multiple organs demonstrate TMA. sC5b9 upper limit of normal is 244 ng/dL. Nephrotic range proteinuria is ≥2 mg/mg. Abbreviations: unrelated donor (URD), bone marrow transplant (BMT), peripheral blood stem cell (PBSC), hemophagocytic lymphohistiocytosis (HLH), myelodysplastic syndrome (MDS), acute b- lymphoblastic leukemia (B-ALL), neuroblastoma (NB), respiratory distress (RD), respiratory failure (RF), diffuse alveolar hemorrahage (DAH), urine protein to creatinine (UPC), lactate dehydrogenase (LDH), upper limit of normal (ULN), hypertension (HTN), sc5b-9 upper limit of normal 244 ng/dL.
All patients who were clinically diagnosed with TA-TMA while alive received eculizumab (n=6). Five received therapy prior to the identification of pTA-TMA, and 1 was diagnosed with TA-TMA on pulmonary biopsy and began treatment shortly after diagnosis. The median time from clinical diagnosis of TA-TMA to eculizumab therapy was 2.5 days (range 0 to 11 days). Patient #1 achieved a complete hematologic response after 30 weeks and complete organ response after 36 weeks of treatment (25 doses of eculizumab) and sildenafil. Patient #2 had a complete hematologic response to eculizumab and partial organ response; he was extubated to nasal cannula after 6 doses of eculizumab and sildenafil. Unfortunately, 3 months after resolution of TA-TMA, he had recurrence of microangiopathy and respiratory distress in the setting of RSV pneumonia. He was restarted on eculizumab though developed DAH and progressed to respiratory failure without any evidence of hematologic or organ response to second therapy of eculizumab. He died after receiving 4 doses and was thought to have recurrent pTA-TMA though family declined autopsy, so there was no tissue to confirm relapsed pTA-TMA. Patient # 10 received eculizumab and had a hematologic and organ response. She later developed respiratory failure in the setting of relapse and pulmonary infections, though had no clinical evidence of TA-TMA at the time of death. All had documented evidence of appropriate complement blockade (total complement <5% of normal limit). The remaining three (60%) had no hematologic or organ response to eculizumab. The median number of doses of eculizumab given in the 3 non-responders was 14 (range 7 to 15).
Only two patients had pulmonary hypertension (PH) detected on ECHO; one diagnosed at the time of respiratory insufficiency and the other later in the course of illness after a lung biopsy. The remaining eight patients had serial ECHOs but none with evidence of PH, right ventricular dilation, or reduced diastolic or systolic function. Three patients were treated with sildenafil, notably one for the treatment of pTA-TMA prior to the diagnosis on ECHO. Two of these three had evidence of response and were extubated, though one of these had recurrent systemic TA-TMA and ultimately died. The remaining patient had ongoing adenoviral pneumonitis and progressive respiratory failure despite interventions.
Risks factors for pTA-TMA were evaluated including mismatched HCT, preceding or concurrent GVHD, and pulmonary infections or inflammatory conditions. Six of nine allo-HCT were from HLA-mismatched donors and 5 of nine allo-HCT patients had concurrent graft versus host disease (4 acute, 1 chronic late-acute) at the time of onset of pulmonary compromise. Pulmonary infection was confirmed in 8/10 patients (6 viral, 2 fungal and 2 bacterial) and two patients were diagnosed with culture-negative sepsis. The autologous patient had cryptogenic organizing pneumonia, a pulmonary inflammatory response. No patients had COVID-19 infections prior to or during their HCT course. Only one patient (10%) is alive at last follow up. He is well without active pulmonary compromise after treatment with eculizumab and sildenafil. The remaining 9 died a median of 52 days after respiratory symptoms (range 4–440), and a median of 126 days post HCT (range 13–947).
Pathology
Vascular changes were present in all tissue samples, though associated pathologic findings were heterogenous. Arterioles had fibrointimal, sclerotic or myxoid infiltrate resulting in thickened walls in small and medium size arteries (Figure 1A and 1B). Microthrombi were visualized in arterioles in 70% of patients. Pulmonary vein microthrombi were appreciated in 2 patients with severe diffuse alveolar damage. It was unclear by the reviewing pathologists if venous thrombi were due to severe parenchymal disease or TA-TMA. Red cell fragments in thickened, abnormal arterioles were present in 20% of patients (Figure 1A). Intimal and endothelial injury were also evident (Figure 1B&C). One patient who was diagnosed with TMA much later than the other patients on day 716 post HCT had very distinctive pathology with intimal arteritis appreciated in all arteries reviewed. Arteries had thickened walls, partial obstruction of the vessel, and an associated perivascular infiltrate (Figure 1C&D). Evidence of systemic TA-TMA outside of pulmonary arterioles was appreciated in a pleural vessel wall (Figure 1E). All patient samples had extensive parenchymal changes. Diffuse alveolar hemorrhage was the most common parenchymal finding (n=9) (Figure 1a–d). Two had a plasmocytic infiltrate and two had organizing pneumonia on histology.
Figure 1:
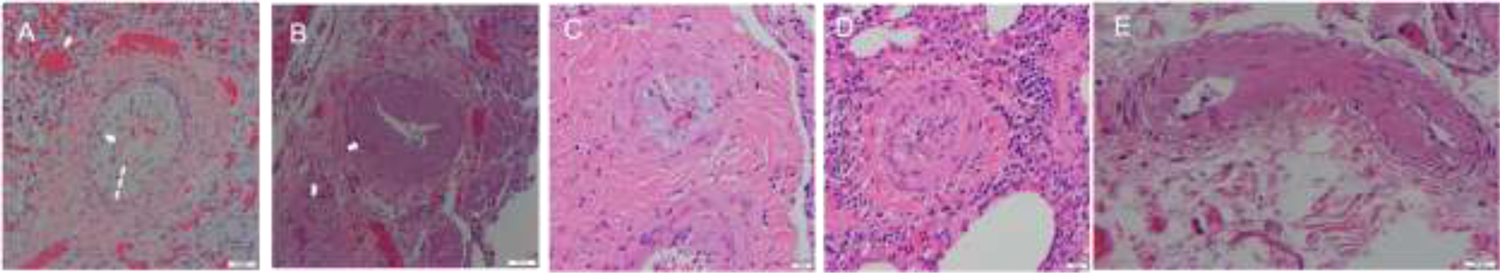
Histopathology of Pulmonary Lesions in TA-TMA
A. Classic features of TMA are present. Acute endothelial injury is associated with subendothelial intimal myxoid widening. Red cell fragments are present in the thickened intimal space (indicated with dashed line) and the lumen is narrowed (thin arrow). The parenchyma of the lung demonstrates diffuse alveolar damage and hemorrhage (indicated in a thick arrow).
B. A small vessel near the hilum has a markedly thickened wall with intimal fibrosis and a narrowed lumen.
C. Similar to A, there is marked subendothelial myxoid widening with near occlusion of the lumen.
D. This case had vascular changes and endothelial injury with an unusual finding of intimal arteritis. There is a prominent perivascular lymphocytic infiltrate around the vessel. The intima is thickened and there are lymphocytes along the intimal surface. The lumen is markedly narrowed.
E. This systemic pleural vessel has a narrowed lumen associated with a hyalinized and thickened wall. Intimal thickening with slight myxoid change and few red cell fragments are also present.
Discussion:
Pulmonary manifestations of TA-TMA are rarely described and poorly understood. We contribute to the literature by describing the clinical course, coincident transplant diagnoses, and endothelial and microvascular pulmonary changes in 10 children with severe pTA-TMA confirmed by histology. Notably, only 30% of these patients had confirmed pulmonary involvement prior to death, consistent with underreporting of this manifestation due the lack of tissue, ability to obtain lung biopsy. All patients presented with respiratory insufficiency after HCT in the setting of TA-TMA, either diagnosed in real time or appreciated post-mortem. In contrast to other reports, pulmonary symptoms occurred soon after clinical diagnosis of TA-TMA except for patients with concurrent pneumonia, in whom hypoxia and increased work of breathing began before the clinical diagnosis of TA-TMA. While most patients developed pTA-TMA in the first year after HCT, one outlier was present in our series in the setting of active uncontrolled chronic GVHD. Inflammation appeared to be contributory factor to pTA-TMA, with 89% of our allo HCT recipients receiving mismatched HCT and/or experiencing GVHD, and the one autologous HCT patient experiencing pTA-TMA in the setting of cryptogenic organizing pneumonia. Collectively, these data support that clinicians should have a high suspicion for pTA-TMA in the setting of systemic TA-TMA, respiratory insufficiency, mismatched transplant, GVHD, or pulmonary inflammation.
A prior case series equated clinical diagnosis of pTA-TMA with pulmonary hypertension detected on echocardiogram in the setting of clinically diagnosed systemic TA-TMA12. While all patients in this series had tissue evidence of pulmonary vasculopathy, only 2/10 (20%) of these patients demonstrated PH via echocardiogram. This was not because of infrequent investigation; all had an ECHO at time of TA-TMA diagnosis and 9/10 were evaluated by echocardiogram many times during their illness. Pulmonary artery systolic pressure is traditionally estimated on ECHO using a derivation of the tricuspid regurgitation velocity added to right atrial pressure. PH protocols obtain multiple additional measurements that can help support evidence for pulmonary HTN. Studies have demonstrated only moderate precision of this ECHO calculations to pulmonary artery systolic pressure measured invasively with catheterization27–29. Transthoracic echocardiogram may lack sensitivity to detect mild PH and cardiac catheterization is rarely performed in children post HCT to confirm this diagnosis. Recognizing the limitations of the test, our data support pursuing ECHOs with special attention to PH in patients with systemic TA-TMA and pulmonary insufficiency.
Hypoxia can also affect pulmonary arterial pressures leading to transient pulmonary hypertension, challenging to diagnose on intermittent ECHOs. Notably, sildenafil treatment was used in 3/10 cases for pTA-TMA (1 of whom had no evidence of PH on ECHO at initiation) and 2/3 of these patients responded and could be extubated. We hypothesize that these patients may have early evidence of pulmonary hypertension insufficient to detect on ECHO and didn’t survive long enough to develop irreversible disease. The vasculopathy appreciated on histology may support consideration of sildenafil or nitric oxide in early stages of disease, even in the absence of detectable PH on ECHO. Collectively, our data highlights the importance of considering pulmonary TA-TMA diagnosis in patients with respiratory decline with known TA-TMA.
Most patients (8/10) had concurrent pulmonary infections with biopsy proven pulmonary TA-TMA, with the remaining two patients received treatment for presumed infections. While TA-TMA can be a multi-organ systemic disease, tropism for organ involvement is not understood. Our data support that pulmonary infections may be a risk factor for pTA-TMA, for which there is biologic rationale as some infections are known to invade the vessel endothelium inciting local damage, including viruses and fungi. Infections associated with endothelial damage include COVID-19, CMV, adenovirus, EBV, candida pneumonia, and aspergillus; many of which were identified in our cohort. In addition, while most complement proteins linked to TA-TMA are liver derived, pulmonary alveolar type II epithelial and bronchiolar epithelial cells can synthesize and secrete complement proteins, suggesting that lung injury may further drive the complement dysregulation in pTA-TMA30,31. Complement dysregulation has also been linked to acute lung injury, with high concentrations of C5a, contributing to neutrophil recruitment to, and amplifying of, local pulmonary injury through neutrophil extracellular trap formation32,33. Thus, it is possible that infections may induce lung tissue sources of complement activation that propagate endothelial injury in pTA-TMA. While we could not examine for local lung tissue or systemic complement activation in these patients, this merits further investigation and may have novel therapeutic implications.
Previous studies have demonstrated improved survival in patients with high-risk TA-TMA (elevated sC5b-9 and proteinuria) who are treated with eculizumab24. Most of our patients had progressive respiratory failure despite treatment with eculizumab, though concurrent infection and GVHD were present, both known risk factors for mortality with TA-TMA2,6,34. For two of these patients, progressive infection was present even at the time of death, which may contribute to the lack of response to complement blockade. It is also possible that the combination of infection and pTA-TMA begins a cascade of alveolar damage that is difficult to reverse despite optimal therapy for infections and TA-TMA. Alternatively, terminal complement inhibition with eculizumab may not be the best therapy for pTA-TMA despite published reports of success in systemic TA-TMA24. While terminal complement activation is a key driver of tissue damage in systemic TA-TMA, it is possible that other complement pathways proximal to C5 may be responsible for acute lung injury, which could be less responsive to monotherapy with C5 inhibitor, eculizumab. In addition to complement blockade, other therapeutic approaches including defibrotide, rituximab, steroids, and plasma pheresis are described in TA-TMA, though their impact on pulmonary manifestations are unknown. This case series highlights the need for investigation of novel agents for pulmonary TA-TMA.
Like prior literature, outcomes were poor in this case series; 9/10 children died despite 6 being treated with eculizumab shortly after clinical diagnosis of TA-TMA. The surviving patient was on combination sirolimus and tacrolimus for GVHD when TA-TMA developed and was changed to monotherapy with TA-TMA diagnosis. Otherwise there were no discernable differences in risk factors or clinical characteristics of the surviving patient and the others. One of our patients died after recurrent TA-TMA in the setting of a new viral infection and DAH, supporting our prior data that while TA-TMA recurrence is rare, it does occur35. This highlights the need for continued screening of TA-TMA in patients after treatment, particularly in the setting of new infections or GVHD.
Histologic examination in this case series was performed to ensure accurate diagnosis of pTA-TMA as other clinically associated signs of endothelial injury can be nonspecific including DAH, pulmonary effusions, and pulmonary hypertension. All patients met clinical criteria for TA-TMA or had TA-TMA evident in other vessels on histology. The classic diagnostic findings of TA-TMA are thickened arterioles, red cell fragments in the vasculature, and microthrombi in arterioles. We describe a variety of endothelial and microvascular changes in patients with available pulmonary tissue. All patients shared features of vascular injury on histopathology though the vessels involved varied by patient, including small and medium sized arterioles. Some patients had classic red cell extravasation and microthrombi of the arterioles, but not all. Some also had plasmacytic infiltration, which could potentially offer opportunities for targeted therapies though this has not been tested to our knowledge. Some variation in histology may be explained by capturing tissue at different points in the evolution of the disease, i.e. at biopsy shortly after presentation (patient #1) versus postmortem after weeks of multiorgan failure (patient # 6). In two patients, vascular injury and microthrombi involved both arteries and veins in the setting of severe lung injury, sharing features of classically described as pVOD. Pulmonary TA-TMA and pVOD, both rare HCT complications, could be on a spectrum of endothelial diseases in the lungs as previously suggested, and should prompt future studies for validation36.
Parenchymal changes were more heterogenous, though the most common feature was diffuse alveolar hemorrhage and alveolar damage. Diffuse alveolar hemorrhage (DAH) is another rare pulmonary complication of HCT associated with a dismal prognosis. The pathophysiology of DAH post HCT is unclear, though infections and TA-TMA are known risk factors. There is emerging evidence that damage of the pulmonary endothelium play a central role in pulmonary diseases by resulting in increased permeability resulting in edema, a proinflammatory and pro-thrombotic phenotype37. The impact of endothelial damage at the level of the pulmonary vasculature in pTA-TMA, particularly in combination with infections are unknown. Pulmonary manifestations of other TMAs also appear to be rare, though there are reports of pulmonary hypertension in cobalamin deficiency associated TMA38, atypical hemolytic uremic syndrome39,40, and thrombotic thrombocytopenia purpura41. By including only patients with available tissue, this study selected for the most severely ill patients, and precluded evaluation of incidence. While the incidence of systemic TA-TMA in our at-risk cohort is about 20–25%, our 10 tissue proven cases would suggest that severe pTA-TMA could complicate ~ 2% of TA-TMA patients. However, this is likely an underestimate of pulmonary involvement as we captured only the most ill patients. Of note, while 9 patients had DAH on histology, only 2 were clinically noted to have DAH. These data support a recent report that DAH may be an under-recognized complication of TA-TMA14 and collectively suggest that there may be missed opportunities for DAH supportive care measures.
Our data were limited by few centers and small sample size with a predominance of tissue obtained later in the disease process. An additional limitation of this study is the lack of specific objective diagnostic criteria for TA-TMA and pulmonary TA-TMA. We have applied clinical criteria to diagnose TA-TMA and utilized lung pathology to confirm the pulmonary manifestation, which is a gold standard. However, endothelial damage is common to many post-HCT complications. Because our screening practice for TA-TMA includes exclusion of other thrombotic microangiopathies (e.g. ADAMTS13 activity and disseminated intravascular coagulation panels), these are unlikely confounding diagnoses. However, other post-HCT pulmonary processes also include endothelial damage as a key contributor, including idiopathic pneumonia syndrome (IPS) and acute respiratory distress syndrome (ARDS)36,42,43. Because most patients had an identified infection, IPS was not thought to contribute to vascular or parenchyma changes in these patients. However, 80% (n=8) of samples were obtained in patients who retrospectively met clinical criteria for acute respiratory distress syndrome (ARDS).
ARDS often results from infections and aberrant inflammation, and can also be initiated after radiation or chemotherapy, all risk factors for the genesis of pTA-TMA.44 We are likely capturing one pathway that can incite ARDS. While vascular changes and thrombosis can be features of ARDS and thus may raise the query of whether pTA-TMA can be deemed a discrete entity, there are several features of our histology that contradict this hypothesis. Because two of our cohort were biopsied prior to ventilation, and all but one within 3 weeks of ventilation, the likelihood that ventilation-induced injury alone led to the vascular findings is unlikely. During the early phases of ARDS (within 3 weeks), the acute exudative, organizing, and late proliferative phases occur, characterized by endothelial injury with splaying of the junctions and fluid extravasation, marked hyaline membrane changes with pneumocyte injury, and later lobular necrosis and fibrosis, features largely not observed on the histology of our cohort45. Further, while microthrombi and infarcts can accompany the late phase (3–4 weeks) after ventilation which are also key components of pTA-TMA, this could reflect both processes, and only represents 10% of our described cases. While our data support that pTA-TMA could lead to ARDS, it does not support that pTA-TMA simply captures ARDS vascular changes. That said, this interplay between ARDS and pTA-TMA merits more investigation, especially with pathologic findings and biomarker studies. Future specific biomarkers might one day be able to identify pTA-TMA with certainty and could be valuable to enhance diagnosis, understanding of disease, and treatment approaches.
In summary, we describe 10 patients with TA-TMA, severe pulmonary manifestations and endothelial and arterial changes after HCT and highlight coincident clinical findings that may be risk factors. Histopathology demonstrated endothelial, microvascular, and parenchyma injury with alveolar damage and hemorrhage a common feature, which was often not clinically appreciated. Our data support that pulmonary hypertension on echocardiogram is a less common feature and while supportive should not be considered necessary for the diagnosis and treatment of pTA-TMA. Providers caring for patients with TA-TMA should maintain a high level of suspicion of pulmonary involvement in children, particular in children with HLA-mismatched transplants, concurrent pulmonary infection, and GVHD or pulmonary inflammation. Eculizumab and sildenafil may be beneficial though outcomes for severe pTA-TMA are poor, with many deaths attributed to advanced disease or GVHD. pTA-TMA remains a severe and fatal complication after HCT and merits further study in large, prospective cohorts of children and adults to evaluate the proposed risk factors, findings, and effective treatments, to improve outcomes.
Highlights.
We report pulmonary vascular changes in 10 children with TA-TMA.
Only 2/10 children had evidence of pulmonary hypertension on echocardiogram.
Nine had diffuse alveolar hemorrhage on histology.
Outcomes were poor despite early therapy with eculizumab.
COI
SC: Agios, Alexion, Daichi Sankyo, Novartis, Takeda – consultancy; Alexion, Global Blood Therapeutics – research funding. No other authors have funding to report
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dandoy CE, Rotz S, Alonso PB, et al. A pragmatic multi-institutional approach to understanding transplant-associated thrombotic microangiopathy after stem cell transplant. Blood Adv. 2020;5(1):1–11. doi: 10.1182/bloodadvances.2020003455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schoettler M, Lehmann LE, Margossian S, et al. Risk factors for transplant-associated thrombotic microangiopathy and mortality in a pediatric cohort. Blood Adv. 2020;4(11):2536–2547. doi: 10.1182/bloodadvances.2019001242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jodele S, Davies SM, Lane A, et al. Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: a study in children and young adults. Blood. 2014;124(4):645 LP–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jodele S, Dandoy CE, Myers K, et al. High-dose Carboplatin/Etoposide/Melphalan increases risk of thrombotic microangiopathy and organ injury after autologous stem cell transplantation in patients with neuroblastoma. Bone Marrow Transplant. 2018. doi: 10.1038/s41409-018-0159-8 [DOI] [PubMed] [Google Scholar]
- 5.Wall SA, Zhao Q, Yearsley M, et al. Complement-mediated thrombotic microangiopathy as a link between endothelial damage and steroid-refractory GVHD. Blood Adv. 2018;2(20):2619–2628. doi: 10.1182/bloodadvances.2018020321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kraft S, Bollinger N, Bodenmann B, et al. High mortality in hematopoietic stem cell transplant-associated thrombotic microangiopathy with and without concomitant acute graft-versus-host disease. Bone Marrow Transplant. 2018. doi: 10.1038/s41409-018-0293-3 [DOI] [PubMed] [Google Scholar]
- 7.Li A, Wu Q, Davis C, et al. Transplant-Associated Thrombotic Microangiopathy Is a Multifactorial Disease Unresponsive to Immunosuppressant Withdrawal. Biol Blood Marrow Transplant. 2019;25(3):570–576. doi: 10.1016/j.bbmt.2018.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schoettler M, Stenger EO, Spencer K, et al. Sickle cell disease is a risk factor for transplant-associated thrombotic microangiopathy in children. Blood Adv. September 2022:bloodadvances.2022008058. doi: 10.1182/bloodadvances.2022008058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hildebrandt GC, Chao N. Endothelial cell function and endothelial-related disorders following haematopoietic cell transplantation. Br J Haematol. 2020;190(4):508–519. doi: 10.1111/bjh.16621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pagliuca S, Michonneau D, Sicre de Fontbrune F, et al. Allogeneic reactivity–mediated endothelial cell complications after HSCT: a plea for consensual definitions. Blood Adv. 2019;3(15):2424–2435. doi: 10.1182/bloodadvances.2019000143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jodele S. Complement in Pathophysiology and Treatment of Transplant-Associated Thrombotic Microangiopathies. Semin Hematol. 2018;55(3):159–166. doi: 10.1053/j.seminhematol.2018.04.003 [DOI] [PubMed] [Google Scholar]
- 12.Jodele S, Hirsch R, Laskin B, Davies S, Witte D, Chima R. Pulmonary Arterial Hypertension in Pediatric Patients with Hematopoietic Stem Cell Transplant-Associated Thrombotic Microangiopathy. Biol Blood Marrow Transplant. 2013;19(2):202–207. doi: 10.1016/j.bbmt.2012.08.022 [DOI] [PubMed] [Google Scholar]
- 13.Dandoy CE, Hirsch R, Chima R, Davies SM, Jodele S. Pulmonary hypertension after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2013;19(11):1546–1556. doi: 10.1016/j.bbmt.2013.07.017 [DOI] [PubMed] [Google Scholar]
- 14.Agarwal S, Cortes-Santiago N, Scheurer ME, et al. Diffuse alveolar hemorrhage: An underreported complication of transplant associated thrombotic microangiopathy. Bone Marrow Transplant. 2022;57(6):889–895. doi: 10.1038/s41409-022-01644-3 [DOI] [PubMed] [Google Scholar]
- 15.Srivastava A, Gottlieb D, Bradstock KF. Diffuse alveolar haemorrhage associated with microangiopathy after allogeneic bone marrow transplantation. Bone Marrow Transplant. 1995;15(6):863–867. http://europepmc.org/abstract/MED/7581082. [PubMed] [Google Scholar]
- 16.Siami K, Kojouri K, Swisher KK, Selby GB, George JN, Laszik ZG. Thrombotic Microangiopathy After Allogeneic Hematopoietic Stem Cell Transplantation: An Autopsy Study. Transplantation. 2008;85(1). https://journals.lww.com/transplantjournal/Fulltext/2008/01150/Thrombotic_Microangiopathy_After_Allogeneic.5.aspx. [DOI] [PubMed] [Google Scholar]
- 17.Warren M, Jodele S, Dandoy C, et al. A complete histologic approach to gastrointestinal biopsy from hematopoietic stem cell transplant patients with evidence of transplant-associated gastrointestinal thrombotic microangiopathy. Arch Pathol Lab Med. 2017;141(11):1558–1566. doi: 10.5858/arpa.2016-0599-RA [DOI] [PubMed] [Google Scholar]
- 18.Yamada R, Nemoto T, Ohashi K, et al. Distribution of Transplantation-Associated Thrombotic Microangiopathy (TA-TMA) and Comparison between Renal TA-TMA and Intestinal TA-TMA: Autopsy Study. Biol Blood Marrow Transplant. 2020;26(1):178–188. doi: 10.1016/j.bbmt.2019.08.025 [DOI] [PubMed] [Google Scholar]
- 19.Lusco MA, Fogo AB, Najafian B, Alpers CE. AJKD Atlas of Renal Pathology: Thrombotic Microangiopathy. Am J Kidney Dis. 2016;68(6):e33–e34. doi: 10.1053/j.ajkd.2016.10.006 [DOI] [PubMed] [Google Scholar]
- 20.Losito A, Pittavini L C C \. Thrombotic microangiopathic nephropathy, pulmonary hypertension and nephromegaly: case report of a patient treated with endothelin receptor antagonist. Clin Nephrol. 2012;77(2):164–170. doi: 10.5414/CN106829 [DOI] [PubMed] [Google Scholar]
- 21.Koestenberger M, Friedberg MK, Nestaas E, Michel-Behnke I, Hansmann G. Transthoracic echocardiography in the evaluation of pediatric pulmonary hypertension and ventricular dysfunction. Pulm Circ. 2016;6(1):15–29. doi: 10.1086/685051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.HACKMAN RC, MADTES DK, PETERSEN FB, CLARK JG. PULMONARY VENOOCCLUSIVE DISEASE FOLLOWING BONE MARROW TRANSPLANTATION1. Transplantation. 1989;47(6). https://journals.lww.com/transplantjournal/Fulltext/1989/06000/PULMONARY_VENOOCCLUSIVE_DISEASE_FOLLOWING_BONE.14.aspx. [DOI] [PubMed] [Google Scholar]
- 23.Zinter MS, Melton A, Sabnis AJ, et al. Pulmonary veno-occlusive disease in a pediatric hematopoietic stem cell transplant patient: a cautionary tale. Leuk Lymphoma. 2018;59(6):1494–1497. doi: 10.1080/10428194.2017.1382697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jodele S, Dandoy CE, Lane A, et al. Complement blockade for TA-TMA: lessons learned from a large pediatric cohort treated with eculizumab. Blood. 2020;135(13):1049–1057. doi: 10.1182/blood.2019004218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Major-Monfried H, Renteria AS, Pawarode A, et al. MAGIC biomarkers predict long-term outcomes for steroid-resistant acute GVHD. Blood. 2018;131(25):2846–2855. doi: 10.1182/blood-2018-01-822957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group Report. Biol Blood Marrow Transplant. 2015;21(3):389–401.e1. doi: 10.1016/j.bbmt.2014.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D’Alto M, Romeo E, Argiento P, et al. Accuracy and precision of echocardiography versus right heart catheterization for the assessment of pulmonary hypertension. Int J Cardiol. 2013;168(4):4058–4062. doi: 10.1016/j.ijcard.2013.07.005 [DOI] [PubMed] [Google Scholar]
- 28.Rich JD, Shah SJ, Swamy RS, Kamp A, Rich S. Inaccuracy of Doppler Echocardiographic Estimates of Pulmonary Artery Pressures in Patients With Pulmonary Hypertension: Implications for Clinical Practice. Chest. 2011;139(5):988–993. doi: 10.1378/chest.10-1269 [DOI] [PubMed] [Google Scholar]
- 29.Fisher MR, Forfia PR, Chamera E, et al. Accuracy of Doppler Echocardiography in the Hemodynamic Assessment of Pulmonary Hypertension. Am J Respir Crit Care Med. 2009;179(7):615–621. doi: 10.1164/rccm.200811-1691OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varsano S, Kaminsky M, Kaiser M, Rashkovsky L. Generation of complement C3 and expression of cell membrane complement inhibitory proteins by human bronchial epithelium cell line. Thorax. 2000;55(5):364–369. doi: 10.1136/thorax.55.5.364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strunk RC, Eidlen DM, Mason RJ. Pulmonary alveolar type II epithelial cells synthesize and secrete proteins of the classical and alternative complement pathways. J Clin Invest. 1988;81(5):1419–1426. doi: 10.1172/JCI113472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bosmann M, Ward PA. Role of C3, C5 and anaphylatoxin receptors in acute lung injury and in sepsis. Adv Exp Med Biol. 2012;946:147–159. doi: 10.1007/978-1-4614-0106-3_9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chakraborty S, Winkelmann VE, Braumüller S, et al. Role of the C5a-C5a receptor axis in the inflammatory responses of the lungs after experimental polytrauma and hemorrhagic shock. Sci Rep. 2021;11(1):2158. doi: 10.1038/s41598-020-79607-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gavriilaki E, Sakellari I, Batsis I, et al. Transplant-associated thrombotic microangiopathy: Incidence, prognostic factors, morbidity, and mortality in allogeneic hematopoietic cell transplantation. Clin Transplant. 2018;32(9):e13371. doi: 10.1111/ctr.13371 [DOI] [PubMed] [Google Scholar]
- 35.Schoettler M, Lehmann L, Li A, Ma C, Duncan C. Thrombotic microangiopathy following pediatric autologous hematopoetic cell transplantation: a report of significant end organ dysfunction in eculizumab treated survivotrs. Biol Blood Marrow Transplant. 2019;In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williams KM. Noninfectious complications of hematopoietic cell transplantation. Hematol Am Soc Hematol Educ Progr. 2021;2021(1):578–586. doi: 10.1182/hematology.2021000293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huertas A, Guignabert C, Barberà JA, et al. Pulmonary vascular endothelium: The orchestra conductor in respiratory diseases. Eur Respir J. 2018;51(4). doi: 10.1183/13993003.00745-2017 [DOI] [PubMed] [Google Scholar]
- 38.Kömhoff M, Roofthooft MT, Westra D, et al. Combined Pulmonary Hypertension and Renal Thrombotic Microangiopathy in Cobalamin C Deficiency. Pediatrics. 2013;132(2):e540–e544. doi: 10.1542/peds.2012-2581 [DOI] [PubMed] [Google Scholar]
- 39.Noris M, Caprioli J, Bresin E, et al. Relative Role of Genetic Complement Abnormalities in Sporadic and Familial aHUS and Their Impact on Clinical Phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844 LP–1859. doi: 10.2215/CJN.02210310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sellier-Leclerc A-L, Fremeaux-Bacchi V, Dragon-Durey M-A, et al. Differential Impact of Complement Mutations on Clinical Characteristics in Atypical Hemolytic Uremic Syndrome. J Am Soc Nephrol. 2007;18(8):2392 LP–2400. doi: 10.1681/ASN.2006080811 [DOI] [PubMed] [Google Scholar]
- 41.Mastrobattista JM, Ramin SM, Gilstrap LC. Thrombotic thrombocytopenic purpura in pregnancy. Prim Care Update Ob Gyns. 2000;7(4):168–171. doi: 10.1016/S1068-607X(00)00040-8 [DOI] [PubMed] [Google Scholar]
- 42.Tamburro RF, Cooke KR, Davies SM, et al. Pulmonary Complications of Pediatric Hematopoietic Cell Transplantation. A National Institutes of Health Workshop Summary. Ann Am Thorac Soc. 2020;18(3):381–394. doi: 10.1513/AnnalsATS.202001-006OT [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cooke KR, Jannin A, Ho V. The Contribution of Endothelial Activation and Injury to End-Organ Toxicity following Allogeneic Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant. 2008;14(1):23–32. doi: 10.1016/j.bbmt.2007.10.008 [DOI] [PubMed] [Google Scholar]
- 44.Tomashefski JF. PULMONARY PATHOLOGY OF ACUTE RESPIRATORY DISTRESS SYNDROME. Clin Chest Med. 2000;21(3):435–466. doi: 10.1016/S0272-5231(05)70158-1 [DOI] [PubMed] [Google Scholar]
- 45.Castro CY. ARDS and Diffuse Alveolar Damage: A Pathologist’s Perspective. Semin Thorac Cardiovasc Surg. 2006;18(1):13–19. doi: 10.1053/j.semtcvs.2006.02.001 [DOI] [PubMed] [Google Scholar]