

Abstract
The COVID-19 pandemic has affected over 114 million people and has resulted in >2.5 million deaths so far. Some people have greater susceptibility which influences both SARS-CoV-2 infectivity and COVID-19 severity. Smoking is associated with increased ACE-2, the receptor for SARS-CoV-2, which facilitates its entry through the lung. However, despite the widespread use of e-cigarettes, also known as ‘vaping’, little is known regarding the effects of vaping on ACE-2 expression and how this affects SARS-CoV-2 infection. In addition, the added effect of nicotine in the vapor is also unknown. Thus, we tested whether vaping induces ACE-2 expression in the mouse lung. BALB/c mice exposed to e-cigarette vapor (±nicotine) resulted in a significant increase in peribronchiolar inflammation and influx of immune cells into the airways. Vapor increased monocyte chemoattractant protein-1, interleukin 1β, and KC levels in bronchoalveolar lavage fluid in both sexes, which were further enhanced by nicotine (whereas increase in interleukin 6 was sex and nicotine independent). The reduction in basal inspiratory capacity with vapor exposure occurred independent of sex or nicotine. The increase in methacholine-induced airway hyper-responsiveness was independent of sex; however, in female mice it was only significant in the nicotine-exposed group. Lung ACE-2 expression was increased in male mice in a nicotine-dependent manner as compared with female mice. Collectively, while vaping (±nicotine) induced airway inflammation and impaired lung function, the induction of lung ACE-2 occurred to a significantly greater degree in males exposed to vapor containing nicotine as compared with females. Thus, via these effects on ACE-2 expression in the lungs and airways, vaping itself may facilitate SARS-CoV-2 entry into the airways.
INTRODUCTION
The COVID-19 pandemic has affected over 114 million people and has resulted in >2.5 million deaths, according to Johns Hopkins University.1 In the USA alone, over 28.7 million people have been infected, leading to over 500,000 deaths to date. This 2019 novel coronavirus (CoV) is structurally similar to the 2003 severe acute respiratory syndrome (SARS) and the 2012 Middle East respiratory syndrome (MERS) CoV, both of which caused two large-scale pandemics in the recent past.2 COVID-19 is caused by the recently identified SARS-CoV-2, a positive-sense, single-stranded, enveloped, large RNA virus of the Coronaviridae family.2 3 SARS-CoV-2 is highly transmissible and can spread readily from human to human, causing acute and severe respiratory failure, and in many cases followed by death.3 4 This scenario appears to be even more likely in patients with underlying health conditions and cardiopulmonary comorbidities.5
Although new data are still emerging, COVID-19 mortality rates appear to be higher in the male population.6 7 The mechanism for this gender-based difference is not fully understood; however, sex steroids can potentiate factors that can facilitate viral entry into the host cell and therefore may contribute to this pathological disparity, including the higher proportion of male smokers.8 9 There may be an intriguing connection between smoking and the endocrine subsystem known as the renin–angiotensin system (RAS) in the lungs. RAS includes both ACE-1 and ACE-2 axes relevant to cardiopulmonary pathophysiology.10 RAS plays an important role in respiratory infections and inflammation. Notably, ACE-2 is the only experimentally confirmed host cell cognate receptor for SARS-CoV (also known as SARS-CoV-1) which binds the viral spike (S) protein.11 The S-protein is found on the surface of SARS-CoV-1 and SARS-CoV-2 and regulates both cross-species and human-to-human CoV transmission.12 13
The binding of SARS-CoV-2 S-protein (a glycoprotein) and ACE-2 receptor is a critical step for virus entry. Human cells expressing ACE-2 (but not human dipeptidyl peptidase-4 or aminopeptidase N) showed enhanced SARS-CoV-2 entry.14 The binding efficiency of SARS-CoV-2 S-protein and ACE-2 is 10-fold to 20-fold higher than that of ACE-2 and the SARS-CoV-1 S-protein, as evidenced by the cryogenic electron microscopy structure of the SARS-CoV-2 S-protein in the prefusion conformation.15 Activation of the S-proteins is required for cellular entry, and two host cell enzymes called furin and transmembrane serine protease 2 (TMPRSS2) are necessary factors for S-protein priming.16 17
Smokers and patients with chronic obstructive pulmonary disease (COPD) have higher expression of ACE-2 in the airway epithelium, type 2 pneumocytes, tissue macrophages, and ciliated airway epithelial cells,18–20 with some evidence that its expression may vary with sex and age.20 21 Because the percentage of men who smoke and/or vape is significantly higher globally, this could be an important factor linking the observed greater COVID-19 mortality in the male population. Smoking, therefore, becomes an important risk factor because it leads to an upregulation of the ACE-2 receptor, thus facilitating SARS-CoV-2 entry into lung resident cells (such as the epithelium).
However, nothing is known regarding e-cigarette use (‘vaping’) and whether it also has similar effects. Because the percentage of men who smoke or vape is significantly higher globally, this could be an important factor linking their greater COVID-19 mortality. It remains to be determined whether other external factors, such as vaping, can enhance the severity or transmissibility of the disease.22 Therefore, we hypothesized that vaping (like cigarette smoking) will similarly induce lung and airway ACE-2 expression, and more specifically that this will vary according to sex and nicotine exposure. Addressing this question will allow us to understand viral host dynamics and the likely detrimental role of vaping when the host is also exposed to SARS-CoV-2.
MATERIALS AND METHODS
Animals, e-cigarette vaping, and lung mechanics
Subchronic mouse exposure model was used to deliver e-cigarette vapor by a KangerTech device (Shenzhen, China), as shown in figure 1A, using an automated exposure system (CH Technologies, USA). BALB/c mice 7–8 weeks old (5 male and 5 female; Palamur Biosciences) were exposed for a total of 30 min (with a 3 s puff duration and 30 s puff interval that generates 55 mL of puff volume: the Coresta protocol) two times per day for 21 days ± nicotine (0 and 18 mg/mL; VapeEmpire), while control animals were exposed to room air. Twenty-four hours after the last exposure, basal inspiratory capacity (IC) and airway responsiveness to increasing concentrations of inhaled methacholine (MCh, 1–50 mg/mL) were measured using the flexiVent system with the latest flexiWare software (Scireq, Canada).23 24
Figure 1.
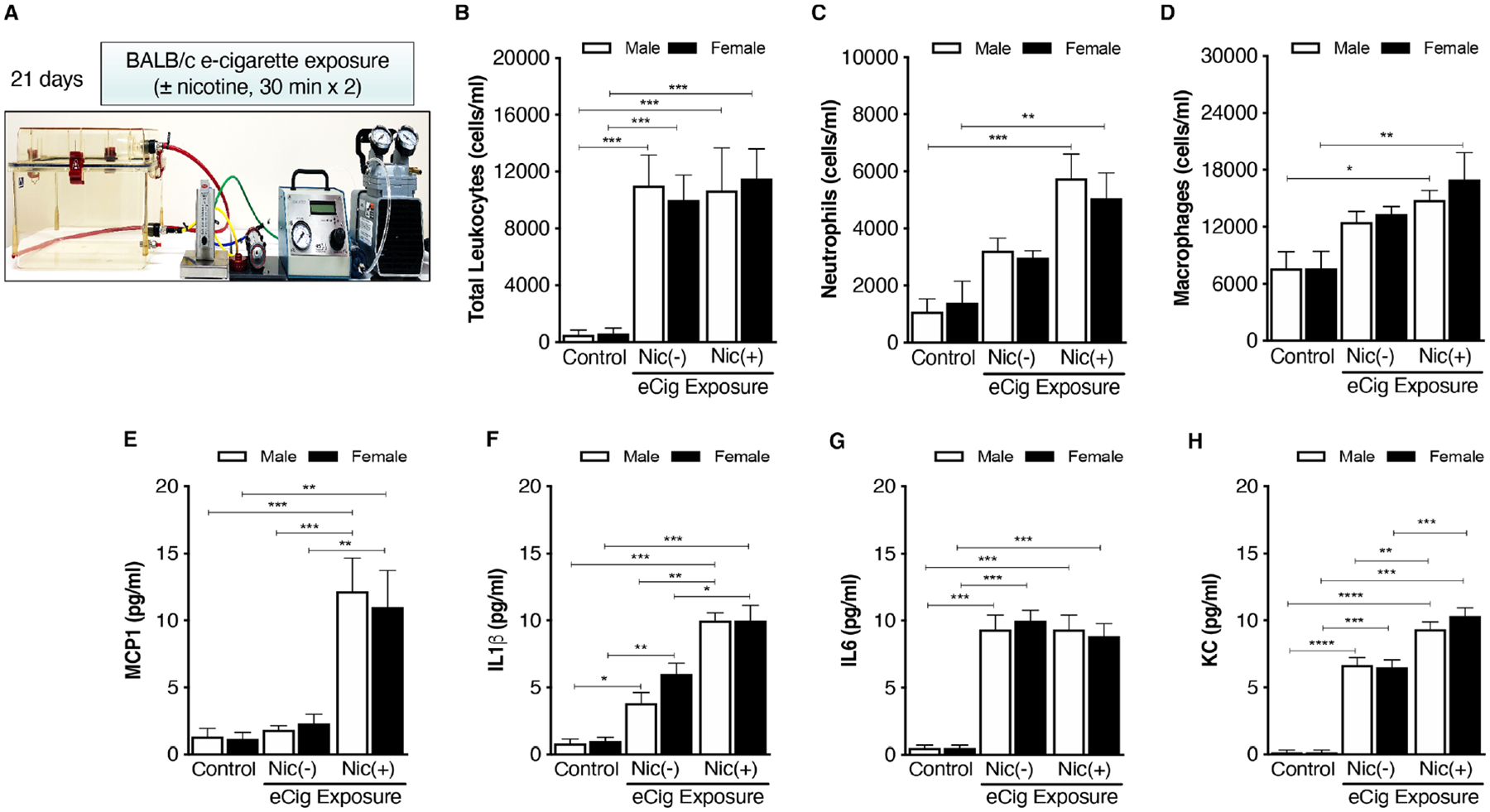
(A) Both male and female mice were exposed to e-cigarette with or without nicotine for 21 days, 2×30 min per day using highly controlled e-cigarette exposure set-up (CH Technologies, New Jersey, USA). 24 hours after the last challenge, lung tissue was isolated, inflated, and fixed with formalin. (B) Total cells were counted in BAL fluid. Cell differential was carried out in BAL fluid for neutrophils (C) and macrophages (D). BAL fluid cytokines were measured for MCP-1 (E), IL-1B (F), IL-6 (G), and KC (H). Data represent at least five animals per group; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 using one-way analysis of variance with Bonferroni correction. BAL, bronchoalveolar lavage; eCig, e-cigarette; IL, interleukin; MCP-1, monocyte chemoattractant protein-1; Nic, nicotine.
BAL inflammatory cell counts and multiplex array analysis
Bronchoalveolar lavage (BAL) fluid (2×0.5 mL) was collected to assess total immune cell influx, perform cell differentials, and measure cytokine levels. Interleukin (IL) 1β, monocyte chemoattractant protein-1 (MCP-1), IL-6, and KC (equivalent to mouse IL-8) levels in BAL fluid were measured using multiplex cytokine array as described previously.23 24
Preparation of lung lysates
Mouse lung (right) was stored at −80°C for further analysis. Frozen lung tissues in TPER cocktail (tissue protein extraction reagent; Thermo Scientific) were slowly thawed on ice and were homogenized using a polytron. The lysate was centrifuged (760 g, 5 min) and the supernatant stored at −80°C for subsequent protein assay and immunoblot analyses.
Immunoblotting
After determining protein concentration, proteins were separated using immunoblotting. Separated proteins were transferred to a nitrocellulose membrane, followed by blocking with 3% bovine serum albumin, and then incubated with ACE-2 antibody (1:1000, #AF3437; R&D Systems, USA) overnight at 4°C, followed by washing and incubation with secondary antibody (1:5000). Membranes were visualized by horseradish peroxidase (HRP)-based chemiluminescence, and densitometry was performed by ImageJ software using glyceraldehyde 3-phosphate dehydrogenase as a loading control.
Real-time PCR
Frozen lung tissues were used for RNA isolation using RNeasy Mini Kit (Qiagen), followed by complementary DNA (cDNA) synthesis from 1 μg of RNA (Takara Bio). Reverse transcription-PCR was performed using 100 ng of cDNA, mouse ACE-2 primers (forward: 5’-ACC CTT CTT ACA TCA GCC CTA CTG-3’; reverse: 5’-TGT CCA AAA CCT ACC CCA CAT AT-3’),25 and 12.5 μL of PowerUP SYBR Green Master Mix (Thermo Scientific). The reaction mixture was placed in one well of a 96-well plate, and the total reaction volume was brought to 25 μL with diethyl pyrocarbonate-treated water. Quantitative real-time PCR was performed at 50°C for 2 min and 95°C for 10 min and was run for 40 cycles at 95°C for 15 s and 61°C for 1 min in an Applied Biosystems machine (QuantStudio 5). The cycle threshold for PCR amplification needed to detect fluorescence was then determined for each unknown cDNA sample. ACE-2 mRNA levels were normalized to β-actin levels in each experiment.
Lung histology
After collecting BAL fluid, the right lung lobe was harvested for immunoblotting, while the left lobe was inflated with formalin, fixed, processed, and embedded in paraffin for analyses. Sections were cut (5 μm) using HM325 Rotary Microtome (Thermo Scientific, USA). H&E (Sigma-Aldrich) was used to assess structural integrity and tissue inflammation. Peribronchial inflammation and the degree of total cell infiltration (inflammation score) were graded by two independent blinded readers using a semiquantitative scoring system,23 24 using our standard laboratory approach. Peribronchial inflammation was graded on a scale of 0–4, with 0 for absent, 1 for slight, 2 for mild, 3 for moderate, and 4 for severe, while the degree of cell infiltration was scored 0–3, with 0 for no cells, 1 for few cells, 2 for moderate influx, and 3 for extensive influx of cells. Immunostaining and optical density for ACE-2 were performed by computer-assisted image analysis using ImageJ.24
Immunohistochemistry and image analysis
Sections were identified with a hydrophobic Dako Pen (Agilent, USA) and endogenous tris-buffered saline, 0.1% Tween 20 detergent (TBST) peroxidase activity was quenched with incubation in 3% H2O2 for 10 min after heated antigen epitome retrieval,24 followed by washes and incubation in Dako serum-free protein block (Agilent) for 10 min. Sections were then washed in TBST and incubated for 40 min with the mouse polyclonal anti-ACE-2 antibody (1:150, AF3437; R&D Systems). Bound antibodies were elaborated with HRP-labeled EnVision+ secondary antibody (K4011; Agilent) incubation for 30 min. The primary antibody was replaced by a species-appropriate, isotype-matched immunoglobulin (Mouse Immunoglobulin Fraction, X0936; Agilent) for a negative control. After washing sections were visualized with the addition of liquid 3,3’-diaminobenzidine (Agilent) and incubated for 10 min, while the nuclei were counterstained with Mayer’s hematoxylin blued with ammoniated water and dehydrated and cleared through graded ethanol solutions and xylene. Sections were mounted with Permount (Fisher Scientific, USA). Computer-assisted image analysis was performed with an Olympus BX51 upright epifluorescence microscope (Olympus, Japan) and ImageJ software.24
Statistical analysis
Where relevant one-way or two-way analysis of variance was used with Bonferroni multiple comparisons test. All data are expressed as mean±SEM. The Prism V.8 software was used for analyses (GraphPad, USA) and p<0.05 was considered statistically significant.
RESULTS
Exposure to e-cigarette vapor increased cellular inflammation in the lung
Subchronic (21-day) e-cigarette vapor exposure caused a significant increase in the influx of immune cells into the airways of both male and female mice (figure 1B). Increased airway inflammation was predominantly driven by neutrophils (figure 1C) and macrophages (figure 1D) when compared with their respective controls in a nicotine-dependent manner. No statistically significant difference was observed in immune cell influx between male and female mice.
We then measured key markers of chemoattraction, proliferation, and differentiation in BAL fluid. The statistical comparison of ‘nicotine dependence’ (or not) was determined within the nicotine exposure groups, and additional comparisons relative to negative controls are also reported in each figure. MCP-1 levels were significantly increased in the e-cigarette vapor group (figure 1E) in a nicotine-dependent manner in both sexes. For MCP-1, nicotine exposure was required to increase MCP-1 levels above controls in both sexes. The increase in IL-1β level was similarly nicotine-dependent in both sexes (figure 1F). Conversely, levels of IL-6 (a pleiotropic cytokine) increased in both male and female mice but independent of nicotine exposure (figure 1G). Levels of KC (a neutrophil chemoattractant) increased in a nicotine-dependent manner in both sexes (figure 1H), similar to MCP-1 and IL-1β.
We then assessed lung inflammation by H&E staining (×20; figure 2). Exposure to e-cigarette vapor caused a significant increase in peribronchiolar inflammation in a nicotine-independent manner in males. Of note, when compared with their respective controls, inflammation on average was greater in males as compared with females (figure 2G).
Figure 2.
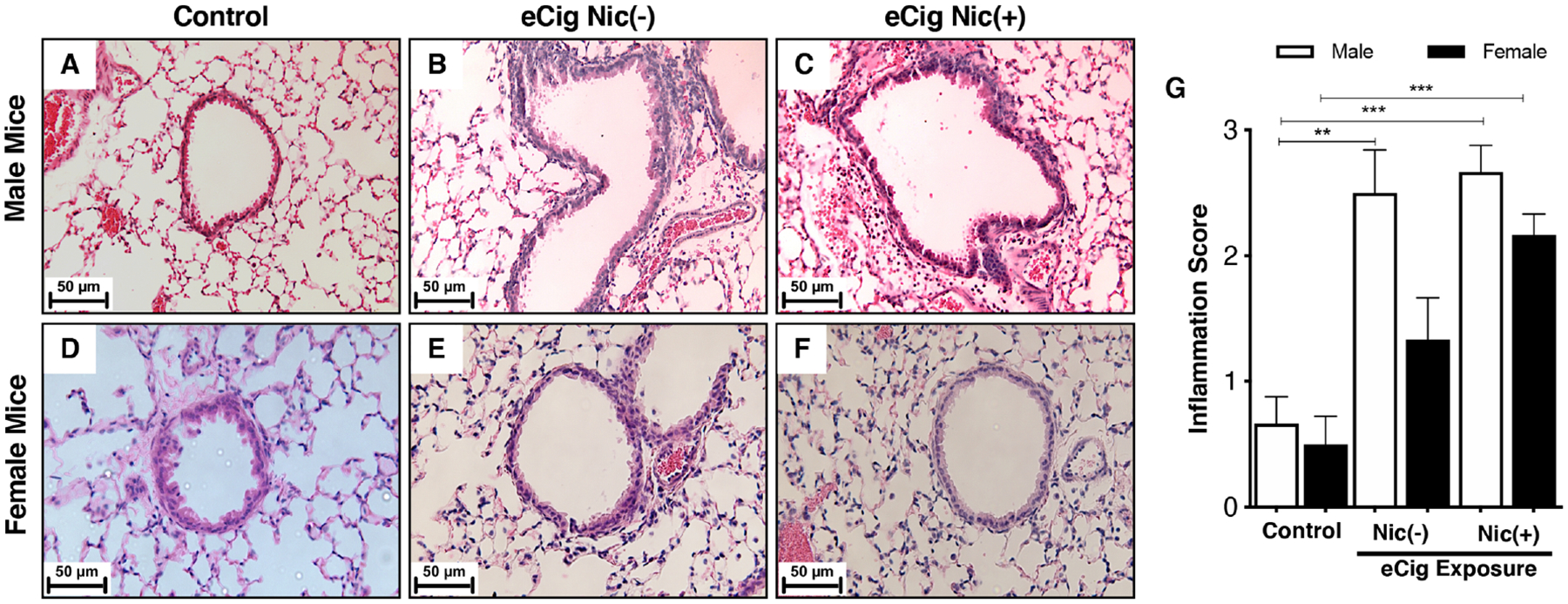
H&E staining of male and female mouse lungs showing saline-exposed animals with minimal infiltration of immune cells (A, D), infiltration of immune cells in the absence of nicotine (eCig Nic(−)) (B, E) and in the presence of nicotine (eCig Nic(+)) (C, F), and peribronchiolar infiltration of immune cells. The degree of inflammation was quantified using a scoring system based on inflammation score as described in the Materials and methods section (G). Data represent at least five animals per group; **p<0.01, ***p<0.001, using one-way analysis of variance with Bonferroni correction. eCig, e-cigarette; Nic, nicotine.
Effect of subchronic e-cigarette exposure on lung mechanics
To understand whether subchronic e-cigarette exposure can alter lung physiology, we measured basal IC after 21-day e-cigarette exposure using flexiVent system (Scireq). As shown in figure 3A,B, there was a significant reduction in basal IC in both sexes after 21 days of e-cigarette exposure when compared with control, and this was independent of nicotine content in the vapor. No significant difference was observed between the two e-cigarette groups (with and without nicotine) in both sexes.
Figure 3.
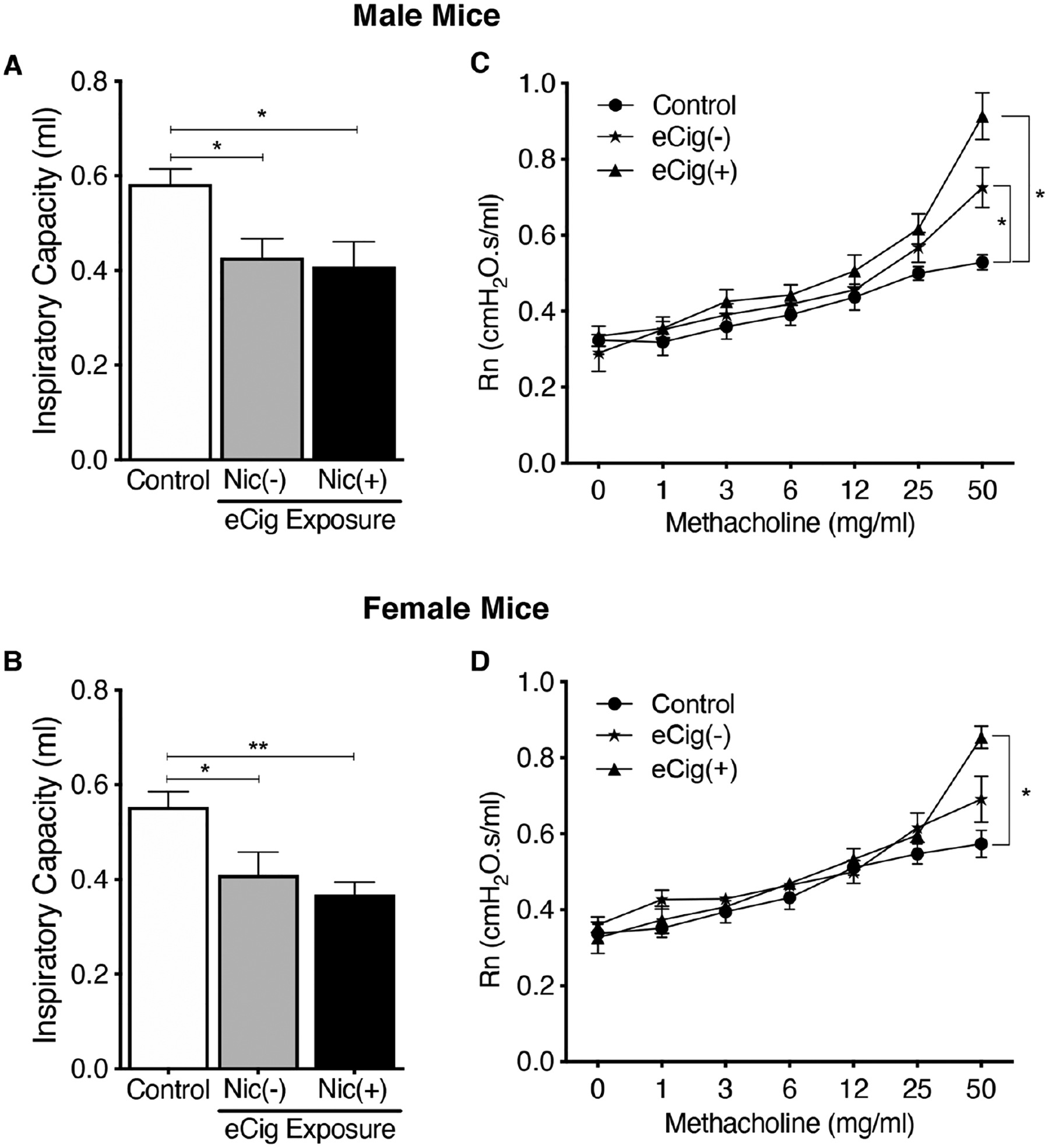
Lung physiology was measured using a flexiVent small animal ventilator. Inspiratory capacity (IC) was measured at baseline (A, B) and airway resistance (Rn) was measured with increasing methacholine (MCh) concentrations (C, D). IC was reduced in both sexes with (eCig Nic(+)) and without (eCig Nic(−)) nicotine when compared with their respective controls. Rn increased in male mice at 50 mg/mL both in the presence and absence of nicotine in the vapor (C), while in females Rn only increased with nicotine coexposure at MCh 50 mg/mL. Data represent at least five animals per group; *p<0.05, **p<0.01, using one-way ANOVA (A, B) and two-way ANOVA (C, D) with Bonferroni correction. ANOVA, analysis of variance; eCig, e-cigarette; Nic, nicotine.
We further measured airway hyper-responsiveness (AHR) to inhaled MCh at increasing concentrations (figure 3C,D). In male mice, at an MCh concentration of 50 mg/mL, there was a significant increase in airway resistance (Rn) in both e-cigarette (±nicotine) groups as compared with control (figure 3C). This increase was greatest in the e-cigarette nicotine group (eCig(+)) as compared with no nicotine (eCig(−)); however, there was no statistically significant difference in the mean Rn between eCig(−) and eCig(+) at this MCh dose. In female mice, at an MCh concentration of 50 mg/mL, there was a significant increase in Rn only in the eCig(+) group as compared with control (figure 3D).
Effect of chronic e-cigarette exposure on lung ACE-2 expression
We measured ACE-2 expression in lung homogenates obtained from male and female mice. Exposure to e-cigarette increased lung ACE-2 protein expression in a nicotine-dependent manner in male but not in female mice (figure 4A,B). These results were further strengthened by a similarly significant increase in ACE-2 mRNA in a nicotine-dependent manner in male but not in female mice (figure 4C). These results suggest that not only does vaping induce ACE-2 lung expression, but in addition nicotine further drives ACE-2 expression in male mice in a sex-specific manner.
Figure 4.
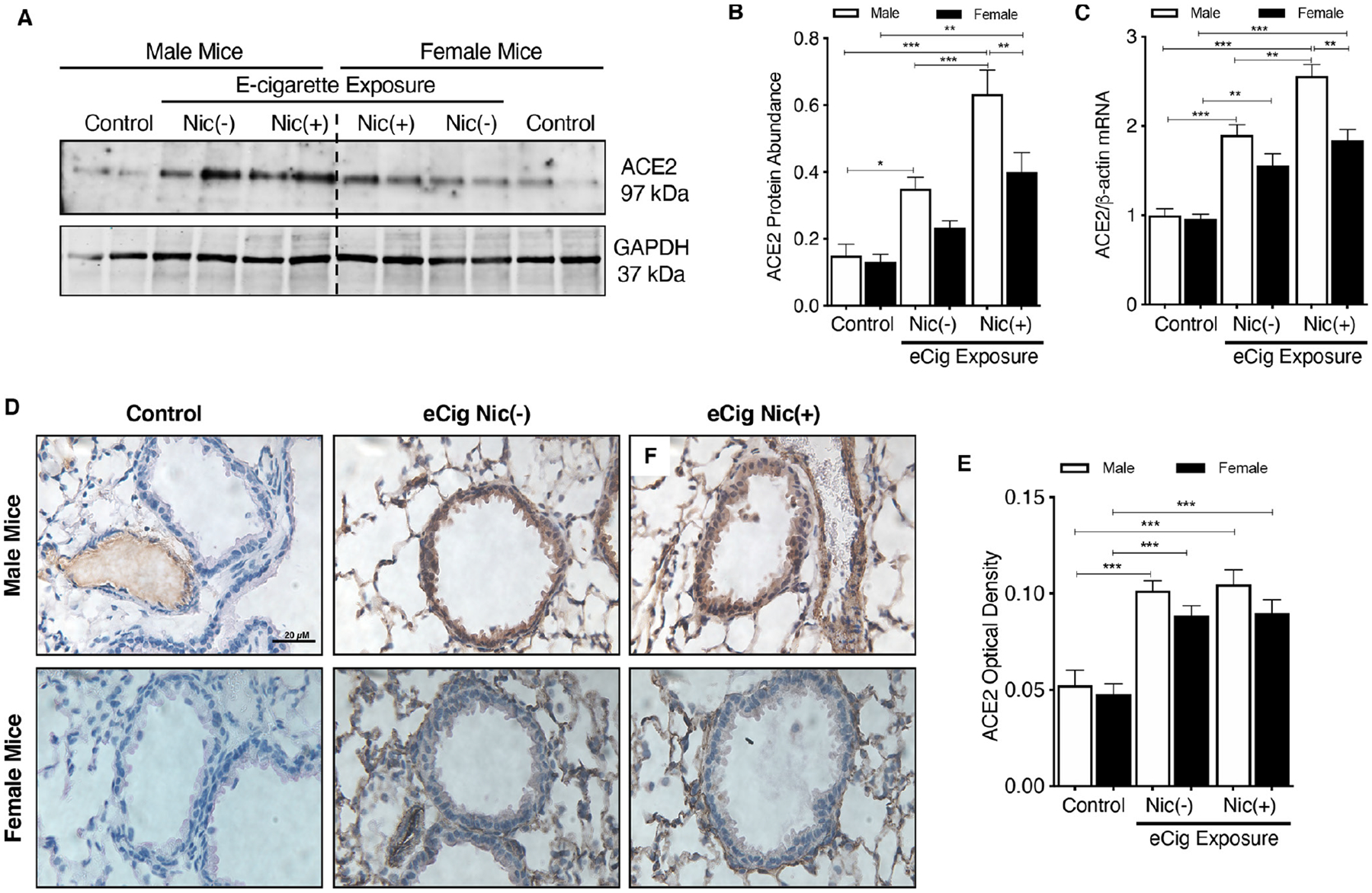
Representative western blots for ACE-2 protein (A) with densitometric analysis (B) and mRNA (C) from lung tissues of male and female mice exposed to e-cigarette vapor (±nicotine). Lung ACE-2 immunostaining (D) after e-cigarette exposure in male (upper panel) and female (lower panel) mice. (E) ACE-2 optical density of lung tissue (D) was quantified. Data represent five animals per group; *p<0.05, **p<0.01, ***p<0.001, using one-way analysis of variance with Bonferroni correction.
We also carried out immunostaining to assess ACE-2 localization in lung tissues including airways. As shown on the immunohistochemistry images (figure 4D), e-cigarette vapor exposure significantly increased ACE-2 expression in both male and female mouse lungs when compared with their respective controls (figure 4E), but this increase was independent of nicotine content within the e-cigarette vapor.
DISCUSSION
From the 2003 SARS outbreak we learned that SARS-CoV engages ACE-2 as its entry receptor16 and can significantly upregulate the expression of ACE-2 to facilitate its entry into the host cell.26 The same mechanism has been confirmed for SARS-CoV-2.27 Compared with SARS-CoV and MERS, SARS-CoV-2 is transmitted more readily from human to human even via normal breathing, yet the underlying mechanisms for this high transmissibility remain unknown.3 4 Thus, given these observations and the known mechanisms of host–viral interactions, attention has turned to the role of host cell ACE-2 receptors.
Early data indicate that COVID-19 mortality rates are higher in men7; however, we do not yet fully understand the demographic differences and susceptibility factors in various age groups. Emerging reports from China suggest that elderly men had higher hospitalization and mortality rates due to COVID-19 infection, where the smoking rates in men are as high as 60%.7 These figures are highly alarming as COVID-19 is not only affecting the elderly but also the younger population. Accumulating evidence, including from our own research, indicates that vaping causes negative health outcomes, including the recently reported EVALI (e-cigarette vapor-induced lung injury) and earlier reports of ‘vapor lung’.28 29 Thus, other external risk factors (besides smoking) such as vaping may also influence the epidemiological patterns of transmissibility, morbidity, and mortality in COVID-19.
Here, we show that e-cigarette vapor exposure increases airway inflammation, impairs lung function, and significantly upregulates ACE-2 expression in the lung. Our results further indicate that vaping itself not only induces airway and lung ACE-2 expression, but that this occurs in a sex-dependent manner primarily driven by nicotine coexposure, where ACE-2 expression is significantly higher in male mice. We found that both ACE-2 protein and mRNA expression are increased in male mice as compared with female mice, and this was specifically augmented by nicotine coexposure. Our observations are supported by recent findings by Kalidhindi et al,9 where males had significantly greater baseline ACE-2 expression in airway smooth muscle (ASM) cells as compared with females, and where stimulation with testosterone significantly upregulated ACE-2 expression in ASM cells from both males and females, further supporting the idea of possible sex-based differences in COVID-19.
While for some of the BAL fluid cytokines we reported that nicotine coexposure further increased cytokine levels in both sexes, increases in ACE-2 expression showed a clear sex-dependent increase in male mice driven by nicotine content in e-cigarette vapor. These observations suggest that nicotine-containing vapor further augments vapor-induced ACE-2 expression in the lungs of male mice. Although this will require additional confirmatory evidence, our initial observations suggest that, like cigarette smokers,7 people who vape could potentially have an increased risk of developing COVID-19 mediated by increased ACE-2 expression in their airways and lungs.
As would be expected, lung function (ie, IC and AHR) was also adversely affected by e-cigarette vapor exposure. MCh caused a significant increase in Rn in both sexes; however, in males it was nicotine-independent, while in females it was nicotine-dependent. The added effects of nicotine in vapor clearly have differential effects in different cellular responses, tissue compartments, and physiological outcomes. While our data show no obvious correlation(s) between the effects of sex and nicotine exposure on ACE-2 expression and lung function, additional research is needed to address this further, especially given the physiological role of ACE-2 in regulating lung inflammation.
RAS contributes directly to cardiopulmonary pathophysiology.30 31 ACE-2 synthesizes angiotensin (ANG) 1–7, which is a proresolution molecule in the lungs to help resolve acute inflammation,32 and this molecule may also be upregulated to mitigate smoke-induced and/or nicotine-induced damage. Lung ACE-2 expression is increased with smoking20 and in patients with COPD,18 19 and this increase in ACE-2 expression could be (at least in part) due to a feedback mechanism designed to resolve inflammation. Conversely, nicotine is known to influence RAS by upregulating the ACE/ANG-1/ANG-2 type 1 receptor axis while also downregulating the compensatory ACE-2/ANG 1–7/Mas receptor axis.33 This apparent paradox in ACE-2 expression could be related to the difference(s) between nicotine in isolation and the combined effects of the numerous noxious constituents in cigarette smoke or e-cigarette vapor.
Our results clearly show that e-cigarette vapor induced ACE-2 expression in murine lungs and that the added presence of nicotine in the vapor further augmented ACE-2 levels. However, it is not clear how nicotine induces ACE-2 expression in the airway cells. One potential mechanism could be through activation of nicotinic acetylcholine receptors (nAChR) because lung resident cells, such as bronchial epithelial cells, alveolar epithelial cells (type 2), and lung fibroblasts, express nAChR, specifically the α7 subtype.34 Russo and colleagues35 recently demonstrated that nicotine through α7-nAChR promoted downstream signaling (phospho-Akt and phospho-p44/42), which is required for nicotine-dependent ACE-2 expression in human primary airway epithelial cells. Nicotine may also increase SARS-CoV-2 entry into the lung cells.8 Similarly, we do not know by what mechanism(s) non-nicotine-containing e-cigarette vapor can induce ACE-2 expression in the lungs.22
While male sex appears to be an important variable with respect to smoking and vapor exposure, could there be a link between the younger population getting COVID-19 and vaping? This is yet another important question for the scientific community to investigate.22 Our experiments did not address the role of age; however, like sex, age is a vital biological and epidemiological variable worthy of investigation. Given the popularity of vaping around the world, especially in the younger adult and teenage populations, and its public health implications, this becomes a rather critical connection to investigate.
Despite our findings in this animal model, it remains to be seen whether vaping increases the risk of developing COVID-19 in human populations. Understanding the impact of this prevalent risk factor will allow the scientific community to not only formulate better therapeutic strategies, but may also help prevent COVID-19. Collectively, and if validated in forthcoming human studies, our findings can spur further research that could impact public health. Given the growing crisis of the COVID-19 global pandemic and our findings, urgent research is needed to validate our findings in human tissues and the affected populations.
Our study has some important limitations and we must exercise caution in how we interpret our mouse ACE-2 (mACE-2) data with respect to human ACE-2 (hACE-2) biology and the risk of developing COVID-19. Previous investigations in SARS-CoV-1 showed that certain inbred mouse strains are prone to SARS-CoV-1 infection.36 37 However, the same is not true for SARS-CoV-2 infection, suggesting possible inefficient binding of S-protein to the ACE-2 receptor in mouse species as compared with humans.16 27 Indeed, the SARS-CoV-2 S-protein binding domains of hACE-2 and mACE-2 differ, and this difference could explain the resistance of outbred wild-type mouse strains to SARS-CoV-2 infection.38
While we recognize that mACE-2 does not provide similar viral receptor entry as hACE-2 for the S-protein,13 27 39–41 our results are informative to direct similar research efforts in human-derived lung tissues to further test and validate our findings, the implication being that increased hACE-2 in patients as a result of vaping is predicted to enhance S-protein:ACE-2 binding, leading to increased viral entry and therefore a higher risk of developing COVID-19. Further, this biological effect may be modified by sex, thus disproportionately affecting males. In addition, we did not evaluate the role of host proteases (eg, TMPRSS2), which are necessary for SARS-CoV-2 viral entry in human cells.27 42
In summary, while vaping (±nicotine) caused airway inflammation and impaired lung function, the induction of mouse lung ACE-2 occurred to a significantly greater degree in males exposed to vapor containing nicotine as compared with females. If vaping in the human population similarly induces the expression of pulmonary ACE-2, then vaping itself may be an underappreciated risk factor for SARS-CoV-2 infection. Further research in relevant human airway and lung tissues is needed to further validate our findings.
Significance of this study.
What is already known about this subject?
Cigarette smoke-induced damage leads to long-term health problems including greater risk of developing chronic diseases and infections.
ACE-2 expression is increased in smokers and patients with chronic obstructive pulmonary disease.
ACE-2 facilitates SARS-CoV-2 entry through the airway epithelium.
There may be sex-based differences related to the development of COVID-19.
What are the new findings?
This study confirms the dangers of vaping in lung physiology, causing inflammation and increased airflow obstruction.
We further show that vaping increases ACE-2 expression in the lung, which is further enhanced by nicotine exposure in male mice.
These results highlight sex-based differences in vapor-induced lung ACE-2 expression that may be relevant to human SARS-CoV-2 susceptibility and infection.
How might these results change the focus of research or clinical practice?
Smoking is a known risk factor for developing infections including viruses, and emerging data suggest that vaping may be no different.
Further, the dangers of vaping were established during the 2019 EVALI (e-cigarette vapor-induced lung injury) epidemic in the USAnited States.
The increased prevalence of vaping especially in the younger and male populations potentially poses an even greater risk during the COVID-19 pandemic.
If vaping in the human population similarly induces the expression of pulmonary ACE-2, then vaping itself may be an under-appreciated risk factor for SARS-CoV-2 infection.
Identifying preventable risks such as e-cigarette use and vaping, with subsequent validation in the human population, may help us prevent and/or mitigate SARS-CoV-2 infection and the development of COVID-19.
Funding
The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Footnotes
Competing interests AAZ is an Associate Editor for the Journal of Investigative Medicine.
Ethics approval Animal studies were approved by the Institutional Animal Ethics Committee (IAEC), North East Technical Educational Society Institute of Pharmaceutical Sciences (NIPS), Guwahati, India (approval ID: NITS/NIPER/PC/2019-03-02).
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Our data are not in a public repository; however, they can be made available upon reasonable request.
REFERENCES
- 1.Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis 2020;20:533–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 2020;382:727–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo Y-R, Cao Q-D, Hong Z-S, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak - an update on the status. Mil Med Res 2020;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shang J, Ye G, Shi K, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020;581:221–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishiga M, Wang DW, Han Y, et al. COVID-19 and cardiovascular disease: from basic mechanisms to clinical perspectives. Nat Rev Cardiol 2020;17:543–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dudley JP, Lee NT. Disparities in age-specific morbidity and mortality from SARS-CoV-2 in China and the Republic of Korea. Clin Infect Dis 2020;71:863–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai H Sex difference and smoking predisposition in patients with COVID-19. Lancet Respir Med 2020;8:e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olds JL, Kabbani N. Is nicotine exposure linked to cardiopulmonary vulnerability to COVID-19 in the general population? Febs J 2020;287:3651–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalidhindi RSR, Borkar NA, Ambhore NS, et al. Sex steroids skew ACE2 expression in human airway: a contributing factor to sex differences in COVID-19? Am J Physiol Lung Cell Mol Physiol 2020;319:L843–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuba K, Imai Y, Penninger JM. Angiotensin-converting enzyme 2 in lung diseases. Curr Opin Pharmacol 2006;6:271–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jia HP, Look DC, Shi L, et al. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J Virol 2005;79:14614–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou P, Yang X-L, Wang X-G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020;579:270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wan Y, Shang J, Graham R, et al. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol 2020;94:e00127–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol 2020;5:562–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wrapp D, Wang N, Corbett KS, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020;367:1260–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li W, Moore MJ, Vasilieva N, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003;426:450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walls AC, Park Y-J, Tortorici MA, et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020;181:281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brake SJ, Barnsley K, Lu W, et al. Smoking upregulates angiotensin-converting enzyme-2 receptor: a potential adhesion site for novel coronavirus SARS-CoV-2 (Covid-19). J Clin Med 2020;9:841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leung JM, Yang CX, Tam A, et al. ACE-2 expression in the small airway epithelia of smokers and COPD patients: implications for COVID-19. Eur Respir J 2020;55:2000688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H, Rostami MR, Leopold PL, et al. Expression of the SARS-CoV-2 ACE2 Receptor in the Human Airway Epithelium. Am J Respir Crit Care Med 2020;202:219–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie X, Xudong X, Chen J, et al. Age- and gender-related difference of ACE2 expression in rat lung. Life Sci 2006;78:2499–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McAlinden KD, Eapen MS, Lu W, et al. COVID-19 and vaping: risk for increased susceptibility to SARS-CoV-2 infection? Eur Respir J 2020;56:2001645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma P, Yi R, Nayak AP, et al. Bitter taste receptor agonists mitigate features of allergic asthma in mice. Sci Rep 2017;7:46166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McAlinden KD, Deshpande DA, Ghavami S, et al. Autophagy activation in asthma airways remodeling. Am J Respir Cell Mol Biol 2019;60:541–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doobay MF, Talman LS, Obr TD, et al. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol 2007;292:R373–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang PH, Cheng Y. Increasing host cellular Receptor—Angiotensin-Converting enzyme 2 (ACE2) expression by coronavirus may facilitate 2019-nCoV infection. bioRxiv preprint 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020;181:271–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crotty Alexander LE, Ware LB, Calfee CS, et al. E-Cigarette or Vaping product Use-associated lung injury: developing a research agenda. An NIH workshop report. Am J Respir Crit Care Med 2020;202:795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matthys A, Zeki AA. “Vapor Lung”: Organizing Pneumonia Due to Electronic Cigarette Use in a Patient with Severe Eosinophilic Asthma. J Invest Med 2019;201:A6684. [Google Scholar]
- 30.Ferrario CM. The renin-angiotensin system: importance in physiology and pathology. J Cardiovasc Pharmacol 1990;15 Suppl 3:S1–5. [PubMed] [Google Scholar]
- 31.Nicholls MG, Richards AM, Agarwal M. The importance of the renin-angiotensin system in cardiovascular disease. J Hum Hypertens 1998;12:295–9. [DOI] [PubMed] [Google Scholar]
- 32.Magalhaes GS, Barroso LC, Reis AC, et al. Angiotensin-(1–7) Promotes Resolution of Eosinophilic Inflammation in an Experimental Model of Asthma. Front Immunol 2018;9:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oakes JM, Fuchs RM, Gardner JD, et al. Nicotine and the renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol 2018;315:R895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cardinale A, Nastrucci C, Cesario A, et al. Nicotine: specific role in angiogenesis, proliferation and apoptosis. Crit Rev Toxicol 2012;42:68–89. [DOI] [PubMed] [Google Scholar]
- 35.Russo P, Bonassi S, Giacconi R, et al. COVID-19 and smoking: is nicotine the hidden link? Eur Respir J 2020;55:2001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts A, Paddock C, Vogel L, et al. Aged BALB/c mice as a model for increased severity of severe acute respiratory syndrome in elderly humans. J Virol 2005;79:5833–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumaki Y, Salazar AM, Wandersee MK, et al. Prophylactic and therapeutic intranasal administration with an immunomodulator, Hiltonol® (Poly IC:LC), in a lethal SARS-CoV-infected BALB/c mouse model. Antiviral Res 2017;139:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bao L, Deng W, Huang B, et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 2020;583:830–3. [DOI] [PubMed] [Google Scholar]
- 39.Li Y, Zhou W, Yang L, et al. Physiological and pathological regulation of ACE2, the SARS-CoV-2 receptor. Pharmacol Res 2020;157:104833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.South AM, Diz DI, Chappell MC. COVID-19, ACE2, and the cardiovascular consequences. Am J Physiol Heart Circ Physiol 2020;318:H1084–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 2020;395:1054–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hou YJ, Okuda K, Edwards CE, et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell 2020;182:429–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data relevant to the study are included in the article or uploaded as supplementary information. Our data are not in a public repository; however, they can be made available upon reasonable request.