

Abstract
γδ T cells are potent anti-cancer effectors with the potential to target tumours broadly, independent of patient-specific neoantigens or HLA background1–5. γδ T cells can sense conserved cell stress signals prevalent in transformed cells2,3, although the mechanisms behind the targeting of stressed target cells remain poorly characterized. Vγ9Vδ2 T cells – the most abundant subset of human γδ T cells4 – recognize a protein complex containing butyrophilin 2A1 (BTN2A1) and BTN3A16–8, a widely expressed cell surface protein that is activated by phosphoantigens abundantly produced by tumour cells. Here we combined genome-wide CRISPR screens in target cancer cells to identify pathways that regulate γδ T cell killing and BTN3A cell surface expression. The screens showed previously unappreciated multilayered regulation of BTN3A abundance on the cell surface and triggering of γδ T cells through transcription, post-translational modifications, and membrane trafficking. In addition, diverse genetic perturbations and inhibitors disrupting metabolic pathways in the cancer cells, particularly ATP-producing processes, were found to alter BTN3A levels. This induction of both BTN3A and BTN2A1 during metabolic crises was dependent on AMP-activated protein kinase (AMPK). Finally, small molecule activation of AMPK in a cell line model and in patient-derived tumour organoids led to increased expression of the BTN2A1-BTN3A complex and increased Vγ9Vδ2 TCR-mediated killing. This AMPK-dependent mechanism of metabolic stress-induced ligand upregulation deepens our understanding of γδ T cell stress surveillance and suggests new avenues available to enhance γδ T cell anti-cancer activity.
γδ T cells exhibit both innate and adaptive immune functions in various cancer and infection settings1–5. The ability of γδ T cell receptor (γδTCR) clones to recognize diverse tumour types and the lack of restriction to antigen presenting molecules lend this cell type to potential autologous and allogeneic cell therapy approaches2,4,5. Such broad anti-cancer activity by individual γδTCR clones sets them apart from the highly specific αβTCRs expressed by conventional αβ T cells. γδ T cells have been shown to sense conserved cell stress signals prevalent in transformed or other perturbed cells in both TCR-dependent and -independent fashions2,3,7–13. However, the mechanisms governing stress-regulated interactions between γδ T cells and their diseased target cells are unclear.
Vγ9Vδ2 T cells have an invariant specificity for the BTN2A1-BTN3A1-BTN3A2 complex6–8,14, which is broadly expressed in both healthy and cancerous human tissues (The Human Protein Atlas15), although BTN3A1 (CD277) can be expressed at higher levels in malignant tumours16. Most of the relevant human components of this interaction – Vγ9/Vδ2 TCR chains, BTN3A1, and BTN3A2 – have no orthologs in rodent models. The BTN2A1-3A1-3A2 complex is activated upon engagement of the BTN3A1 intracellular binding pocket by phosphoantigen metabolite intermediates of the mevalonate pathway (Extended Data Fig. 1a)17, allowing BTN2A1 to bind directly an invariant germline-encoded region of Vγ9 and activate Vγ9Vδ2 T cells7,8. A hyperactive mevalonate pathway, which feeds into cholesterol and other biosynthesis pathways, is a hallmark of many transformed cells owing to their heightened proliferative needs18. However, other parameters regulating the interaction between Vγ9Vδ2 T cells and cancer cells remain largely unknown.
Here, we systematically characterized the functional molecular determinants of cancer cell interactions with Vγ9Vδ2 T cells by combining two genome-wide CRISPR screens. These screens allowed for a focused examination of the interface between γδ T cells and tumour cells, which is part of the complex network of interactions among cancer cells, γδ T cells, the rest of the immune system, and the tumour microenvironment.
Vγ9Vδ2 T cell co-culture screen
To identify the genes regulating killing of cancer cells by human Vγ9Vδ2 T cells, we used CRISPR to create a genome-wide pool of knockout (KO) Daudi cancer cells (Burkitt’s lymphoma) constitutively expressing Cas9 (Daudi-Cas9) and co-cultured them with expanded primary Vγ9Vδ2 T cells from healthy donors. We treated Daudi-Cas9 cells with zoledronate (ZOL) for 24 hours prior to the γδ T cell co-culture. While Daudi cells are naturally targeted by Vγ9Vδ2 T cells, ZOL further enhances the Vγ9Vδ2 TCR interaction with the butyrophilin complex by upregulating intracellular levels of phosphoantigen (Extended Data Fig. 1a, b). We compared the frequency of single guide RNAs (sgRNAs) in surviving Daudi-Cas9 cells after the co-culture relative to the baseline KO cell library to identify target cell genes that either enhance or diminish their killing by Vγ9Vδ2 T cells (Fig. 1a). The screen hits (false discovery rate [FDR] < 0.05) were largely consistent among the 3 donors (Extended Data Fig. 1c–e, Supplementary Tables 1 and 2), with some variability as expected in cell-cell interaction screens. Confidence in key hits was bolstered further by consistent enrichment or depletion of multiple sgRNAs targeting the same genes (Fig. 1b).
Figure 1. Vγ9Vδ2 T cell co-culture with genome-wide KO library of Daudi cells reveals killing-evasion and -enhancement KOs.
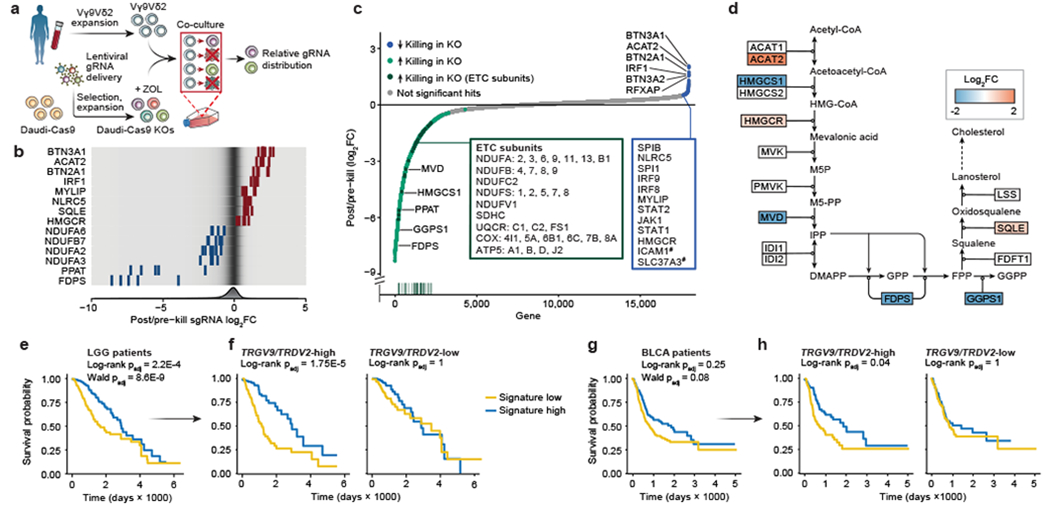
(a) Vγ9Vδ2 T cell co-culture screen with a genome-wide KO library of Daudi-Cas9 cells (ZOL = zoledronate). (b) Enrichment or depletion of individual single guide RNAs (sgRNA) for a selection of significant hits, overlaid on a gradient showing distribution of all sgRNAs (fold change [FC]). (c) All 18,010 genes ranked from negative to positive enrichment of Daudi-Cas9 KOs that change killing. Boxes show a subset of significant hits. Vertical line plot shows rank positions of electron transport chain (ETC) subunits listed in the green box. False-discovery rate (FDR) < 0.05, except #FDR<0.1. (d) Schematic of the enrichment and depletion of mevalonate pathway KOs. Shading indicating log2FC shown only for significant hits (FDR<0.05). (e-h) Survival of (e) low grade-glioma (LGG) (n=529) and (g) bladder urothelial carcinoma (BLCA) patients (n=433) split by the co-culture screen gene signature expression levels, (f, h) including after splitting each patient cohort by TRGV9/TRDV2 expression levels. (a-c) n=3 human PBMC donors; enrichment and statistics calculated by the MAGeCK algorithm. (e-h) Log-rank test (Kaplan-Meier survival analysis) and (e, g) Wald test (Cox regression), adjusted (padj) with Benjamini-Hochberg multiple comparisons correction.
Expected regulators of cancer cell-Vγ9Vδ2 T cell interactions were identified successfully, with increased survival among the following KOs: (1) components of the butyrophilin complex (BTN2A1, BTN3A1, BTN3A2); (2) mevalonate pathway enzymes, ACAT2 and HMGCR, that are upstream of phosphoantigen synthesis; (3) SLC37A3, a transporter of ZOL into the cytosol19; (4) NLRC5, a confirmed transactivator of BTN3A1-3 genes20; and (5) ICAM1, a surface protein important for Vγ9Vδ2 T cell recognition of target cells21 (Fig. 1c, d). Similarly, we observed reduced survival of KOs that would be expected to have increased phosphoantigen concentrations in the mevalonate pathway (FDPS, GGPS1) (Fig. 1b–d).
Gene Set Enrichment Analysis (GSEA) highlighted various metabolic pathways whose KOs led to decreased survival in co-culture, with a particularly strong enrichment for OXPHOS, the tricarboxylic acid (TCA) cycle, and purine metabolism KEGG pathways (Fig. 1c, Extended Data Fig. 1f). These pathways are essential for maintaining a proper ATP balance. We observed a striking enrichment of structural subunits of Complexes I – V of the electron transport chain (ETC) driving OXPHOS (Fig. 1c). Additionally, KO of various type I interferon (IFN-I) signalling components (IRF1, IRF8, IRF9, JAK1, STAT1, STAT2) enhanced Daudi-Cas9 cell survival upon co-culture (Fig. 1c). Of note, certain enriched KEGG gene sets have considerable overlap among their constituent genes, especially the overlap between purine and pyrimidine metabolism gene sets and between OXPHOS and Parkinson’s disease gene sets (Extended Data Fig. 2g, Supplementary Table 3). Nonetheless, strong enrichment of hits in specific pathways, especially ones that jointly contribute toward cell energy state maintenance (Fig. 1c, Extended Data Fig. 1f), further enhanced our confidence in the screen results.
We wanted to assess the relevance of our hits beyond the Daudi cell line co-culture system and test if they accord with human clinical data. We generated a gene signature based on co-culture screen results, measured gene signature scores for tumours across 33 cancer types in The Cancer Genome Atlas (TCGA), and correlated the scores with patient survival. The strongest correlation was observed in low-grade glioma (LGG) tumours (Extended Data Fig. 2a). LGG patients whose tumours exhibited high gene signature scores (high/low expression of positive/negative regulators of γδ T cell killing, respectively) had significantly better overall survival compared to those with low signature scores (Fig. 1e). Notably, 204 of the 1040 genes in the signature individually correlated with survival in the LGG cohort (Extended Data Fig. 2b, Supplementary Table 4). Gene signatures derived from various enriched pathways also significantly correlated with survival outcomes in LGG patients, with the TCA Cycle KEGG pathway signature showing the strongest (negative) correlation (Extended Data Fig. 3c, f, Supplementary Table 5).
We examined if the association of the co-culture genetic signature with patient survival depends on the presence of γδ T cells in patient tumours. We split the 529 LGG patients into two groups according to their TRGV9 (Vγ9) and TRDV2 (Vδ2) transcript abundance in the tumours and found that the survival advantage conferred by high signature levels is seen only in the subset of LGG patients with high Vγ9Vδ2 T cell infiltration (Fig. 1f). Among the 1040 genes in the signature, significant correlation with survival was observed with 142 genes in TRGV9/TRDV2-high LGG patients and only 8 genes in TRGV9/TRDV2-low patients (Extended Data Fig. 2d, Supplementary Table 4). Gene pathway signatures also preferentially correlated with survival outcomes in TRGV9/TRDV2-high LGG patients, with the TCA Cycle KEGG pathway signature again showing the strongest (negative) correlation (Extended Data Fig. 2e, g, Supplementary Table 5). A similar pattern was found in the bladder urothelial carcinoma (BLCA) cohort with 433 patients, in which the co-culture screen signature correlated with survival only in the subset of patients with high TRGV9/TRDV2 expression levels (Fig. 1g, h). Importantly, the significant signature score correlation with survival was not observed when the LGG and BLCA cohorts were subset based on expression of the αβ T cell receptor constant regions (TRAC and TRBC) (Extended Data Fig. 2h, i). These data suggest that the co-culture screen identified cancer gene pathways that shape the effects of Vγ9Vδ2 T cells in patients.
We hypothesized that many of the co-culture screen hits could affect Vγ9Vδ2 T cell killing by regulating BTN3A1 levels. Indeed, public data from healthy tissues show that expression of key hits and gene ontology pathways are correlated and gene ontology pathways were correlated strongly with BTN3A1 expression (Extended Data Fig. 3a, b). As expected, the upstream mevalonate pathway hit ACAT2 showed no such correlation (Extended Data Fig. 3b). Notably, the vast majority of OXPHOS genes negatively correlated to BTN3A1 expression across healthy immune tissue (Extended Data Fig. 3c), suggesting that BTN3A1 levels could be affected by the cellular energy state.
BTN3A expression screen
To understand systematically gene pathways that alter BTN3A abundance, we performed a complementary genome-wide CRISPR screen to identify regulators of BTN3A surface levels. Using a genome-wide KO pool of Daudi-Cas9 cells, we sorted BTN3Ahigh and BTN3Alow cells (top and bottom 25% of combined surface expression of BTN3A1, BTN3A2, and BTN3A3, which have similar ectodomains equally recognized by available anti-BTN3A antibody clones22) and determined which KOs regulate BTN3A levels at the cell surface (Fig. 2a). The hits were correlated strongly across three replicates (Extended Data Fig. 4a–c, Supplementary Tables 6 and 7). Comparing hits from the BTN3A and the co-culture screens, we identified concordant hits whose KOs either (1) conferred a survival advantage against T cells and downregulated BTN3A, or (2) conferred a survival disadvantage against T cells and upregulated BTN3A (Fig. 2b). Of the significant hits (FDR<0.01) in the BTN3A expression screen, 74% were concordant with the co-culture screen (FDR<0.05), indicating that regulators of BTN3A surface expression strongly influence Vγ9Vδ2 T cell killing of target cells. For example, a number of transcriptional regulator KOs that conferred a survival advantage in the co-culture screen showed downregulated BTN3A expression, such as NLRC5, IRF1, IRF8, IRF9, SPI1, and SPIB (Fig. 1c, 2c). Overall, we observed a strong correlation between the effect sizes for gene KO hits that protected against Vγ9Vδ2 T cell killing and downregulated BTN3A (Pearson’s r = 0.77) and a moderate correlation for hits that enhanced T cell killing and upregulated BTN3A (Pearson’s r =0.51) (Extended Data Fig. 4d). These correlations suggested that the effects of many hits in the co-culture screen may largely depend on their effects on BTN3A levels.
Figure 2. Systematic discovery of BTN3A surface expression regulators.
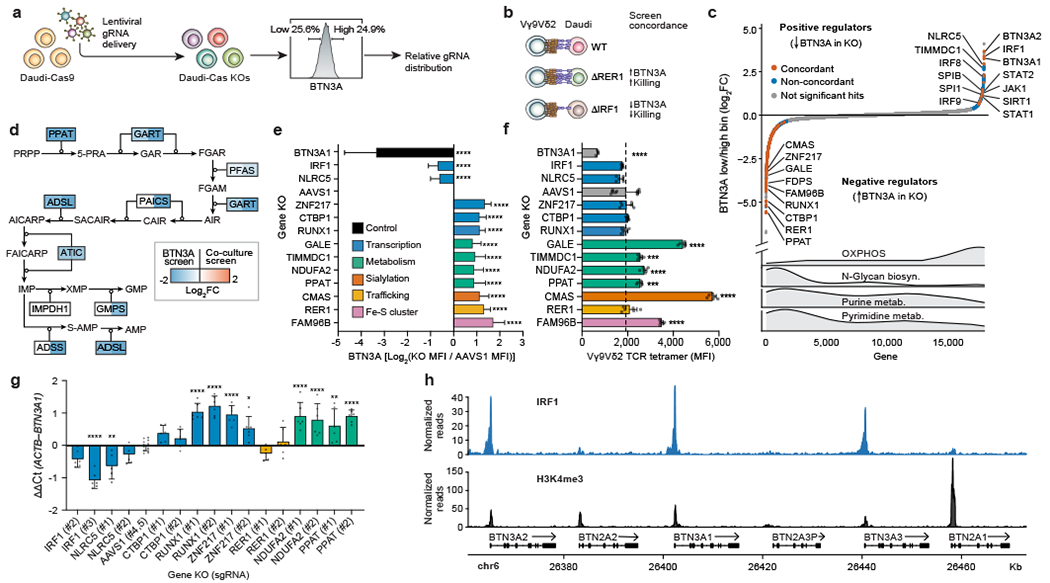
(a) Genome-wide KO screen for surface expression of BTN3A. Top and bottom 25% based on BTN3A surface staining were sorted for downstream analysis. (b) Screen concordance with examples of hits concordant between the two screens. (c) All 18,010 genes ranked from negative to positive enrichment of Daudi-Cas9 KOs in BTN3Alow relative to BTN3Ahigh cells. Concordant and non-concordant hits highlighted (BTN3A screen FDR<0.01, co-culture screen FDR<0.05). Distribution of selected KEGG gene sets shown below. (d) Depletion of purine biosynthesis pathway KOs across both screens. Shading indicating log2FC shown only for significant hits (FDR<0.05). (e) Surface BTN3A median fluorescence intensity (MFI) at 13 days post-transduction, normalized to BTN3A MFI in AAVS1 controls and log2-transformed. Two distinct KOs per gene deletion, except BTN3A1 (one KO). Combined data from three separate experiments, each individually normalized. AAVS1 n=36, BTN3A1 n=9, all other deletions n=18. (f) Vγ9Vδ2 TCR tetramer staining fluorescence (MFI) at 13 days post-transduction. Data from one experiment. AAVS1 n=12, BTN3A1 n=3, all other deletions n=6. (g) qPCR data for BTN3A1 transcripts normalized to ACTB transcripts. n=5-6, AAVS1 n=12, data combined from two independent experiments. (h) CUT&RUN data for IRF1 binding and H3K4me3 chromatin modification at the butyrophilin locus. n=3 per condition. (e-g) One-way ANOVA with Dunnett’s multiple comparisons test. Mean ± SD. p<0.0001 (****), p<0.001 (***), p<0.01 (**), p<0.05 (*).
It is well recognized that the mevalonate pathway regulates Vγ9Vδ2 TCR–BTN2A1/3A1 interactions through modulation of phosphoantigen levels, but it was not previously thought to regulate BTN3A surface abundance. Our screen revealed increased surface staining of BTN3A among cells with deletions of the mevalonate pathway enzyme FDPS (Fig. 2c). Confirming this result, we observed a substantial and dose-dependent increase in BTN3A median fluorescence intensity (MFI) upon FDPS inhibition (Extended Data Fig. 4e). Further confirming this result, Daudi-Cas9 FDPS KO cells showed consistent upregulation of BTN3A MFI compared to the CRSIPR cutting control KO of AAVS1, a safe harbour site (Extended Data Fig. 4f).
The BTN3A expression screen revealed regulation by specific metabolic pathways (Fig. 2c, Extended Data Fig. 4g). Concordant enrichment between the two screens was observed for pathways whose KOs upregulated BTN3A surface expression and reduced survival in the Vγ9Vδ2 T cell co-culture: de novo purine biosynthesis (Fig. 2d), iron-sulphur (Fe-S) cluster formation, N-glycan biosynthesis, and sialylation (Extended Data Fig. 5b–d). The purine biosynthesis pathway produces IMP, GMP, and AMP nucleotides, the latter of which is important in maintaining proper energy homeostasis both by regulating AMP-activated protein kinase (AMPK) activity and by being regenerated into ATP (Fig. 2d). Meanwhile, Fe-S clusters are produced for certain mitochondrial and cytosolic proteins (Extended Data Fig. 5b), such as PPAT (purine biosynthesis) and OXPHOS Complexes I, II, and III23. Finally, in the original study identifying the cognate ligand of Vγ9Vδ2 TCRs, BTN2A1 itself and the global negative regulator of glycosylation, SPPL3, resulted in the highest disruption of Vγ9Vδ2 TCR-ligand interactions7,24. Our findings further suggest that decreased N-linked glycosylation and sialylation increase BTN3A surface staining and increases γδ T cell killing of target cells (Extended Data Fig. 5c, d).
Similar to the co-culture gene signature analysis of TCGA data, the BTN3A screen gene signature also had the strongest correlation with LGG patient survival. This signature correlated with a more prominent survival advantage when the tumours exhibited either high Vγ9Vδ2 T cell or high αβ T cell infiltration (Extended Data Fig. 6), suggesting that this signature could contain genes relevant to both T cell types. Previously, γδ T cell infiltration was reported to correlate with better survival in different brain cancer types and LGG in particular25. Our findings suggest that LGG patient survival could be affected by activity of cancer cell genes that regulate BTN3A expression.
To validate a subset of the identified BTN3A regulators, we generated one BTN3A1 KO and two distinct KOs for every other screen hit. For each target, the effects on BTN3A MFI were consistent between the two distinct KOs. As expected, deletion of NLRC5 had a marked effect on BTN3A levels. We also confirmed our discovery that IRF1 is similarly required to maintain BTN3A levels and that the transcription factors ZNF217, CtBP1, and RUNX1 negatively regulate BTN3A surface expression (Fig. 2e, Extended Data Fig. 7a). Interestingly, CtBP1 – a metabolic sensor whose transcriptional regulation depends on the cellular NAD+/NADH ratio26 – was the top ranked KO among cells with upregulated BTN3A in the CRISPR screen (Supplementary Table 6). We also confirmed increased BTN3A surface abundance after disruption of various cell processes: sialylation (CMAS), Fe-S cluster formation (FAM96B), and control of egress out of the endoplasmic reticulum (ER) of properly assembled multiprotein complexes (RER1)27 (Fig. 2e, Extended Data Fig. 7a). The latter provides a mechanistic explanation for earlier findings that BTN3A1-BTN3A2 heterodimeric complexing and retention in the ER is important for proper downstream function at the plasma membrane and that BTN2A1 and BTN3A depend on each other for trafficking to the plasma membrane28,29. Finally, we confirmed that metabolic perturbations through deletions in galactose catabolism (GALE), de novo purine biosynthesis (PPAT), and OXPHOS Complex I (NDUFA2, TIMMDC1) can increase surface BTN3A abundance (Fig. 2e). We also determined that GALE, NDUFA2, PPAT, CMAS, and FAM96B KOs showed consistently higher Vγ9Vδ2 TCR (clone G115) tetramer binding, with particularly high tetramer staining in sialylation (CMAS) KOs (Fig. 2f, Extended Data Fig. 7b). While Vγ9Vδ2 TCRs have never been shown to directly bind to BTN3A, they are known to have invariant binding through a germline encoded region of Vγ9 to BTN2A17,8. Increased Vγ9Vδ2 TCR tetramer binding would imply either increased surface abundance of BTN2A1 or increased binding affinity to BTN2A1 due to perturbed post-translational modifications.
To help determine the mechanism by which some of the validated hits regulate the butyrophilin complex, we measured BTN2A1, BTN3A1, and BTN3A2 transcript levels in a subset of the Daudi-Cas9 KOs. As expected for a protein trafficking regulator, RER1 KOs did not affect transcript levels. Among transcription factors, we confirmed that ablation of IRF1 and NLRC5 reduced BTN3A1/2 transcript levels and ablation of ZNF217 and RUNX1 increased BTN3A1/2 transcript levels. Unexpectedly, KO of metabolic factors NDUFA2 (OXPHOS) and PPAT (purine biosynthesis) increased BTN3A1/2 transcript levels (Fig. 2g, Extended Data Fig. 7c). Only two of the tested KOs showed statistically significant changes in BTN2A1 transcript levels (Extended Data Fig. 7d).
To characterize more deeply the mechanisms of IRF1 and ZNF217, we performed Cleavage Under Targets and Release Using Nuclease (CUT&RUN) to determine their genome-wide binding sites. We discovered that the positive regulator IRF1 occupied BTN3A1, BTN3A2, and BTN3A3 promoter regions associated with H3K4me3, a histone modification found at transcriptional start sites (Fig. 2h). Minimal CUT&RUN enrichment for IRF1 was observed at the BTN2A1 and BTN2A2 promoter regions (Fig. 2h), consistent with the lack of BTN2A1 transcriptional changes in Daudi-Cas9 IRF1 KO cells (Extended Data Fig. 7d). This binding pattern in the butyrophilin locus was consistent with publicly available ChIP-seq data in K562 cells stably expressing IRF1 (Extended Data Fig. 8a). Sites of CUT&RUN enrichment for the negative regulator ZNF217 overlapped with those for the positive regulator IRF1 at the BTN3A1/2/3 promoter regions, suggesting that the two factors directly regulate BTN3A genes and possibly influence each other’s regulatory activity (Extended Data Fig. 8b–d). Notably, ZNF217 is reported to form a complex with the negative regulator CtBP130. These data collectively indicate that the individual subunits of the BTN2A1-3A1-3A2 complex are differentially regulated, as IRF1 and ZNF217 exert direct transcriptional control specifically over BTN3A1/2 genes and have little effect on BTN2A1 transcription. Furthermore, we confirmed that both Daudi KOs of IRF1 had increased survival in a co-culture with Vγ9Vδ2 T cells (Extended Data Fig. 8e).
Surprisingly, most of the subunits comprising the five ETC complexes driving OXPHOS were significant hits with discordant effects in our two screens (Extended Data Fig. 5a). However, validation results for Complex I KOs (NDUFA2, TIMMDC1) contradicted the BTN3A screen results (Fig. 2c, e–g) and were instead concordant with the co-culture screen findings (Fig. 1c) and in agreement with the negative correlation between OXPHOS genes and BTN3A1 in healthy human immune samples (Extended Data Fig. 3c). These data suggested a complex relationship between OXPHOS and BTN3A expression that could depend on exogenous culture conditions, given the different requirements of a high-coverage genome-wide screen and culturing individual KOs. Given how strong the OXPHOS signatures were in both screens and the observed transcriptional changes in BTN3A1/2 among OXPHOS KOs, we delved further into the effects of this metabolic pathway on BTN3A.
AMPK activation upregulates BTN3A
We tested whether the energy state imbalance via deficient ATP production or the redox state imbalance due to an elevated NADH/NAD+ ratio in the OXPHOS KOs (NDUFA2, TIMMDC1) was causing BTN3A expression changes (Fig. 3a). When cells were cultured in glutamine-containing medium lacking glucose and pyruvate, increasing glucose levels resulted in upregulated BTN3A surface expression in OXPHOS KOs (TIMMDC1, NDUFA2), with a much lower effect in AAVS1 KO control cells (Fig. 3b). No such effect was observed in cells grown in increasing levels of pyruvate, which should alleviate the redox imbalance by depleting excess NADH during the conversion of pyruvate to lactate (Extended Data Fig. 9a). These results suggested a strong link between the ATP levels in the OXPHOS KOs and their BTN3A expression. This dependence on glucose levels may also help explain the OXPHOS signature divergence between the two screens, which likely were performed under distinct nutrient conditions due to markedly different cell concentrations and the presence of highly proliferative T cells in the co-culture screen.
Figure 3. Metabolic regulation of BTN3A.
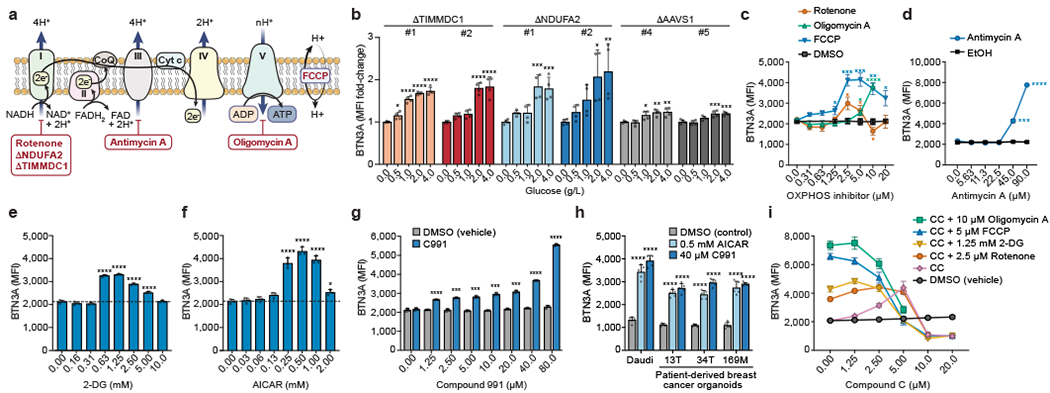
(a) Schematic of OXPHOS, inhibitor targets, and genetic KOs. (b) Surface BTN3A MFI in Daudi-Cas9 KOs cultured in different glucose concentrations for 3 days in RPMI (no glucose, no pyruvate). Normalized to cells grown without glucose (0 g/L). (c, d) Surface BTN3A MFI in WT Daudi-Cas9 cells cultured with vehicles (DMSO, ethanol) or OXPHOS inhibitors of (c) Complex I (rotenone), Complex V (oligomycin A), mitochondrial membrane potential (FCCP), and (d) Complex III (antimycin A) for 72 hours in complete RPMI. (e-g) Surface BTN3A MFI in WT Daudi-Cas9 cells cultured with (e) 2-DG, (f) AICAR, and (g) Compound 991 (C991) or equivalent amount of DMSO (vehicle) for 72 hours in complete RPMI. (h) Surface BTN3A MFI in patient-derived breast cancer organoids and Daudi cells cultured for 3 days with pamidronate and AICAR, C991, or DMSO. (i) Surface BTN3A MFI in WT Daudi-Cas9 cells co-treated with an OXPHOS/glycolysis inhibitor and increasing amounts of Compound C (CC, AMPK inhibitor). (b) n=4 per condition, data combined from two independent experiments, each individually normalized. (c) n=4 per condition, data combined from two independent experiments. (d) n=3 per condition, representative data from one of two experiments. (e, f) n=3 per condition, representative data from one of three independent experiments. (g) n=3 per condition, representative data from one of two independent experiments. (h) n=5, data combined from two independent experiments. (i) n=3 per condition, representative data from one of three independent experiments. (b, e, f, h) One-way ANOVA comparison to the zero or control treatment condition with Dunnett’s multiple comparisons test. (c) Two-tailed unpaired Student’s t test with FDR adjustment for the tested concentrations (1.25-20 μM). (d, g) Two-tailed unpaired Student’s t test with Bonferroni correction. (b-i) Mean ± SD. p<0.0001 (****), p<0.001 (***), p<0.01 (**), p<0.05 (*).
We examined the effects of OXPHOS inhibitors on BTN3A expression. Complex I inhibition (rotenone) caused a BTN3A upregulation at two lower doses and a downregulation at one higher dose. Strikingly, inhibiting Complex III, Complex V/ATP synthase, or uncoupling ATP synthesis from the ETC led to the highest BTN3A upregulation (Fig. 3c, d). Furthermore, wildtype (WT) Daudi-Cas9 cells treated with glycolysis-blocking 2-deoxy-D-glucose (2-DG) showed upregulated BTN3A levels (Fig. 3e), consistent with GSEA identification of glycolysis as negatively regulating BTN3A in the screen (Extended Data Fig. 4g). These data indicate that a cell undergoing an energy crisis changes BTN3A expression. The dose-dependent variable effects of Complex I inhibition on BTN3A expression mirror the variable results observed with Complex I KOs (NDUFA2, TIMMDC1) in the screen and the validations, suggesting that inhibiting the complex most distal from ATP synthesis has more complicated effects on BTN3A regulation.
We examined the role of several stress sensors that detect nutrient and OXPHOS deprivation31,32. Inhibition of mTOR, inhibition of the integrated stress response (ISR), and activation of ISR had generally negligible effects on BTN3A surface expression (Extended Data Fig. 9b). Meanwhile, AICAR-mediated indirect activation of AMPK, which senses elevated AMP:ATP ratios that occur during an energy crisis, led to a marked increase in surface BTN3A (Fig. 3f). Treatment with metformin, another indirect AMPK agonist, moderately increased surface BTN3A (Extended Data Fig. 9c). Using two direct agonists of AMPK, the highly potent Compound 991 (C991) and the less potent A-769662, we confirmed this AMPK-mediated upregulation of surface BTN3A in WT Daudi-Cas9 cells (Fig. 3g, Extended Data Fig. 9d). Patient-derived breast cancer organoids treated with AICAR or C991 also had significantly increased surface BTN3A (Fig. 3h).
Confirming that the effects of small molecule agonists and inhibitors on BTN3A are AMPK-dependent, increasing amounts of the AMPK inhibitor Compound C diminished BTN3A induction by AICAR (Extended Data Fig. 9e), OXPHOS inhibition (rotenone, oligomycin A, FCCP), or glycolysis inhibition (2-DG) (Fig. 3i). Additionally confirming the dependence on AMPK, we showed that BTN3A upregulation by OXPHOS inhibitors and C991 was blunted in Daudi-Cas9 AMPKα1 KO and AMPKα1/α2 double KO cells (Extended Data Fig. 9f). Note, none of the seven genes encoding for the three AMPK subunits (α, β, γ) was a significant hit in either CRISPR screen (Supplementary Tables 1 and 6). We confirmed that deletion of AMPK without an accompanying cell stress is not sufficient to change BTN3A expression; however, AMPK is required for proper BTN3A induction in stressed cells (Extended Data Fig. 9f). These results show that cancer cells undergoing an energy crisis upregulate BTN3A expression through an AMPK-dependent process, which can be phenocopied by directly activating AMPK.
AMPK activation upregulates BTN2A1
We examined the effects of AMPK on the BTN2A1-BTN3A complex as a whole. C991 and AICAR treatment of control Daudi-Cas9 AAVS1 KO cells increased Vγ9Vδ2 TCR tetramer staining six-fold and two-fold, respectively (Fig. 4a), with similar effects observed in WT Daudi-Cas9 cells (Extended Data Fig. 10a). As expected, BTN3A1 KO cells had a modest reduction in TCR tetramer staining while BTN2A1 KO cells had only background levels of Vγ9Vδ2 TCR tetramer staining (Fig. 4a, Extended Data Fig. 10b). These results demonstrate that the increased Vγ9Vδ2 TCR tetramer staining in AICAR or C991-treated cells depend on increased BTN2A1 expression. C991 treatment transcriptionally upregulated BTN2A1, BTN3A1, and BTN3A2, explaining the high Vγ9Vδ2 TCR tetramer staining that would require increased abundance of the butyrophilin complex (Extended Data Fig. 10c).
Figure 4. Regulation of BTN2A1 and Vγ9Vδ2 TCR-mediated killing of cancer cells.
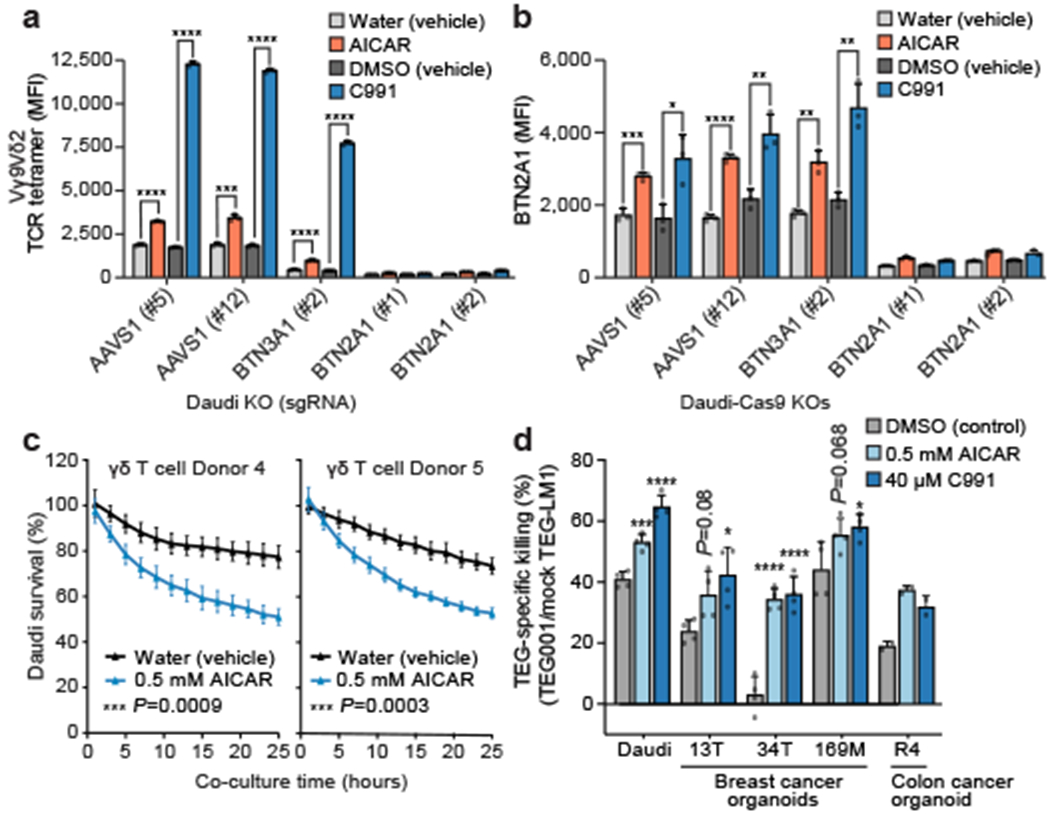
(a, b) (a) Vγ9Vδ2 TCR tetramer staining MFI and (b) surface BTN2A1 MFI for Daudi-Cas9 KOs treated with 80 μM C991 (DMSO), DMSO (vehicle), 0.5 mM AICAR (aqueous), or water (vehicle) for 72 hours. (c) Survival of eGFP+ Daudi cells treated for 3 days with AICAR or water prior to co-culture (E:T 1:1) with primary Vγ9Vδ2 T cells, normalized to Daudi cells cultured without T cells. Measured by real-time quantitative live-cell imaging. (d) TEG-mediated killing of patient-derived cancer organoids and Daudi cells after 4 days of treatment with 10 μM pamidronate and AICAR, C991, or DMSO. Cancer cells were co-cultured with TEG001 or mock TEG-LM1 and 10 μM pamidronate for 2 days. (a) n=3 per condition, representative data from one of four independent experiments. Two-tailed unpaired Student’s t test. (b) n=3, representative data from one of two independent experiments. Two-tailed unpaired Student’s t test. (c) n=4 per condition, representative data from one of two independent experiments. Repeated measure two-way ANOVA with Geisser-Greenhouse correction. (d) n=4 per condition, data combined from two independent experiments (R4 organoid: n=2 per condition, data from one experiment, no statistical significance tests provided). One-way ANOVA comparison to the DMSO treatment condition with Dunnett’s multiple comparisons test. (a-d) Mean ± SD. p<0.0001 (****), p<0.001 (***), p<0.01 (**), p<0.05 (*).
Using a novel anti-BTN2A1 monoclonal antibody, we confirmed that BTN2A1 is upregulated on AMPK agonist-treated Daudi-Cas9 cells (Fig. 4b). BTN2A1 surface expression was not reduced in BTN3A1 KO cells, while background staining in BTN2A1 KO cells was only slightly above background (Fig. 4b, Extended Data Fig. 10d). Although BTN2A1 and BTN2A2 have 87% ectodomain homology7, we designed CRISPR sgRNAs that specifically target BTN2A1. Therefore, the upregulated staining is specifically due to increased surface levels of BTN2A1. Interestingly, cell surface abundance of EphA2, a ligand of an unrelated Vγ9Vδ1 TCR MAU clone, was also previously shown to be upregulated by AMPK activation12, suggesting a common mechanism of engaging various human γδ T cell subsets.
AMPK promotes Vγ9Vδ2 TCR-induced killing
Since AMPK activation leads to cell surface upregulation of the components required for productive Vγ9Vδ2 TCR engagement – BTN2A1 and BTN3A – we tested the ability of the small molecule agonists to increase Vγ9Vδ2 TCR-mediated killing of treated cancer cells. Daudi cells that were treated with AICAR prior to co-culture with primary Vγ9Vδ2 T cells had significantly reduced survival compared to vehicle-treated Daudi cells (Fig. 4c). We did not observe significant Vγ9Vδ2 T cell-mediated cytotoxicity of AICAR or vehicle-treated Daudi cells when an antagonistic anti-BTN3A antibody (clone 103.2) was present during the co-culture (Extended Data Fig. 10e). Because clone 103.2 blocks butyrophilin-induced activation of Vγ9Vδ2 T cells [Ref. 6], AICAR-mediated sensitization to Vγ9Vδ2 T cell cytotoxicity is specifically mediated through the BTN3A-containing complex.
To extend the applicability of our findings, we tested Vγ9Vδ2 TCR-dependent killing of patient-derived breast and colon cancer organoids. The organoids were treated with pamidronate (an FDPS inhibitor like ZOL) alone or with AICAR or C991. Organoids were then co-cultured with TEG cells (αβ T cells engineered to express specific Vγ9Vδ2 TCR clones) in the presence of pamidronate33. We compared killing of organoid cells by TEG001 (highly reactive Vγ9Vδ2 TCR clone) and TEG-LM1 (non-functional mutated G115 Vγ9Vδ2 TCR clone) cells34,35. AMPK agonist treatment of tumour organoids and Daudi cells increased killing by TEG001 cells (Fig. 4d). In a previous report, breast cancer organoids 13T and 169M were found to be the most sensitive to TEG001 killing, while 34T was most insensitive to TEG001 cell targeting among the tested organoids36. AMPK agonist treatment of the insensitive 34T organoid increased its TEG001 susceptibility to the range of previously sensitive organoids 169M and 13T, which became even better targets after these treatments (Fig. 4d). These TEG cell assays allowed us to examine how AMPK activation in cancer cells affects their interaction with the Vγ9Vδ2 TCR. In addition, with TEG001 cells currently in a clinical trial (NTR6541), our data suggest that AMPK activation could be explored as an addition to the therapy regimen.
Discussion
The BTN2A1-3A1-3A2 cell surface complex is known to be activated by phosphoantigens of the mevalonate pathway binding intracellularly to BTN3A1, allowing BTN2A1 to engage Vγ9Vδ2 TCRs. Previous models of Vγ9Vδ2 T cell-target cell interactions have focused largely on phosphoantigen abundance as the main factor controlling the strength of this interaction. Here, we uncover that cell surface BTN2A1 and BTN3A levels themselves are regulated by pathways sensing the cellular metabolic state, conveying signals to surveilling Vγ9Vδ2 T cells that the target cell could be transformed or stressed. Our study reveals multilayered regulation of BTN3A abundance and/or accessibility through transcriptional regulators, glycosylation and sialylation, iron-sulphur cluster formation, trafficking, metabolic sensing, and various metabolic pathways.
We identified AMPK as a critical regulator of BTN2A1 and BTN3A expression in cells undergoing an energy crisis, thereby providing a mechanism of stress-mediated regulation of a key γδ T cell-cancer cell interaction. Importantly, we show that AMPK activation in tumour cells increases their killing by T cells expressing the Vγ9Vδ2 TCR (Fig. 5). Without the phosphoantigen activation, BTN3A1 has been shown to inhibit αβ T cell activity by disrupting the immunological synapse16. Therefore, AMPK-induced upregulation of BTN3A1 could have immunosuppressive effects when it is not accompanied either by elevated phosphoantigen levels or agonistic anti-BTN3A monoclonal antibodies, a therapeutic modality that is currently being developed and tested in a clinical trial37. A recent study also showed that AMPK activation within Vγ9Vδ2 T cells promotes their anti-tumoral activity and reverses T cell dysfunction induced by a high glucose environment38, suggesting that AMPK agonist treatment could promote Vγ9Vδ2 T cell killing of tumour cells from both sides of the cell-cell interaction.
Figure 5. Schematic of the BTN2A1-BTN3A1-BTN3A2 complex regulation.
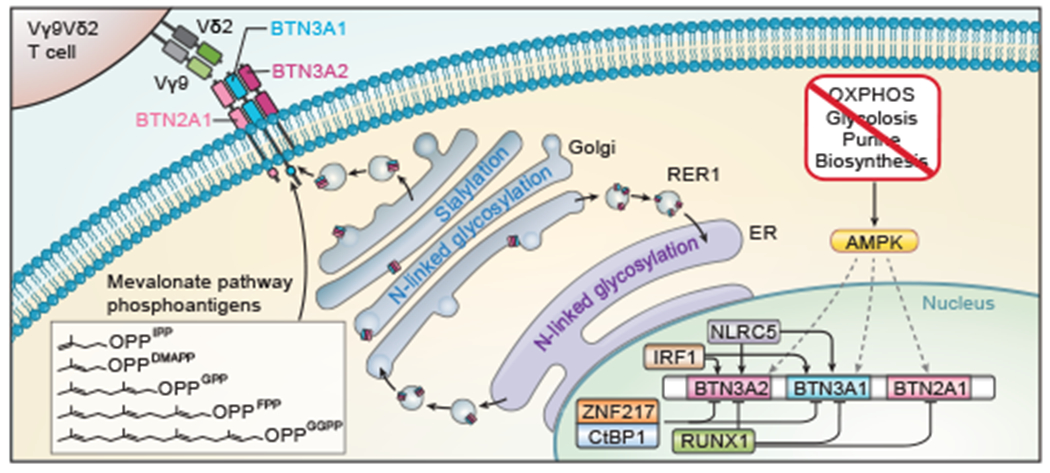
Model of the butyrophilin complex regulation uncovered in context of known activity by the mevalonate pathway and NLRC5.
The immune system is able to monitor cellular and tissue homeostasis and intervene upon encounter with dysregulated states that exceed normal homeostatic boundaries39. We present a specific mechanism by which such surveillance is carried out by human γδ T cells. Our findings indicate that BTN3A expression is modulated both by conventional interferon-responsive signalling, as well as by metabolic stress signals. One of the first identified hallmarks of cancer cells was metabolic reprogramming in the form of aerobic glycolysis (Warburg effect)40, which inefficiently produces ATP compared to OXPHOS. In addition, OXPHOS can be disrupted by hypoxic conditions often found in tumour microenvironments41. Given that productive engagement of Vγ9Vδ2 TCRs requires both excess levels of the mevalonate pathway phosphoantigens and sufficient expression of the BTN2A1-3A1-3A2 complex, we propose that γδ T cell surveillance allows them to detect transformed cells exhibiting the combination of a hyperactive mevalonate pathway and decreased OXPHOS activity. Our screens reveal a cellular logic determining how cell stresses help trigger cancer cell interactions with Vγ9Vδ2 T cells. Understanding this mechanism opens new possibilities in regard to enhancement of Vγ9Vδ2 T cell activity in patients with cancer and more effective induction of their activity against cancer cells in a state of metabolic stress.
Methods
Cancer-T cell Co-Culture Screen
Human Improved Genome-wide Knockout CRISPR Library (Addgene Pooled Library #67989 from Kosuke Yusa; 90,709 gRNAs targeting 18,010 genes)42 was transformed into Endura ElectroCompetent E. coli cells (Lucigen) following the manufacturer’s instructions. Briefly, 9 transformations were performed for appropriate coverage (1 transformation per ~10,000 sgRNA). For each transformation, 2 μL of library DNA was mixed with the cells. The mixture was loaded into a 1.0-mm cuvette and electroporated (1800 V, 10 μF, 600 Ohms) in a Gene Pulser Xcell (Bio-Rad). Electroporated cells were rescued with 975 μL of Recovery Medium (Lucigen) and incubated at 37°C with agitation for 1 hour. Transformed cells were grown overnight at 30°C in 150 mL Luria broth (LB) with ampicillin. Appropriate transformation efficiency and library coverage (2250-fold) was confirmed by plating various dilutions of the transformed cells on LB agar plates with ampicillin. Library diversity was measured by PCR amplifying (3 min at 98°C; 15 cycles of 10 sec at 98°C, 10 sec at 62°C, and 25 sec at 72°C; 5 min at 72°C) around the gRNA site with reactions made up of 10 ng DNA template, 25 μL NEBNext Ultra II Q5 Master Mix (NEB), 1 μL Read1-Stagger equimolar primer mix (10 μM) (NxTRd1.Stgr0-7 primers), 1 μL Read2-TRACR primer (10 μM), and water bringing the total volume to 50 μL. The PCR product was used in a second PCR reaction with the same PCR conditions and a reaction mix consisting of 1 μL of PCR product (1:20 dilution), 25 μL NEBNext Ultra II Q5 Master Mix, 1 μL P7.i701 (10 μL) primer, and 1 μL P5.i501 (10 μM) primer, and water bringing the total volume to 50 μL43.The final PCR product was treated with SPRI purification (1.0X), quantified on the NanoDrop, and sequenced on the MiniSeq using a MiniSeq High Output Reagent Kit (75 cycles) (Illumina). Distribution of gRNAs in the library was analysed using the MAGeCK algorithm44. Relevant primers mentioned throughout these methods are listed in Supplementary Table 8.
The genome-wide knockout CRISPR library was packaged into lentivirus using HEK293T cells (Takara Bio). In a 15-cm TC-treated dish, about 16 hours before transfection, 12 million cells were seeded in 25 mL of DMEM containing high-glucose and GlutaMAX (Gibco) supplemented with 10% FBS, 100 U/mL Penicillin-Streptomycin (Sigma-Aldrich), 10 mM HEPES (Sigma-Aldrich), 1% MEM Non-essential Amino Acid Solution (Millipore Sigma), and 1 mM sodium pyruvate (Gibco). HEK293T cells were transfected with 17.8 μg gRNA transfer plasmid library, 12 μg pMD2.G (Addgene plasmid # 12259), and 22.1 μg psPAX2 (Addgene plasmid # 12260) using the FuGENE HD transfection reagent (Promega) following the manufacturer’s protocol. Twenty-four hours after transfection, old media was replaced with fresh media supplemented with ViralBoost Reagent (Alstem). Cell supernatant was collected 48 hours after transfection, centrifuged at 300xg (10 min, 4°C), and transferred into new tubes. Four volumes of the supernatant were mixed with 1 volume of Lentivirus Precipitation Solution (Alstem) and incubated overnight at 4°C. Lentivirus was pelleted at 1500xg (30 min, 4°C), resuspended in 1/100th of the original volume in cold PBS, and stored at −80°C.
Daudi-Cas9 cells (gift from Benjamin Gewurz)45,46 were cultured in RPMI 1640 (Gibco) supplemented with 10% FBS, 2 mM L-glutamine (Lonza), and 100 U/mL Penicillin-Streptomycin. Cells were confirmed to be negative for mycoplasma with a PCR method. For two weeks prior to lentiviral gRNA delivery, Daudi-Cas9 cells were cultured in complete RPMI supplemented with 5 μg/ml blasticidin (Thermo Fisher) (cRPMI+Blast). On the day of lentiviral transduction, 250 million Daudi-Cas9 cells were resuspended in cRPMI+Blast at 3 million cells/mL, supplemented with 4 μg/mL Polybrene (Sigma-Aldrich), and aliquoted into 6-well plates (2.5 mL per well). Each well of cells received 6.25 μL of lentiviral genome-wide KO CRISPR library, and the plates were centrifuged at 300xg for 2 hours at 25°C. After the centrifugation, the cells were rested at 37°C for 6 hours, the media was replaced with cRPMI+Blast with cells seeded at 0.3x106 cells/mL, and the cells were cultured at 37°C for 3 days. Three days after transduction, Daudi-Cas9 cells were diluted to 0.3x106 cells/mL and treated with 5 μg/mL puromycin (Thermo Fisher). At this time point, the infection rate was determined to be 21% by staining cells with the 7-AAD viability dye (BioLegend) in FACS buffer (PBS, 0.5% bovine serum albumin [Sigma], 0.02% sodium azide) and assessing levels of BFP+ cells on the Attune NxT flow cytometer (Thermo Fisher). The general flow cytometry gating strategy for Daudi cells is provided in Supplementary Figure 1a. After four days of antibiotic selection, Daudi-Cas9 cells were placed in complete RPMI without blasticidin or puromycin. Puromycin-selected cells were >90% BFP+, as measured by flow cytometry following a viability stain. From this point onwards, Daudi-Cas9 cells were passaged every 2 to 3 days, maintaining at least 45x106 cells at each passage to retain sufficient knockout library diversity (>495X coverage per gRNA in the genome-wide knockout library)45. For 24 hours prior to the co-culture with expanded γδ T cells, genome-wide knockout library Daudi-Cas9 cells were treated with 50 μM of zoledronate (Sigma-Aldrich).
Residual cells in leukoreduction chambers of Trima Apheresis from de-identified donors following informed consent (Vitalant, San Francisco, CA) were used as the source of primary cells for the co-culture screen, under protocols approved by the University of California San Francisco Institutional Review Board (IRB) and the Vitalant IRB. Primary human peripheral blood mononuclear cells (PBMCs) were isolated using Lymphoprep (STEMCELL) and SepMate-50 PBMC Isolation Tubes (STEMCELL). To expand Vγ9Vδ2 T cells, PBMCs were resuspended in cRPMI with 100 U/mL human IL-2 (AmerisourceBergen) and 5 μM zoledronate. Zoledronate artificially elevates phosphoantigen levels by inhibiting a downstream step of the mevalonate pathway (Extended Data Fig. 1a) in the bystander PBMCs that then can trigger Vγ9Vδ2 T cell activation. PBMC cultures were supplemented with 100 U/mL IL-2 at 2, 4, and 6 days after seeding the cultures. After 8 days of Vγ9Vδ2 T cell expansion, γδ T cells were isolated following the manufacturer’s instructions using a custom human γδ T cell negative isolation kit without CD16 and CD25 depletion (STEMCELL). Isolated γδ T cells were confirmed to be >97% Vγ9Vδ2 TCR+ by flow cytometry using APC-conjugated anti-Vγ9 TCR (clone B3, 1:50 dilution) and Pacific Blue-conjugated anti-Vδ2 TCR (clone B6, 1:50 dilution) antibodies (BioLegend). Both Daudi-Cas9 cells and isolated γδ T cells were resuspended at 2 million cells/mL in cRPMI. For each donor, T cells and Daudi-Cas9 cells were mixed at effector-to-target (E:T) ratios of 1:2 and 1:4. Given the donor-to-donor variability in γδ T cell cytotoxicity, for each of the three T cell donors we chose the E:T ratio resulting in comparable Daudi cell survival across all donors during the analysis stage. Cultures were supplemented with 5 μM zoledronate and 100 U/mL IL-2. Surviving Daudi-Cas9 cells were harvested after 24 hours of co-culturing with γδ T cells. Using the manufacturer’s depletion protocol, the cell mixture was treated with the EasySep Human CD3 Positive Isolation Kit II (STEMCELL). Daudi-Cas9 cells were cultured in cRPMI+Blast for 4 days after isolation from the T cell co-culture and frozen down as cell pellets, which were used to generate sequencing libraries. The final library was sequenced using a NovaSeq 6000 S1 SE100 kit (Illumina).
BTN3A Expression Screen
Daudi-Cas9 cells were edited with the genome-wide knockout CRISPR library as described above. The screen was performed with 3 replicates of Daudi-Cas9 cell pools, each starting with 250 million cells, that were kept entirely separate after the lentiviral transduction step. All the replicates had an infection rate of 23-25%. Per replicate, 180 million Daudi-Cas9 cells were stained with the 7-AAD (Tonbo) viability dye and the Alexa Fluor 647-conjugated anti-BTN3A1 antibody (clone BT3.1, 1:40 dilution) (Novus Biologicals) 14 days after lentiviral transduction. Live BTN3A-high (top ~25%) and BTN3A-low (bottom ~25%) Daudi-Cas9 cells were sorted using FACSAria II, FACSAria III, and FACSAria Fusion (BD Biosciences) cell sorters. Each sorted population had between 12 and 23 million cells. Cell pellets were frozen and used to generate sequencing libraries. The final library was sequenced using a NovaSeq 6000 S4 PE150 kit (Illumina).
Next-Generation Sequencing Library Preparation
Cell pellets were lysed overnight at 66°C in 400 μL of cell lysis buffer (1% SDS, 50 mM Tris, pH 8, 10 mM EDTA) and 16 μL of sodium chloride (5 M), with 2.5 million cells per 416-μL lysis reaction. 8 μL of RNase A (10 mg/mL, Qiagen) was added to the cell lysis solution and incubated at 37°C for 1 hour. 8 μL of Proteinase K (20 mg/mL, Ambion) was then added and incubated at 55°C for 1 hour. 5PRIME Phase Lock Gel – Light tubes (Quantabio) were prepared by spinning the gel at 17,000xg for 1 minute. Equal volumes of the cell lysis solution and Phenol:Chloroform:Isoamyl alcohol (25:24:1, saturated with 10 mM Tris, pH 8.0, 1 mM EDTA, Sigma) were added to a 5PRIME Phase Lock Gel – Light tube. The tubes were vigorously inverted and centrifuged (17,000xg, 5 min, room temperature). The aqueous layer containing the genomic DNA above the gel was poured into DNA LoBind tubes (Eppendorf). 40 μL of sodium acetate (3 M), 1 μL of GenElute-LPA (Sigma-Aldrich), and 600 μL of isopropanol were added, and the solution was vortexed and frozen at −80°C. Once thawed, the solution was centrifuged at 17,000xg for 30 minutes at 4°C. After discarding the supernatant, the DNA pellet was washed with fresh room temperature ethanol (70%) and mixed by inverting the tube. The solution was then centrifuged at 17,000xg for 5 minutes at 4°C. The supernatant was removed and the DNA pellet was left to air dry for 15 minutes. The DNA Elution Buffer (Zymo Research) was added to the DNA pellet and incubated for 15 minutes at 65°C to resuspend the genomic DNA.
A two-step PCR method was used to amplify and index the genomic DNA samples for Next Generation Sequencing (NGS). For the first PCR reaction, 10 μg of genomic DNA was used per 100-μL reaction (0.75 μL of Ex Taq polymerase, 10 μL of 10x ExTaq buffer, 8 μL of dNTPs, 0.5 μL of Read1-Stagger equimolar primer mix (100 μM) (NxTRd1.Stgr0-7 primers), and 0.5 μL of Read2-TRACR primer (100 μM)) to amplify the integrated gRNA. The PCR #1 program was 5 min at 95°C; 28 cycles of 30 sec at 95°C, 30 sec at 53°C, 20 sec at 72°C; 10 min at 72°C. The PCR product solution was treated with SPRI purification (1.0X), and the DNA was eluted in 100 μL of water. To index the samples, 2 μL of purified PCR product (1:20 dilution) was used in a 50-μL PCR reaction containing 25 μL of Q5 Ultra II 2X MasterMix (NEB), 1.25 μL of Nextera i5 indexing primer (10 μM) (P5.i501-508 primers), and 1.25 μL of Nextera i7 indexing primer (10uM) (P7.i701-708 primers). The PCR #2 program was 3 min at 98°C; 10 cycles of 10 sec at 98°C, 10 sec at 62°C, 25 sec at 72°C; 2 min at 72°C. The final PCR product was treated with SPRI purification (0.7X), including two washes in 80% ethanol. DNA was eluted in 15 μL of water. The concentration was determined using a Qubit fluorometer (Thermo Fisher), and the library size was confirmed by gel electrophoresis and Bioanalyzer (Agilent). All indexed samples were pooled in equimolar amounts and analysed by NGS.
Analysis of Genome-Wide CRISPR Screens
A table of individual guide abundance in each sample was generated using the count command in MAGeCK (version 0.5.8)3. The MAGeCK test command was used to identify differentially enriched sgRNA targets between the low and high bins or the pre-killing and post-killing conditions. For the co-culture killing screen, all genes with an FDR-adjusted p-value < 0.05 were considered significant. For the BTN3A screen, all genes with an FDR-adjusted p-value < 0.01 were considered significant. Gene set enrichment analysis (GSEA) for both screens was performed using GSEA (version 4.1.0 [build: 27], UCSD and Broad Institute)47,48 using a ranked list of genes with their log-fold change values. The following GSEA settings were used: 1000 permutations, No Collapse, gene sets database C2.CP.KEGG.7.4. Both the web interface and the R package (version 1.0.0) of Correlation AnalyzeR49 were used to determine the pairwise and gene set-wide BTN3A1 expression correlations in publicly available samples provided by the ARCH4 repository50.
sgRNA Plasmids and Lentivirus
To make sgRNA plasmids for arrayed validation studies, we cloned individual sgRNAs into the pKLV2-U6gRNA5(BbsI)-PGKpuro2ABFP-W vector (Addgene plasmid # 67974 from Kosuke Yusa), generally following the depositing lab’s “Construction of gRNA expression vectors V2015-8-25” protocol. Briefly, the vector was digested with BbsI-HF (New England Biolabs [NEB]), run on a 1% agarose gel, and gel extracted. For each sgRNA, oligo pairs with appropriate overhangs were annealed using T4 Polynucleotide Kinase (NEB) and T4 DNA Ligase Reaction Buffer (NEB). Annealed inserts and the linearized vector were ligated using the T4 DNA Ligase (NEB) and transformed into MultiShot StripWell Stbl3 E. coli competent cells (Invitrogen) that were grown on Lysogeny broth (LB) agar Carbenicillin plates at 37°C overnight. Single colonies were grown out in ampicillin-containing LB and screened for the correct sgRNA insert by Sanger sequencing PCR amplicons of the insert site. Successful clones were grown and processed with a Plasmid Plus Midi Kit (Qiagen), with the DNA product serving as the transfer plasmid during lentiviral packaging. Pelleted lentivirus was resuspended in RPMI 1640. Collected lentivirus was titrated for optimal transduction in Daudi-Cas9 cells and used to generate single gene Daudi-Cas9 KOs. All sgRNAs used during the study are listed in Supplementary Table 9.
Arrayed CRISPR sgRNA KO
To generate single gene Daudi-Cas9 KOs for BTN3A expression screen validation (Fig. 2e–g), we resuspended cells at 3 million cells/mL in cRPMI with 4 μg/mL Polybrene. Daudi-Cas9 cells were aliquoted at 150 μL per well into 96-well V-bottom plates. 10 μL of virus diluted for optimal transduction was added to the cells, with 3 replicates per sgRNA (6 replicates per AAVS1 sgRNA #4 and #5). The plates were centrifuged at 300xg for 2 hours at 25°C. After the centrifugation, the cells were rested at 37°C for 6 hours, pelleted, resuspended at 750,000 cells/mL in fresh cRPMI, and cultured at 37°C for 3 days. Three days after transduction, Daudi-Cas9 cells were diluted to 0.3x106 cells/mL and treated with 5 μg/mL puromycin (Thermo Fisher). After four days of antibiotic selection, Daudi-Cas9 cells were placed in cRPMI without puromycin. From this point onwards, Daudi-Cas9 cells were passaged every 2 to 3 days. Cells were collected at 13 days post-transduction to assess frequency of indels in the CRISPR target site for each of the KOs. Further analysis described below confirmed that edited cells had disruptive insertions/deletions (indels) in >90% of the cells. At the same time point, we analysed the cells for BTN3A expression by flow cytometry. BFP+ (lentivirally induced) Daudi-Cas9 KO cells were blocked with Human TruStain FcX (Fc receptor blocking solution, 1:80 dilution) (BioLegend) in FACS buffer for 20 min at 4°C. Blocked cells were stained for 30 min at 4°C with 7-AAD viability dye (1:150 dilution) and either APC-conjugated anti-CD277 antibody (clone BT3.1, 1:50 dilution) (Miltenyi Biotec) or APC-conjugated IgG1 isotype control antibody (Miltenyi Biotec, 1:50 dilution, anti-KLH, clone IS5-21F5) in FACS buffer. Stained and washed cells were analysed on the Attune NxT flow cytometer. No appreciable signal was detected in the APC channel when cells were stained with the isotype control antibody.
Daudi-Cas9 FDPS, AMPKα1, AMPKα1/α2, and accompanying AAVS1 KO cells were generated by the same method through the end of puromycin selection. Daudi-Cas9 FDPS KO and accompanying AAVS1 KO cells were stained for 30 min at 4°C with 7-AAD viability dye (1:150 dilution) and APC-conjugated anti-CD277 antibody (clone BT3.1, 1:50 dilution) prior to analysis on the Attune NxT flow cytometer. Daudi-Cas9 AMPKα1, AMPKα1/α2, and accompanying AAVS1 KO cells were used in experiments involving metabolic inhibitors and agonists.
CRISPR Genotyping Primers
To determine indel frequency among arrayed Daudi-Cas9 KO cells used for BTN3A expression screen validation (Fig. 2e–g), we generated an indexed NGS library of amplicons around the CRISPR cut sites of the various knockouts. Primers to generate amplicons around the CRISPR genomic target site were designed with CRISPOR (version 4.8)51 with the options “--ampLen=250 --ampTm=60”. To analyse the NGS genotyping data, adapter sequences were trimmed from fastq files using cutadapt (version 2.8)52 using default settings keeping a minimum read length of 50 bp. Insertions and deletions at each CRISPR target site were then calculated using CRISPResso2 (version 2.0.42)53 with the options “--quantification_window_size 3” and “--ignore_substitutions”.
Pooled CRISPR Genotyping for Arrayed KOs
Approximately 50,000 cells from appropriate samples were pelleted (300xg, 5 min) and resuspended in 50 μL of QuickExtract DNA Extraction Solution (Lucigen). Samples were run on a thermocycler according to the following protocol (QuickExtract PCR): 10 min at 65°C, 5 min at 95°C, hold at 12°C. Samples were stored at −20°C until further steps. The PCR reaction for each sample consisted of 5 μL of the extracted DNA sample, 1.25 μL of 10 μM pre-mixed forward and reverse primer solution, 12.5 μL of Q5 High-Fidelity 2X Master Mix (NEB), and 6.25 μL of molecular biology grade water. The samples were then run on a thermocycler according to the following PCR #1 program: 3 min at 98°C; 15 cycles of 20 sec at 94°C, 20 sec at 65°C-57.5°C with a 0.5°C decrease per cycle, 1 min at 72°C; 20 cycles of 20 sec at 94°C, 20 sec at 58°C, 1 min at 72°C; 10 min at 72°C, hold at 4°C. The PCR product was stored at −20°C until further steps. PCR #1 products were indexed in PCR #2 reaction: 1 μL of PCR #1 product (diluted 1:200), 2.5 μL of 10 μM forward indexing primer, 2.5 μL of 10 μM reverse indexing primer, 12.5 μL of Q5 High-Fidelity 2X Master Mix (NEB), and 6.5 μL molecular biology grade water. PCR reactions were run on a thermocycler according to the following program: 30 sec at 98°C; 13 cycles of 10 sec at 98°C, 30 sec at 60°C, 30 sec at 72°C; 2 min at 72°C, hold at 4°C. PCR #2 product was stored at −20°C until further steps. PCR #2 product was pooled, SPRI purified (1.1X), and eluted in water. The final library was sequenced using a NovaSeq 6000 SP PE150 kit (Illumina).
Sanger Sequencing Genotyping
Daudi-Cas9 NLRC5 (gRNA #2) KOs were genotyped by Sanger sequencing. Approximately 50,000 cells were pelleted (300xg, 5 min) and resuspended in 50 μL of QuickExtract DNA Extraction Solution. Samples were run on a thermocycler according to the QuickExtract PCR program. Samples were stored at −20°C until further steps. The PCR reaction for each sample consisted of 1 μL of the QuickExtract DNA sample, 0.75 μL of 10 μM forward primer, 0.75 μL of 10 μM reverse primer, 12.5 μL of KAPA HiFi HotStart ReadyMix PCR Kit (Roche Diagnostics), and 10 μL molecular biology grade water. The samples were amplified on a thermocycler according to the following protocol: 3 min at 95°C; 35 cycles of 20 sec at 98°C, 15 sec at 67°C, 30 sec at 72°C; 5 min at 72°C, hold at 4°C. The amplified products were analysed using Sanger sequencing and knockout efficiencies were assessed using the TIDE (Tracking of Indels by Decomposition) algorithm54.
RT-qPCR of Daudi KOs and AICAR/991-Treated Cells
For measurements on Daudi-Cas9 KOs, samples were collected at 13 days after lentiviral transduction. For measurements on drug-treated WT Daudi-Cas9 cells, 180 μL of Daudi-Cas9 cells were seeded in a round-bottom 96-well plate at 275,000 cells/mL. All surrounding edge wells were filled with 200 μL of sterile PBS or water. With 4 replicates per treatment, cells were treated with 20 μL of AICAR (final concentration 0.5 mM), Compound 991 (final concentration 80 μM), DMSO, or water. The cells were collected for RT-qPCR measurements after 72 hours of incubation. RNA was extracted from approximately 70,000 cells per sample using the Quick-RNA 96 Kit (Zymo Research) or Direct-zol RNA Microprep Kit (Zymo) according to the manufacturer’s protocol without the optional on-column DNase I treatment. According to the manufacturer’s protocol, 1 μL of RNA was immediately processed using the Maxima First Strand cDNA Synthesis Kit for RT-qPCR with the dsDNase treatment (Thermo Fisher). Two cDNA synthesis reactions, in addition to a reverse transcriptase minus (RT-) negative control reaction, were performed for each biological replicate. RNA template minus (RNA-) negative controls were performed as well. cDNA samples were stored at −20°C until they were used for RT-qPCR. To perform the RT-qPCR, the two cDNA samples per biological replicate were pooled and diluted 1:1 in molecular biology grade water. Negative controls were diluted the same way. According to the manufacturer’s protocol, 3 μL of diluted cDNA and negative controls were used for the RT-qPCR reactions using the PrimeTime Gene Expression Master Mix (Integrated DNA Technologies [IDT]) including a reference dye. RT-qPCR for each biological replicate was performed in triplicate along with the RT- control for each biological replicate, the RNA- controls, and no cDNA template negative controls. None of the negative controls showed target amplification. Samples were run on the QuantStudio 5 Real-Time PCR System (384-well, Thermo Fisher) according to the following program: 3 min at 95°C; 40 cycles of 5 sec at 95°C, 30 sec at 60°C. BTN2A1, BTN3A1, BTN3A2, and ACTB loci were amplified using the PrimeTime Standard qPCR Probe Assay (IDT) resuspended with 500 μL IDTE Buffer (IDT) (Supplementary Table 10). Ct values across the three technical replicates for each sample were assessed for significant outliers resulting from technical failures (any samples in triplicate with a standard deviation above 0.2 were assessed) and subsequently averaged. The following calculations were performed: ΔCt = CtACTB – CtTarget; ΔΔCt = ΔCt(KO or treatment) – average(ΔCt(control)). Individual control ΔCt measurements were used to determine standard deviation of the control ΔΔCt. AAVS1 KO served as the control for qPCR measurements across Daudi KOs, and vehicle controls (DMSO, water) were used for measurements in Daudi cells treated with AICAR and Compound 991.
Glucose and Pyruvate Dose Response
190 μL of Daudi-Cas9 KO cells were seeded at 250,000 cells/mL in round-bottom 96-well plates in glucose-free cRPMI (+glutamine, +foetal calf serum, +penicillin/streptomycin, –glucose, –pyruvate) (Fisher Scientific). 10 μL of glucose (Life Tech) or sodium pyruvate (Gibco) at various concentrations were added to the cells. Plate edge wells were filled with 200 μL of sterile water or PBS. The cells were grown at 37°C for 72 hours, stained with APC-conjugated anti-human CD277 antibody (clone BT3.1, 1:50 dilution) (Miltenyi Biotec) and 7-AAD (1:150 dilution) (Tonbo) in FACS buffer, and analysed on the Attune NxT flow cytometer.
Agonist and Inhibitor Treatment
180 μL of Daudi-Cas9 cells were seeded at 275,000 cells/mL in cRPMI in round-bottom 96-well plates. 20 μL of zoledronate, rotenone (MedChemExpress), oligomycin A (Neta Scientific), FCCP (MedChemExpress), antimycin A (Neta Scientific), AICAR (Sigma), 2-deoxy-D-glucose (2-DG) (Sigma), Compound 991 (Selleck Chemical), A-769662 (Sigma), metformin hydrochloride (MedChemExpress), ethanol (vehicle), or DMSO (vehicle, at dilutions matching the treatment) at various concentrations were added to the cells. Plate edge wells were filled with 200 μL of sterile water or PBS. The cells were grown at 37°C for 72 hours, stained with APC-conjugated anti-human CD277 antibody (clone BT3.1, 1:50 dilution) (Miltenyi Biotec) and 7-AAD (1:150 dilution) (Tonbo), and analysed on the Attune NxT flow cytometer.
190 μL of Daudi-Cas9 AAVS1 and PPAT KO cells were seeded at 250,000 cells/mL in round-bottom 96-well plates. Cells received 10 μL of DMSO (vehicle) or one of the following compounds at a final concentration of 10 μM: sephin1 (APExBIO), ISRIB (MedChemExpress), guanabenz acetate (MedChemExpress), Sal003 (MedChemExpress), salubrinal (MedChemExpress), raphin1 acetate (MedChemExpress), and rapamycin (MilliporeSigma). Edge wells were filled with 200 μL of sterile PBS or water. After being cultured for 72 hours, the cells were stained with APC-conjugated anti-human CD277 antibody (clone BT3.1, 1:50 dilution) (Miltenyi Biotec) and 7-AAD (1:150 dilution) (Tonbo), and analysed on the Attune NxT flow cytometer.
180 μL of Daudi-Cas9 AMPKα1 (PRKAA1) KO, AMPKα1/α2 (PRKAA1/2) double KO, and AAVS1 KO cells were seeded at 275,000 cells/mL in cRPMI in round-bottom 96-well plates. 20 μL of antimycin A, FCCP, Compound 991, DMSO (vehicle), ethanol (vehicle), or cRPMI (no treatment) were added to the cells. The cells were grown at 37°C for 72 hours, stained with APC-conjugated anti-human CD277 antibody (clone BT3.1, 1:50 dilution) (Miltenyi Biotec) and 7-AAD (1:150 dilution) (Tonbo), and analysed on the Attune NxT flow cytometer.
Compound C Dose Response in Combination with AICAR or OXPHOS Inhibition
170 μL of Daudi-Cas9 cells were seeded at 292,000 cells/mL in cRPMI in round-bottom 96-well plates. 10 μL of Compound C (Abcam) were added to all the cells at various concentrations. At indicated concentrations, 20 μL of rotenone, oligomycin A, FCCP, 2-DG, AICAR, or cRPMI (control) were added to the wells that received Compound C. 10 μL of DMSO at dilutions matching Compound C and 20 μL of cRPMI were added to the DMSO-only vehicle control wells. Plate edge wells were filled with 200 μL of sterile water or PBS. The cells were grown at 37°C for 72 hours, stained with APC-conjugated anti-human CD277 antibody (clone BT3.1, 1:50 dilution) (Miltenyi Biotec) and 7-AAD (1:150 dilution) (Tonbo), and analysed on the Attune NxT flow cytometer.
γδTCR Tetramer Production
The G115 Vγ9Vδ2 TCR clone tetramer was generated using the following methods. The G115 γ-chain sequence55 was cloned into the pAcGP67A vector with a C-terminal acidic zipper, and the G115 δ-chain sequence55 was cloned into the pAcGP67A vector with a C-terminal AviTag followed by a basic zipper. Zippers stabilized the TCR complex. The TCR was expressed in the High Five baculovirus insect-cell expression system and purified via affinity chromatography over a Ni-NTA column. TCRs were biotinylated and biotinylation was confirmed using a TrapAvidin SDS-PAGE assay. The G115 TCR was then further purified using size-exclusion chromatography (Superdex200 100/300 GL column, GE Healthcare) and purity was confirmed via SDS-PAGE. Tetramers were generated by incubating biotinylated TCR with streptavidin conjugated to the PE fluorophore.
The DP10.7 Vγ4Vδ1 TCR that recognizes CD1d-sulfatide was used as a control tetramer56. The DP10.7 Vγ-chain sequence was cloned into the pAcGP67A vector with the human TCRβ-chain constant region (Supplementary Table 11) followed by a C-terminal basic zipper and histidine tag, and the DP10.7 Vδ-chain sequence was cloned into the pAcGP67A vector with the human TCRα-chain constant region (Supplementary Table 11) followed by a C-terminal AviTag, an acidic zipper, and a histidine tag. The DP10.7 TCR was expressed, purified, and tetramerized with Streptavidin-PE using the same methods as described above.
γδTCR Tetramer and BTN2A1 Staining
Baseline staining with the Vγ9Vδ2 TCR (clone G115) tetramer of Daudi-Cas9 KO cells was performed at 13 and 14 days post-lentiviral transduction (Fig. 2f, Extended Data Fig. 7b). WT and KO Daudi-Cas9 cells were analysed after being cultured for 72 hours with 0.5 mM AICAR, 80 μM Compound 991, DMSO (vehicle control at the concentration matching Compound 991), water, or nothing. For TCR tetramer staining, cells were washed (300xg, 5 min) in 200 μL FACS buffer containing human serum (PBS, 10% human serum AB [GeminiBio], 3% FBS, 0.03% sodium azide), and stained with 7-AAD (1:150 dilution) on ice in the dark for 20 min. After the first stain, the cells were pelleted (300xg, 5 min) and stained with 168.3 nM PE-conjugated Vγ9Vδ2 TCR (clone G115) or PE-conjugated Vγ4Vδ1 TCR (clone DP10.7) tetramer for 1 hour in the dark at room temperature. Following the tetramer stain, cells were thoroughly washed three times in 200 μL FACS buffer containing human serum (400xg, 5 min).
For BTN2A1 staining, cells were blocked with Human TruStain FcX in FACS buffer for 20 min at 4°C. Blocked cells were stained for 30 min at 4°C with 17.7 nM anti-BTN2A1 antibody (clone 2A1.4) or IgG1κ isotype control (clone P3.6.2.8.1) (Thermo Fisher). Following two washes, cells were stained with a PE-conjugated rat anti-mouse IgG1 secondary antibody (clone RMG1-1, 1:200 dilution) (BioLegend) and 7-AAD (1:150 dilution) in the dark, for 20 min, at 4°C. Washed and stained cells were analysed on the Attune NxT flow cytometer. The anti-BTN2A1 antibody was made by the laboratories of Erin Adams and Anthony Kossiakoff (University of Chicago).
T cells Engineered to Express a Defined γδTCR
T cells engineered to express the tumour-reactive Vγ9Vδ2 TCR clone 5 (TEG001) and mock T-cells engineered to express a mutant Vγ9Vδ2 TCR with abrogated function (TEG-LM1) were produced as previously described33,34,36. In short, Phoenix-Ampho cells were transfected with retroviral vector pMP71 containing both Vγ9 and Vδ2 TCR chains separated by a T2A, ribosomal-skipping sequence, using the FuGENE HD transfection reagent (Promega). Next, human PBMCs from healthy donors were activated using 50 IU/mL IL-2 (Proleukin) and 30 ng/mL anti-CD3 antibody (clone OKT3) (Miltenyi Biotec) and transduced twice with the viral supernatant containing 50 IU/mL IL-2 and 6 mg/mL of Polybrene (Sigma-Aldrich). CD4/8+ MACS selected T cells transduced with the TCR were expanded by stimulating with anti-CD3/CD28 Dynabeads (500,000 beads per million cells) (Thermo Fisher) and 50 IU/mL IL-2. Thereafter, T cells were depleted of non-engineered T cells via magnetic-activated cell sorting (MACS) via αβ TCR depletion (Miltenyi Biotec). After MACS isolation, TEG001 and TEG-LM1 cells were stimulated every two weeks using the rapid expansion protocol as described before33.
Patient-Derived Organoid and Daudi Killing Assay and BTN3A staining
Patient-derived breast and colon cancer organoids were cultured in BME (basement membrane extract) (Corning) in 12-well plates as previously described57,58. In short, advanced DMEM (Thermo Fisher) was supplemented with 1% Penicillin-Streptomycin (Thermo Fisher), 10% heat-inactivated foetal calf serum, 10 mM HEPES (Thermo Fisher), GlutaMAX (Thermo Fisher), and multiple growth factors for either breast cancer organoids57 or the colon cancer organoid58. These patient-derived organoids (PDOs) were recovered between passage 5-30 from the BME by incubation with TrypLE Express before using the PDOs for T cell co-cultures. Before administration of T cells, organoid suspensions were filtered through a 70-μm strainer. Daudi cells (ATCC) were cultured in RPMI 1640 (Thermo Fisher) supplemented with 1% Penicillin-Streptomycin, 10% heat-inactivated foetal calf serum, 10 mM HEPES, and GlutaMAX.
For 4 days prior to the co-culture, Daudi cells were cultured in complete RPMI and PDOs were cultured in BME for 4 days in the presence of 10 μM pamidronate and either 500 μM AICAR (Merck) or 40 μM Compound 991 (TargetMol). In control conditions, pamidronate was combined only with DMSO. Afterwards, 50,000 TEG001 or TEG-LM1 cells were administered for a 2-day co-culture in the presence of 10 μM pamidronate. With the anticipated proliferation rate of PDO cells, this allows for an E:T ratio of 1:1 at the start of the co-culture. Cells were strained and stained using a FITC-conjugated CD3 antibody (clone UCHT1) (BioLegend) to distinguish T cells from tumour cells. The flow cytometry gating strategy is provided in Supplementary Figure 1b. Furthermore, BTN3A expression on the tumour cells was determined by flow cytometry using a PE-conjugated anti-BTN3A antibody (clone 849203) (R&D Systems) before T cell administration at day-3. T cell specific killing was determined by quantification of the total tumour load after the two-day co-culture and calculated using the following formula:
Daudi Killing Assays
Vγ9Vδ2 T cells were expanded from healthy donor PBMCs for 8 days as described above. PBMCs were isolated from fresh peripheral blood leukopaks (STEMCELL), under an informed consent and protocols approved by an IRB. Vγ9Vδ2 T cells were isolated following the manufacturer’s instructions using a custom human γδ T cell negative isolation kit without CD16 and CD25 depletion (STEMCELL).
In the presence of 5 μM ZOL, BFP+ Daudi-Cas9 AAVS1 and IRF1 KO cells were co-cultured with Vγ9Vδ2 T cells expanded for 8 days. Control Daudi-Cas9 KO cells were cultured without T cells. 25,000 target cells were mixed with 50,000 T cells in a round-bottom 96-well plate. After 24 hours, the cells were washed in FACS buffer at 4°C and stained for 30 min at 4°C with 7-AAD (1:150 dilution) and APC-conjugated anti-Vγ9 (clone B3, 1:50 dilution). Washed and stained cells were analysed on the Attune NxT flow cytometer. Daudi cells were identified as live and BFP+.
For 3 days, Daudi-fLuc-eGFP cells (Imanis Life Sciences) were cultured at 250,000 cells/mL in complete RPMI and 0.5 mM AICAR or water (vehicle control). For each condition, 10,000 Daudi-fLuc-eGFP cells were co-cultured with 10,000 Vγ9Vδ2 T cells in a flat-bottom 96-well plate in complete RPMI supplemented with 100 U/mL IL-2. Control Daudi-fLuc-eGFP cells were cultured without T cells. Additional control wells were cultured with 10 μg/mL antagonistic anti-BTN3A antibody (clone 103.2). Daudi-fLuc-eGFP cells were quantified longitudinally by real-time quantitative live-cell imaging using an Incucyte S3 instrument and accompanying software (Sartorius).
To compare killing of Daudi-fLuc-eGPF cells at different E:T ratios, 10,000 Daudi-fLuc-eGFP cells were co-cultured with different amounts of T cells with 100 U/mL IL-2 and with or without 5 μM ZOL. Control Daudi-fLuc-eGFP cells were cultured without T cells. Daudi-fLuc-eGFP cells were quantified longitudinally using an Incucyte S3 instrument and accompanying software (Sartorius).
CUT&RUN
Cleavage Under Targets and Release Using Nuclease (CUT&RUN) preceded by nuclei isolation was performed according to the manufacturer’s protocol with the CUTANA™ ChIC/CUT&RUN Kit (EpiCypher). In brief, 500,000 WT Daudi-Cas9 cells per reaction were washed with PBS before nuclear isolation using the EpiCypher recommended lysis buffer consisting of 20 mM HEPES pH 7.9 (Sigma-Aldrich), 10 mM KCl (Sigma-Aldrich), 0.1% Triton X-100 (Sigma-Aldrich), 20% glycerol (Sigma-Aldrich), 1 mM MnCl2 (Sigma-Aldrich), 1X cOmplete Mini-Tablet (Roche), and 0.5 mM spermidine (Sigma-Aldrich). Per reaction, washed cells were resuspended in 100 μL cold nuclear extraction buffer and incubated on ice for 10 minutes. Following lysis, per reaction, nuclei were pelleted and resuspended in 100 μL nuclear extraction buffer. Isolated nuclei were used as input for CUT&RUN beginning with binding to activated conA beads. After adsorption of nuclei to beads, permeabilization was performed with 0.01% digitonin-containing buffer. Per reaction, 500 ng of polyclonal rabbit IgG antibody (kit reagent, EpiCypher) or antibody against H3K4me3 (kit reagent, EpiCypher), IRF1 (clone D5E4) (Cell Signaling), or ZNF217 (Invitrogen, Cat.# 720352) were added. All reactions were performed in triplicate, except the IgG reaction, which was performed in duplicate. Following antibody binding, pAG-MNase addition, and chromatin cleavage, 0.5 ng of the provided E. coli DNA was added to each sample. DNA purification was performed with the provided spin columns and buffers. Purified DNA was prepared into sequencing libraries using the CUTANA™ CUT&RUN Library Prep Kit (EpiCypher) according to the manufacturer’s protocol.
Pooled libraries were sequenced on a NextSeq 500 with a Mid Output v2.5 kit (150 cycles) (Illumina) and paired end sequencing. Bcl2fastq (version 2.19) was run with --minimum-trimmed-read-length 8 to generate fastqs. CUT&RUN data analysis was performed according to an established analysis pipeline with the provided settings unless specified differently59. In brief, paired fastqs were trimmed using cutadapt (version 1.18) prior to merging of technical replicates. Bowtie2 (version 2.2.5) was used to align the trimmed fastqs to the GRCh38 human genome assembly using settings --local --very-sensitive --no-mixed --no-discordant --phred33 --dovetail -I 10 -X 700 -p 8 -q and E. coli (EMBL accession U00096.2) with settings --local --very-sensitive --no-overlap --no-dovetail --no-mixed --no-discordant --phred33 -I 10 -X 700 -p 8 -q. Aligned sam files were converted to bam files with samtools (version 1.9) view and from bam to bed with bedtools (version 2.30.0) bamtobed -bedpe. Bed files were filtered to include only paired reads of less than 1000 bp with the command awk ‘$1==$4 && $6-$2 < 1000 {print $0}’ samplename.bed before generating normalized bedgraph files using bedtools (version 2.30.0) genomecov -bg -scale $scale_factor; where the scale_factor is equal to 1000000 divided by the number of aligned E. coli reads. Peak calling was performed with SEACR (version 1.3)60 using normalized bedgraph files as the input to identify peaks above an IgG background using the stringent and non options. All CUT&RUN data visualization was performed in RStudio (version 1.4.1717) with the Sushi package (version 1.3).
Pathway Visualization
Pathway data visualizations were generated using Cytoscape (version 3.9.0)61 and the WikiPathways app (version 3.3.7)62. Glycan glyphs for the N-glycan pathway were generated using GlycanBuilder2 (version 1.12.0)63 in SNFG format64, and were incorporated into the pathway in Cytoscape using the RCy3 package (version 2.14.0)65. OXPHOS subunits encoded by mitochondrial genes are not included in the visualization because mitochondrial genes are not included in the genome-wide CRISPR library. All pathway visualizations were based on WikiPathways models66: the mevalonate pathway was adapted from WP4718 [www.wikipathways.org/instance/WP4718] and WP197 [https://www.wikipathways.org/instance/WP197]; the purine biosynthesis pathway was adapted from WP4224 [www.wikipathways.org/instance/WP4224]; the OXPHOS pathway was adapted from WP111 [www.wikipathways.org/instance/WP111]; the iron-sulphur cluster biogenesis pathway corresponds to WP5152 [www.wikipathways.org/instance/WP5152]; the sialylation pathway corresponds to WP5151 [www.wikipathways.org/instance/WP5151]; N-glycan biosynthesis pathway was based on WP5153 [www.wikipathways.org/instance/WP5153].
Generation of Co-culture and BTN3A Regulator Screen Signatures
TCGA bulk RNA-seq and survival data from 11,093 patients across 33 cancer types were obtained using the R package TCGAbiolinks, and matched normal samples were removed. The signature was generated using genes with significant fold change (FDR<0.01) in the co-culture screen or the BTN3A screen. TCGA samples were scored using the level of the signature adopting a previously described strategy67. A sample’s signature level was estimated as the Pearson correlation between normalized gene expression of signature genes and the screen score of signature genes: . We used −log10(Padj)× sign(Fold Change) as the screen score of each gene. The expression of a signature gene was normalized within the TCGA sample by dividing its average across all 11,093 samples. Furthermore, for biological pathways which were enriched in our co-culture screen hits, pathway signature levels were estimated by limiting the comparison to genes that overlapped between the co-culture screen signature and the pathway and calculating the score in the same way.
Signature Survival Associations
The Cox proportional hazard model was used to check associations of signature or gene expression with patient survival:
is the hazard function (defined as the risk of death across patients per unit time), and is the baseline hazard function at time is patients’ screen signature levels or gene expression levels. The coefficient of survival association () and its significance (Wald’s test) were determined using the R-package “Survival”. To show the association of survival with a signature using a Kaplan-Meier plot, we divided TCGA samples into two groups using the median of the signature levels across samples within a given cancer type and compared the survival between the two groups. The significance of survival difference was estimated using a log-rank test. For each screen signature’s results, all p-values were corrected for multiple hypothesis testing to account for other signatures that were tested in parallel (Supplementary Table 12).
To test the dependence of the survival association with the signatures on the presence or absence of γδ T cells, we used the average expression (transcripts per million) of TRGV9 (Vγ9) and TRDV2 (Vδ2) genes in a sample as its Vγ9Vδ2 T cell transcript abundance. We also used the average expression of TRAC (TCRα constant region) and TRBC (TCRβ constant region) genes in a sample as its αβ T cell transcript abundance. The likely interaction of a screen signature with TRGV9/TRDV2 transcript abundance or TRAC/TRBC transcript abundance was estimated using Cox regression with the following model:
Where is the signature level and is the TRGV9/TRDV2 transcript abundance or the TRAC/TRBC transcript abundance in TCGA samples. To show the interactions using Kaplan-Meier plots, we divided TCGA samples into four groups using the median signature levels and median TRGV9/TRDV2 transcript abundance or TRAC/TRBC transcript abundance. For each screen signature’s results, all p-values were corrected for multiple hypothesis testing to account for other signatures that were tested in parallel (Supplementary Table 12).
ChIP-Seq Analysis
The following paired-end IRF1 ChIP-Seq fastq files were downloaded from ENCODE68,69: IRF1 K562 IP rep 1 (ENCFF031RWN, ENCFF031WIT), IRF1 K562 IP rep 2 (ENCFF602NSL, ENCFF071PWE), IRF1 K562 input (ENCFF285JYI, ENCFF420KFM). The fastq files were mapped to the GRCh38 human genome assembly with the nf-core ChIP-Seq pipeline (version v1.2.2)70 using default settings to generate normalized bigWig files scaled to 1 million mapped reads. ChIP-Seq coverage at the BTN locus was visualized using the pyGenomeTracks package (version 3.6)71.
Software
Plots were generated in ggplot2 in R (versions 4.0.2 and 4.2.2), as well as in Prism 9 (GraphPad). Flow cytometry data were analysed in FlowJo (version 10.8.1, Beckton Dickinson). Figures were compiled in Illustrator (version 27.0, Adobe). The OXPHOS schematic was adapted from “Electron Transport Chain” by BioRender.com (2021), retrieved from https://app.biorender.com/biorender-templates.
Data Availability
The sequencing datasets for the two screens and CUT&RUN are available in the NCBI Gene Expression Omnibus (GEO) repository (co-culture screen: GSE192828; BTN3A screen: GSE192827; CUT&RUN: GSE226931). Publicly available paired-end IRF1 ChIP-Seq fastq files were downloaded from ENCODE68,69: IRF1 K562 IP rep 1 (ENCFF031RWN, ENCFF031WIT), IRF1 K562 IP rep 2 (ENCFF602NSL, ENCFF071PWE), IRF1 K562 input (ENCFF285JYI, ENCFF420KFM). Also, publicly available TCGA data were utilized for this study (https://www.cancer.gov/tcga).
Code Availability
Computer code used to analyse the TCGA dataset has been deposited on Zenodo (10.5281/zenodo.8011891).
Extended Data
Extended Data Figure 1. Mevalonate pathway effects, co-culture screen consistency, and gene set enrichment analysis (GSEA).
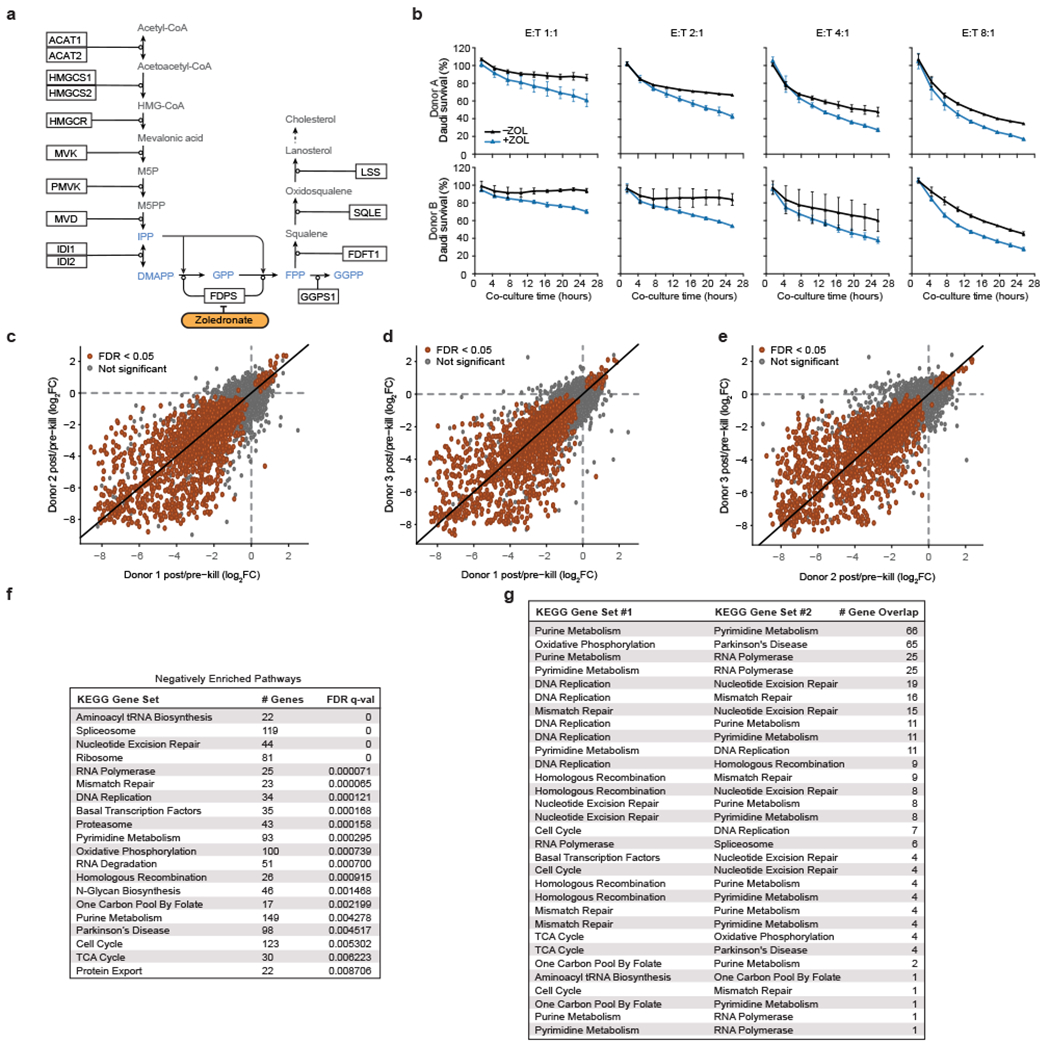
(a) Schematic of the mevalonate pathway, adapted from WikiPathways. Phosphoantigens highlighted in blue. (b) Survival of eGFP+ Daudi cells co-cultured with primary Vγ9Vδ2 T cells at different effector-to-target (E:T) ratios with or without zoledronate (ZOL). Cells were quantified using real-time quantitative live-cell imaging (Incucyte). Survival was normalized to Daudi cells cultured without T cells. Mean ± SD. n=3 per condition. (c-e) Pairwise comparisons of log2(fold change [FC]) of screen results among the three healthy human PBMC donors. (f) Number of genes contained within each negatively enriched KEGG gene set after filtering out genes that were not in the screen dataset, FDR q-values, and (g) number of genes found in two KEGG gene sets.
Extended Data Figure 2. Gene, gene set, and signature TCGA analysis.
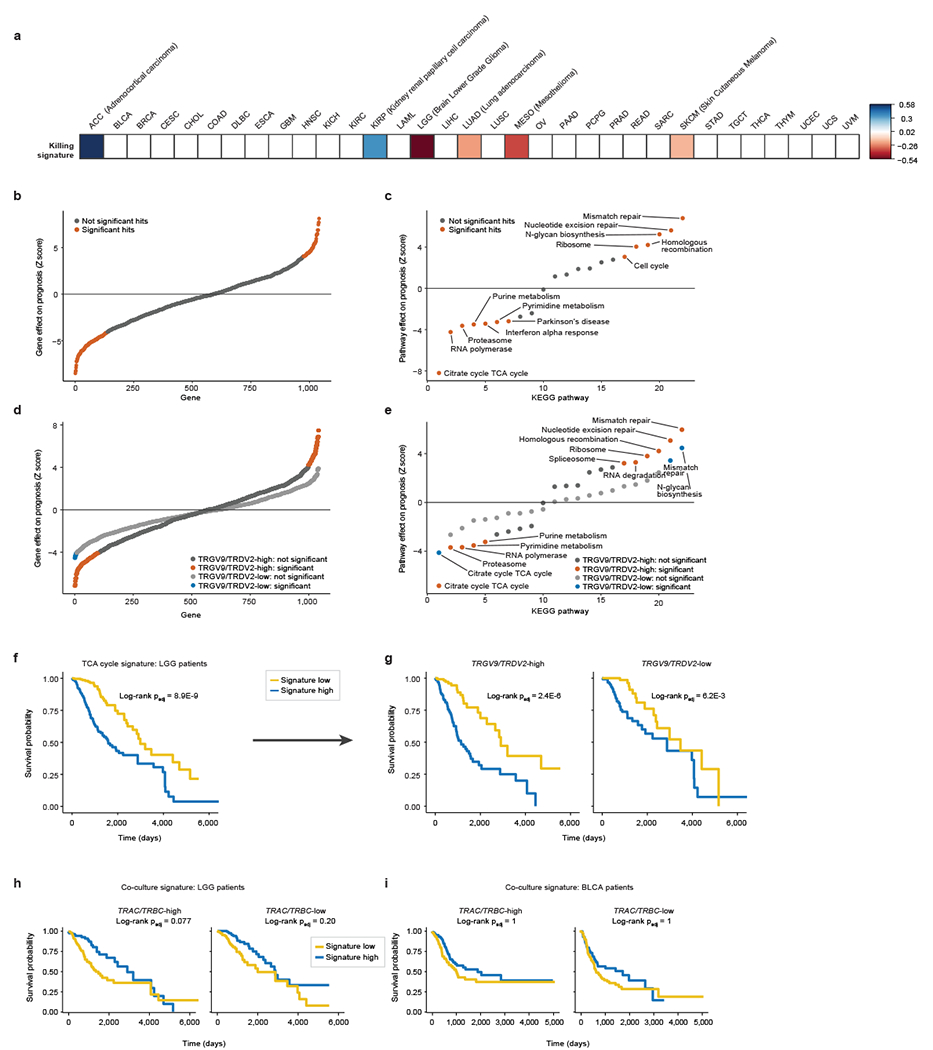
(a) Heatmap of the hazard ratios (natural log-transformed) associated with the co-culture screen gene signature in TCGA patients for 33 cancer types (a positive log-ratio indicates a worse prognosis and a negative one indicates a protective effect of the gene signature). The co-culture screen gene signature was scaled to mean=0, SD=1. Values shown only for cancer types with significant survival and signature association in patient tumours, as determined by a Wald test with Benjamini-Hochberg multiple comparisons correction (two-sided padj < 0.05). (b, d) Correlation of tumour gene expression and survival in the low-grade glioma (LGG) patient cohort, (b) with the entire cohort and (d) with the cohort split according to TRGV9/TRDV2 tumour transcript abundance. Patients with high and low expression of every given gene were compared across the 1040 genes in the co-culture screen signature. Positive Wald test Z score indicates a positive correlation with survival, and negative Z score indicates a negative correlation with survival. (c, e) Correlation of KEGG pathway-derived and type I interferon response pathway-derived signature scores and survival in the low-grade glioma (LGG) patient cohort, (c) with the entire cohort and (e) with the cohort split according to TRGV9/TRDV2 tumour transcript abundance. Patients with high and low pathway signature scores were compared. (f, g) Survival of (f) all LGG patients and (g) TRGV9/TRDV2-high or TRGV9/TRDV2-low LGG patients split by high and low expression of the TCA cycle pathway signature. (h, i) Survival of TRAC/TRBC-high/low (h) LGG and (i) BLCA patients split by high and low expression of the co-culture screen gene signature. (b-e) Significance was determined by a Wald test (Cox regression) with Bonferroni multiple comparisons correction (two-sided padj<0.05). (f-i) Log-rank test (Kaplan-Meier survival analysis), adjusted (padj) with Benjamini-Hochberg multiple comparisons correction. (c, e-g) Pathway signature levels were estimated by limiting the comparison to genes that overlapped between the co-culture screen hits signature and the pathway.
Extended Data Figure 3. Correlation of expression between BTN3A1 and screen hits across thousands of samples.
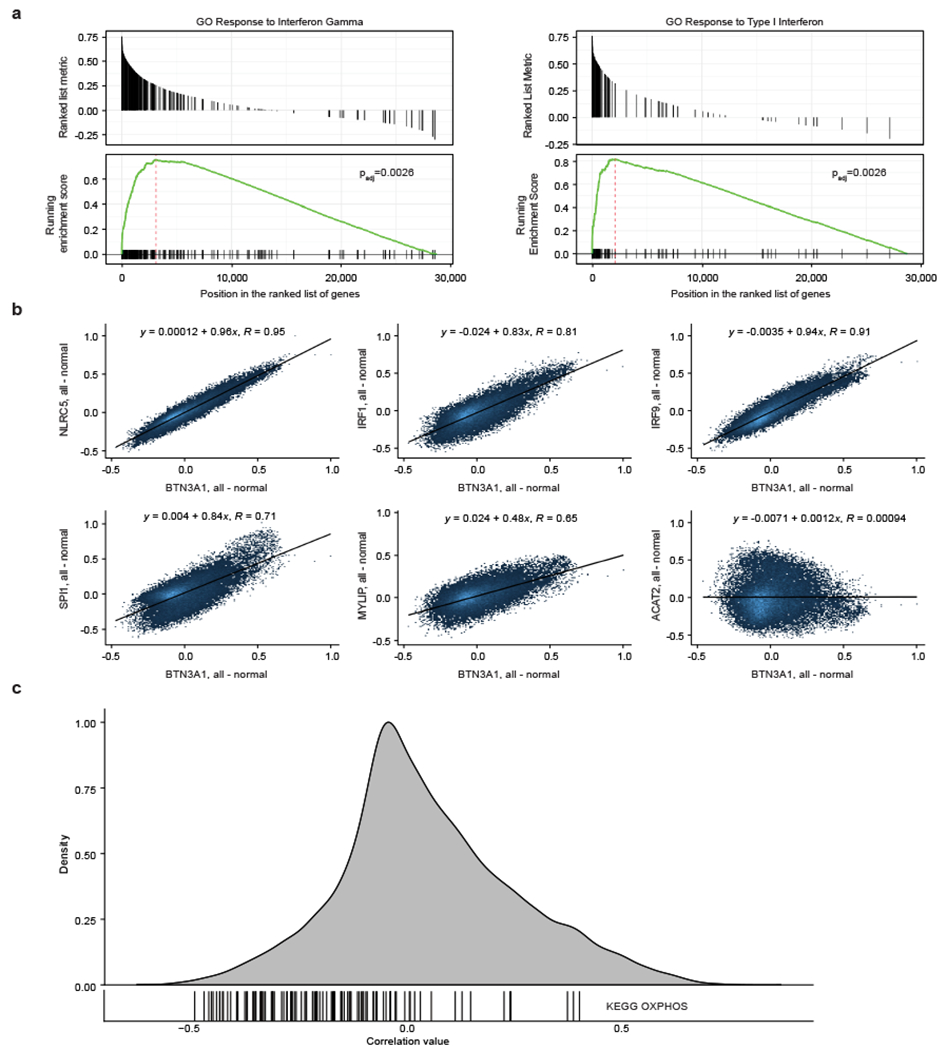
(a) BTN3A1 expression correlation with gene ontology (GO) pathways across thousands of healthy samples (all tissues combined) collated by Correlation AnalyzeR. To determine pathways that correlate with BTN3A1, genome-wide Pearson correlations for BTN3A1 are used as a ranking metric in the GSEA algorithm, which determines the padj-value. (b) Pairwise correlations between expression of BTN3A1 and shown genes across thousands of healthy samples (all tissues combined) (Correlation AnalyzeR). (c) Each vertical line indicates the correlation between expression of BTN3A1 and one of the genes in the KEGG oxidative phosphorylation (OXPHOS) gene set, overlaid over a density plot of the BTN3A1 pairwise correlations with genes across the entire human genome. Data from healthy immune tissues (Correlation AnalyzeR).
Extended Data Figure 4. BTN3A expression screen consistency, cross-screen correlation, FDPS validation, and GSEA.
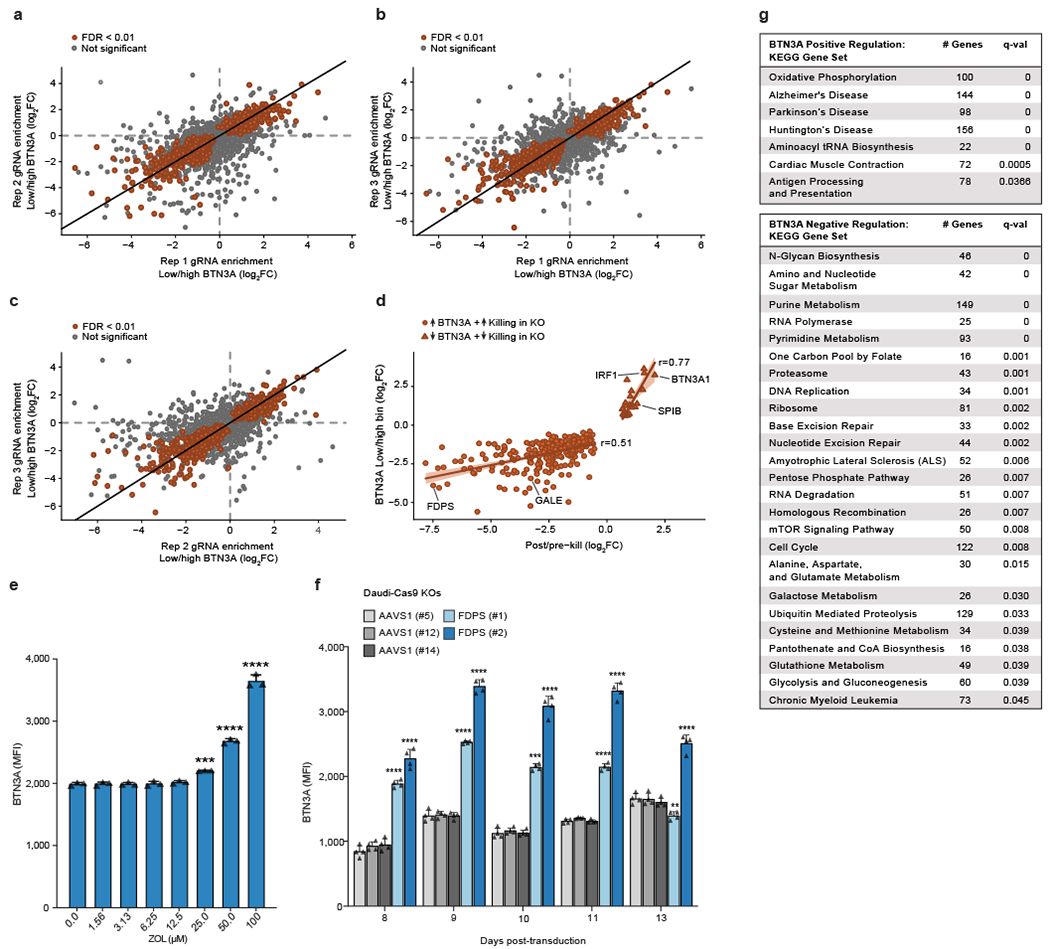
(a-c) Pairwise comparisons of significant (FDR<0.01) and not significant results among the three replicates (Rep) of Daudi-Cas9 cell populations used for the BTN3A expression screen. (d) Correlation of screen effect sizes (LFC) among concordant hits separated into positive and negative regulators of BTN3A surface expression. Linear regression line with a 95% confidence interval is shown. (e) Surface BTN3A staining on live Daudi-Cas9 cells treated for 72 hours with zoledronate (ZOL), an inhibitor of FDPS. n=3 per ZOL dose, representative data from one of three independent experiments. One-way ANOVA comparison to no treatment with Dunnett’s multiple comparisons test. (f) Surface BTN3A staining on live Daudi-Cas9 FDPS KO or control AAVS1 KO cells at indicated days after lentiviral sgRNA transduction. n=4 for each KO. One-way ANOVA comparison to Daudi-Cas9 AAVS1 (#5) KO cells with Dunnett’s multiple comparisons test. (e, f) Mean ± SD. p<0.0001 (****), p<0.001 (***), p<0.01 (**). (g) GSEA of KEGG gene sets that positively or negatively regulate surface BTN3A expression. Number of genes contained within each KEGG gene set after filtering out genes that were not in the screen dataset and FDR q-values are shown.
Extended Data Figure 5. Pathway Enrichment Visualization.
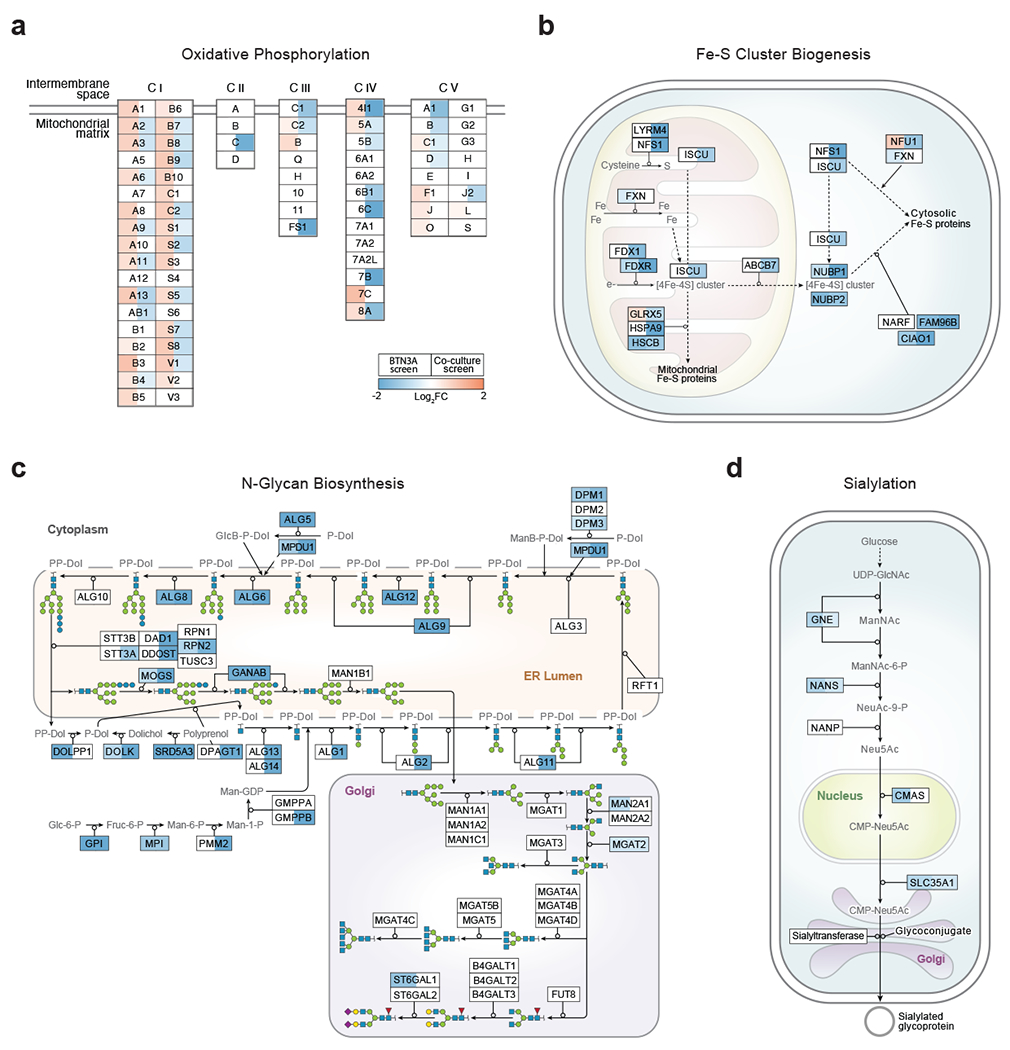
(a-d) Schematics of the depletion and enrichment of KOs within the (a) oxidative phosphorylation, (b) iron-sulphur (Fe-S) cluster biogenesis, (c) N-glycan biosynthesis, (d) and sialylation pathways across both screens. Shading indicating log2FC shown only for significant hits (FDR<0.05). All pathways were adapted from WikiPathways. (a) OXPHOS subunits are shown with abridged names without accompanying prefixes (C I: NDUF; C II: SDH; C III: UQCR, C IV: COX; C V: ATP5) (e.g., B4 in CI is NDUFB4). Subunits encoded by mitochondrial genes are not included in the visualization.
Extended Data Figure 6. BTN3A expression screen gene signature.
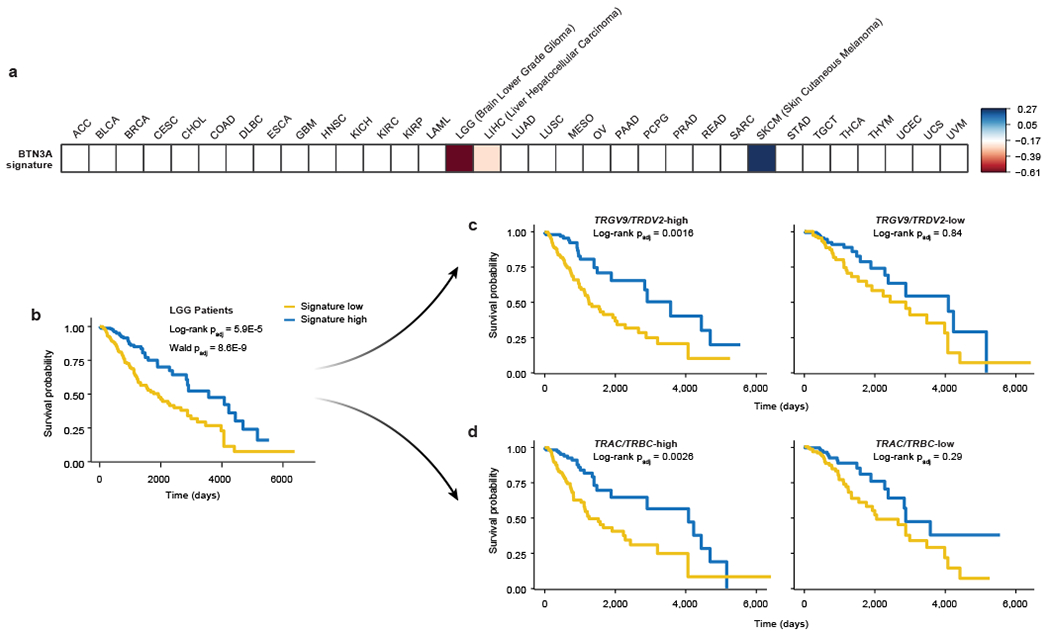
(a) Heatmap of the hazard ratios (natural log-transformed) associated with the BTN3A screen gene signature in TCGA patients for 33 cancer types (a positive log-ratio indicates a worse prognosis and a negative one indicates a protective effect of the gene signature). The BTN3A screen gene signature was scaled to mean=0, SD=1. Values shown only for cancer types with significant survival and signature association in patient tumours, as determined by a Wald test with Benjamini-Hochberg multiple comparisons correction (two-sided padj < 0.05). (b-d) Survival of (b) total, (c) TRGV9/TRDV2-high/low, or (d) TRAC/TRBC-high/low LGG patients split by high and low expression of the BTN3A expression screen gene signature. Log-rank test (Kaplan-Meier survival analysis) and Wald test (Cox regression), adjusted (padj) with Benjamini-Hochberg multiple comparisons correction.
Extended Data Figure 7. Effects of screen hit KOs on Vγ9Vδ2 TCR tetramer staining and BTN3A2 and BTN2A1 expression.
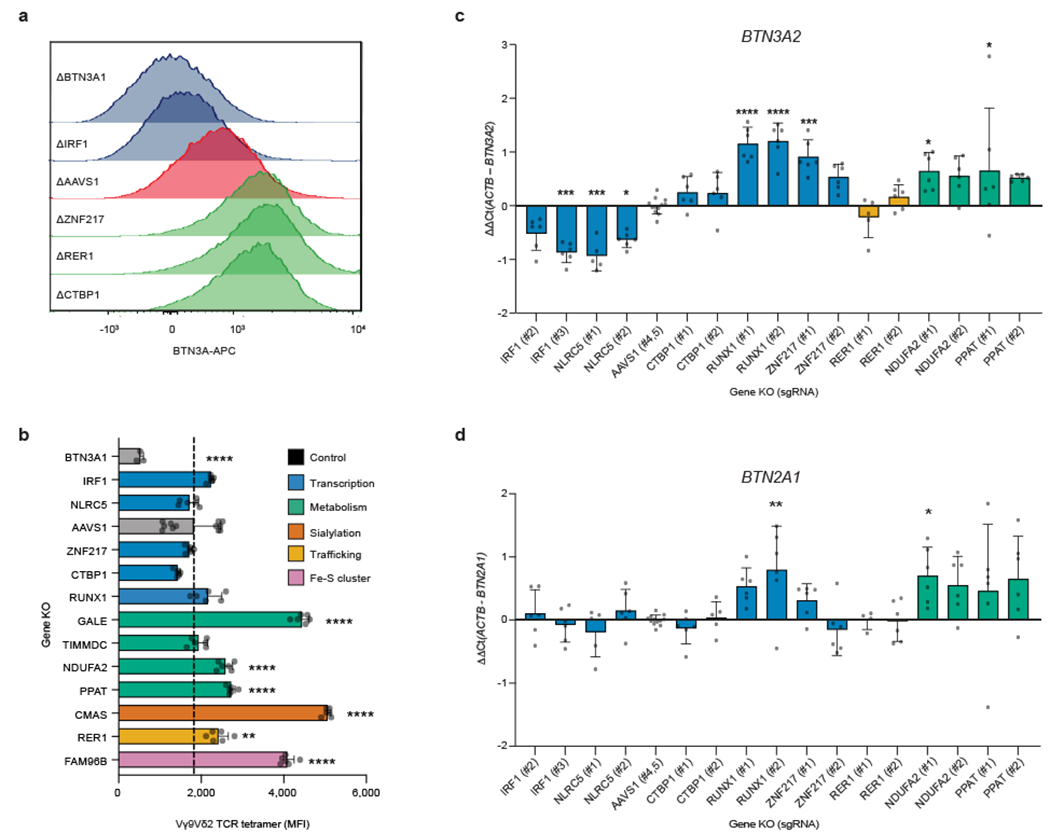
(a) Representative histograms of surface BTN3A fluorescence for a subset of single gene Daudi-Cas9 KOs and the AAVS1 control. (b) G115 clone Vγ9Vδ2 TCR tetramer staining fluorescence (MFI) at 14 days after lentiviral sgRNA transduction. Data from one experiment. AAVS1 KO n=12, BTN3A1 KO n=3, all other deletions n=6. (c, d) qPCR data for (c) BTN3A2 and (d) BTN2A1 transcripts normalized to ACTB transcripts. n=5-6 (except RER (#1) KO n=4 for BTN2A1), AAVS1 KO n=12, data combined from two independent experiments. (b-d) One-way ANOVA with Dunnett’s multiple comparisons test. Mean ± SD. p<0.0001 (****), p<0.001 (***), p<0.01 (**), p<0.05 (*).
Extended Data Figure 8. IRF1 and ZNF217 CUT&RUN, ChIP-Seq, and Vγ9Vδ2 T cell killing.
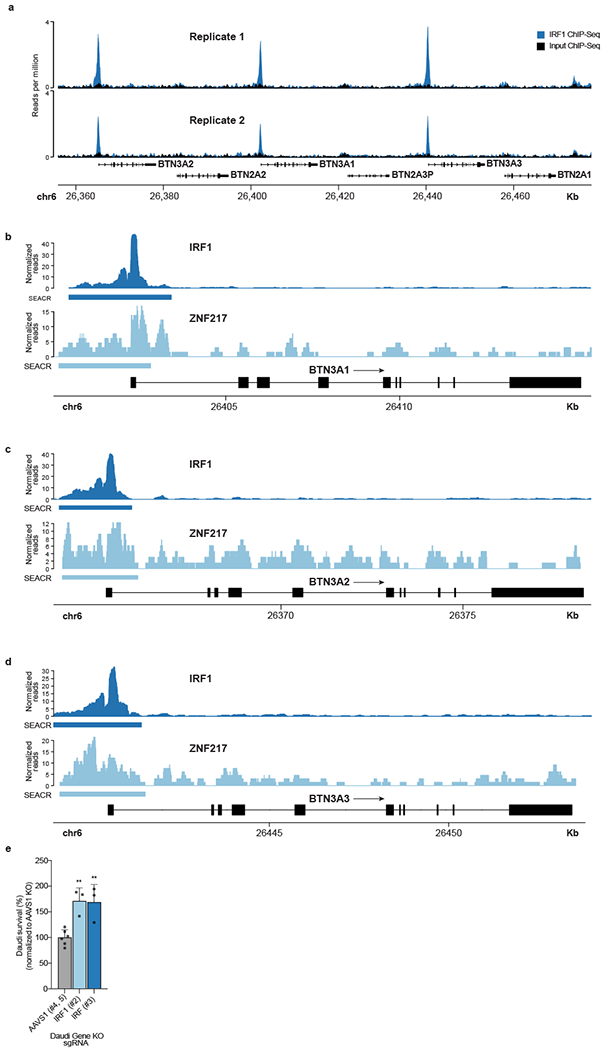
(a) Publicly available IRF1 ChIP-Seq for the butyrophilin locus in K562 cells stably expressing C-terminal eGFP-tagged IRF1 (ENCODE). (b-d) CUT&RUN data for IRF1 and ZNF217 binding at promoters in (b) BTN3A1, (c) BTN3A2, and (d) BTN3A3 loci in WT Daudi-Cas9 cells. n=3 per condition. The algorithm SEACR calls peaks and verifies them above a stringent background signal threshold. (e) Daudi-Cas9 KO survival after 24-hour co-culture with expanded Vγ9Vδ2 T cells in the presence of ZOL at an E:T ratio of 2:1. For each γδ T cell donor, Daudi survival is calculated relative to Daudi cells cultured without T cells and normalized to the Daudi-Cas9 AAVS1 KO control cell survival. Combined data from three donors and two independent experiments. AAVS1 KO n=6, IRF1 KO n=3. One-way ANOVA comparison to AAVS1 KO cells with Dunnett’s multiple comparisons test. Mean ± SD. p<0.01 (**).
Extended Data Figure 9. Metabolic effects on surface BTN3A expression.
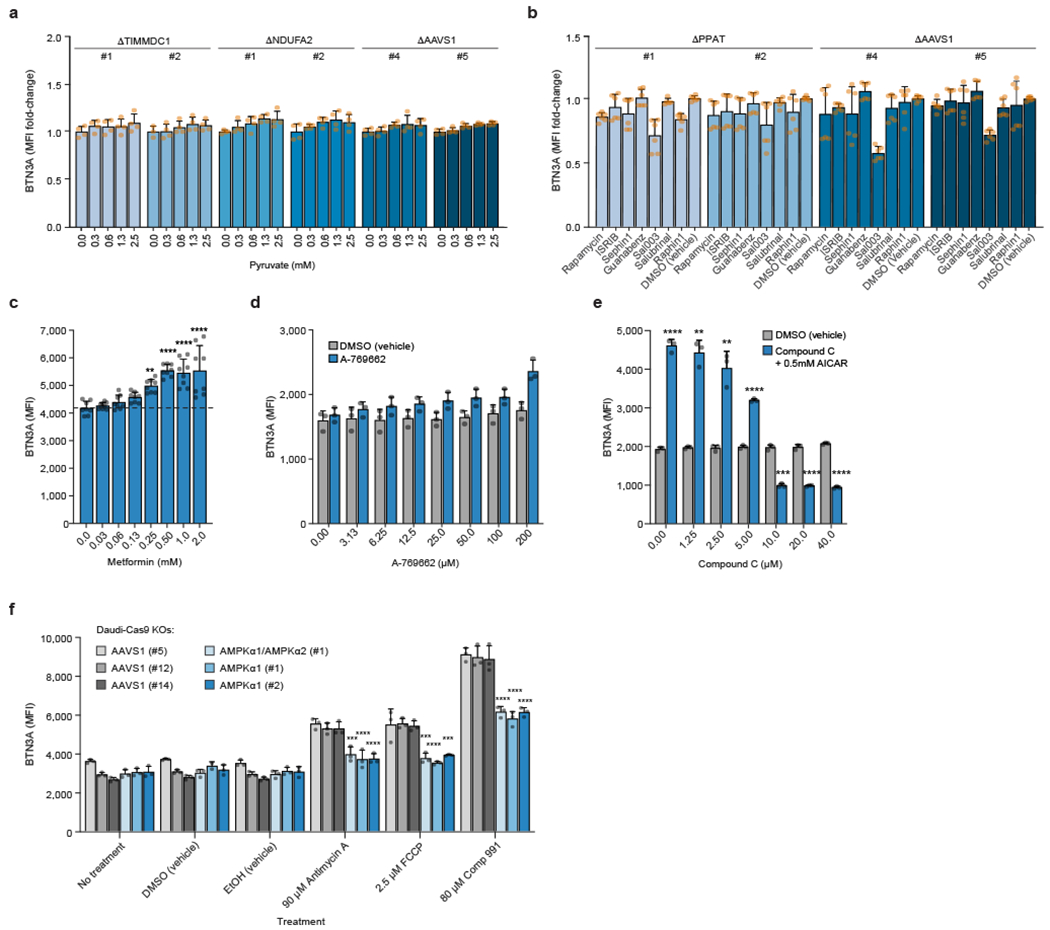
(a) Surface BTN3A MFI in Daudi-Cas9 KOs cultured in different pyruvate concentrations for 3 days in RPMI (no glucose, no pyruvate). Normalized to cells grown without pyruvate (0 mM). (b-d, f) Surface BTN3A MFI in Daudi-Cas9 cells treated for 72 hours with (b) an mTOR inhibitor (rapamycin), an ISR inhibitor (ISRIB), ISR agonists (guanabenz, Sal003, salubrinal, raphin1, sephin1), and DMSO (vehicle) (KO cells); (c) metformin (WT cells); (d) A-769662 compared to equivalent amounts of DMSO (vehicle) (WT cells); or (f) the shown compounds (KO cells). (e) Surface BTN3A MFI in WT Daudi-Cas9 cells co-treated with AICAR and increasing amounts of Compound C (AMPK inhibitor) or DMSO (vehicle). (a) n=4 per condition (n=3, TIMMDC1 (#2) at 0 mM), data combined from two independent experiments, each individually normalized. (b) n=6 per condition (except n=5 for AAVS1 (#5) with guanabenz and for PPAT (#1) with salubrinal) , data combined from two independent experiments, each individually normalized to DMSO (vehicle)-treated cells. (c) n=8 per condition, data combined from two independent experiments. One-way ANOVA comparison to cells that received no treatment with Dunnett’s multiple comparisons test. (d) n=3 per condition, representative data from one of two independent experiments. (e) n=3 per conditions, representative data from one of two independent experiments. Two-tailed unpaired Student’s t test with Bonferroni correction. (f) n=3 per condition (n=2 for AMPKα1 (#1) treated with DMSO), representative data from one of two independent experiments. One-way ANOVA comparison to AAVS1 (#5) KO cells with Dunnett’s multiple comparisons test. (a-f) Mean ± SD. p<0.0001 (****), p<0.001 (***), p<0.01 (**), p<0.05 (*), p>0.05 (N.S.).
Extended Data Figure 10. Isotype Control Staining and BTN3A1 blocking.
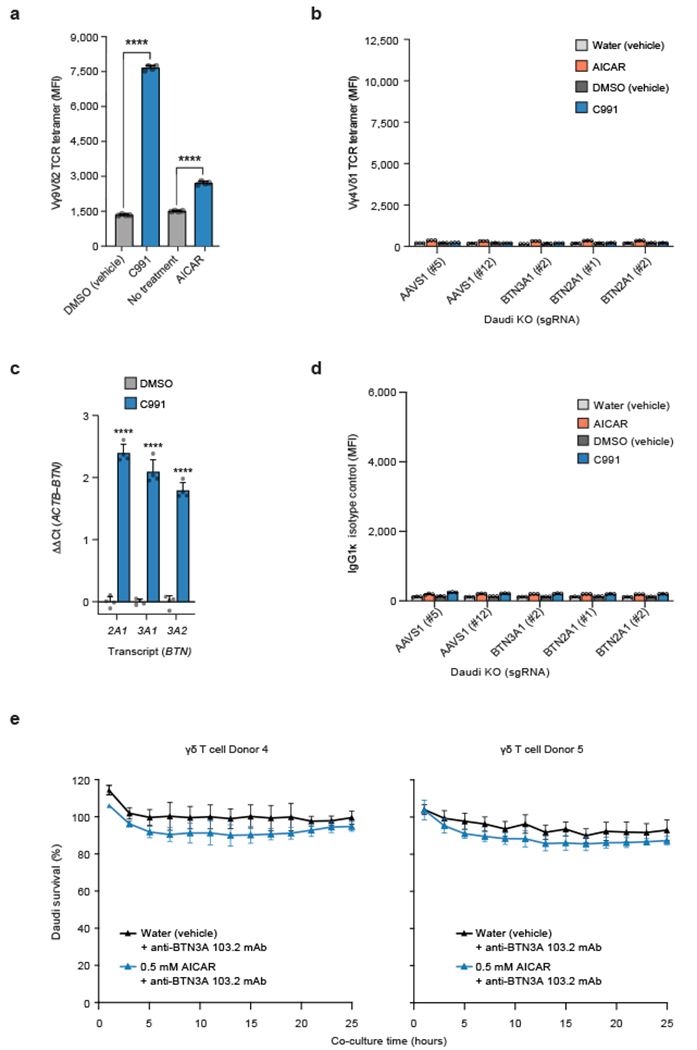
(a) G115 clone Vγ9Vδ2 TCR tetramer staining MFI of WT Daudi-Cas9 cells treated with 80 μM C991 (DMSO), DMSO (vehicle), 0.5 mM AICAR (aqueous), or without treatment for 72 hours. Two-tailed unpaired Student’s t test. (b) Vγ4Vδ1 TCR (clone DP10.7) tetramer staining fluorescence (MFI) of Daudi-Cas9 KO cells treated with 80 μM C991 (DMSO), DMSO (vehicle), 0.5 mM AICAR (aqueous), or water for 72 hours. This staining with a tetramer of an irrelevant γδTCR clone defines the background for Vγ9Vδ2 TCR tetramer staining in Figure 4a. (c) qPCR data for BTN2A1, BTN3A1, and BTN3A2 transcripts in Daudi-Cas9 cells treated with C991, internally normalized to ACTB transcripts and normalized to DMSO (vehicle)-treated cells. Two-tailed unpaired Student’s t test. (d) IgG1κ isotype control staining in Daudi-Cas9 KO cells treated with 80 μM Compound 991 (DMSO), DMSO (vehicle), 0.5 mM AICAR (aqueous), or water (vehicle) treatment for 72 hours. (e) Survival of eGFP+ Daudi cells treated for 3 days with AICAR or water prior to co-culture (E:T 2:1) with primary Vγ9Vδ2 T cells in the presence of an anti-BTN3A antibody (clone 103.2). Cells were quantified using real-time quantitative live-cell imaging (Incucyte). Survival was normalized to Daudi cells cultured without T cells. (a) n=4 per condition, representative data from one of two independent experiments. (b) n=3 per condition, representative data from one of two independent experiments. (c) n=4 per condition, representative data from one of three independent experiments. (d) n=3, representative data from one of two independent experiments. (e) n=4 per condition. (a-e) Mean ± SD. p<0.0001 (****).
Source Data files provided for Figures 2, 3, 4, and Extended Data Figures 1, 4, 7, 8, 9, and 10.
Supplementary Material
Acknowledgements
We thank members of the Marson laboratory, Isha Jain, Stacie Dodgson, Sarah Pyle, Tami Tolpa, and the Gladstone Flow Cytometry Core for providing valuable input and technical expertise. We also thank Ben Gewurz (Harvard Medical School) for kindly sharing Daudi-Cas9 cells. M.R.M. was a Cancer Research Institute (CRI) Irvington Fellow supported by CRI and was funded by the Human Vaccines Project Michelson Prize for Human Immunology. J.W.F. was funded by an NIH grant (R01HG008140). A.R. was supported by an NIH training grant (T32GM007281). M.M.A. is supported by an NSF GRFP grant (2038436). M.O. was supported by Astellas Foundation for Research on Metabolic Disorder, Chugai Foundation for Innovative Drug Discovery Science (C-FINDs), and Mochida Memorial Foundation for Medicine and Pharmaceutical Research. K.A.T. was supported by the Gladstone PUMAS program, funded by an NIH grant (5R25HL121037). J.K. and Z.S. were supported by Oncode-PACT and Dutch Cancer Society grants (KWF 11393, 12586, 13043). E.J.A. is funded by an NIH grant (R01AI155984). The Marson laboratory has received funds from the Cancer Research Institute (CRI) Lloyd J. Old STAR grant, The Cancer League, the Innovative Genomics Institute (IGI), the Simons Foundation, and the Parker Institute for Cancer Immunotherapy (PICI). We thank the Hubrecht Organoid Technology (HUB) for providing patient-derived breast cancer organoids and Jeanine M. L. Roodhart (Department of Medical Oncology, University Medical Center Utrecht, Utrecht University, Utrecht, The Netherlands) for providing the patient-derived colon cancer organoid. The Gladstone Flow Cytometry Core is supported by the James B. Pendleton Charitable Trust. Some of the results shown here are based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Competing Interests Declaration
A.M. is a co-founder of Arsenal Biosciences, Spotlight Therapeutics, and Survey Genomics, serves on the boards of directors at Spotlight Therapeutics and Survey Genomics, is a board observer (and former member of the board of directors) at Arsenal Biosciences, is a member of the scientific advisory boards of Arsenal Biosciences, Spotlight Therapeutics, Survey Genomics, NewLimit, Amgen, Lightcast, and Tenaya, owns stock in Arsenal Biosciences, Spotlight Therapeutics, NewLimit, Survey Genomics, PACT Pharma, Lightcast, and Tenaya, and has received fees from Arsenal Biosciences, Spotlight Therapeutics, NewLimit, Survey Genomics, Tenaya, Lightcast, 23andMe, PACT Pharma, Juno Therapeutics, Trizell, Vertex, Merck, Amgen, Genentech, AlphaSights, Rupert Case Management, Bernstein, and ALDA. A.M. is an investor in and informal advisor to Offline Ventures and a client of EPIQ. J.W.F. was a consultant for NewLimit, is an employee of Genentech, and has equity in Roche. The Marson laboratory has received research support from Juno Therapeutics, Epinomics, Sanofi, GlaxoSmithKline, Gilead, and Anthem. J.K. is a shareholder of Gadeta B.V.. J.K. and Z.S. are inventors on patents with γδTCR related topics. A.M. and M.R.M. are inventors on patent applications that have been filed based on the findings described here.
Footnotes
Supplementary Information
Supplementary Information is available for this paper.
References
- 1.Silva-Santos B, Serre K & Norell H γδ T cells in cancer. Nat. Rev. Immunol 15, 683–691 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Sebestyen Z, Prinz I, Déchanet-Merville J, Silva-Santos B & Kuball J Translating gammadelta (γδ) T cells and their receptors into cancer cell therapies. Nat. Rev. Drug Discov 19, 169–184 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Raverdeau M, Cunningham SP, Harmon C & Lynch L γδ T cells in cancer: a small population of lymphocytes with big implications. Clin. Transl. Immunol 8, e01080 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silva-Santos B, Mensurado S & Coffelt SB γδ T cells: pleiotropic immune effectors with therapeutic potential in cancer. Nat. Rev. Cancer 19, 392–404 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mensurado S, Blanco-Domínguez R & Silva-Santos B The emerging roles of γδ T cells in cancer immunotherapy. Nat. Rev. Clin. Oncol 20, 178–191 (2023). [DOI] [PubMed] [Google Scholar]
- 6.Harly C. et al. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human γδ T-cell subset. Blood 120, 2269–2279 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rigau M. et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by γδ T cells. Science 367, eaay5516 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Karunakaran MM et al. Butyrophilin-2A1 Directly Binds Germline-Encoded Regions of the Vγ9Vδ2 TCR and Is Essential for Phosphoantigen Sensing. Immunity 52, 487–498.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groh V, Steinle A, Bauer S & Spies T Recognition of stress-induced MHC molecules by intestinal epithelial γδ T cells. Science 279, 1737–1740 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Strid J, Sobolev O, Zafirova B, Polic B & Hayday A The Intraepithelial T Cell Response to NKG2D-Ligands Links Lymphoid Stress Surveillance to Atopy. Science 334, 1293–1297 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Girardi M. et al. Regulation of cutaneous malignancy by γδ T cells. Science 294, 605–609 (2001). [Google Scholar]
- 12.Harly C. et al. Human γδ T cell sensing of AMPK-dependent metabolic tumor reprogramming through TCR recognition of EphA2. Sci. Immunol 6, eaba9010 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Marlin R. et al. Sensing of cell stress by human γδ TCR-dependent recognition of annexin A2. Proc. Natl. Acad. Sci. U.S.A 114, 3163–3168 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chien Y, Meyer C & Bonneville M γδ T cells: first line of defense and beyond. Annu. Rev. Immunol 32, 121–155 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Uhlén M. et al. Tissue-based map of the human proteome. Science 347, 1260419–1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Payne KK et al. BTN3A1 governs antitumor responses by coordinating αβ and γδ T cells. Science 369, 942–949 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sandstrom A. et al. The intracellular B30.2 domain of butyrophilin 3A1 binds phosphoantigens to mediate activation of human Vγ9Vδ2 T cells. Immunity 40, 490–500 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mullen PJ, Yu R, Longo J, Archer MC & Penn LZ The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 16, 718–731 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Yu Z. et al. Identification of a transporter complex responsible for the cytosolic entry of nitrogen-containing bisphosphonates. eLife 7, e36620 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dang AT et al. NLRC5 promotes transcription of BTN3A1-3 genes and Vγ9Vδ2 T cell-mediated killing. iScience 24, 101900 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corvaisier M et al. Vγ9Vδ2 T cell response to colon carcinoma cells. J. Immunol 175, 5481–5488 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Palakodeti A et al. The Molecular Basis for Modulation of Human Vγ9Vδ2 T Cell Responses by CD277/Butyrophilin-3 (BTN3A)-specific Antibodies. J. Biol. Chem 287, 32780–32790 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheftel A, Stehling O & Lill R Iron–sulfur proteins in health and disease. Trends Endocrinol. Metabolism 21, 302–314 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Voss M et al. Shedding of glycan-modifying enzymes by signal peptide peptidase-like 3 (SPPL3) regulates cellular N-glycosylation. EMBO J. 33, 2890–2905 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park JH et al. Tumor hypoxia represses γδ T cell-mediated antitumor immunity against brain tumors. Nat. Immunol 22, 336–346 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Blevins MA, Huang M & Zhao R The Role of CtBP1 in Oncogenic Processes and Its Potential as a Therapeutic Target. Mol. Cancer Ther 16, 981–990 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Annaert W & Kaether C Bring it back, bring it back, don’t take it away from me – the sorting receptor RER1. J. Cell Sci 133, jcs231423 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Cano CE et al. BTN2A1, an immune checkpoint targeting Vγ9Vδ2 T cell cytotoxicity against malignant cells. Cell Rep. 36, 109359 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Vantourout P et al. Heteromeric interactions regulate butyrophilin (BTN) and BTN-like molecules governing γδ T cell biology. Proc. Natl. Acad. Sci. U.S.A 115, 1039–1044 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quinlan KGR et al. Specific Recognition of ZNF217 and Other Zinc Finger Proteins at a Surface Groove of C-Terminal Binding Proteins. Mol. Cell Biol 26, 8159–8172 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mick E et al. Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell. eLife 9, e49178 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herzig S & Shaw RJ AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Bio 19, 121–135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marcu-Malina V et al. Redirecting αβ T cells against cancer cells by transfer of a broadly tumor-reactive γδT-cell receptor. Blood 118, 50–59 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Gründer C et al. γ9 and δ2CDR3 domains regulate functional avidity of T cells harboring γ9δ2TCRs. Blood 120, 5153–5162 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Straetemans T et al. GMP-Grade Manufacturing of T Cells Engineered to Express a Defined γδTCR. Front. Immunol 9, 1062 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dekkers JF et al. Uncovering the mode of action of engineered T cells in patient cancer organoids. Nat. Biotechnol 41, 60–69 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gassart AD et al. Development of ICT01, a first-in-class, anti-BTN3A antibody for activating Vγ9Vδ2 T cell–mediated antitumor immune response. Sci. Transl. Med 13, eabj0835 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Mu X et al. Glucose metabolism controls human γδ T-cell-mediated tumor immunosurveillance in diabetes. Cell Mol. Immunol 19, 944–956 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meizlish ML, Franklin RA, Zhou X & Medzhitov R Tissue Homeostasis and Inflammation. Annu. Rev. Immunol 39, 1–25 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Warburg O, Posener K & Negelein E Ueber den Stoffwechsel der Tumoren. Biochem. Z 152, 319–344 (1924). [Google Scholar]
- 41.Heiden MGV, Cantley LC & Thompson CB Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 324, 1029–1033 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 42.Tzelepis K et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 17, 1193–1205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shifrut E et al. Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 175, 1958–1971.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li W et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma Y et al. CRISPR/Cas9 Screens Reveal Epstein-Barr Virus-Transformed B Cell Host Dependency Factors. Cell Host Microbe 21, 580–591.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang S et al. CRISPR/Cas9-Mediated Genome Editing in Epstein-Barr Virus-Transformed Lymphoblastoid B-Cell Lines. Curr. Protoc. Mol. Biol 121, 31.12.1–31.12.23 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Mootha VK et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Subramanian A et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller HE & Bishop AJR Correlation AnalyzeR: functional predictions from gene co-expression correlations. BMC Bioinformatics 22, 206 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lachmann A et al. Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun 9, 1366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Concordet J-P & Haeussler M CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011). [Google Scholar]
- 53.Clement K et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brinkman EK, Chen T, Amendola M & van Steensel B Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168–e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davodeau F et al. Close correlation between Daudi and mycobacterial antigen recognition by human gamma delta T cells and expression of V9JPC1γ/V2DJCδ-encoded T cell receptors. J. Immunol 151, 1214–1223 (1993). [PubMed] [Google Scholar]
- 56.Bai L et al. The majority of CD1d-sulfatide-specific T cells in human blood use a semiinvariant Vδ1 TCR. Eur. J. Immunol 42, 2505–2510 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sachs N et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 172, 373–386.e10 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Pleguezuelos-Manzano C et al. Establishment and Culture of Human Intestinal Organoids Derived from Adult Stem Cells. Curr. Protoc. Immunol 130, e106 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng Y, Ahmad K & Henikoff S CUT&Tag Data Processing and Analysis Tutorial. Protocol.io (2020). [Google Scholar]
- 60.Meers MP, Tenenbaum D & Henikoff S Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenet. Chromatin 12, 42 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shannon P et al. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kutmon M, Lotia S, Evelo CT & Pico AR WikiPathways App for Cytoscape: Making biological pathways amenable to network analysis and visualization. F1000Res. 3, 152 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsuchiya S et al. Implementation of GlycanBuilder to draw a wide variety of ambiguous glycans. Carbohyd. Res 445, 104–116 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Varki A et al. Symbol Nomenclature for Graphical Representations of Glycans. Glycobiology 25, 1323–1324 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gustavsen JA, Pai S, Isserlin R, Demchak B & Pico AR RCy3: Network biology using Cytoscape from within R. F1000Res. 8, 1774 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martens M et al. WikiPathways: connecting communities. Nucleic Acids Res. 49, D613–D621 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang P et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med 24, 1550–1558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dunham I et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luo Y et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 48, D882–D889 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ewels PA et al. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol 38, 276–278 (2020). [DOI] [PubMed] [Google Scholar]
- 71.Lopez-Delisle L et al. pyGenomeTracks: reproducible plots for multivariate genomic datasets. Bioinformatics 37, 422–423 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing datasets for the two screens and CUT&RUN are available in the NCBI Gene Expression Omnibus (GEO) repository (co-culture screen: GSE192828; BTN3A screen: GSE192827; CUT&RUN: GSE226931). Publicly available paired-end IRF1 ChIP-Seq fastq files were downloaded from ENCODE68,69: IRF1 K562 IP rep 1 (ENCFF031RWN, ENCFF031WIT), IRF1 K562 IP rep 2 (ENCFF602NSL, ENCFF071PWE), IRF1 K562 input (ENCFF285JYI, ENCFF420KFM). Also, publicly available TCGA data were utilized for this study (https://www.cancer.gov/tcga).