

Abstract
This study aimed to determine the prevalence and clinical features of Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) caused by pathogenic mutations in the Phospholamban (PLN) gene. The study included 170 patients who had a confirmed diagnosis of ARVC and underwent PLN genetic screening using next-generation sequencing. The findings of this study provide valuable insights into the association between PLN mutations and ARVC, which can aid in the development of more effective diagnostic and treatment strategies for ARVC patients. Out of the patients evaluated, six had a rare pathogenic mutation in PLN with the same p.R14del variant. Family screening revealed that heterozygous carriers of p.R14del exhibited a definite ARVC phenotype. In clinical studies, individuals with the p.R14del mutation experienced a similar rate of malignant arrhythmia events as those with classic desmosome mutations. After adjusting for covariates, individuals with PLN mutations had a two point one seven times greater likelihood of experiencing transplant-related risks compared to those who did not possess PLN mutations (95% CI 1.08–6.82, p = 0.035). The accumulation of left ventricular fat and fibers is a pathological marker for ARVC patients with p.R14del mutations. In a cohort of 170 Chinese ARVC patients, three point five percent of probands had the PLN pathogenic variant (p.R14del) and all were female. Our data shows that PLN-related ARVC patients are at high risk for ventricular arrhythmias and heart failure, which requires clinical differentiation from classic ARVC. Furthermore, carrying the p.R14del mutation can be an independent prognostic risk factor in ARVC patients.
Supplementary Information
The online version contains supplementary material available at 10.1007/s43657-023-00126-w.
Keywords: Arrhythmogenic right ventricular cardiomyopathy, Phospholamban, Left ventricular involvement, Heart failure, Risk stratification
Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a hereditary cardiomyopathy characterized by progressive death of ventricular myocytes, myocardial atrophy and replacement by fibroadipose tissue, which is mostly inherited in autosomal dominant form (Corrado et al. 2017b, 2020). ARVC is relatively rare, with a prevalence of about 1:5000 to 1:2000, more men than women, and the age of onset is more than 20 to 50 years old (Castanos Gutierrez et al. 2016; Choudhary et al. 2016). Clinically, frequent malignant arrhythmia events and progressive ventricular structural dysfunction are the main manifestations (Bhonsale et al. 2017; Stadiotti et al. 2017). It is one of the main causes of sudden cardiac death in athletes and young people, accounting for about 20% of sudden arrhythmic death in young patients (Calkins et al. 2017; Corrado et al. 2017a).
With the development of molecular genetics and the progress of gene detection technology in the 21 century (Fu et al. 2022), ARVC pathogenic gene mutations have been discovered successively, which has established the status of ARVC as hereditary cardiomyopathy (Hoorntje et al. 2017). At present, 16 genes have been detected to be associated with ARVC, including genes encoding desmosomal proteins: Plakophilin two (PKP2), Desmoplakin (DSP), Desmoglein two (DSG2), Desmocollin two (DSC2), Junction Plakoglobin (JUP); and non-desmosomal proteins: Transmembrane Protein 43 (TMEM43), Lamin A/C (LMNA), Desmin (DES), Catenin Alpha three (CTNNA3), Transforming Growth Factor Beta three (TGFB3), Titin (TTN), Sodium Voltage-Gated Channel Alpha Subunit five (SCN5A), Cadherin two (CDH2), Filamin C (FLNC), Ryanodine Receptor two (RYR2), Phospholamban (PLN) (Corrado et al. 2017b, a; Corrado et al. 2020; Quarta et al. 2011). Most of them are inherited according to the Mendelian law of autosomal dominant inheritance. Desmosomal gene mutations accounted for 50% to 60% of ARVC patients, among which the most common gene was PKP2 (10% to 45%), followed by DSP (10% to 15%), DSG2 (7% to 10%), DSC2 (2%), JUP (< 1%) (Gandjbakhch et al. 2018). But, less than 10% had non-desmosomal mutations, and up to 25% had compound heterozygous mutations (Quarta et al. 2011). With the development of molecular genetics and pathological imaging technology, the genotype–phenotype correlation study based on ARVC cohort concluded that ARVC patients with desmosome gene mutations mainly have right ventricular accumulation and classic ARVC phenotype (Chen et al. 2019a, 2019c). In addition, ARVC non-desmosome carriers have diverse clinical manifestations, are geographically concentrated, and their clinical phenotype and genotype overlap with other cardiomyopathy, especially dilated cardiomyopathy (DCM), Brugada syndrome, and hypertrophic cardiomyopathy (HCM) (Murray et al. 2018). Thus, the relationship between mutations in genes encoding non-desmosomal proteins and ARVC pathogenicity remains to be explored.
PLN is a small phosphorylated protein in the sarcolemmic cardiac reticulum (SR) that is a master regulator of the cardiac sarcoplasmic/endoplasmic reticulum Ca2+-dependent ATPase 2a (SERCA2a) activity and calcium cycling (Kranias and Hajjar 2012). The SERCA2a mediates calcium uptake by SR and initiates relaxation, which is closely related to myocardial systolic function and ventricular remodeling (Jiang et al. 2022). Natural mutations in the human PLN gene have been reported in a variety of inherited cardiomyopathies. For example, the PLN heterozygous mutation R9C was first identified in a DCM family (Schmitt et al. 2003). The PLN heterozygous mutation T116G co-segregates with the HCM phenotype (Haghighi et al. 2003). The third mutation is a deletion of amino acid 14 in PLN (p.R14del), heterozygous carriers of which develop left ventricular dilatation, decreased cardiac function and ventricular arrhythmias, and death in middle age (Haghighi et al. 2006). Interestingly, this PLN mutation was detected in approximately 15% of DCM patients and 12% of ARVC patients in the Netherlands (van der Zwaag et al. 2012). However, the prevalence of these mutations in ARVC and the clinical features of ARVC patients with PLN mutations are not yet well understood.
All in all, recent studies have suggested that PLN components may play a role in the development of ARVC, which expands its genetic basis. However, in order to accurately estimate their contribution, prevalence, and clinical characteristics, these findings need to be validated in a large number of patients. This study aimed to investigate the relationship between PLN genotype and clinical phenotype in 170 ARVC patients, analyzing the variation spectrum in both desmosome and non-desmosome components. The results of this study provide a foundation for genetic counseling and precision medicine in ARVC.
Materials and Methods
Study Population
This study collected retrospective clinical information on ARVC patients who visited the Fuwai Hospital of the Chinese Academy of Medical Sciences between January 2003 and December 2019. Patients who met the following criteria were enrolled in this study: (1) met the diagnostic criteria revised by the ARVC Task Force in 2010 (Muona et al. 2015); (2) had complete clinical information recorded, including demographic data, physical examination, medical history, family history, serological tests, electrocardiography, echocardiography, cardiac magnetic resonance imaging, myocardial biopsy and treatment plans, etc.; (3) underwent genetic testing. In the end, 170 unrelated ARVC probands who met the inclusion criteria were enrolled in this study (Fig. S1).
The clinical investigation was carried out in accordance with the principles of the Declaration of Helsinki and was approved by the ethics committee review board of the Fuwai Hospital. All participants provided written informed consent prior to inclusion in the study.
Inclusion of Clinical Information and Follow-up
The clinical information gathered included symptoms such as dyspnoea, palpitation, presyncope, syncope, abdominal distension, and bilateral oedema, as well as the patient's implantable cardioverter defibrillator (ICD), medication history, and radiofrequency current catheter ablation (RFCA) procedures. This information was obtained by reviewing the patient's chief complaints and medical history during hospital visits, as well as their symptoms at the time of admission and subsequent treatment plans. For the purpose of this study, medication history was determined based on the patient's adherence to the use of Class III antiarrhythmic drugs such as sotalol or amiodarone, Class Ic antiarrhythmic drugs like propafenone, or β blockers including propranolol or metoprolol. In addition, the New York Heart Association (NYHA) and echocardiographic results of transplant patients were evaluated prior to heart transplantation (HT). The echocardiographic results included assessing for left ventricular (LV) or right ventricular (RV) dilation or dyskinesia, as well as measuring left ventricle end diastolic dimension (LVEDD), right ventricle end diastolic dimension (RVEDD), and left ventricle ejection fraction (LVEF). For non-transplant patients, the most recent available records were used for these results.
Follow-up information was obtained by reviewing hospital medical records and conducting phone consultations. The study had two main endpoints: end-stage heart failure leading to heart transplant and major arrhythmic cardiovascular events (MACE), which included sudden cardiac death (SCD), survival after SCD, ventricular fibrillation, sustained ventricular tachycardia, or arrhythmic syncope (Saguner et al. 2014).
Genetic and Bioinformatic Analysis of Next-Generation Sequencing Data
Genetic analysis of 16 ARVC-related genes (PKP2, DSP, DSG2, DSC2, JUP, TMEM43, LMNA, DES, CTNNA3, TGFB3, TTN, SCN5A, CDH2, FLNC, RYR2 and PLN) was performed on 170 ARVC probands using whole-genome sequencing (WGS), whole-exome sequencing (WES), and targeted next-generation sequencing (NGS). Most patients (98.8%) underwent WES (n = 42, 24.7%) and targeted NGS (n = 126, 74.1%), whereas only a few patients (n = 2, 1.2%) underwent WGS analysis. WGS was performed using TruSeq Nano DNA HT Sample Preparation Kit (Illumina). WES libraries were obtained using either Agilent SureSelect XT Human All Exon V four/five/six kit or TruSeq DNA Exome Kit (Illumina). Sequencing was performed on NextSeq 500, HiSeq 2000, or HiSeqX10 platforms (Illumina). Targeted NGS was performed through a dedicated custom Cardio panel designed using SureDesign software (Agilent Technologies). SureSelectQXT kit was used for transposase based library preparation and target enrichment, while 4200 TapeStation instruments (Agilent Technologies) were used for quality control of fragment size and purity of DNA libraries. The NGS library was sequenced in the NextSeq 500 or HiSeq 2000 Illumina platform.
As shown in Fig. 1a pipeline, the FASTQ files obtained by sequencing were firstly controlled by Trim_galore and Fastqc software, and then Bam files were obtained by using Bwa and Samtools based on the hg19 version of the human reference genome. After that, the pipeline used GATK software's Markduplicates, ApplyBQSR, HaplotypeCaller, CombineGVCFs and VariantRecalibrator commands in turn to complete Bam files reduplications, quality correction, SNP and InDel mutation detection and false positive correction to obtain VCF files. Finally, generated VCF files were further annotated with ANNOVAR (http://www.openbioinformatics.org/annovar/) to obtain genetic information that could distinguish variant types: pathogenic (P), likely pathogenic (LP), uncertain significance (VUS), likely benign (LB) and benign (B), according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al. 2015). Carrying P/LP mutations was defined as having a positive result in genetic testing. Additionally, all mutations were confirmed by Sanger sequencing.
Fig. 1.

Screening process and sequence results of PLN variant a NGS analysis pipeline diagram, fq fastq, QC quailty control, vcf the variant call format, bam binary alignment map, bqsr base quality score recalibration, vqsr variant quality score recalibration, b sanger sequence diagrams of six PLN probands, c family cascade screening of proband 1
Pathological Examination and Qualitative Pathological Analysis
The gross and microscopic examination of the transplanted hearts were performed by two pathologists without knowledge of clinical and genetic data. Six parts of the transplanted hearts were collected from each case. The LV region was composed of the anterior free wall of the left ventricle (ALV), lateral free wall of LV (LLV), and posterior free wall of the LV (PLV). The anterior free wall of RV (ARV) (right ventricular outflow tract) and posterior free wall of the RV (PRV) (sub-tricuspid valve region) constituted the RV region. The interventricular septum (IVS) was also included. The specimen was fixed in 10% formalin for histological examination. The tissue samples were stained with Masson's three primary colors, and the slides were scanned into digital images.
Before quantitative analysis, Adobe Photoshop CC 2018 (Adobe Systems, San Jose, CA) was used to remove epicardial adipose tissue, including blood vessels and pericardium. When endocardium thickening was detected, the endocardium was removed from the analysis of fibrosis. Evaluate myocardial, fibrotic, and adipose tissue using Image Pro Plus (version 6.0; Media Controllers, Rockville, MD). The LV myocardium, fibrosis, and adipose tissue ratios were averaged over ALV, LLV, and PLV, while the RV ratios were averaged over ARV and PRV.
Statistical Analysis
Clinical variables were expressed as the mean ± standard deviation for continuous variables and as counts and frequencies (%) for categorical variables. The independent samples t-test, in combination with Levene's test, was used to compare two groups of continuous variables. If necessary, categorical variables were compared using the chi-square test or Fisher's exact analysis. Cumulative degrees of freedom from birth to clinical outcome were determined by the Kaplan–Meier method and differences were assessed by the log-rank test. Univariate and multivariate proportional hazards (COX) regression analyses were used to estimate transplant risk factors affecting patients. Reported probability p values are two-way and p value < 0.05 were considered statistically significant. SPSS statistical software (version 25, IBM Corp., Armonk, NY, USA) was used for analysis.
Results
DNA Mutation Spectrum of the 170 ARVC Probands
This study enrolled 170 patients diagnosed with ARVC at Fuwai Hospital. Data for analysis was obtained through NGS genome sequencing and variant analysis. The study found that 33.5% of patients had positive genotypes, with the majority of these being desmosomal variants. Specifically, PKP2 was present in 13.5% of patients and DSG2 in 9.4% of patients. Among them, six unrelated probands were found to carry a pathogenic variant of the non-desmosomal gene PLN, while not carrying P/LP variants of the 15 other ARVC-related genes. In Fig. 1b, the results of Sanger sequencing validation for six individuals (3.5%) at this locus were displayed. All six individuals had a heterozygous deletion variant, which caused a frameshift mutation by removing three bases (GAA) from one DNA strand. The variant is named according to Human Genome Variation Society (HGVS) nomenclature as NM_002667: c.36_38delGAA, p.R14del. After successful cascade screening in the proband 1's family, it was discovered that two half-sisters with different fathers were diagnosed with ARVC. They were found to carry the same maternal heterozygous genotype variant p.R14del as their mother (the proband) (Fig. 1c).
The variant was analyzed using T-coffee software and found to be conserved across different species (Fig. S2a). Further analysis using PROVEAN software suggests that this variant may result in the deletion of ARG arginine after PLN translation, which could impair its normal function and contribute to the development of ARVC (Fig. S2b).
Clinicopathological Differences Between PLN Patients and Desmosome-Mutated ARVC Patients
A detailed summary of functional impairments and structural changes, repolarization abnormalities, depolarization/conduction abnormalities, and arrhythmias in the six PLN probands were summarized in Table 1. Then, Table S1 showed the evidence for 23 PKP2, 16 DSG2, and six PLN probands diagnosed with ARVC. Among the 170 ARVC probands in this study, 113 (66.5%) were found to be negative for genetic testing, including 48 who received HT (none variants-HT, NV-HT). To study the clinical characteristics of six patients with PLN, we selected 23 PKP2, 16 DSG2, and 48 genetically negative but transplanted patients as control groups. We then compared the clinical characteristics between these groups. As shown in Table 2, there were two major characteristics.
Table 1.
Phenotypic characteristics of six PLN probands
| PLN Probands | Family screening | Dysfunction and structural alterations | Repolarization abnormalities | Depolarization/conduction abnormalities | Arrhythmias |
|---|---|---|---|---|---|
| Proband 1 | YES | Regional RV dyskinesia, PLAX RVOT = 30 mm | TWI in V1-3 without CRBBB | TAD of QRS > 55 ms | VT of LBBB with superior |
| Proband 2 | – | Regional RV dyskinesia, PLAX RVOT = 30 mm | – | TAD of QRS > 55 ms | – |
| Proband 3 | – | – | – | – | Sustained VT of LBBB |
| Proband 4 | – | – | TWI in V1-3 without CRBBB | TAD of QRS > 55 ms | VT of LBBB with superior |
| Proband 5 | – | Regional RV dyskinesia, PLAX RVOT = 29 mm | TWI in V1-4 with CRBBB | Epsilon wave in V1-3 | VT of LBBB with superior |
| Proband 6 | – | – | – | Epsilon wave in V1-3 | VPB > 500 times/h |
CRBBB complete right bundle branch block, LBBB left branch bundle block morphology, PLAX parasternal long-axis view in echocardiography, RVOT right ventricular outflow tract, TAD terminal activation duration, TWI T wave inversion, VT ventricular tachycardia, VPB ventricular premature beat
Table 2.
Characteristics of patients with PLN variants, DSG2 variants or PKP2 variants and NV-HT patients
| Variables | PLN (N = 6) | PKP2 (N = 23) | DSG2 (N = 16) | NV-HT (N = 48) | p-value (PLN vs PKP2) | p-value (PLN vs DSG2) | p-value (PLN vs NV-HT) |
|---|---|---|---|---|---|---|---|
| Age of onset (years) | 35.17 ± 8.86 | 33.65 ± 14.58 | 30.75 ± 13.80 | 34.13 ± 14.46 | 0.811 | 0.478 | 0.865 |
| Male, n (%) | 0 (0) | 18 (78) | 9 (56) | 30 (63) | 0.001 | 0.046 | 0.005 |
| Dyspnoea, n (%) | 3 (50) | 5 (22) | 8 (50) | 28 (58) | 0.305 | 1.000 | 1.000 |
| Palpitation, n (%) | 5 (83) | 22 (96) | 9 (56) | 33 (69) | 0.377 | 0.351 | 0.657 |
| Presyncope, n (%) | 6 (100) | 19 (83) | 11 (69) | 29 (60) | 0.553 | 0.266 | 0.080 |
| Syncope, n (%) | 4 (67) | 13 (57) | 5 (31) | 14 (29) | 1.000 | 0.178 | 0.087 |
| Abdominal distension, n (%) | 0 (0) | 0 (0) | 3 (19) | 16 (33) | – | 0.532 | 0.163 |
| Bilateral oedema, n (%) | 3 (50) | 3 (13) | 3 (19) | 21 (44) | 0.083 | 0.283 | 1.000 |
| MACE, n (%) | 4 (67) | 21 (91) | 13 (81) | 29 (60) | 0.180 | 0.585 | 1.000 |
| ICD, n (%) | 1 (17) | 9 (39) | 5 (31) | 12 (25) | 0.633 | 0.634 | 1.000 |
| RFCA, n (%) | 0 (0) | 11 (48) | 2 (13) | 7 (15) | 0.058 | 1.000 | 1.000 |
| NYHA, n (%) | < 0.001 | 0.043 | 0.461 | ||||
| I | 0 (0) | 9 (39) | 8 (50) | 0 (0) | |||
| II | 0 (0) | 12 (52) | 1 (6) | 6 (13) | |||
| III | 3 (50) | 1 (4) | 6 (38) | 28 (58) | |||
| IV | 3 (50) | 1 (4) | 1 (6) | 14 (29) | |||
| RV dilation, n (%) | 3 (50) | 18 (78) | 15 (94) | 43 (90) | 0.305 | 0.046 | 0.036 |
| RV dyskinesia, n (%) | 3 (50) | 16 (70) | 11 (69) | 39 (81) | 0.633 | 0.624 | 0.116 |
| RVEDD (mm) | 26.83 ± 6.11 | 35.67 ± 8.55 | 40.31 ± 11.29 | 37.83 ± 11.46 | 0.030 | 0.012 | 0.026 |
| LV dilation, n (%) | 5 (83) | 0 (0) | 3 (19) | 17 (35) | < 0.001 | 0.011 | 0.036 |
| LV dyskinesia, n (%) | 6 (100) | 3 (13) | 4 (25) | 19 (40) | < 0.001 | 0.003 | 0.007 |
| LVEDD (mm) | 57.75 ± 6.50 | 43.68 ± 4.72 | 45.56 ± 7.06 | 52.06 ± 8.14 | < 0.001 | 0.006 | 1.118 |
| LVEF (%) | 25.75 ± 8.02 | 60.20 ± 11.06 | 53.75 ± 12.74 | 38.75 ± 11.13 | < 0.001 | 0.001 | 0.008 |
The bold and italicized value indicates statistical significance. ICD implantable cardioverter defibrillator, RFCA radiofrequency current catheter ablation, LVEDD left ventricle end diastolic dimension, NYHA New York Heart Association, RVEDD right ventricle end diastolic dimension, LVEF left ventricle ejection fraction
First, all PLN patients were females, and the gender distribution was significantly different from that of PKP2, DSG2, and NV-HT (p-values of 0.001, 0.046, and 0.005, respectively). The other major characteristic was that PLN patients had poorer cardiac function and more obvious LV involvement before transplantation: 32 PLN patients (87%) were classified as NYHA III-IV, which was significantly higher than the proportions for PKP2 and DSG2 (p-values of < 0.001 and 0.043, respectively); the proportions of LV dilation (83%) and LV dyskinesia (100%) in PLN patients were significantly higher than those in PKP2, DSG2, and NV-HT patients (p-values of LV dilation were < 0.001, 0.011, and 0.036, respectively; p-values of LV dyskinesia were < 0.001, 0.003, and 0.007, respectively); the LV dilation in PLN patients was more obvious, with LVEDD (57.75 mm ± 6.50 mm) significantly greater than those of DSG2 and PKP2 patients (p-values of < 0.001 and 0.006, respectively); the LVEF of PLN patients was the lowest (25.7% ± 8.02%). On the contrary, for the RV, the proportion of RV dilation in PLN patients was significantly lower than that in DSG2 and NV-HT patients (p-values of 0.046 and 0.036, respectively), and the degree of dilation was the lowest in PLN patients (RVEDD = 26.83 mm ± 6.11 mm).
Digital Quantitative Evaluation Fibrosis and Fat Infiltration
We carried out pathological quantitative analysis of the transplanted heart specimens from PLN transplant patients (n = 6), PKP2 transplant patients (n = 2), DSG2 transplant patients (n = 5), and NV-HT patients (n = 48). As shown in Fig. 2a, the main histological features of these patients were myocardial loss and fibrofatty infiltration. Firstly, for PLN transplant patients, the percentage of myocardial tissue in LV was significantly lower than that in RV and IVS (56.5% ± 11.6% vs 86.7% ± 7.0% and 56.5% ± 11.6% vs 77.9% ± 18.6%, p = 0.004 and p = 0.042 respectively) (Fig. 2b), indicating that LV involvement was predominant in PLN patients. Then, we compared these four groups cross-sectionally and found that for LV, although there was no statistical significance, the myocardial percentage of PLN was the lowest and the fibrosis and fat percentage were the highest (56.6% ± 11.7%, 19.9% ± 8.2% and 23.6% ± 12.9% respectively) (Fig. 2c). Secondly, for RV, the myocardial percentage in PLN was significantly higher than that in DSG2 and NV-HT (77.8% ± 18.6% vs 32.1% ± 6.6% and 77.8% ± 18.6% vs 45.6% ± 26.8%, p = 0.005 and p = 0.006 respectively), and the fat component was significantly lower than that in NV-HT (14.5% ± 19.4% vs 38.8.7% ± 25.9%, p = 0.034) (Fig. 2e). This indicates that the RV of PLN patients is relatively less affected.
Fig. 2.

Analysis of pathological features of transplant patients a Masson map of six site pathology of transplant patients. The top arrow indicates fibrosis and the bottom arrow indicates fatty infiltration. b Qualitative pathological analysis was used to quantify the degree of fibrosis and adipose invasion at different sites (LV, IVS, and RV) in PLN transplant patients. c–e Comparison of fat, fibrosis and Myocardium proportions at different sites (LV, IVS and RV) in patients carrying different variants (PLN, PKP2, DSG2 and NV-HT). *p < 0.05; **p < 0.01
Patients with PLN Mutations Experience Faster Progression of Heart Failure
The study utilized Kaplan–Meier (KM) survival curves to assess the cumulative incidence rate of MACE events from birth. The results indicated that there was no significant difference in survival between PLN carriers and patients with PKP2, DSG2, and NV-HT (as shown in Fig. 3a). When analyzing the occurrence of HT events after the diagnosis of ARVC, it was found that the transplant survival rate of PLN patients was significantly lower compared to PKP2 and DSG2 groups. The median survival time of the PLN group was the shortest, at 72 weeks (Fig. 3b). After that, to assess whether carrying a PLN mutation is a predictive factor for transplantation risk, we analyzed all patients with mutations in PLN, PKP2, DSG2, and NV-HT in this study and developed a COX regression model (refer to Table 3). According to the results of the univariate COX regression analysis, gender and carrying a PLN mutation were identified as independent predictors of transplantation risk (HR = 0.53, p = 0.021; HR = 3.62, p = 0.004 respectively). After adjusting for covariates, it was discovered that individuals with PLN mutations had a two point one seven times greater likelihood of experiencing transplant-related risks compared to those who did not possess PLN mutations (95% CI 1.08–6.82, p = 0.035).
Fig. 3.
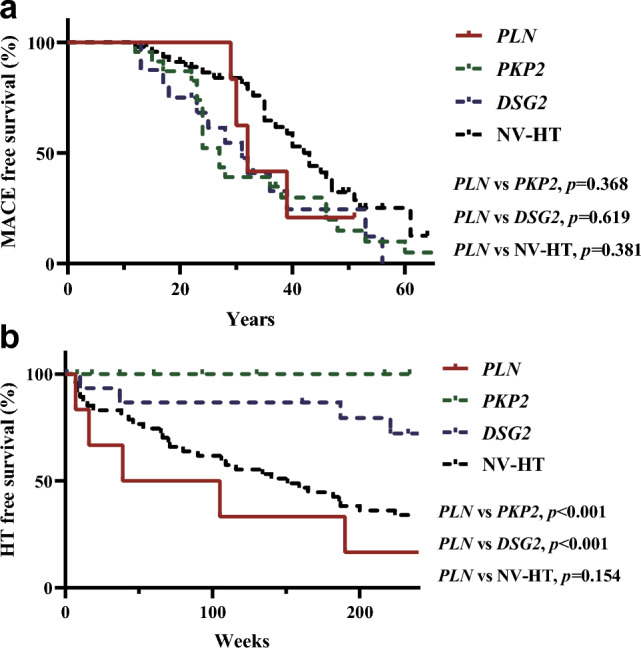
Survival analysis a From birth to endpoint follow-up, with the endpoint being MACE events. b From diagnosis of ARVC to endpoint follow-up, with the endpoint being HT
Table 3.
COX regression models were used to assess the risk of transplantation in ARVC patients
| Variables | Univariate analysis | Multivariate analysis | ||
|---|---|---|---|---|
| HR (95% CI) | p-value | HR (95% CI) | p-value | |
| Age of onset | 1.01 (0.99–1.03) | 0.297 | ||
| Male | 0.53 (0.31–0.91) | 0.021 | 0.62 (0.35–1.09) | 0.096 |
| Whether with p.R14 del | 3.62 (1.52–8.64) | 0.004 | 2.17 (1.08–6.82) | 0.035 |
| Dyspnoea | 1.43 (0.86–2.39) | 0.143 | ||
| Palpitation | 0.74 (0.43–1.29) | 0.289 | ||
| Presyncope | 0.64 (0.37–1.11) | 0.115 | ||
| Syncope | 0.69 (0.41–1.19) | 0.190 | ||
| Abdominal distension | 1.49 (0.86–2.57) | 0.157 | ||
| Bilateral oedema | 1.60 (0.97–2.65) | 0.068 | ||
| ICD | 0.56 (0.32–1.00) | 0.052 | ||
| Medication history | 1.11 (0.65–1.89) | 0.699 | ||
| RFCA | 0.51 (0.24–1.08) | 0.077 | ||
The bold and italicized value indicates statistical significance
Discussion
This study discovered that in the Chinese population with ARVC, the frequency of PLN variation was three point five percent. Furthermore, all of the variations were p.R14del heterozygous. In a large Greek pedigree, the Kranias lab originally described the PLN p.R14del mutation during the early stages of their research (Haghighi et al. 2006). This mutation was found to co-segregate with a phenotype of cardiomyopathy and sudden cardiac death. Subsequently, it is demonstrated that the PLN p.R14del mutation is frequently observed in patients with arrhythmogenic right ventricular cardiomyopathy (12%) or DCM (15%) in the Netherlands (van der Zwaag et al. 2012). Based on data analysis of PLN p.R14del, a founder mutation was identified in the northern region of the Netherlands in the year 1400 A.D. (van der Zwaag et al. 2013). Currently, there are 1500 carriers with the same haplotype identified in the Netherlands, along with several additional families in European countries, the United States, and Canada. It is worth noting, this finding suggests that the prevalence of PLN mutations in the Chinese cohort differs from that in the European cohort, as the mutations were more randomly distributed in the former. Our study included six probands carrying PLN mutations who were independent individuals with no blood relationship. These individuals were from various regions of China. In addition, family screening of proband 1 showed that the p.R14del heterozygous genotype co-segregated with the ARVC phenotype. A noteworthy observation is that all the patients who tested positive for PLN were female. This could be due to varying degrees of mutation tolerance caused by different metabolic environments between men and women. This is because the protein affected by PLN, SERCA2a, is closely related to ATP and metabolism (Engelhardt et al. 2004; Heinis et al. 2013).
To enhance the understanding of clinical characteristics of patients with PLN, control groups comprising classic PKP2 patients, DSG2 patients, and gene-negative patients who underwent HT were selected. PKP2 patients were specifically chosen as they have the most prevalent type of mutation in ARVC, which predominantly presents as right-dominant type (Marcus et al. 1982; Towbin et al. 2019). While we have previously reported a bi-ventricular phenotype in Asians for DSG2 patients (Chen et al. 2019b). The clinical feature results indicate that PLN patients had poor LV function prior to transplantation, while their right ventricle was relatively less affected. To further confirm this, transplanted hearts from PLN, PKP2, DSG2, and NV-HT patients were analyzed using digital pathology quantitative analysis to compare myocardial composition and fibrofatty content in different regions. The results showed that the LV myocardium in PLN patients was severely replaced by fibrofatty tissue compared to the IVS and RV. Additionally, the degree of myocardial replacement in the RV was lower than that of DSG2 and NV-HT. These findings explain why PLN patients have worse left ventricular cardiac function.
Our follow-up study has shown that early detection and medical treatment of PKP2 patients have resulted in favorable outcomes and high survival rates. The rate of medical treatment, RFCA, or ICD implantation in PKP2 patients is 88%, and the transplant survival rate in PKP2 patients from birth to 60 years of age remains above 75%. However, patients with PLN typically develop heart failure either at or prior to the onset of malignant arrhythmias or when drug therapy is necessary. Therefore, the presence of PLN mutation in patients with ARVC warrants attention. Early identification of PLN mutation and family risk groups in ARVC patients is crucial in formulating heart failure intervention measures and preventing the occurrence of MACE events.
This study has some limitations that should be considered. Firstly, the sample size for PLN is small, and further subgroup analysis is not possible. Additionally, all PLN cases are female, which may introduce bias due to the small sample size. Therefore, more PLN probands are needed to confirm the conclusions. Furthermore, the reason why PLN is different from PKP2 and DSG2 in being a left-dominant ARVC subtype may require further experimental verification. For example, exploring the mechanism of fibrofatty formation using single-cell transcriptomics or spatial transcriptomics techniques could provide a foundation for the clinical translation of treatments for PLN heart failure patients.
Conclusions
Total and for the first time, we are the first to summarize the prevalence and clinical phenotypic characteristics of PLN mutations based on an ARVC cohort in the Chinese population. And clearly pointed out that ARVC patients with PLN mutations are at high risk of heart failure and heart transplantation, and more attention should be paid to early genetic counseling and genetic screening. Pathological evidence from transplantation suggests that this early onset heart failure is due to the predominance of left ventricular involvement in patients with PLN mutations.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We gratefully appreciate the numerous sample donors for making this work possible.
Abbreviations
- ARVC
Arrhythmogenic right ventricular cardiomyopathy
- PLN
Phospholamban
- DCM
Dilated cardiomyopathy
- HCM
Hypertrophic cardiomyopathy
- SERCA2a
Sarcoplasmic/endoplasmic reticulum Ca2+-dependent ATPase 2a
- ICD
Implantable cardioverter defibrillator
- RFCA
Radiofrequency current catheter ablation
- NYHA
The New York Heart Association
- HT
Heart transplantation
- NV-HT
None variants-heart transplantation
- LV
Left ventricular
- RV
Right ventricular
- LVEDD
Left ventricle end diastolic dimension
- RVEDD
Right ventricle end diastolic dimension
- LVEF
Left ventricle ejection fraction
- MACE
Major arrhythmic cardiovascular events
- SCD
Sudden cardiac death
- P
Pathogenic
- LP
Likely pathogenic
- VUS
Uncertain significance
- LB
Likely benign
- B
Benign
- ACMG
American College of Medical Genetics and Genomics
- ALV
Anterior free wall of the left ventricle
- LLV
Lateral free wall of left ventricular
- PLV
Posterior free wall of the left ventricular
- ARV
Anterior free wall of right ventricular
- PRV
Posterior free wall of the right ventricular
- IVS
The interventricular septum
- HGVS
Human genome variation society
- COX
Proportional hazards
Authors’ Contributions
Conceived and designed the study: JS, XH and HM; Performed the experiments: MB, XC and ZS; Analyzed the data: HM, XH and MX; Prepared the manuscript: JS, XH and HM.
Funding
This work was Supported by the National Natural Science Foundation for Distinguished Young Scholars of China (Grant No. 82125004), the Shenzhen Science and Technology Innovation Commission (Grant No. JCYJ20220818103414030), the National Natural Science Foundation of China (Grant No. 82300397), the key project of Shenzhen Basic Research Program (Natural Science Foundation of Shenzhen, Grant No. 20220241), and the Program for Guangdong Introducing Innovative and Enterpreneurial Teams (Grant No. 2019ZT08Y481).
Data Availability
Data relating to this article are available in the article itself or in its Supplementary material online.
Declarations
Conflict of interest
The authors have no conflicts of interest to declare in relation to this study.
Ethical approval
All clinical investigations were conducted in accordance with the principles expressed in the Declaration of Helsinki. The study protocol was approved by the Ethics Committee Review Board of Fuwai Hospital.
Consent to participate
All the participants provided written informed consent.
Consent to publication
The participant's consent to have their data published.
Footnotes
Han Mo and Xiumeng Hua have contributed equally to this work.
References
- Bhonsale A, Te Riele A, Sawant AC, et al. Cardiac phenotype and long-term prognosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia patients with late presentation. Heart Rhythm. 2017;14(6):883–891. doi: 10.1016/j.hrthm.2017.02.013. [DOI] [PubMed] [Google Scholar]
- Calkins H, Corrado D, Marcus F. Risk stratification in arrhythmogenic right ventricular cardiomyopathy. Circulation. 2017;136(21):2068–2082. doi: 10.1161/CIRCULATIONAHA.117.030792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanos Gutierrez SL, Kamel IR, Zimmerman SL. current concepts on diagnosis and prognosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Thorac Imaging. 2016;31(6):324–335. doi: 10.1097/RTI.0000000000000171. [DOI] [PubMed] [Google Scholar]
- Chen K, Rao M, Guo G, et al. Recessive variants in plakophilin-2 contributes to early-onset arrhythmogenic cardiomyopathy with severe heart failure. Europace. 2019;21(6):970–977. doi: 10.1093/europace/euz026. [DOI] [PubMed] [Google Scholar]
- Chen L, Rao M, Chen X, et al. A founder homozygous DSG2 variant in East Asia results in ARVC with full penetrance and heart failure phenotype. Int J Cardiol. 2019;274:263–270. doi: 10.1016/j.ijcard.2018.06.105. [DOI] [PubMed] [Google Scholar]
- Chen L, Song J, Chen X, et al. A novel genotype-based clinicopathology classification of arrhythmogenic cardiomyopathy provides novel insights into disease progression. Eur Heart J. 2019;40(21):1690–1703. doi: 10.1093/eurheartj/ehz172. [DOI] [PubMed] [Google Scholar]
- Choudhary N, Tompkins C, Polonsky B, et al. Clinical presentation and outcomes by sex in arrhythmogenic right ventricular cardiomyopathy: findings from the north american arvc registry. J Cardiovasc Electrophysiol. 2016;27(5):555–562. doi: 10.1111/jce.12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrado D, Basso C, Judge DP. Arrhythmogenic cardiomyopathy. Circ Res. 2017;121(7):784–802. doi: 10.1161/CIRCRESAHA.117.309345. [DOI] [PubMed] [Google Scholar]
- Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2017;376(1):61–72. doi: 10.1056/NEJMra1509267. [DOI] [PubMed] [Google Scholar]
- Corrado D, van Tintelen PJ, McKenna WJ, et al. Arrhythmogenic right ventricular cardiomyopathy: evaluation of the current diagnostic criteria and differential diagnosis. Eur Heart J. 2020;41(14):1414–1429. doi: 10.1093/eurheartj/ehz669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt S, Hein L, Dyachenkow V, et al. Altered calcium handling is critically involved in the cardiotoxic effects of chronic beta-adrenergic stimulation. Circulation. 2004;109(9):1154–1160. doi: 10.1161/01.CIR.0000117254.68497.39. [DOI] [PubMed] [Google Scholar]
- Fu F, Tao X, Jiang Z, et al. Identification of germline mutations in east-asian young never-smokers with lung adenocarcinoma by whole-exome sequencing. Phenomics. 2022 doi: 10.1007/s43657-022-00062-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandjbakhch E, Redheuil A, Pousset F, et al. Clinical diagnosis, imaging, and genetics of arrhythmogenic right ventricular cardiomyopathy/dysplasia: JACC state-of-the-art review. J Am Coll Cardiol. 2018;72(7):784–804. doi: 10.1016/j.jacc.2018.05.065. [DOI] [PubMed] [Google Scholar]
- Haghighi K, Kolokathis F, Pater L, et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111(6):869–876. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi K, Kolokathis F, Gramolini AO, et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci USA. 2006;103(5):1388–1393. doi: 10.1073/pnas.0510519103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinis FI, Andersson KB, Christensen G, et al. Prominent heart organ-level performance deficits in a genetic model of targeted severe and progressive SERCA2 deficiency. PLoS One. 2013;8(11):e79609. doi: 10.1371/journal.pone.0079609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoorntje ET, Te Rijdt WP, James CA, et al. Arrhythmogenic cardiomyopathy: pathology, genetics, and concepts in pathogenesis. Cardiovasc Res. 2017;113(12):1521–1531. doi: 10.1093/cvr/cvx150. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Li X, Guo T, et al. Ranolazine rescues the heart failure phenotype of PLN-deficient human pluripotent stem cell-derived cardiomyocytes. Stem Cell Reports. 2022;17(4):804–819. doi: 10.1016/j.stemcr.2022.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012;110(12):1646–1660. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus FI, Fontaine GH, Guiraudon G, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65(2):384–398. doi: 10.1161/01.cir.65.2.384. [DOI] [PubMed] [Google Scholar]
- Muona M, Berkovic SF, Dibbens LM, et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet. 2015;47(1):39–46. doi: 10.1038/ng.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray B, Hoorntje ET, Te Riele A, et al. Identification of sarcomeric variants in probands with a clinical diagnosis of arrhythmogenic right ventricular cardiomyopathy (ARVC) J Cardiovasc Electrophysiol. 2018;29(7):1004–1009. doi: 10.1111/jce.13621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarta G, Muir A, Pantazis A, et al. Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: impact of genetics and revised task force criteria. Circulation. 2011;123(23):2701–2709. doi: 10.1161/CIRCULATIONAHA.110.976936. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saguner AM, Ganahl S, Baldinger SH, et al. Usefulness of electrocardiographic parameters for risk prediction in arrhythmogenic right ventricular dysplasia. Am J Cardiol. 2014;113(10):1728–1734. doi: 10.1016/j.amjcard.2014.02.031. [DOI] [PubMed] [Google Scholar]
- Schmitt JP, Kamisago M, Asahi M, et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003;299(5611):1410–1413. doi: 10.1126/science.1081578. [DOI] [PubMed] [Google Scholar]
- Stadiotti I, Catto V, Casella M, et al. Arrhythmogenic cardiomyopathy: the guilty party in adipogenesis. J Cardiovasc Transl Res. 2017;10(5–6):446–454. doi: 10.1007/s12265-017-9767-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin JA, McKenna WJ, Abrams DJ, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019;16(11):e301–e372. doi: 10.1016/j.hrthm.2019.05.007. [DOI] [PubMed] [Google Scholar]
- van der Zwaag PA, van Rijsingen IA, Asimaki A, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012;14(11):1199–1207. doi: 10.1093/eurjhf/hfs119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zwaag PA, van Rijsingen IA, de Ruiter R, et al. Recurrent and founder mutations in the Netherlands-Phospholamban p Arg14del mutation causes arrhythmogenic cardiomyopathy. Neth Heart J. 2013;21(6):286–293. doi: 10.1007/s12471-013-0401-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data relating to this article are available in the article itself or in its Supplementary material online.