

Abstract
Dendritic cell (DC)-targeted vaccination is a new mode of antigen delivery that relies on the use of monoclonal antibodies (mAb) to target antigen to specific DC subsets. The neonatal Fc receptor (FcRn) is a non-classical Fc receptor that binds to immunoglobulin G (IgG) in acidified endosomes and controls its intracellular transport and recycling. FcRn is known to participate in the antigen presentation of immune complexes, however its contribution to DC-targeted vaccination has not previously been examined. Here we have investigated the role of FcRn in antigen presentation using antigen conjugated to IgG mAb which target specific DC receptors, including DEC205 and Clec9A expressed by the conventional DC 1 (cDC1) subset. We show that FcRn is expressed at high levels by cDC1, both at steady-state and following activation and plays a significant role in MHC I cross-presentation and MHC II presentation of antigens that are targeted to cDC1 via mAb specific for DEC205. This effect of FcRn is intrinsic to cDC1 and FcRn impacts the efficacy of anti-DEC205-mediated vaccination against B cell lymphoma. In contrast, FcRn does not impact presentation of antigens targeted to Clec9A and does not regulate presentation of cell-associated antigen. These data highlight a new and unique role of FcRn in controlling the immunogenicity of anti-DEC205-based vaccination, with consequences for exploiting this pathway to improve DC-targeted vaccine outcomes.
Subject terms: Antigen presentation, Tumour immunology
Introduction
Dendritic cells (DCs) are unique in their ability to capture and present antigen by major histocompatibility complex (MHC) molecules to T cells, making them an attractive cellular platform for vaccine development. DC-targeted vaccination is a strategy that relies on delivering the vaccine antigen directly to DCs in situ via conjugation to immunoglobulin G (IgG) monoclonal antibody (mAb) specific to DC surface receptors1,2. Several receptors have been explored for DC targeting1,2. These include the C-type lectins DC-SIGN and mannose receptor (MR), both expressed by various myeloid cells including monocytes, macrophages and DCs. Antigen delivered to DC-SIGN and MR are efficiently processed for MHC I cross-presentation to CD8+ T cell and MHC II presentation to CD4+ T cells3. Specific DC subsets can also be targeted for vaccination. In this case, antigen is delivered using mAb specific for receptors that possess restricted expression patterns to a particular DC subset of interest. The most widely studied examples are C-type lectin receptors DEC205 and C-type lectin domain family 9 member A (Clec9A), and X-C motif chemokine receptor 1 (XCR1). DEC205, Clec9A and XCR1 are expressed at high levels by conventional DC 1 (cDC1) and few other cell types. cDC1 are a desirable cell type to target for vaccination given their ability to undertake efficient MHC I cross-presentation4,5. DEC205 is a receptor for type B oligonucleotides6, while Clec9A, also known as DC natural killer lectin group receptor 1 (DNGR1), binds to F-actin exposed by necrotic cells7–9. XCR1 is expressed by murine and human cDC1 and recognizes XCR1 ligand XCL1, also known as lymphotactin. DC-targeted vaccination using mAb specific for DEC205, Clec9A and XCR1 induces cytotoxic T cell immunity10–14 that contributes to tumor eradication in prophylactic and therapeutic settings15–17. In addition, DEC205 and Clec9A-targeted vaccine antigen is efficiently processed for MHC II presentation, resulting in efficient CD4+ T cell responses10,11,13,18. In contrast to cDC1 receptors, dendritic cell inhibitory receptor 2 (DCIR2), also known as Clec4A4, is a C-type lectin highly expressed by mouse cDC2, a subset specialized in MHC II antigen presentation. DCIR2-targeted vaccination induces poor CD8+ T cell responses but generates potent CD4+ T cell immunity and robust antibody responses14,19,20. DC-targeted vaccines against MR and DEC205 are now used in clinical treatment21–23, and efforts to further boost their efficacy is of major interest.
The neonatal Fc receptor (FcRn) is an Fc binding receptor that belongs to the MHC I superfamily, forming a heterodimer encompassing a heavy α chain and a β-2-microglobulin chain24. FcRn is predominantly located in endosomal compartments where it interacts with two ligands, IgG and serum albumin via independent binding sites25. This receptor-ligand interaction is pH dependent, with relatively strong binding affinity below pH 6.5 but low affinity at neutral pH26. FcRn protects IgG and albumin from intracellular catabolism by transporting ligands in transport carriers for release at the cell surface and diverting them away from proteases-containing late endosomes/lysosomes27. Another function of FcRn is to deliver IgG from mother to neonates across the intestinal and/or placental epithelia by transcytosis28,29. In DCs, FcRn interacts with endocytosed IgG immune complexes and shuttles them to intracellular compartments that enable MHC I and MHC II presentation for priming of CD8+ and CD4+ T cell responses30–32.
Given the role for FcRn in intracellular IgG trafficking and presentation of IgG-associated antigen, we investigated whether FcRn contributes to immune responses downstream of IgG mAb-based DC-targeted vaccines, with a focus on DEC205 and Clec9A-mediated vaccine targeting. Here we show that FcRn plays a critical role in MHC I cross-presentation and MHC II presentation of DEC205-targeted, but not Clec9A-targeted antigen. Our data highlight a critical role for FcRn in settings of DC-targeted vaccination that can be exploited to boost their efficacy.
Results
FcRn expression by splenic DCs
We first examined FcRn expression in primary mouse cDCs compared to other immune cell types. Total cellular FcRn was detected by western blot for bone marrow-derived macrophages, cDC1, cDC2 and B cells. Low levels of FcRn were detected in CD8+ T cells whereas CD4+ T cells expressed very little (Fig. 1A). cDC1 and cDC2 express comparable levels of toll-like receptor (TLR)933,34 and both subsets are activated by the TLR9 agonist CpG oligodeoxynucleotides (CpG)34–36 (Supplementary Fig. 1). Activation of cDC1 or cDC2 by exposure to CpG did not notably alter FcRn levels for either subset (Fig. 1B). To evaluate FcRn in human DCs, we used the Human DC Atlas37,38. FCGRT expression was expressed by both human blood-derived cDC1 and cDC2 without stimulation and ex vivo incubation of DCs with bacterial or viral stimuli did not alter FCGRT expression for either subset (Supplementary Fig. 2).
Fig. 1. FcRn is expressed by DC subsets.

A Immune cell populations were purified from C57BL/6 mice and analyzed by immunoblotting for FcRn expression. Representative of two independent experiments. M: Pre-stained protein marker, with the molecular weight indicated in kDa. B FcRn expression in spleen cDC1 and cDC2 was analyzed by immunoblotting either in resting conditions or following in vitro activation with CpG for 18 h at 37 °C. M: Pre-stained protein marker, with the molecular weight indicated in kDa. C Serum levels of the different IgG subtypes, IgA, IgE and IgM in WT and Fcgrt−/− mice. Each symbol represents an individual mouse. ****P < 0.0001, *P < 0.05 by Student’s t test. ns, not significant. D The number of spleen cDC1 and cDC2 in WT and Fcgrt−/− mice was measured. Each symbol represents an individual mouse. ns not significant.
We next characterized the phenotype of Fcgrt−/− mice. In line with previous reports27, Fcgrt−/− mice developed hypogammaglobulinemia, as evidenced by the absence of IgG1, IgG2c, IgG3 and a strong reduction in IgG2b levels in the serum. In contrast, IgA, IgE and IgM were detected at normal levels (Fig. 1C). Both cDC1 and cDC2 subsets were present at normal numbers in the absence of FcRn (Fig. 1D).
FcRn is required for effective tumor immunity in response to DEC205 and Clec9A-targeted vaccination
The effectiveness of DC receptor-targeted vaccines in eliminating B cell lymphoma was examined using a mouse model. This model consists of Eμ-myc transformed lymphoma cells that express the antigen ovalbumin (OVA)39. Prophylactic vaccination is effective in suppressing the growth of Eμ-myc-OVA lymphoma, as shown previously when OVA-coated splenocytes adjuvanted with lipopolysaccharide (LPS) were used as a vaccine39. Here, DC vaccines were designed to target whole OVA to DC receptors DEC205 and Clec9A through a genetic fusion of the protein to the Fc region of rat IgG2a specific to the respective receptor. Murine FcRn binds rat IgG2a with high affinity40. WT mice were intravenously immunized with either DEC205 or Clec9A-targeted OVA adjuvanted with LPS. Five days later, mice were inoculated with B cell lymphoma and the number of B cell lymphoma cells in the spleen evaluated 4 days later (Fig. 2A). Vaccination with either DEC205 or Clec9A-targeted OVA elicited a significant reduction in tumor cell number, indicating successful anti-tumor therapy for this mode of immunization (Fig. 2B). A role for FcRn was evaluated by undertaking the same analysis in Fcgrt−/− mice. In the absence of FcRn, the ability to eradicate tumors was impaired and there was no significant reduction in tumor cell numbers when mice were immunized with DEC205-targeted OVA, while only a mild reduction in tumor load occurred in response to Clec9A-targeted OVA (Fig. 2B). A second measurement of successful tumor immunization was included, whereby evidence of tumor immunoediting in response to vaccination was assessed. In the B cell lymphoma model used here, green fluorescent protein (GFP) is linked by an internal ribosome entry site to the tumor antigen OVA39. Therefore, the tumor GFP signal provides a surrogate read out of OVA expression and is a useful marker of tumor antigen expression. In WT mice, DEC205 and Clec9A-targeted OVA immunization results in a significant reduction in tumor GFP. This is likely due to the tumors reducing OVA expression to avoid anti-OVA CD8+ T cells elicited by the vaccination regime. In contrast, tumors in Fcgrt−/− mice had higher GFP signals, while a significant reduction in GFP signal was still evident compared to tumours in unvaccinated mice. This suggests that in the absence of FcRn there was less pressure on the tumor to edit OVA following immunization (Fig. 2C). Taken together, this data suggests that FcRn plays a pivotal role in promoting the anti-tumor response elicited by DEC205 and/or Clec9A-directed DC-targeted vaccination.
Fig. 2. FcRn impacts tumor immunotherapy.
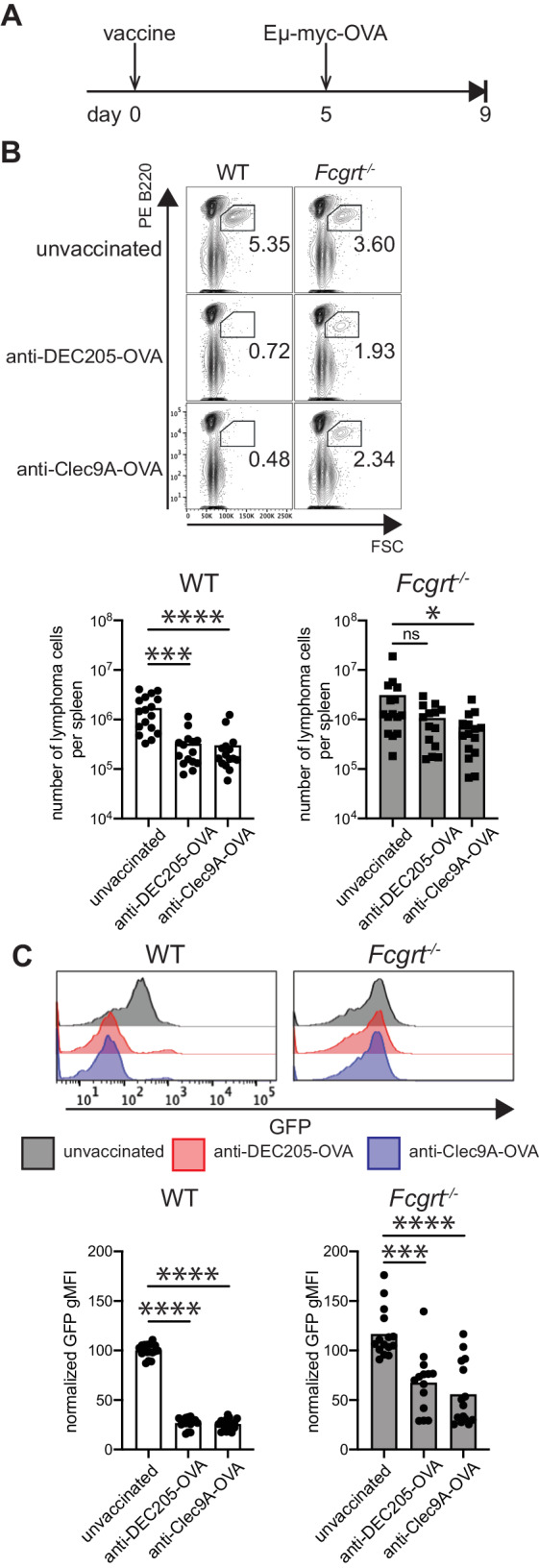
A WT and Fcgrt−/− mice were immunized with anti-DEC205-OVA or anti-Clec9A-OVA mAb together with LPS. After five days, mice were inoculated with Eμ-myc–GFP-OVA tumor cells and four days later, spleens were collected and analyzed by flow cytometry. B The contour plots show FSC and B220 expression in whole splenocytes for each group, with the gate indicating the presence of the tumour. The number of tumor cells per spleen is indicated in the histogram. C Representative GFP profiles of lymphoma cells for each group. GFP expression level was quantified and shown in the accompanying histogram. Data are pooled from 5 independent experiments with each symbol representing an individual mouse. *P < 0.05. ***P < 0.001, ****P < 0.0001 by Krustal-Wallis with Dunn’s test for multiple comparisons. ns not significant.
FcRn regulated MHC I and MHC II antigen presentation in response to DEC205-targeted DC vaccination
We next examined how FcRn participates in DEC205-targeted DC vaccination. Surface DEC205 was unaltered on splenic cDC1 and cDC2 in the absence of FcRn (Fig. 3A). This finding indicates that similar amounts of DEC205-targeted OVA antigen is delivered to cDCs in both WT and Fcgrt−/− mice. Next, the capacity of Fcgrt−/− mice to present DEC205-targeted antigen by MHC I and MHC II molecules was examined. To do this, Cell Trace Violet (CTV)-labelled OT-I or OT-II T cells were transferred into WT and Fcgrt−/− mice. One day later, mice were immunized with DEC205-targeted OVA intravenously and the number of dividing T cells in the spleen was evaluated by flow cytometry after three days. In the absence of FcRn, DEC205-targeted OVA elicited significantly reduced numbers of divided OT-I, and significantly more divided OT-II cells (Fig. 3B, for gating see Supplementary Fig. 3A). Presentation of OVA-coated splenocytes, used as mAb-independent cell-associated antigen, to OT-I and OT-II was not impacted by FcRn deficiency (Supplementary Fig. 4). To determine if the reduction in MHC I and MHC II antigen presentation was intrinsic to FcRn-deficient cDCs, WT and Fcgrt−/− mice were intravenously immunized with DEC205-targeted OVA. Twenty hours later cDC1 were sorted to purity by flow cytometry and equal numbers of WT or Fcgrt−/− cDC1s incubated in the presence of CTV labelled OT-I or OT-II T cells. Fcgrt−/− cDC1 displayed a significantly reduced capacity to stimulate OT-I and OT-II cells in response to in vivo targeting with DEC205-targeted OVA (Fig. 3C, for gating see Supplementary Fig. 3B). Similarly, cDC2, that express low surface level of DEC205 (Fig. 3A), were significantly impaired in their ability to activate OT-I and OT-II following OVA targeting to DEC205 (Supplementary Fig. 5). This suggests reduced MHC I presentation, and in contrast to in vivo adoptive transfer experiments, reduced MHC II presentation in response to in vivo targeting with DEC205-targeted OVA.
Fig. 3. FcRn alters MHC I and MHC II presentation following DEC205-targeted DC vaccination.

A Flow cytometry analysis of surface DEC205 expression in WT and Fcgrt−/− spleen cDCs. Dashed histograms: fluorescence minus one (FMO) background. Data are representative of one out of three analyzed mice. B CTV-labelled OT-I and OT-II T cells and CFSE-labelled C57BL/6 mouse splenocytes were adoptively transferred into WT and Fcgrt−/− mice. One day later, mice were injected with anti-DEC205-OVA mAb. Spleens were harvested 64 h later and analyzed by flow cytometry. The CTV histograms show OT-I and OT-II proliferation in each group of mice. The number of dividing OT-I and OT-II T cells per spleen was calculated and shown as mean ± SD. Data are pooled from four independent experiments, with 3–4 mice per group. ****P < 0.0001, ***P < 0.001 by unpaired Student’s t-test. C WT and Fcgrt−/− mice received anti-DEC205-OVA mAb and 20 h later, spleens were harvested and cDC1 purified. Increasing numbers of cDC1 were co-incubated with CTV-labelled OT-I or OT-II at 37 °C for 60 h or 84 h, respectively. The number of dividing OT-I and OT-II cells was quantified by flow cytometry. Data are pooled from two independent experiments, each one done in triplicate. *P < 0.05, by Mann-Whitney test with Holm-Sidak correction for multiple comparisons. D WT and Fcgrt−/− mice were injected with anti-DEC205-Eα46-72 or PBS. Twenty hours later, spleens were harvested. I-Ab-Eα52-68 peptide complexes (pMHC II) at the surface of cDC1 were measured by flow cytometry. Histograms are representative of one mouse per group. Data are pooled from two independent experiments with each symbol representing an individual mouse. ***P < 0.001 by unpaired Student’s t-test. ns not significant.
To examine if FcRn impacted presentation of DC-targeted antigen other than OVA, the epitope Eα46-72 was targeted to DEC205 and the presence of Ea52-68 peptide-loaded I-Ab MHC II molecules at the surface of cDC1 examined using the Yae mAb41. WT or Fcgrt−/− mice were injected intravenously with DEC205-targeted Eα46-72 and cDC isolated from spleen 20 h later. Significantly reduced MHC II presentation of DEC205-targeted Eα52-68 was detected at the surface of Fcgrt−/− compared to WT cDC1 (Fig. 3D). To further confirm this finding, the Fcgrt gene was deleted in the cDC1 cell line MutuDCs42 using CRISPR/Cas9 (Supplementary Fig. 6A). Following delivery of the Eα46-72 peptide to DEC205 in vitro, Fcgrt−/− MutuDCs were significantly impaired in the MHC II presentation of Eα52-68 (Supplementary Fig. 6B). Altogether, this data show that FcRn is an important contributor to the response elicited by anti-DEC205-targeted vaccination by promoting MHC I cross-presentation and MHC II presentation of the delivered antigen.
The role of FcRn in DEC205-targeted DC vaccination is DC-intrinsic
One possibility for defects in antigen presentation following DC-targeted vaccination in vivo, is that FcRn is required to promote circulating levels of the DC-targeted IgG vaccine. To address this, we used mixed bone marrow chimeras where WT mice were lethally irradiated and reconstituted with a 1:1 ratio of CD45.1 WT or CD45.2 Fcgrt−/− bone marrow. This created a scenario where CD45.1 WT or Fcgrt−/− CD45.2 DCs are both in a WT environment with similar levels of circulating DC-targeted IgG vaccine. Six weeks after reconstitution, mice were immunized with anti-DEC205-OVA and 20 h later CD45.1+ WT or Fcgrt−/− cDC1 were isolated from spleens and incubated with CTV labelled OT-I or OT-II. In comparison to WT, Fcgrt−/− cDC1 displayed significantly reduced MHC I and MHC II presentation of DEC205-targeted OVA (Fig. 4). This rules out that differences in vaccine half-life were responsible for the defects in Fcgrt−/− cDC presentation of DEC205-targeted antigen and highlights an intrinsic role of FcRn in DCs during anti-DEC205-targeted vaccination.
Fig. 4. FcRn intrinsically regulate the presentation on antigen targeted to DEC205.

A WT mice were lethally irradiated and reconstituted with a mix of CD45.1+ WT BM and Fcgrt−/− (CD45.2+) BM. After six weeks, mixed BM chimera mice were injected with anti-DEC205-OVA mAb and 20 h later, spleens were harvested and cDC1 purified. Increasing numbers of cDC1 were co-incubated with CTV-labelled OT-I or OT-II at 37 °C for 60 h or 84 h, respectively. B The number of dividing OT-I and OT-II cells was quantified by flow cytometry. Data are pooled from three independent experiments, each one done in triplicate. *P < 0.05 by by Mann-Whitney test with Holm-Sidak correction for multiple comparisons.
FcRn does not regulate MHC I and MHC II antigen presentation in response to Clec9A-targeted DC vaccination
Next, we examined how FcRn impacts anti-Clec9A-targeted OVA immunization. Like DEC205, surface Clec9A on cDC1 was not altered by the deficiency in FcRn (Fig. 5A). In contrast to anti-DEC205-OVA mAb, MHC I and MHC II antigen presentation following immunization with anti-Clec9A-OVA mAb was not significantly impacted by the absence of FcRn. This was the case as measured by T cell responses following adoptive OT-I and OT-II T cell transfer into WT and Fcgrt−/− mice (Fig. 5B), extrinsic isolation and culture of WT or Fcgrt−/− cDC1 from anti-Clec9A-OVA immunized mice (Fig. 5C) and analysis of Fcgrt−/− cDC1 compared to WT cDC1 isolated from WT mice reconstituted with 1:1 ratio of WT and Fcgrt−/− bone marrow (Fig. 5D). In contrast, MHC II presentation of Clec9A-targeted Eα46--72 peptide by cDC1 was significantly reduced in the absence of FcRn (Fig. 5E). Therefore, in contrast to anti-DEC205-targeted vaccination, FcRn does not have a prominent role in regulating the presentation of antigen targeted via Clec9A but can participate under some conditions.
Fig. 5. FcRn does not impact MHC I and MHC II presentation following Clec9A-targeted DC vaccination.

A Flow cytometry analysis of Clec9A expression in WT and Fcgrt−/− spleen cDCs. Dashed histograms: FMO background. Data are representative of one out of three mice analyzed. B CTV-labelled OT-I and OT-II T cells and CFSE-labelled C57BL/6 mouse splenocytes were adoptively transferred into WT and Fcgrt−/− mice. One day later, mice were injected with anti-Clec9A-OVA mAb. Spleens were harvested 64 h later and analyzed by flow cytometry. CTV histograms are representative of OT-I and OT-II proliferation in each group. and the number of dividing OT-I and OT-II T cells per spleen is shown as mean ± SD. Data are pooled from two independent experiments with 3–4 mice per group. ns, not significant by unpaired Student’s t-test. C WT and Fcgrt−/− mice received anti-Clec9A-OVA mAb and 20 h later, spleens were harvested and cDC1 purified. Increasing numbers of cDC1 were co-incubated with CTV-labelled OT-I or OT-II at 37 °C for 60 h or 84 h, respectively. The number of dividing OT-I and OT-II cells was quantified by flow cytometry. Data are pooled from two independent experiments, each one done in triplicate and analysed by Mann-Whitney test with Holm-Sidak correction. D C57BL/6 mice were lethally irradiated and reconstituted with a mix of CD45.1+ BM and Fcgrt−/− (CD45.2+) BM. Mixed BM chimera mice were then treated as in (C) and the number of dividing OT-I and OT-II cells was measured. Data are pooled from three independent experiments, each one done in triplicate, and analysed by Mann-Whitney test with Holm-Sidak correction. E WT and Fcgrt−/− mice were injected with anti-Clec9A-Eα46-72 or PBS. Twenty hours later, spleens were harvested. I-Ab-Eα52-68 peptide complexes (pMHC II) at the surface of cDC1 were measured by flow cytometry. Histograms are representative of one mouse per group. Data are pooled from two independent experiments with each symbol representing an individual mouse. ***P < 0.001 by Student’s t-test. ns not significant.
FcRn is required for CTL immunity to DEC205 but not Clec9A-targeted DC vaccination
Elimination of Eμ-myc-OVA B cell lymphoma in response to DC-targeted vaccination likely involves the generation of anti-OVA cytotoxic T lymphocytes (CTL). We therefore examined whether FcRn is required for eliciting CTL in response to anti-DEC205 or Clec9A-OVA mAb. WT or Fcgrt−/− mice were intravenously immunized with DC-targeted anti-DEC205 or Clec9A-OVA mAb and six days later the presence of OVA-specific CTL detected by the intravenous injection of OVA257-264 peptide-pulsed, or not, target cells. In the absence of FcRn, immunization with anti-DEC205-OVA mAb elicited a significant reduction in CTL lysis compared to WT mice (Fig. 6A, for gating see Supplementary Fig. 7A). In contrast, anti-Clec9A-OVA mAb elicited similar CTL in both WT and Fcgrt−/− mice (Fig. 6B). CTL immunity elicited by OVA-coated splenocytes was not impacted by FcRn-deficiency (Supplementary Fig. 7B). Hence, these results demonstrate that priming a robust CTL response following DC-targeted vaccination requires FcRn when delivering antigen to DEC205, but not Clec9A.
Fig. 6. FcRn is required for CTL in response to DEC205, but not Clec9A-targeted DC vaccination.
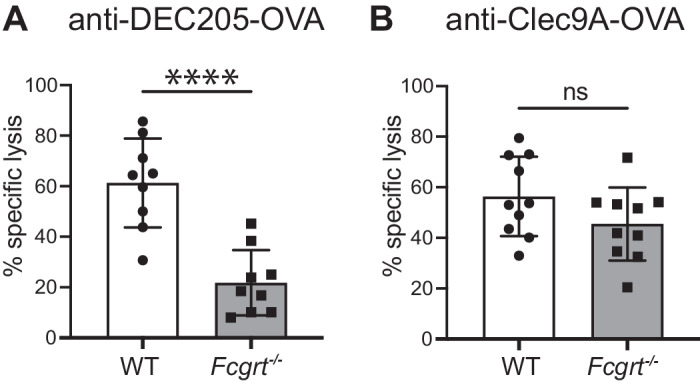
WT and Fcgrt−/− mice were vaccinated with anti-DEC205-OVA mAb (A) or anti-Clec9A-OVA mAb (B), supplemented with LPS as adjuvant. Six days later, mice received an equal number of CTVhi (pulsed with OVA257-261 peptide) and CTVlo (unpulsed) target cells. 36–42 h later, spleens were harvested, and the percentage lysis of CTVhi cells was measured by flow cytometry. Data are pooled from two independent experiments with each symbol representing an individual mouse. **** P < 0.0001 by Student’s t-test. ns not significant.
Discussion
DC-targeted vaccination using antigen-conjugated DC-receptor specific IgG mAb allows efficient and precise delivery of vaccine antigen to DC subsets specialized in antigen presentation and T cell priming. The success of this immune therapy relies on the intracellular trafficking pathways inside DCs, that will dictate whether, or not, the vaccine antigen and accompanying targeting IgG can access a compartment that is favourable for antigen presentation. Here we show that mAb-conjugated antigen targeted to DEC205 requires FcRn, a receptor of emerging therapeutic interest43, for efficient MHC I cross-presentation and MHC II presentation. In contrast, Clec9A-targeted antigen is presented via a mechanism that is largely independent of FcRn.
Our study highlights a role for FcRn in cDC1. Previously, FcRn was identified as critical for MHC I cross-presentation of immune complex-derived antigen in cDC2, but not cDC130,31. In these systems, the alkaline pH of the cDC1 cross-presentation compartment was not permissive for FcRn binding. In contrast, when antigen is delivered via anti-DEC205 mAb, it accesses late endosomes44,45 with a luminal pH that is sufficiently acidic for FcRn binding to the Fc domain of the targeting mAb. In addition to cDC1, other antigen presenting cell populations that express DEC205 may also use FcRn to effectively present DEC205-targeted antigen. For example, B cells which express FcRn, have high surface expression of DEC20546 and respond to DEC205 mAb-targeting in vivo by presenting the associated antigen in the context of MHC II47,48. Langerhans cells and a subset of dermal DCs also exhibit high surface levels of DEC205. After subcutaneous immunization, these skin DCs capture and transport anti-DEC205 mAb to the draining lymph nodes for presentation of the conjugated antigen to T cells49,50. It remains to be determined if FcRn regulates the presentation of DEC205-targeted antigen by these cell types.
FcRn impacts immunization with mAb-conjugated antigen to DEC205, but not Clec9A. Why is this the case? Both DEC205 and Clec9A are expressed at high levels by cDC1, an antigen presenting cell type that we observe expresses high levels of FcRn. Therefore, the different roles for FcRn likely stem from differences in the intracellular trafficking properties of the two receptors. DEC205 and Clec9A exhibit similar plasma membrane internalization patterns in cDC1, with rapid and high surface turnover51. Furthermore, we have previously shown that antigen presentation outcomes downstream of antigen targeting to DCs does not correlate with the amount of antigen being endocytosed51. Therefore, the difference in requirements for FcRn between antigen targeting to DEC205 versus Clec9A cannot be explained by a different antigen load reaching the endosomal compartment. Inside the cell, however, the receptor pathways diverge. Anti-DEC205 mAbs traffic to late endosomes in immature and mature bone marrow-derived DCs44,45 and the highly acidic lumen of these compartments may favour FcRn binding to the Fc-domain of anti-DEC205 IgG. In contrast, Clec9A mAbs transit through early and recycling endosomes9,52, compartments without an acidic lumen that may not permit FcRn-IgG binding. Another difference between the two receptors is that mAb binding to Clec9A induces spleen tyrosine kinase (Syk) phosphorylation and endosomal rupture52. This has not been reported for DEC205. Therefore, endosomes with antigen-conjugated anti-Clec9A mAb may rupture instead of routing to FcRn-containing endosomes. It is also possible that another binding partner of anti-Clec9A mAb controls its intracellular trafficking in cDC1. Regardless, the impaired presentation of DEC205-targeted antigen when FcRn is absent is likely due to altered trafficking and processing. In cDC2, FcRn directs captured immune complexes to endosomes that are enriched with components of the cross-presentation machinery. These include the translocon Sec61, which mediates the transfer of antigen from the endosomal lumen to the cytosol53, and transporter associated with antigen processing 1 (TAP1), which reimports proteasomal generated peptides into the endosome30. Interestingly, DEC205-targeted antigen is also cross-presented via a TAP-dependent mechanism10,54, suggesting that FcRn may divert anti-DEC205 mAb to similar cross-presenting compartments in cDC1.
Currently, two anti-DEC205 targeting mAbs are in clinical trials. CDX-1401 is a human IgG1 conjugated to the tumor antigen NY-ESO1 that has been evaluated in six phase I and phase II trials2,21,22. Another DEC205-specific mAb, DCVax-001 has been conjugated to the human immunodeficiency virus (HIV) antigen Gag p24 and has been tested in a phase I clinical trial55. Although these vaccines have shown promising results with robust B cell and T cell immunity, their efficiency may be further enhanced by harnessing FcRn to extend their half-life and activity. Several therapeutic mAb have been modified in their Fc domain to introduce mutations near the key FcRn-binding residues in order to increase the IgG-FcRn binding affinity at low pH. For example, inserting the LS (M428L, N434S) mutation into anti-vascular endothelial growth factor (VEGF) IgG1 Bevacizumab greatly extended mAb half-life by approximately four fold, resulting in increased anti-tumor activity56. YTE (M252Y/S254T/T256E) is another mutation introduced into mAb specific for viral proteins. This modification enhanced the half-life of the anti-respiratory syncytial virus IgG1 Motalizumab up to 100 days post-injection57. This mAb engineering approach represents a straightforward strategy applicable to DEC205-targeted DC vaccination to improve FcRn affinity and immunogenicity.
In conclusion, our study demonstrates the crucial role of FcRn in the efficient presentation of antigen targeted to DEC205 by DCs. We have shown that FcRn mediates the presentation of DEC205-targeted antigen, while Clec9A-targeted antigen presentation is largely independent of FcRn. The differential trafficking and processing of these receptors likely contributes to the distinct roles of FcRn in antigen presentation of DC-targeted mAb. The clinical trials of anti-DEC205 mAb underscore the potential of utilizing FcRn to enhance the efficacy of DC-targeted vaccines. By engineering the Fc domain of these mAb to increase their affinity for FcRn, the half-life and activity of the vaccines can be extended, leading to improved immunogenicity. These findings provide valuable insights into the development of DC-targeted vaccination strategies and highlight the importance of FcRn in modulating immune responses for effective antigen presentation by DC subsets.
Methods
Mice
C57BL/6, Fcgrttm1Dcr (Fcgrt−/−), B6.CH-2bm-1 (BM-1), Ly5.1, OT-I x Ly5.1 and OT-II x Ly5.1 mice were used at 6–12 weeks of age. All mice were bred and maintained in specific pathogen–free conditions at the Melbourne Bioresources Platform at Bio21 Molecular Science and Biotechnology Institute. Experimental procedures were approved by the Animal Ethics Committee of the University of Melbourne (protocols 20150 and 21410). Mice were euthanized by carbon dioxide asphyxiation followed by cervical dislocation.
Isolation of immune cells
For DC purification, spleens were finely chopped and digested in the presence of DNase I (Roche) and collagenase type 3 (Worthington Biochemicals). Intercellular clusters were disrupted by addition of 10 mM Ethylenediaminetetraacetic acid (EDTA). Light density cells were isolated by density gradient separation in 1.077 g/cm3 Nycodenz (Nycomed Pharma). Upper fractions were collected, washed and subject to further enrichment by resuspending in a depletion cocktail containing the following rat anti-mouse mAb specific for: CD3 (KT3-1.1; dilution 1:100), CD90 (T24/31.7; dilution 1:100), red blood cells (Ter119; dilution 1:10), B220 (RA3-6B2; dilution 1:100), Ly6G/Ly6C (RB68C5; dilution 1:50) (all Walter and Eliza Hall Antibody Facility). Cells were incubated with antibodies, washed, and incubated with BioMag anti-rat IgG-coupled magnetic beads (Qiagen). The DC-enriched supernatant was recovered by magnetic separation. Where required, DCs were stained with CD11c-FITC (N418; BioLegend Cat. No. 117306; dilution 1:200), CD8α-APC (YTS169.4; Walter and Eliza Hall Antibody Facility; dilution 1:200), CD11b-PE (M1/70; Walter and Eliza Hall Antibody Facility; dilution 1:800) and sorted to purity by flow cytometry on a Becton Dickinson Influx (Murdoch Children’s Research Institute Flow Cytometry and Imaging facility). cDC1 were defined as CD11c+ CD8α+ CD11b−, cDC2: CD11c+ CD8α− CD11b+. To assess DEC205 and Clec9A expression, cDCs were stained with DEC205-biotin (NLDC-145; Walter and Eliza Hall Antibody Facility; dilution 1:200) followed by streptavidin-APC (BioLegend Cat. No. 405207; dilution 1:500), and Clec9A-APC (7H11; BioLegend Cat. No. 143506; dilution 1:200) and analyzed by flow cytometry. For in vitro activation, cDCs were isolated and incubated overnight in complete RPMI with 0.5 μM type B CpG (Bioneer) at 37 °C or 4 °C. Following activation, cDC subsets were sorted by flow cytometry and analyzed by immunoblotting. Resting and activated cDCs were also stained with CD86-APC (GL1; BioLegend Cat. No. 105012; dilution 1:400), MHC II-Alexa Fluor 700 (M5/114; Walter and Eliza Hall Antibody Facility; dilution 1:400), CD274-APC (10 F.9G2; BioLegend Cat. No. 124312; dilution 1:50) and CD40-FITC (FGK45.5; Walter and Eliza Hall Antibody Facility; dilution 1:100) and analyzed by flow cytometry.
For T cell isolation, single cell suspensions were generated from lymph nodes. Cells were stained with rat anti-mouse mAb specific for: CD11b (M1/70; dilution 1:20), red blood cells (TER119; dilution 1:10), Ly6G (1A8; dilution 1:10), MHC II (M5/114; dilution 1:20), B220 (RA3-6B2; dilution 1:100) and CD4 (GK1.5; dilution 1:10) for CD8+ T cells or CD8α (YTS 169.4; dilution 1:25) for CD4+ cells (all Walter and Eliza Hall Antibody Facility). Cells were washed and incubated with BioMag anti-rat IgG-coupled magnetic beads (Qiagen). After magnetic depletion, the CD4+ or CD8+ T cell-enriched supernatant was recovered. Purity was determined by flow cytometry using TCRVα2-APC (B20.1; Walter and Eliza Hall Antibody Facility; dilution 1:1,600) and CD4-FITC (GK1.5; BioLegend Cat. No. 100406; dilution 1:500) or CD8α-FITC (YTS169.4; Walter and Eliza Hall Antibody Facility; dilution 1:1,600). The proportion of CD4+ TCRVα2+ OT-II cells or CD8α+ TCRVα2+ OT-I cells was > 90%. For B cell isolation, whole-splenocyte suspensions were subjected to gradient centrifugation (at 2237 g) with Ficoll-Paque Plus (GE Healthcare) and subsequent negative depletion using FITC-conjugated mAb against CD4 (GK1.5; dilution 1:300), Ly76 (TER119; dilution 1:300), CD43 (S7; dilution 1:200) (all made by Walter and Eliza Hall Antibody Facility) and magnetic anti-FITC MicroBeads (Miltenyi Biotec). The purity of the B cell preparation was measured by flow cytometry using CD19-FITC (1D3; Walter and Eliza Hall Antibody Facility; dilution 1:800) and B220-PE (RA3-6B2; Beckton Dickinson Cat. No. 553089; dilution 1:200). The proportion of CD19+ B220+ B cells was > 95%.
Bone-marrow-derived macrophages
Bone marrow was isolated from the femur and tibia of 6–12-week-old C57BL/6 mice and non-adherent bone marrow cells cultured for 5–7 days in the presence of conditioned media harvested from L-929 cells as a source of macrophage colony-stimulating factor (M-CSF). Purity was assessed by flow cytometry using CD11b-FITC (M1/70; Walter and Eliza Hall Antibody Facility; dilution 1:800) and F4/80-APC (Walter and Eliza Hall Antibody Facility; dilution 1:800). ~95% of live cells express both CD11b and F4/80.
Immunoblotting
Immune cells were isolated as previously described and were denatured in 1x reducing lithium dodecyl sulfate (LDS) sample buffer (25% (v/v) 4x NuPAGE LDS Sample Buffer (Invitrogen) and 10% (v/v) 10x NuPAGE Reducing Agent (Invitrogen) in distilled water) at 100 °C. Samples were analyzed by sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using 4–12% gradient NuPAGE Bis-Tris polyacrylamide gels (Invitrogen). Proteins were transferred onto Immobilon-P polyvinylidene fluoride (PVDF) membranes (Merck Millipore) and stained with primary antibodies against FcRn (R&D Systems; dilution 1:1,000), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (6C5; Millipore; dilution 1:2000) followed by horseradish peroxidase (HRP)-conjugated secondary antibodies. Membranes were then incubated with Amersham ECL Plus chemiluminescence reagent (GE Healthcare) for 1 min and protein bands were visualized using the ChemiDoc Imaging System (Bio-Rad). Uncropped and unprocessed blots are shown in Supplementary Fig. 8.
Serum antibody analysis
For quantification of total serum antibodies, sandwich enzyme-linked immunosorbent assay (ELISA) was performed by coating high protein-binding 96-well plates (Corning) with 20 µg/ml of goat mAb specific for mouse IgA (Cat. No. 1040-01), IgG1 (Cat. No. 1070-01), IgG2b (Cat. No. 1090-01), IgG2a/c (Cat. No. 1080-01), IgG3 (Cat. No. 1100-01), and IgM (Cat. No. 1020-01) and IgE (Cat. No. 1130-01) (all SouthernBiotech). Coated plates were subsequently incubated with diluted serum or standards, followed by detection with HRP-conjugated polyclonal mAb against mouse IgA (Cat. No. 1040-08; dilution 1:1000), IgG1 (Cat. No. 1070-05; dilution 1:500), IgG2b (Cat. No. 1090-05; dilution 1:500), IgG2a/c (Cat. No. 1080-05; dilution 1:500), IgG3 (Cat. No. 1100-05; dilution 1:500), IgM (Cat. No. 1020-05; dilution 1:500) and IgE (Cat. No. 1130-05; dilution 1:500) (all SouthernBiotech) and 1-Step Ultra3,3’,5,5’-Tetramethylbenzidine ELISA Substrate (Thermo Fisher Scientific). Absorbance at 440 nm was measured with a FLUOstar Omega PlateReader, and 4PL analysis was performed with GraphPad Prism software.
Generating antigen conjugated targeting antibodies
Constructs encoding recombinant DC targeting mAb genetically fused to OVA were generated as previously described in ref. 13. For Eα46-72 targeting, an antigenic sequence (LESIINFEKLTEEFAKFASFEAQGALANIAVDKANLDVMKEKLPGFGDSIE) containing the epitope Eα46-72 (underlined) was genetically fused to the H chain of anti-DEC205 (clone NLDC-145) and anti-Clec9A (clone 10B4) mAb via an alanine linker as described previously58. Targeting mAb were expressed using Freestyle 293-F cells and purified by affinity chromatography using protein G sepharose, as previously described13.
Prophylactic vaccination assay and analysis of killing of Eμ-myc-OVA lymphoma
The Eμ-myc lymphoma expressing GFP-OVA was described previously39. Mice were immunized i.v. with 1 μg of anti-DEC205-OVA or Clec9A-OVA mAb or 20 × 106 OVA-coated splenocytes, all adjuvanted with 1 μg of LPS. Five days later, mice were challenged with 106 Eμ-myc-OVA lymphoma cells and spleens were harvested after four days. Single-cell splenocyte suspensions were treated with red blood cell (RBC) removal buffer, stained with a B220-PE mAb (RA3-6B2; Beckton Dickinson; Cat. No. 553089; dilution 1:200) and analyzed by flow cytometry.
In vivo antigen presentation assay
Single-cell suspensions were generated from lymph nodes of OT-I x Ly5.1or OT-II x Ly5.1 mice and the CD8+ or CD4+ T cells were purified as before. Purified OT-I or OT-II cells were washed with PBS supplemented with 0.1% bovine serum albumin (BSA) and labelled with CTV (Thermo Fisher Scientific). To enable accurate quantification of the number of OT-II or OT-I cells, mice also received transfer of a control population of splenocytes labelled with carboxyfluorescein succinimidyl ester (CFSE, Thermo Fisher Scientific). Single-cell suspensions were treated with RBC removal buffer. Cells were washed with PBS, 0.1% BSA, and labelled with CFSE. Equal numbers of CTV-labelled OT-II or OT-I cells and CFSE-labelled splenocytes were pooled and resuspended in RPMI 1640, 2% (v/v) fetal calf serum (FCS; University of Melbourne Media Preparation Unit) at 1 × 107 cells/ml, respectively, and injected i.v. Twenty-four hours later, mice received 20 × 106 OVA-coated splenocytes, or 0.2 μg anti-DEC205-OVA or anti-Clec9A-OVA mAb via i.v. injection13. For OVA-coated splenocytes, single-cell splenocyte suspensions were treated with RBC removal buffer, washed, and incubated with 10 mg/ml OVA protein (Sigma-Aldrich) in FCS-free RPMI 1640 at 37 °C. Sixty-four hours after immunization, spleens were harvested, single-cell suspensions were generated, and RBCs were lysed. Cells were stained with the following mAb: CD8α-APC (53-6.7; BioLegend Cat. No. 100712; dilution 1:800) or CD4-APC (GK1.5; Walter and Eliza Hall Antibody Facility; dilution 1:800), TCRVα2-PE (B20.1; Walter and Eliza Hall Antibody Facility; dilution 1:800) and Ly5.1-PECy7 (A20.1; BioLegend Cat. No. 110730; dilution 1:400). The number of divided OT-II and OT-I was determined as the number of CD4+ or CD8α+ Ly-5.1+ cells that had undergone CTV dilution. The number of cells was normalized to the number of CFSE-labelled splenocytes recovered.
Ex vivo antigen presentation assay
Mice were immunized i.v. with 1 μg of anti-DEC205-OVA or Clec9A-OVA mAb or anti-DEC205-Eα46-72 peptide mAb. After 22–24 h, splenic cDCs were either examined for Eα52-68–loaded I-Ab surface expression by flow cytometry using biotinylated Eα52-68 mAb (Yae; Thermo Fisher Scientific Cat. No. 13-5741-82; dilution 1:50) and streptavidin-PE (BioLegend Cat. No. 405203; dilution 1:400) or sorted to purity. Sorted cDC1 or cDC2 were cultured in vitro with 5 × 104 CTV-labelled OT-II or OT-I cells in complete medium. Four or five days later, the number of divided OT cells was determined.
In vitro targeting of MutuDCs and analysis of antigen presentation
MuTu DCs stably expressing Cas959 were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM)-GlutaMax™ (Thermo Fisher Scientific) supplemented with 10% (v/v) (FCS), 100 μM β-mercaptoethanol (Thermo Fisher), 100 μg/mL penicillin and 100 μg/mL streptomycin (University of Melbourne Media Preparation Unit) at 37 °C, 10% CO2. To knockout FcRn, the LV04 vector containing the sgRNA was used (Sigma Aldrich). sgRNA sequences were as follows: non-targeting control: 5’ CGCGATAGCGCGAATATATT 3’; FcRn: 5’ CCGTCGGCCCCTCTCCAGG 3’. To produce lentivirus, HEK293T were transfected using polyethylenimine (Polysciences) with sgRNA vector, pMDL (Addgene #12251), pRSV-REV (Addgene #12253), pMD2.G (Addgene #12259). Lentivirus were subsequently used to transduced Cas9+ MuTu DCs by spinfection in the presence of polybrene (Sigma-Aldrich). Transduced cells were then selected by culturing them for three days with puromycin (Thermo Fisher Scientific).
Mixed bone marrow chimera
Recipient mice (C57BL/6) were irradiated twice at 550 cGy (rad), three hours apart before being injected i.v. with 50:50 mixed bone marrow cells from Ly5.1 WT mice and Fcgrt−/− mice. Recipient mice were intraperitoneally injected with anti-Thy1 mAb (T24, Walter and Eliza Hall Antibody Facility) to eliminate radio-resistant host T cells the day after irradiation. Mice were reconstituted for six weeks before analysis.
In vivo CTL killing assay
Mice were injected with 1 μg of anti-DEC205-OVA mAb or anti-Clec9A-OVA mAb, or with 2 × 107 OVA-coated splenocytes, together with 1 µg of LPS as adjuvant. Six days later, single-cell suspensions of target cells (splenocytes) were prepared from spleens of WT mice. Half of the suspension (OVA+ splenocytes pulsed with OVA257-264 labelled with a high concentration of CTV [CTVhi]) was pulsed with 1 μg of OVA257-264 (Worthington Biochemical) and labelled with 5 mM CTV (Thermo Fisher Scientific). The other half of the suspension (OVA splenocytes labelled with a low concentration of CTV [CTVlo]) was labelled with 0.5 mM CTV only. An equal number of OVA+ CTVhi and OVA- CTVlo target cells were pooled, and 10 × 106 target cells were injected i.v; 36–42 h later, spleens were harvested and the percentage of OVA-specific lysis was determined as follows: R = (%CTVlo / %CTVhi). %OVA-specific lysis = [1 - (runprimed / rprimed)]100.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Author contributions
C.M. contributed to the conception, design and execution of the study, interpretation of the results, writing and revising the manuscript. M.P., D.J., X.P.L. contributed to the execution of the study. Z.E. contributed to the revision of the manuscript. I.C. and M.H.L. generated antigen-conjugated mAbs and contributed to the design of the study. P.A.G. and J.A.V. contributed to the design of the study. J.D.M. contributed to the conception and design of the study, interpretation of the work, writing and revising the manuscript.
Data availability
Data from this study will be made available upon request to the corresponding author (Justine D. Mintern, jmintern@unimelb.edu.au).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41541-024-00854-8.
References
- 1.Macri C, Dumont C, Johnston AP, Mintern JD. Targeting dendritic cells: a promising strategy to improve vaccine effectiveness. Clin. Transl. Immunol. 2016;5:e66. doi: 10.1038/cti.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Macri C, Jenika D, Ouslinis C, Mintern JD. Targeting dendritic cells to advance cross-presentation and vaccination outcomes. Semin. Immunol. 2023;68:101762. doi: 10.1016/j.smim.2023.101762. [DOI] [PubMed] [Google Scholar]
- 3.Schetters STT, et al. Mouse DC-SIGN/CD209a as Target for Antigen Delivery and Adaptive Immunity. Front. Immunol. 2018;9:990. doi: 10.3389/fimmu.2018.00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pooley JL, Heath WR, Shortman K. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8- dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J. Immunol. 2001;166:5327–5330. doi: 10.4049/jimmunol.166.9.5327. [DOI] [PubMed] [Google Scholar]
- 5.den Haan JM, Lehar SM, Bevan MJ. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 2000;192:1685–1696. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lahoud MH, et al. DEC-205 is a cell surface receptor for CpG oligonucleotides. Proc. Natl. Acad. Sci. USA. 2012;109:16270–16275. doi: 10.1073/pnas.1208796109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang JG, et al. The dendritic cell receptor Clec9A binds damaged cells via exposed actin filaments. Immunity. 2012;36:646–657. doi: 10.1016/j.immuni.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 8.Sancho D, et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature. 2009;458:899–903. doi: 10.1038/nature07750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zelenay S, et al. The dendritic cell receptor DNGR-1 controls endocytic handling of necrotic cell antigens to favor cross-priming of CTLs in virus-infected mice. J. Clin. Invest. 2012;122:1615–1627. doi: 10.1172/JCI60644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonifaz L, et al. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 2002;196:1627–1638. doi: 10.1084/jem.20021598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonifaz LC, et al. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 2004;199:815–824. doi: 10.1084/jem.20032220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caminschi I, et al. The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood. 2008;112:3264–3273. doi: 10.1182/blood-2008-05-155176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lahoud MH, et al. Targeting antigen to mouse dendritic cells via Clec9A induces potent CD4 T cell responses biased toward a follicular helper phenotype. J. Immunol. 2011;187:842–850. doi: 10.4049/jimmunol.1101176. [DOI] [PubMed] [Google Scholar]
- 14.Dudziak D, et al. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315:107–111. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 15.Liu Z, Zhou H, Wang W, Fu YX, Zhu M. A novel dendritic cell targeting HPV16 E7 synthetic vaccine in combination with PD-L1 blockade elicits therapeutic antitumor immunity in mice. Oncoimmunology. 2016;5:e1147641. doi: 10.1080/2162402X.2016.1147641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silva MO, et al. Antigen Delivery to DEC205(+) Dendritic Cells Induces Immunological Memory and Protective Therapeutic Effects against HPV-Associated Tumors at Different Anatomical Sites. Int. J. Biol. Sci. 2021;17:2944–2956. doi: 10.7150/ijbs.57038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sancho D, et al. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J. Clin. Invest. 2008;118:2098–2110. doi: 10.1172/JCI34584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato Y, et al. Targeting Antigen to Clec9A Primes Follicular Th Cell Memory Responses Capable of Robust Recall. J. Immunol. 2015;195:1006–1014. doi: 10.4049/jimmunol.1500767. [DOI] [PubMed] [Google Scholar]
- 19.Sulczewski FB, et al. Conventional type 1 dendritic cells induce TH 1, TH 1-like follicular helper T cells and regulatory T cells after antigen boost via DEC205 receptor. Eur. J. Immunol. 2020;50:1895–1911. doi: 10.1002/eji.202048694. [DOI] [PubMed] [Google Scholar]
- 20.Sulczewski FB, et al. STAT6 signaling pathway controls germinal center responses promoted after antigen targeting to conventional type 2 dendritic cells. Curr. Res. Immunol. 2021;2:120–131. doi: 10.1016/j.crimmu.2021.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhodapkar MV, et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci. Transl. Med. 2014;6:232ra251. doi: 10.1126/scitranslmed.3008068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Griffiths EA, et al. NY-ESO-1 Vaccination in Combination with Decitabine Induces Antigen-Specific T-lymphocyte Responses in Patients with Myelodysplastic Syndrome. Clin. Cancer Res. 2018;24:1019–1029. doi: 10.1158/1078-0432.CCR-17-1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morse MA, et al. Phase I study utilizing a novel antigen-presenting cell-targeted vaccine with Toll-like receptor stimulation to induce immunity to self-antigens in cancer patients. Clin. Cancer Res. 2011;17:4844–4853. doi: 10.1158/1078-0432.CCR-11-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burmeister WP, Gastinel LN, Simister NE, Blum ML, Bjorkman PJ. Crystal structure at 2.2 A resolution of the MHC-related neonatal Fc receptor. Nature. 1994;372:336–343. doi: 10.1038/372336a0. [DOI] [PubMed] [Google Scholar]
- 25.Chaudhury C, et al. The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J. Exp. Med. 2003;197:315–322. doi: 10.1084/jem.20021829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raghavan M, Bonagura VR, Morrison SL, Bjorkman PJ. Analysis of the pH dependence of the neonatal Fc receptor/immunoglobulin G interaction using antibody and receptor variants. Biochemistry. 1995;34:14649–14657. doi: 10.1021/bi00045a005. [DOI] [PubMed] [Google Scholar]
- 27.Roopenian DC, et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J. Immunol. 2003;170:3528–3533. doi: 10.4049/jimmunol.170.7.3528. [DOI] [PubMed] [Google Scholar]
- 28.Firan M, et al. The MHC class I-related receptor, FcRn, plays an essential role in the maternofetal transfer of gamma-globulin in humans. Int. Immunol. 2001;13:993–1002. doi: 10.1093/intimm/13.8.993. [DOI] [PubMed] [Google Scholar]
- 29.Simister NE, Rees AR. Isolation and characterization of an Fc receptor from neonatal rat small intestine. Eur J Immunol. 1985;15:733–738. doi: 10.1002/eji.1830150718. [DOI] [PubMed] [Google Scholar]
- 30.Baker K, et al. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8-CD11b+ dendritic cells. Proc. Natl. Acad. Sci. USA. 2011;108:9927–9932. doi: 10.1073/pnas.1019037108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baker K, et al. Neonatal Fc receptor expression in dendritic cells mediates protective immunity against colorectal cancer. Immunity. 2013;39:1095–1107. doi: 10.1016/j.immuni.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qiao SW, et al. Dependence of antibody-mediated presentation of antigen on FcRn. Proc. Natl. Acad. Sci. USA. 2008;105:9337–9342. doi: 10.1073/pnas.0801717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luber CA, et al. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity. 2010;32:279–289. doi: 10.1016/j.immuni.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 34.Edwards AD, et al. Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur. J. Immunol. 2003;33:827–833. doi: 10.1002/eji.200323797. [DOI] [PubMed] [Google Scholar]
- 35.Macri C, Fancke B, Radford KJ, O’Keeffe M. Monitoring Dendritic Cell Activation and Maturation. Methods Mol. Biol. 2019;1988:403–418. doi: 10.1007/978-1-4939-9450-2_28. [DOI] [PubMed] [Google Scholar]
- 36.Wilson KR, et al. MHC Class II Ubiquitination Regulates Dendritic Cell Function and Immunity. J. Immunol. 2021;207:2255–2264. doi: 10.4049/jimmunol.2001426. [DOI] [PubMed] [Google Scholar]
- 37.Choi J, et al. Stemformatics: visualize and download curated stem cell data. Nucleic Acids Res. 2019;47:D841–D846. doi: 10.1093/nar/gky1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elahi Z, et al. The Human Dendritic Cell Atlas: An Integrated Transcriptional Tool to Study Human Dendritic Cell Biology. J. Immunol. 2022;209:2352–2361. doi: 10.4049/jimmunol.2200366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prato S, Mintern JD, Lahoud MH, Huang DC, Villadangos JA. Induction of antigen-specific effector-phase tolerance following vaccination against a previously ignored B-cell lymphoma. Immunol Cell Biol. 2011;89:595–603. doi: 10.1038/icb.2010.131. [DOI] [PubMed] [Google Scholar]
- 40.Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int. Immunol. 2001;13:1551–1559. doi: 10.1093/intimm/13.12.1551. [DOI] [PubMed] [Google Scholar]
- 41.Rudensky A, Rath S, Preston-Hurlburt P, Murphy DB, Janeway CA., Jr On the complexity of self. Nature. 1991;353:660–662. doi: 10.1038/353660a0. [DOI] [PubMed] [Google Scholar]
- 42.Fuertes Marraco SA, et al. Novel murine dendritic cell lines: a powerful auxiliary tool for dendritic cell research. Front. Immunol. 2012;3:331. doi: 10.3389/fimmu.2012.00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pyzik, M., Kozicky, L. K., Gandhi, A. K. & Blumberg, R. S. The therapeutic age of the neonatal Fc receptor. Nat. Rev. Immunol. 1–18 (2023). 10.1038/s41577-022-00821-1. [DOI] [PMC free article] [PubMed]
- 44.Platt CD, et al. Mature dendritic cells use endocytic receptors to capture and present antigens. Proc. Natl. Acad. Sci. USA. 2010;107:4287–4292. doi: 10.1073/pnas.0910609107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mahnke K, et al. The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J. Cell Biol. 2000;151:673–684. doi: 10.1083/jcb.151.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Witmer-Pack MD, Swiggard WJ, Mirza A, Inaba K, Steinman RM. Tissue distribution of the DEC-205 protein that is detected by the monoclonal antibody NLDC-145. II. Expression in situ in lymphoid and nonlymphoid tissues. Cell Immunol. 1995;163:157–162. doi: 10.1006/cimm.1995.1110. [DOI] [PubMed] [Google Scholar]
- 47.Gitlin AD, et al. HUMORAL IMMUNITY. T cell help controls the speed of the cell cycle in germinal center B cells. Science. 2015;349:643–646. doi: 10.1126/science.aac4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pasqual G, Angelini A, Victora GD. Triggering positive selection of germinal center B cells by antigen targeting to DEC-205. Methods Mol. Biol. 2015;1291:125–134. doi: 10.1007/978-1-4939-2498-1_10. [DOI] [PubMed] [Google Scholar]
- 49.Flacher V, et al. Skin langerin+ dendritic cells transport intradermally injected anti-DEC-205 antibodies but are not essential for subsequent cytotoxic CD8+ T cell responses. J. Immunol. 2012;188:2146–2155. doi: 10.4049/jimmunol.1004120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Flacher V, et al. Epidermal Langerhans cells rapidly capture and present antigens from C-type lectin-targeting antibodies deposited in the dermis. J. Invest. Dermatol. 2010;130:755–762. doi: 10.1038/jid.2009.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reuter A, et al. Criteria for dendritic cell receptor selection for efficient antibody-targeted vaccination. J. Immunol. 2015;194:2696–2705. doi: 10.4049/jimmunol.1402535. [DOI] [PubMed] [Google Scholar]
- 52.Canton J, et al. The receptor DNGR-1 signals for phagosomal rupture to promote cross-presentation of dead-cell-associated antigens. Nat. Immunol. 2021;22:140–153. doi: 10.1038/s41590-020-00824-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zehner M, et al. The translocon protein Sec61 mediates antigen transport from endosomes in the cytosol for cross-presentation to CD8(+) T cells. Immunity. 2015;42:850–863. doi: 10.1016/j.immuni.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 54.Ngu LN, et al. In vivo targeting of protein antigens to dendritic cells using anti-DEC-205 single chain antibody improves HIV Gag specific CD4(+) T cell responses protecting from airway challenge with recombinant vaccinia-gag virus. Immun. Inflamm. Dis. 2019;7:55–67. doi: 10.1002/iid3.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Derby N, Martinelli E, Robbiani M. Myeloid dendritic cells in HIV-1 infection. Curr. Opin. HIV AIDS. 2011;6:379–384. doi: 10.1097/COH.0b013e3283499d63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zalevsky J, et al. Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 2010;28:157–159. doi: 10.1038/nbt.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robbie GJ, et al. A novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob. Agents Chemother. 2013;57:6147–6153. doi: 10.1128/AAC.01285-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schriek P, et al. Marginal zone B cells acquire dendritic cell functions by trogocytosis. Science. 2022;375:eabf7470. doi: 10.1126/science.abf7470. [DOI] [PubMed] [Google Scholar]
- 59.Wilson KR, et al. MARCH1-mediated ubiquitination of MHC II impacts the MHC I antigen presentation pathway. PLoS One. 2018;13:e0200540. doi: 10.1371/journal.pone.0200540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data from this study will be made available upon request to the corresponding author (Justine D. Mintern, jmintern@unimelb.edu.au).