

Summary
This work identifies the Fragile X-related protein (FXR1) as a reciprocal regulator of HuR target transcripts in vascular smooth muscle cells (VSMC). FXR1 was identified as an HuR interacting protein by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The-HuR-FXR1 interaction is abrogated in RNase-treated extracts, indicating that their association is tethered by mRNAs. FXR1 expression is induced in diseased, but not normal arteries. SiRNA knock down of FXR1 increases abundance and stability of inflammatory mRNAs, while overexpression of FXR1 reduces their abundance and stability. Conditioned media from FXR1 siRNA-treated VSMC enhances activation of naïve VSMC. RNA EMSA and RIP demonstrate that FXR1 interacts with an ARE and a previously uncharacterized element in the 3’UTR of TNFα. FXR1 expression is increased in VSMC challenged with the anti-inflammatory cytokine IL-19, and FXR1 is required for IL-19 reduction of HuR. This suggests FXR1 is an anti-inflammation responsive, HuR counter-regulatory protein that reduces abundance of pro-inflammatory transcripts.
Keywords: RNA stability, AU-rich elements (AREs), HuR, FXR1, inflammation, IL-19, anti-inflammatory
Introduction
Despite nutritional modification and lipid reducing medications, atherosclerotic and other vascular syndromes account for 50% of all mortality and is increasing in the developing world. The injurious effects of pro-inflammatory cytokines resulting in Vascular Smooth Muscle Cell (VSMC) activation and development of multiple vascular diseases are well described (Allahverdian et al., 2014; Ross, 1999). Results from the recent CANTOS trial support the preeminent role of inflammation in vascular disease (Weber and von Hundelshausen, 2017). VSMC respond to and synthesize pro-inflammatory immune modulators (Doran et al., 2008; Hansson and Libby, 2006; Singer et al., 2004), and promulgate recruitment of leukocytes to the lesion (Hansson and Libby, 2006; Libby et al., 1997), leading to a localized vascular inflammatory lesion. In many vascular diseases, VSMC migrate into the intima, where they proliferate and synthesize cytokines and matrix proteins leading to loss of lumen and subsequent tissue ischemia. Resolution of inflammation is a dynamic and tightly regulated process and much attention has been aimed at identification of countervailing mechanisms that modulate inflammatory processes (Fredman and Tabas, 2017; Libby et al., 2014). A better understanding of countervailing mechanisms that modulate inflammatory processes, and identification of proteins and pathways which modulate the VSMC response to injury is key to development of therapeutics to combat multiple vascular diseases.
The regulation of mRNA stability and translation are two levels of post-transcriptional regulation that permit VSMC to rapidly respond to inflammatory stimuli (Barreau et al., 2005). AU-rich elements (AREs) in the 3’UTR of mammalian mRNA appear to be the target sequence for degradation or stabilization of transcripts. Most of the transcripts targeted for rapid degradation encode key regulatory proteins involved in cell growth, inflammation, and other responses to external stimuli (Bakheet et al., 2001). Importantly, most inflammatory cytokines contain conserved or semi-conserved AU-rich elements in their 3’UTR, imparting target specificity for a potential anti-inflammatory modality (Peng et al., 1996). Controlling mRNA decay allows the cell to fine-tune mRNA abundance and translation for rapid adaptation to environmental conditions, especially inflammation (Schoenberg and Maquat, 2012). An essential regulatory protein involved in this process is Human antigen R (HuR), a member of the Elav protein family and one of the best characterized, ARE-recognizing, RNA-binding proteins (RBPs) involved in mRNA stability and regulation of pro-inflammatory gene expression (Doller et al., 2008; Palanisamy et al., 2012). While HuR is ubiquitously expressed, it is activated in response to inflammatory signals to stabilize inflammatory mediators (Doller et al., 2008; Palanisamy et al., 2012). Since most pro-inflammatory transcripts contain AREs in their 3’UTR, this is a crucial and specific mechanism for the initiation and maintenance of the pro-inflammatory phenotype observed in vascular diseases. The exact mechanisms of HuR regulation have yet to be characterized, however they could represent key targets in regulating inflammation (Gallouzi and Steitz, 2001). Even though modulation of mRNA stability has been posited as a possible therapeutic strategy (Eberhardt et al., 2007), surprisingly, there is negligible literature exploring the concept that it could be directly regulated, or possibly reduced by anti-inflammatory stimuli. We posit that the regulation of HuR and other RBPs is a critical, and understudied step in the regulation of vascular inflammation.
We previously reported that IL-19, an anti-inflammatory cytokine, reduced inflammatory transcript mRNA stability in VSMC (Cuneo et al., 2010), and reduced HuR abundance in several cell types (Ellison et al., 2013). In this report, we identify and characterize one protein termed Fragile X-related protein (FXR1), a muscle-enhanced, autosomal homologue of the FMR (Fragile X mental retardation) neural protein, which interacts with HuR in inflammatory, but not basal conditions, a novel finding. We report here that FXR1 expression is induced by IL-19 in VSMC and that modulation of FXR1 regulates ARE-containing transcripts in VSMC. RNA EMSA and RNA immunoprecipitation demonstrate that FXR1 interacts with the canonical AREs and a previously uncharacterized element in the 3’UTR of TNFα. This work implicates FXR1 as a previously unrecognized negative regulator of inflammation, and suggests that IL-19 induction of FXR1 expression is a negative compensatory, counter-regulatory mechanism used by VSMC to respond to and resolve inflammation.
Results
HuR interacts with FXR1.
It is presumed that HuR activity is regulated by interacting proteins (Doller et al., 2008; Gallouzi and Steitz, 2001; Pullmann et al., 2005). HVSMCs were transfected with a Flag-tagged HuR or Flag-tagged empty control vector, then starved for 48 hours in 0.1% FBS before stimulation with TNFα. HuR pull-down was followed by un-biased LC-MS/MS to identify the proteins which immunoprecipitated with HuR. HuR interacting candidates were identified by eliminating any protein with a raw peptide count below ten (Supplemental Table 1). Interacting proteins were also scrutinized in the Contaminant Repository for Affinity Purification (CRAPome) database (Mellacheruvu et al., 2013) to determine the occurrence of proteins in control experiments to eliminate “sticky proteins” that may non-specifically interact with HuR. The final list of proteins which met these criteria were examined for Gene Ontology (GO) annotation. A number of interacting proteins were identified, most involved in various aspects of mRNA processing (Table 1). The last row of Table 1 includes Elav1 (HuR), as it was the bait protein used to perform LC/MS Mass Spectrometry, although nothing is known about HuR and the Flag-tag epitope according to the CRAPome database. Fragile X-related protein (FXR1) was chosen for further study because of the novelty of its interaction; because FXR1 expression is muscle-enhanced (Garnon et al., 2005; Mientjes et al., 2004); and because no literature exists on FXR1 inducibility by inflammatory stimuli or VSMC, making FXR1 a particularly novel target to study in the context of vascular disease. Finally, similar to HuR, FXR1 is presumed to be an RNA binding protein (Adinolfi et al., 1999). FXR1 exists in several isoforms in mouse, and is predicted to have three isoforms in human (Dubé et al., 2000). We were only able to detect isoform 1 in human VSMC by Western blot and transcript-specific qRT-PCR (data not shown). Subsequent experiments utilized isoform 1 to ensure all domains were represented.
Table 1.
Putative HuR-interacting proteins identified by LC-MS/MS ranked in order of percent occurrence by FLAG epitope found in the Contaminant Repository for Affinity Purification (CRAPome) database.
| Protein | − | + | % occurrence FLAG epitope | GO Analysis |
|---|---|---|---|---|
| ATXN2 | 0 | 13 | 0.64 | Poly(A) RNA binding and structural constituent of ribosome |
| HELZ2 | 0 | 50 | 1.28 | Poly(A) RNA binding, ligand-dependent nuclear receptor transcription coactivator activity |
| STAU2 | 0 | 16 | 1.92 | Poly(A) RNA binding, double-stranded RNA binding |
| FAM120A | 0 | 43 | 2.56 | Poly(A) RNA binding |
| TROVE2 | 0 | 18 | 2.56 | RNA binding and U2 snRNA binding. |
| EIF4G3 | 0 | 12 | 5.13 | Poly(A) RNA binding and binding |
| TRIM25 | 0 | 11 | 5.13 | Poly(A) RNA binding and acid-amino acid ligase activity. |
| AGO3 | 0 | 11 | 5.13 | Nucleic acid binding and RNA binding |
| MOV10 | 0 | 26 | 5.77 | RNA binding, helicase activity |
| ZC3HAV1 | 0 | 10 | 5.77 | Poly(A) RNA binding and NAD+ ADP-ribosyltransferase activity |
| FXR1 | 0 | 13 | 6.41 | Nucleic acid binding and RNA binding |
| HNRNPUL2 | 0 | 10 | 6.41 | Poly(A) RNA binding and kinase activity |
| STAU1 | 0 | 16 | 7.69 | Poly(A) RNA binding, double-stranded RNA binding |
| UPF1 | 0 | 45 | 8.33 | Poly(A) RNA binding |
| PRRC2C | 0 | 23 | 8.33 | Poly(A) RNA binding and protein C-terminus binding |
| FUBP3 | 0 | 16 | 8.33 | Nucleic acid binding and RNA binding |
| ATXN2L | 0 | 15 | 8.33 | Poly(A) RNA binding and protein C-terminus binding |
| MYO5A | 0 | 12 | 8.33 | Nucleic acid binding and chromatin binding |
| ELAV1 | 0 | 18 | N/A | Nucleic acid binding and RNA binding |
Columns Flag empty-vector (−) and the Flag-immunoprecipitated (+) provide the raw peptide sequences from VSMCs following IP with anti-Flag antibody. Proteins were identified by LC MS/MS.
Indicates Elav1 was the bait protein used to pull-down proteins for LC MS/MS. CRAPome database provided no additional information about Elav1.
A series of immunoprecipitations were performed in order to confirm the interaction between HuR and FXR1. First, we performed a co-immunoprecipitation of endogenous FXR1 for HuR in hVSMCs which were either serum-starved or serum-starved and stimulated with TNFα for 8 hours (Figure 1A). We also overexpressed FXR1 using a flag-tagged adenovirus (AdFXR1) and concurrently overexpressed HuR also using an adenovirus (AdHuR) in hVSMCs and performed an immunoprecipitation using anti-flag-conjugated beads (Figure 1B). Next, hVSMCs were treated as described, but after serum starvation, they were stimulated with TNFα for 8 hours. Figure 1C shows that the HuR/FXR1 interaction was enhanced in TNFα-stimulated cells. Figure 1D shows that the FXR1/HuR interaction is abrogated by addition of RNAse A, suggesting that the interaction we identified via proteomics may be mediated by RNA tethering. The increased FXR1-HuR interaction observed in TNFα stimulated VSMC may be due to an increase in transcripts that harbor both FXR1 and HuR binding elements.
Figure 1.

FXR1 and HuR interact A. Co-immunoprecipitation of endogenous FXR1 with HuR in unstimulated or TNFα stimulated VSMC. B. HVSMCs transduced with AdenoGFP-control vector, flag-tagged adenoFXR1 and HuR adenovirus, followed by immunoprecipitation by anti-flag-conjugated beads. C. The FXR1-HuR interaction increases in TNFα-stimulated conditions. D. FXR1-HuR interaction is mediated by mRNA. The addition of RNAse A to the immunoprecipitation reaction disrupted TNFa-driven FXR1-HuR interaction. E. HuR and FXR1 co-localization in hVSMC. HuR remained predominantly nuclear while FXR1 localized to the cytoplasm in unstimulated VSMC. Upon stimulation with TNFα, HuR shuttled to the cytoplasm where it co-localized with FXR1. F. High-resolution confocal microscopy of HuR/FXR1 interaction in cytoplasm of TNFα-stimulated hVSMC. G. FXR1 and HuR co-localize to stress granules. HVSMCs were un-treated or stimulated with 20uM Clotrimazole for 45 minutes, then stained for stress granule marker Poly-A-binding protein (PABP), FXR1, HuR, and with DAPI and were imaged using confocal microscopy to determine co-localization, magnification is 630X for E and G, 1260X for F.
We next used confocal microscopy to determine HuR and FXR1 localization under basal and inflammatory conditions in hVSMCs. HuR nucleocytoplasmic shuttling has been reported (Wu et al., 2016), and consistent with the literature, in TNFα stimulated VSMC, HuR translocated from the nucleus to the cytoplasm. FXR1 remained predominantly cytoplasmic in both unstimulated and stimulated conditions. Interestingly, HuR and FXR1 co-localized in the cytoplasm following 8 hour TNFα stimulation, which is consistent with literature showing HuR nucleocytoplasmic translocation upon inflammatory stimuli (Figures 1E, F). RNA processing often occurs in phase-dense structures that form in the cytoplasm of eukaryotic cells in response to environmental stresses. The composition of stress granules suggests that regulation of labile ARE-containing inflammatory transcripts could be occurring there. To further associate a relationship between FXR1 with HuR in RNA processing, we used immunostaining and confocal microscopy to co-localize HuR and FXR1 within punctate stress granules in hVSMCs. Clotrimazole was used to induce stress granule formation, and Poly-A binding protein (PABP) was used as a marker for stress granules (Kedersha et al., 2008). Figure 1G shows that hVSMCs stimulated with 20μM Clotrimazole demonstrated well-defined, punctate co-localization of FXR1 and HuR in stress granules which further suggest a role for FXR1 along with HuR in RNA processing in VSMC.
FXR1 expression is induced in diseased VSMC.
There is no literature describing FXR1 induction in VSMC or models of vascular injury. We examined FXR1 expression in mouse and human atherosclerotic and restenotic tissue and detected inducible FXR1 expression in VSMC in multiple models of vascular injury. Figure 2A indicates that FXR1 expression is increased in neointimal, compared with medial VSMC in carotid artery from ligated mice. Similarly, FXR1 expression is increased in VSMC in atherosclerotic plaque and cap, but much lower in non-diseased medial VSMC in aortic arch from LDLR−/− mice fed an atherogenic diet (Figure 2B). FXR1 expression is negligible in normal, non-diseased arteries from these mice (Supplemental Data Figures 1A and B). Importantly, FXR1 expression is barely detectible in a coronary artery from a non-failing human heart, but expression is enhanced in myofibrous atherosclerotic plaque from a human coronary artery (Figures 2C and D). Figure 2E is dual-color immunohistochemistry showing that FXR1 expression co-localizes in plaque SMC in human coronary artery. Together, these data are the first to suggest that FXR1 induction is a VSMC response to inflammatory stimuli in vivo.
Figure 2.
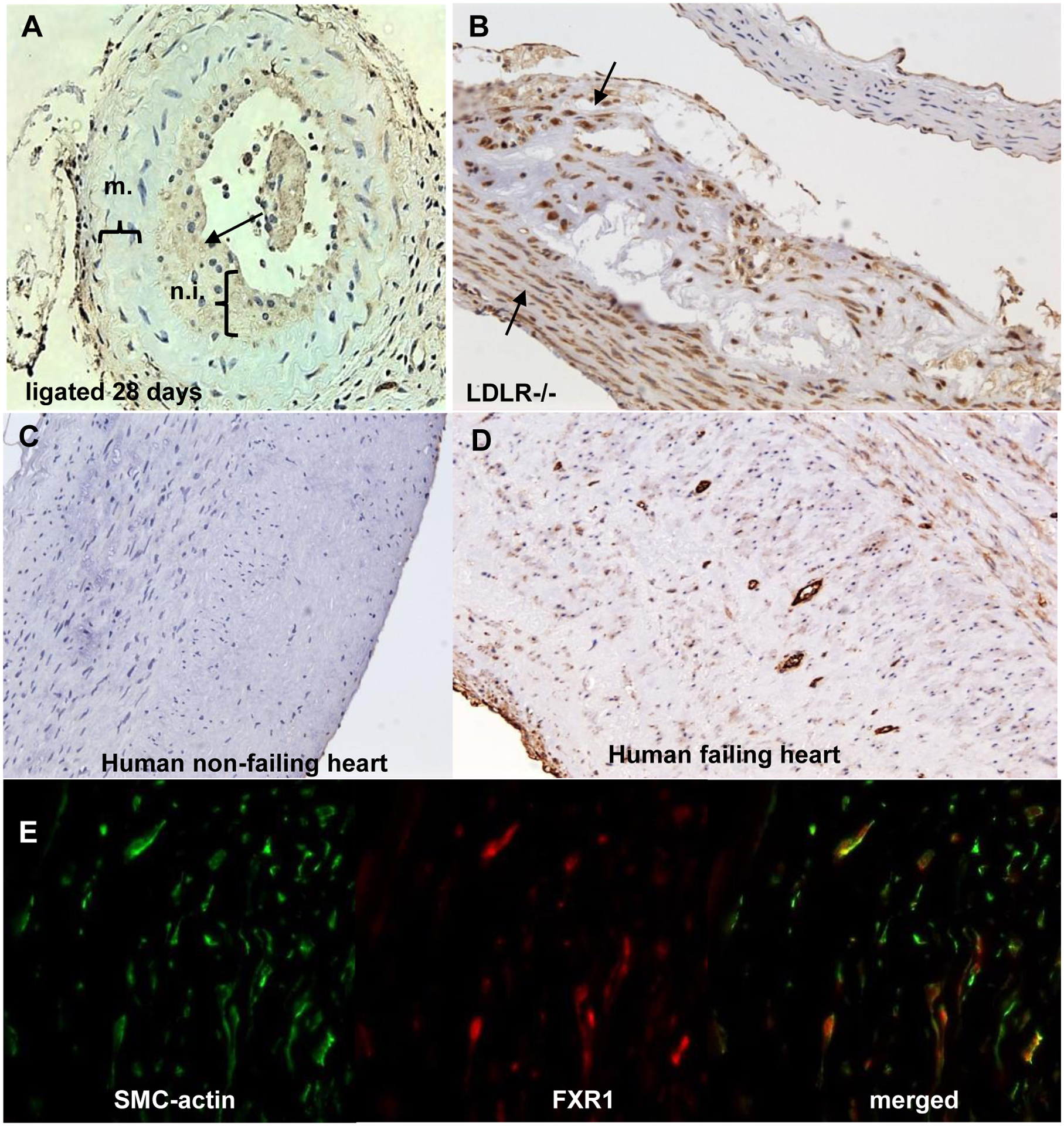
FXR1 protein expression in vascular injury and disease models. A. FXR1 expression in ligated murine carotid artery. Mouse carotid arteries were harvested 28 days after ligation, and immunohistochemistry performed. FXR1 primarily stains in the neointima (n.i.), but not the media (m.) in the ligated artery. B. FXR1 expression in mouse atherosclerotic plaque. Cross section from an LDLR−/− mouse aorta fed a HFD for 12 weeks to develop atherosclerotic plaque. VSMC in plaque and smooth muscle cell cap are enriched for FXR1 expression. C. FXR1 expression in normal human coronary artery from a non-failing heart. D. FXR1 expression in myleofibroid atherosclerotic plaque from a failing human artery. FXR1 expression is enriched in myleofibroid VSMC in the plaque as compared to healthy human control artery. E. Fluorescent co-staining of fibro-atherosclerotic cap from human atherosclerotic plaque using antibody to SMC-actin and FXR1. See also Supplemental Figure 1 for normal mouse artery and negative controls for immunohistochemistry. Magnification is 200X for all.
FXR1 regulates abundance and mRNA stability of pro-inflammatory mRNA and protein.
Literature on FXR1 function is inconsistent, and appears to be cell-type specific. To more definitively link FXR1 function with vascular inflammation, we transfected hVSMC with FXR1-specific siRNA, then stimulated the cells with TNFα. Knockdown of FXR1 resulted in a dramatic and significant increase in abundance of inflammatory transcripts in hVSMC (IL-1 β, ICAM1 and MCP-1 are shown as examples). Interestingly, these transcripts have been previously shown to be stabilized by HuR in other cell types (Aguado et al., 2015; Chen et al., 2006; Krishnamurthy et al., 2010; Wu et al., 2016) (Figure 3A). Correspondingly, Figure 3B shows that siRNA reduction of FXR1 also increases abundance of inflammatory proteins as well as HuR in TNFα-stimulated human VSMC. As FXR1 knock down increased inflammatory transcripts, we reasoned that FXR1 overexpression would reduce abundance of inflammatory mRNA and protein. Adenoviral overexpression of FXR1 decreases abundance of inflammatory mRNA (Figure 3C) and protein (Figure 3D) in a dose-dependent fashion compared with AdGFP control. The siRNA knockdown and overexpression data are complementary and strongly suggest that FXR1 expression may regulate abundance of inflammatory proteins as well as HuR in VSMCs.
Figure 3.
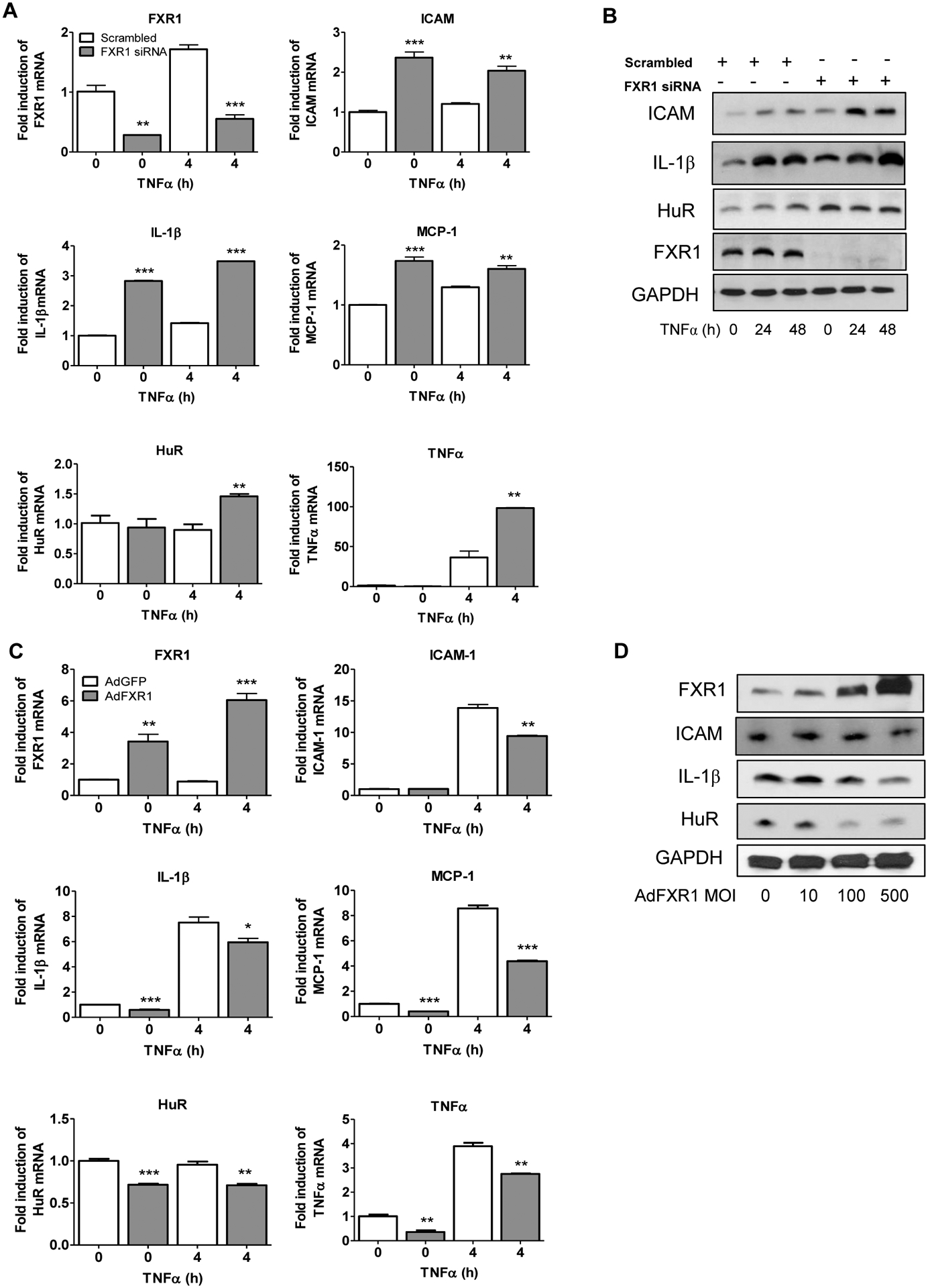
FXR1 regulates abundance of pro-inflammatory mRNA and proteins in VSMC. A. FXR1 knockdown increases mRNA abundance of inflammatory mediators in VSMC. HVSMCs transfected with FXR1 siRNA or scrambled control siRNA, serum starved, then stimulated with TNFα for 4 hours to induce mRNA expression quantitated by qRT-PCR and normalized to GAPDH expression. B. FXR1 knockdown increases protein abundance of inflammatory mediators in VSMC. Cell extracts were prepared from VSMC treated as described above, stimulated with TNFα for 24 hours and proteins identified by western blot analysis. C. Overexpression of FXR1 reduces abundance of pro-inflammatory mRNA and proteins. HVSMCs were transduced with 100 MOI of AdFXR1 or AdGFP, serum-starved, then stimulated with TNFα for 4 hours to induce mRNA expression which was quantitated by qRT-PCR and normalized to GAPDH expression. D. FXR1 overexpression increases protein abundance of inflammatory mediators in VSMC. Cell extracts were prepared from VSMC treated as described above, stimulated with TNFα for 24 hours and proteins identified by western blot analysis. P< *0.05, **0.01, or ***0.001.
FMR1 family members are putative RNA-binding proteins (Adinolfi et al., 1999), and we next determined if modulation of FXR1 would affect mRNA stability. Using the transcription inhibitor actinomycin D in TNFα-stimulated VSMC, we determined that the mRNA stability of ARE-containing transcripts IL-1 β, ICAM1, and HuR are significantly increased when FXR1 is knocked down, and importantly, significantly decreased when FXR1 is over expressed (Figures 4A and 4B). The mRNA stability of PPARα, the expression of which is not regulated by ARE in its 3’UTR, was not affected by FXR1 knock down or overexpression, demonstrating target transcript specificity for FXR1 activity. Of particular importance was the finding that FXR1 appears to have a reciprocal relationship with HuR abundance, suggesting important, possibly competitive roles for these proteins in regulation of mRNA stability. Together, these results suggest a novel function for FXR1 in regulation of mRNA stability and subsequent abundance of pro-inflammatory proteins.
Figure 4.
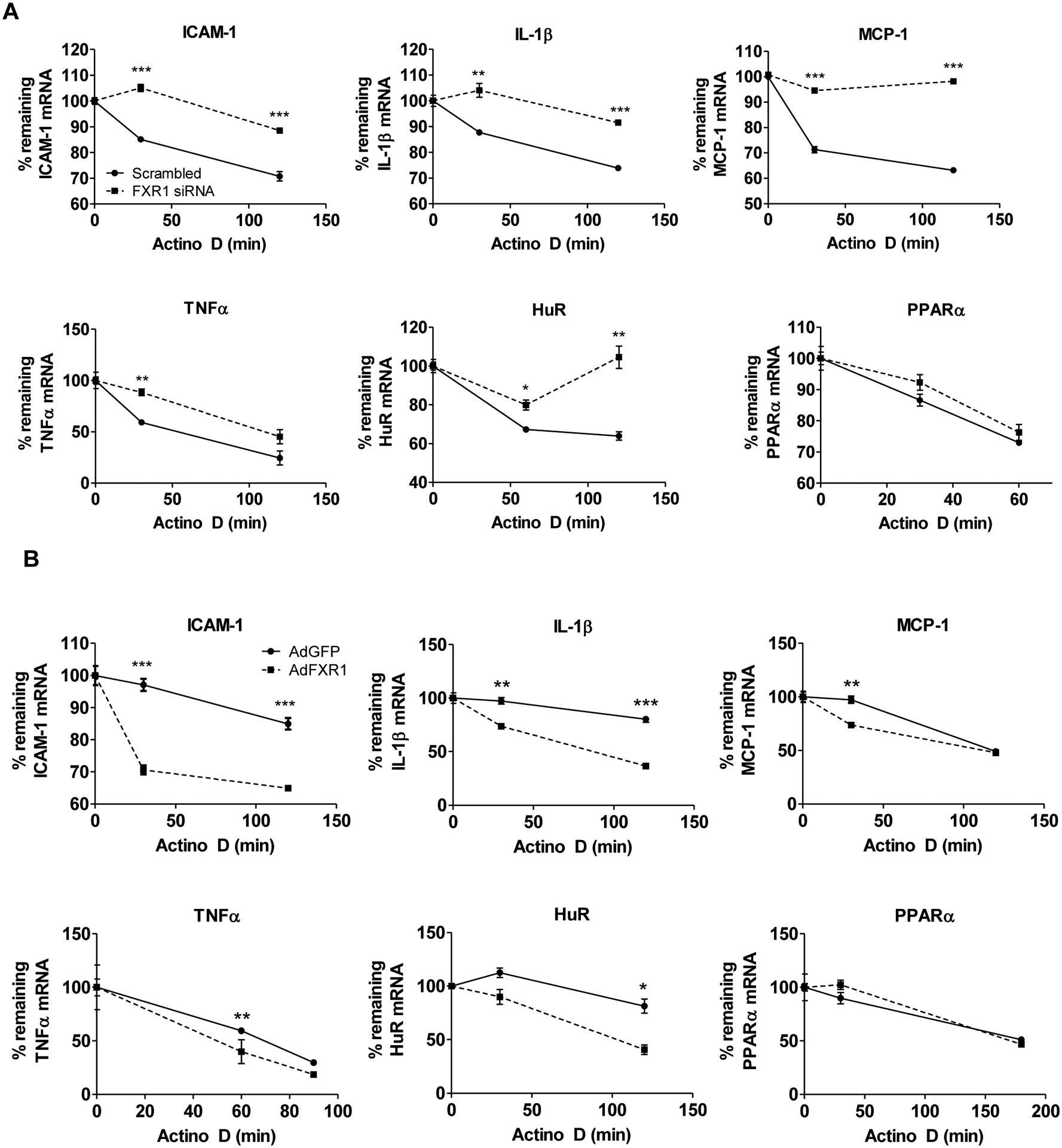
FXR1 regulates mRNA stability of pro-inflammatory mRNA. A. FXR1 knockdown increases inflammatory mRNA stability. HVSMCs were transfected with FXR1 siRNA or scrambled control siRNA, serum starved, and stimulated with TNFα for 4h at which point actinomycin D (10ng/ml) was added to halt transcription. RNA was isolated at indicated times post addition of actinomycin D, and mRNA abundance quantitated by qRT-PCR. Percent mRNA remaining was determined by normalizing each time point to Beta-2-microglobulin. B. FXR1 overexpression reduces inflammatory mRNA stability. HVSMCs were transduced adenoFXR1 and AdenoGFP, serum-starved, and stimulated with TNFα for 4h at which point actinomycin D (10ng/ml) was added. RNA was isolated at the various time points post addition of actinomycin D, and mRNA abundance quantitated by qRT-PCR. Percent mRNA remaining was determined by normalizing each time point to Beta-2-microglobulin (B2M). P< *0.05, **0.01, or ***0.001.
FXR1 regulates VSMC cell proliferation and inflammation through paracrine signaling.
Maladaptive VSMC proliferation is a hallmark of several vascular pathologies, and is driven by inflammatory gene expression (Hansson and Libby, 2006; Libby, 2002; Libby et al., 2014; Ross, 1999). Knockdown of FXR1 in hVSMCs significantly increased cell proliferation compared to scrambled control cells (Figure 5A). Concordantly, VSMC proliferation was significantly decreased in a manner inversely proportional with FXR1 expression, confirming FXR1 can regulate VSMC proliferation (Figure 5B).
Figure 5.

FXR1 regulates VSMC proliferation. A. HVSMCs transfected with FXR1 siRNA or scrambled control siRNA were seeded at 10,000 cells/well and counted at days 3 and 6. B. HVSMCs were transduced with 100 MOI of AdFXR1 or AdGFP, seeded at 10,000 cells/well and counted at day 3 and 6. P< 0.05, 0.01, or 0.001. C. Conditioned media from VSMC in which FXR1 is deleted can induce inflammatory and proliferative responses from naive VSMC. HVSMCs were transfected with scrambled control or FXR1 siRNA, washed, and media collected after 48 hours. Conditioned media from each of these groups was added to serum-starved hVSMC for four hours, RNA isolated and reverse-transcribed for qRT-PCR analysis. D. HVSMCs were seeded at 20,000 cells/well in FXR1 siRNA conditioned media or scrambled control media. Cells were counted at days 3 and 5. P< *0.05, **0.01, or ***0.001.
VSMC paracrine signaling is also a characteristic of many vascular diseases. Since FXR1 appeared to regulate abundance of cytokines, it was important to determine if this participated in paracrine signaling. First, serum-starved VSMC were stimulated with conditioned media collected from VSMC transfected with FXR1 siRNA or scrambled controls for 4 hours, then inflammatory transcript mRNA was quantitated. Figure 5C shows that hVSMCs treated with FXR1 siRNA knockdown conditioned media had increased inflammatory gene expression compared to scrambled control.
Next, using conditioned media from scrambled control or FXR1 siRNA knockdown VSMCs, we performed a proliferation assay to demonstrate the autocrine and paracrine effects on cell growth (Figure 5D). HVSMCs treated with FXR1 siRNA conditioned media had significantly increased proliferation compared to scrambled media control. These data suggest the reduction of FXR1 results in increased cytokine production that has potential autocrine and paracrine effects on other hVSMCs.
FXR1 binds RNA via canonical ARE and non-ARE sequences.
Various complementary methods were used to determine if FXR1 binds mRNA. First, GST and human GST-FXR1 fusion proteins were used in RNA-electro mobility shift assays (cRNA-EMSAs) with a biotinylated probe consisting of a 50bp region of the human TNFα 3’UTR (position 1333–1380). Addition of recombinant FXR1 to this probe suggested it bound to RNA (Figure 6A). The AUUUA monomer (as negative control probe) did not complex with FXR1. Interaction specificity was demonstrated by super-shift of the FXR1-probe complex by addition of anti-FXR1 antibody (Figure 6B). FXR1 binding affinity for this region was calculated, and in our hands, FXR1 has similar affinity to the TNFα probe as HuR (Supplemental Data Fig 2A and B).
Figure 6.
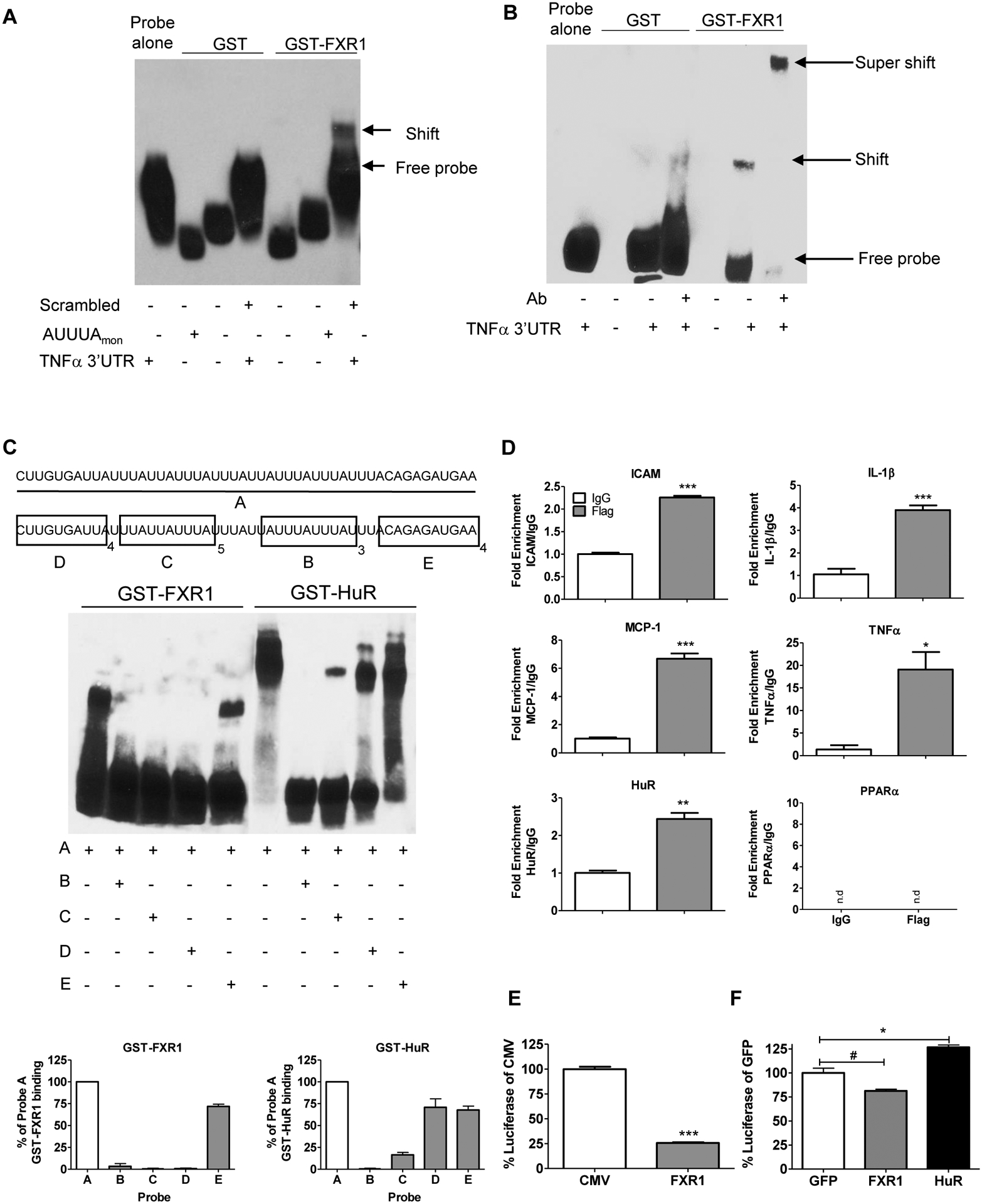
FXR1 binds mRNA. A. RNA-EMSA. Biotinylated RNA probes containing a scrambled control, a 50mer of human TNFα 3’UTR, or (AUUUA)monomer were incubated with GST or GST-FXR1, and membrane blotted to demonstrate a shift indicating a protein-RNA complex. B. GST or FXR1 antibody was added to the EMSA reaction; supershift of the complex demonstrates specificity. C. FXR1 binds ARE and a novel element. Non-biotinylated probes consisting of a 25-mer of (AUUUA)5, a 27-mer of (UUAUUUAUU)3, a 36- mer of (CUUGUGAUU)4, a 40-mer of (CAGAGAUGAA)4 (40 bases) were added to the reaction to prior to the addition of the biotinylated TNFα 50mer, and RNA EMSA performed using GST-FXR1 and GST-HuR. Densitometry of protein-probe complex as a percentage of binding to Probe A for both GST-FXR1 and GST-HuR. D. RNA immunoprecipitation. VSMC were transduced with FLAG-tagged AdFXR1, serum starved for 48 hours, then stimulated with TNFα. RNA-protein complexes were immunoprecipitated with FLAG or IgG control beads. PPARα, lacking ARE in its 3’UTR was not amplified (n.d). E. The TNFα 3’UTR luciferase construct was co-transfected into HEK 293 cells with either a vector control or pFXR1. The cells were seeded in triplicate for 48 hours before harvesting. Luminescence was measured using Infinite M1000 Pro plate reader and graphed as a percentage of GFP luciferase control. F. Adenoviral expression of TNFα 3’UTR luciferase construct was co-transduced into hVSMCs with AdGFP, AdFXR1, or AdHuR and the results were graphed as a percentage of luciferase AdGFP.
To determine the site or sites on the TNFα 3’UTR recognized by FXR1, four probes representing different regions of the 50bp TNFα 3’UTR (probe A) were synthesized; a 25-mer of (AUUUA)5 (probe B), a 27-mer of (UUAUUUAUU)3 (probe C), a 36-mer of (CUUGUGAUU)4 (probe D), a 40-mer of (CAGAGAUGAA)4 (probe E), were added to the cRNA-EMSAs to compete with the biotinylated 50bp TNFα 3’UTR probe. The GST negative control protein did not interact with any probes (Supplemental Figures 3A–B). FXR1 binding with various amounts of cold competitor probes was performed to determine probe input concentrations (Supplemental Figure 4A and B). Figure 6C shows that probes B and C, which contained recognized ARE were capable of competing with the full length probe for FXR1 binding, and nearly ablated the gel shift. Interestingly, probe D, which does not contain a canonical ARE, also successfully competed with the full length TNFα 3’UTR for FXR1 binding, but did not compete with the full length probe for HuR, suggesting an additional, previously unrecognized region on the TNFα 3’UTR recognized by FXR1. These results were quantified using densitometry as a percentage of GST-protein bound to probe A (lower panel, Figure 6C). An additional binding element termed the G quadruplex has been implicated as a binding site for FXR1 (Bechara et al., 2007). Using this element as a cold competitor to the biotinylated TNFα 3’UTR probe, we found the G quadruplex was able to bind FXR1 (Supplemental Figure 4C and D). Using RNA immunoprecipitation (RIP), we determined if FXR1 directly binds RNAs that were shown to be regulated by FXR1. VSMC were transduced with Flag-tagged AdFXR1, serum starved for 48 hours, then stimulated with TNFα for 8 hours. Figure 6D shows that several transcripts were identified as interacting with FXR1 compared to IgG control antibody. Importantly, mRNA transcripts not regulated by AREs in 3’UTR such as PPARα were not amplified. It was possible that FXR1 could compete with HuR for occupancy on 3’UTR of transcripts that contained these regions. A constitutively driven luciferase reporter representing the TNFα 3’UTR and containing ARE tandem repeats was transfected into HEK cells, and also transduced with plasmid encoding FXR1 cDNA or a control empty-vector plasmid. Figure 6E shows that FXR1 reduced luciferase activity, suggesting that FXR1 may compete with HuR for ARE occupancy. We also used an adenovirus expressing the TNFα 3’UTR luciferase construct to perform the experiment in hVSMCs. AdFXR1 and AdHuR were co-transduced with the adeno-expression TNFα 3’UTR luciferase, as well as an AdGFP for control. Figure 6F demonstrates that in hVSMCs, FXR1 over expression reduced TNFα 3’UTR luciferase activity, while HuR was able to increase it, supporting the concept that FXR1 is a negative regulator of inflammatory transcripts.
IL-19 induces FXR1 expression in VSMC.
IL-19 is anti-proliferative for VSMC, and reduces inflammatory transcript mRNA stability (Tian et al., 2008). Stimulation of VSMC with IL-19 significantly induces FXR1 mRNA and protein expression (Figures 7A–C). Long-term treatment of VSMC with IL-19 also reduces HuR protein abundance (Cuneo et al., 2010; Ellison et al., 2013). To determine if FXR1 mediated IL-19 decrease in HuR abundance, VSMCs were transfected with FXR1 siRNA or scrambled control siRNA and serum-starved. VSMC were then treated with IL-19 for various time points and cells extracts were used for western blot analysis (Figure 7D). As reported, IL-19 treatment can reduce HuR protein abundance; however, in the absence of FXR1, IL-19 is unable to reduce HuR, indicating that IL-19 reduction of HuR requires FXR1. To further dissect the mechanism by which IL-19 reduces HuR abundance, we determined that IL-19 is able to reduce HuR mRNA stability in hVSMCs following actinomycin D treatment (Figure 7E). However, in the absence of FXR1, IL-19 was unable to reduce HuR mRNA stability compared to scrambled control, suggesting FXR1 is necessary for IL-19 induced destabilization of HuR (Figure 7F). Since FXR1 is induced by IL-19 and is necessary for IL-19 destabilization and reduction of HuR, we conclude that FXR1 expression is a negative compensatory, counter-regulatory mechanism used by VSMC to respond to and dampen inflammation.
Figure 7.
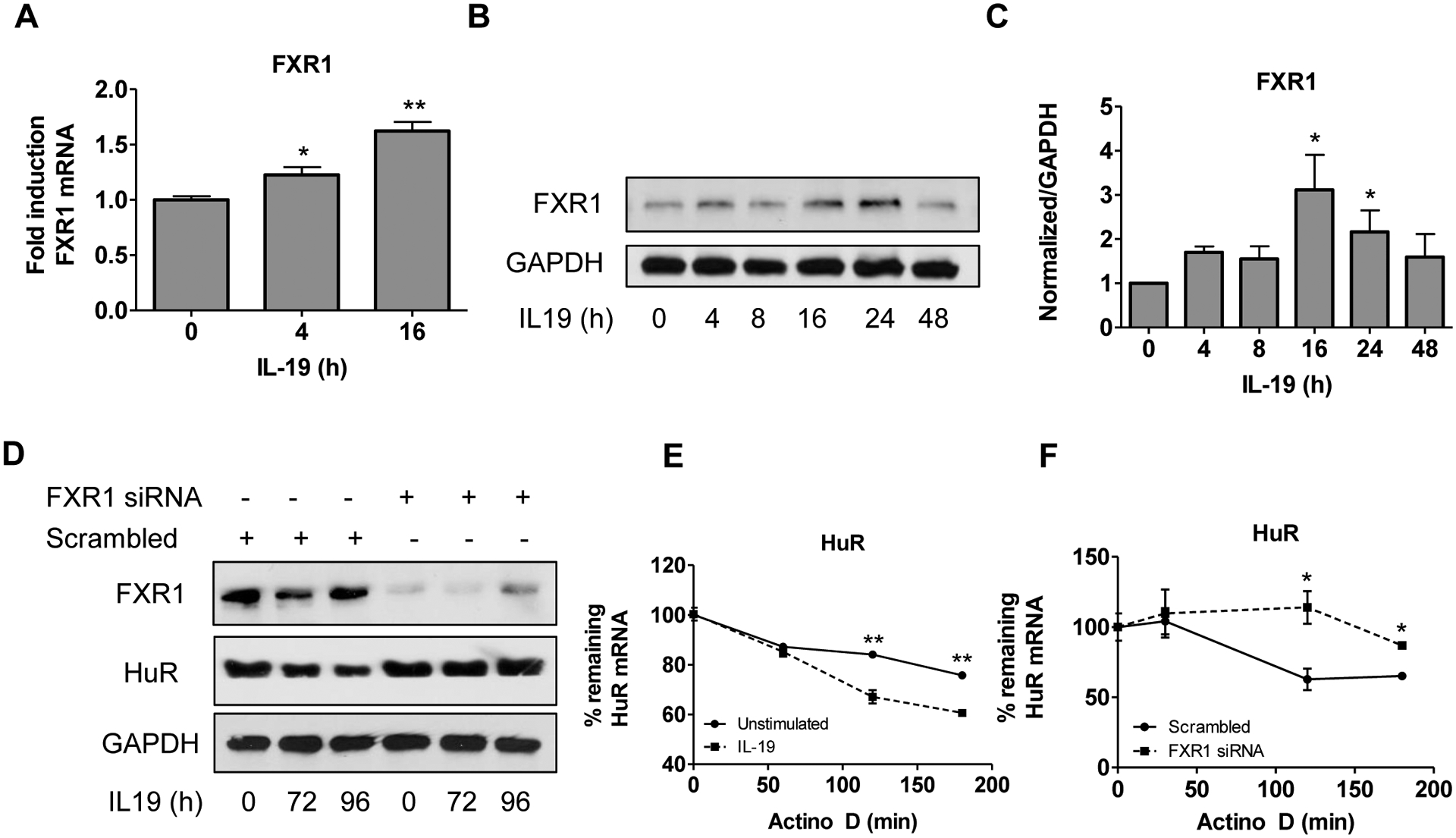
Anti-inflammatory cytokine IL-19 induces expression of FXR1. A. IL-19 induces FXR1 mRNA. HVSMC were serum-starved for 48 hours, then treated with IL-19 for the times indicated, and FXR1 mRNA quantitated by qRT-PCR normalized to GAPDH. B. IL-19 induces FXR1 protein expression. HVSMC were treated as described in “A”, and FXR1 protein detected by western blot analysis. C. Densiometric analysis of FXR1 protein expression in IL-19-treated VSMC normalized to GAPDH. D. FXR1 mediates IL-19 reduction in HuR protein abundance. VSMCs were transfected with FXR1 siRNA or scrambled control siRNA, treated with IL-19 for the times indicated, and extracts blotted to detect FXR1, HuR, and GAPDH proteins. E. IL-19 reduces HuR mRNA stability. HVSMCs were unstimulated or treated with IL-19 for 16 hours prior to the addition of actinomycin D and HuR mRNA abundance was quantitated by qRT-PCR. Percent mRNA remaining was determined by normalizing each time point to B2Microglobulin. F. HVSMCs were transfected with FXR1 siRNA or scrambled control prior to treatment with IL-19 for 16 hours. Actinomycin D was added and HuR mRNA abundance was quantitated by qRT-PCR. Percent mRNA remaining was determined by normalizing each time point to B2M. P< *0.05, **0.01, or ***0.001.
Discussion
The major findings of this study are that FXR1 expression in VSMC reduces mRNA stability and abundance of pro-inflammatory proteins, is induced by the anti-inflammatory cytokine IL-19, and acts as an effector of IL-19 anti-inflammatory activity in VSMCs. This has important implications for resolution of inflammation in general, and in attenuation of severity of vascular inflammatory syndromes such as atherosclerosis, restenosis, and allograft vasculopathy in particular. Investigation into vascular inflammation primarily focuses on the role of immune cells, but in this study we show that a non-immune cell can respond to anti-inflammatory stimuli through post-transcriptional mechanisms. Most inflammatory cytokines contain conserved or semi-conserved AU-rich elements in their 3’UTR (Peng et al., 1996), imparting target specificity to allow the cell to fine-tune mRNA abundance and translation for rapid response to inflammation (Schoenberg and Maquat, 2012). An essential regulatory protein involved in this process is Human antigen R (HuR) (Peng et al., 1996; Schoenberg and Maquat, 2012), and proteins and pathways which regulate HuR may be key targets in regulation of inflammation. We previously reported that IL-19 reduces HuR activity in VSMC, leading to a decrease in mRNA stability in a HuR-mediated, but un-characterized mechanism. A goal of this study was to identify proteins that would interact with HuR and regulate its activity.
An un-biased pull-down experiment using flag-tagged HuR and LC-MS/MS to identify protein constituents of HuR complexes in VSMC uncovered a number of interesting proteins recognized to participate in various aspects of mRNA processing. Studies have extensively characterized the mRNP complexes that form to regulate mRNA transcripts and translation, while the profile of proteins known to directly interact with HuR, particularly in an inflammation responsive fashion, is more limited. Interaction with one candidate identified in our analysis, FXR1, was validated by co-immunoprecipitation and cellular co-localization assays, validating the LC-MS/MS result, and is the first novel finding of this study. FXR1 is an autosomal homolog of Fragile X mental retardation (FMR) protein, the prototypical and best studied member of the FXR (Fragile X-related) family of neuronal proteins (Bardoni et al., 2001). The vast majority of work on FMR1, FXR1, and FXR-related proteins are performed in neurons and focus on their role in cognitive ability; nothing is known about this family of proteins in VSMC or vascular pathophysiology. FXR1 is the only fragile X protein family member significantly expressed in muscle cells (Garnon et al., 2005; Mientjes et al., 2004), and has been described as the “muscle-homologue” of the FMR family (Mientjes et al., 2004). In addition, our data suggests FXR1 is upregulated in VSMCs specifically in inflamed regions of arteries from various and disease states. For these reasons we focused on the role of FXR1 in VSMC for further study.
HuR is characterized as an mRNA stabilizing RBP, while FXR1 function is less understood and appears to be cell-type specific. When we knocked down FXR1 using siRNA in TNFα-stimulated hVSMCs, we were initially surprised to observe that inflammatory transcripts were increased at both the transcript and protein level. Knockdown of FXR1 in VSMC in the presence of actinomycin D demonstrated significantly increased mRNA stability of several pro-inflammatory transcripts, in direct contrast to a study in macrophages showing that absence of FXR1 did not affect TNFα mRNA half-life (Garnon et al., 2005). Stability of PPARα mRNA, a transcript not regulated by AREs in its 3’UTR, was unaffected by FXR1 knockdown. RBPs have the ability to be cell-type specific in their function, especially those that have cell-specific expression (Musunuru, 2003). Since knock down of FXR1 increased inflammatory transcripts, we reasoned that FXR1 overexpression would reduce abundance of inflammatory mRNA and protein. Indeed, adenoviral overexpression of FXR1 significantly decreased abundance of several inflammatory mRNA and proteins. Concordant with knockdown, overexpression did not affect PPARα mRNA stability in any way. This study indicates that FXR1 knockdown results in increased HuR protein abundance mRNA stability, and overexpression results in the opposite, suggesting a previously unrecognized mechanism in regulation of proinflammatory transcripts. HuR abundance is auto regulated by a positive-feedback loop involving HuR interaction with the 3’ UTR of its own mRNA (Dai et al., 2012). These authors suggested that HuR mRNA is destabilized through an ARE-dependent, but unidentified mechanism, which we posit could be FXR1 interaction with the HuR 3’UTR. FXR1 knockdown also increased hVSMC proliferation, and FXR1 overexpression reduced hVSMC proliferation. This proliferative effect may be due to a decrease in autocrine expression of cytokines and growth factors, as VSMC are known to proliferate in an autocrine fashion. Concordantly, conditioned media from FXR1 knockdown VSMCs increased inflammatory cytokine expression and proliferation of naive VSMCs as well. FXR1 expression is critically important, so much so that FXR1 knock out mice are postnatally lethal (Mientjes et al., 2004), and in conjunction with its upregulation in injured arteries, this supports a role for FXR1 in maintenance of the quiescent VSMC phenotype. Overall, siRNA knock down and overexpression data are complementary, and are associated with the presence of ARE in transcript 3’UTR. These data strongly suggest that in VSMC, FXR1 functions as an mRNA de-stability factor to reduce inflammatory transcripts. In this regard, FXR1 appears to function similarly to other destabilizing RBPs such as Tristetraprolin (TTP) and AUF1 (hnRNP D), which function to alter transcript stability of TNFα via AREs in the 3’UTR (Carballo et al., 1998). Because FXR1 expression is enhanced in muscle, is induced by IL-19, an anti-inflammatory stimuli, and can regulate inflammatory protein abundance in VSMC, FXR1 can potentially have key regulatory effects in modulation of vascular inflammatory diseases.
It was plausible that both FXR1 and HuR would bind the same region in 3’UTR. The addition of RNAse A abrogated the FXR1/HuR interaction, suggesting that the FXR1/HuR interaction was indirect, a result of tethering by occupancy on the same mRNA. This would also explain why we observed increased FXR1/HuR interaction in TNFα-stimulated VSMC, as TNFα would induce expression of inflammation-inducible transcripts with AREs in their 3’UTR, thus increasing the availability of transcripts for each protein to bind.
Many assumptions on functions of FXR family members are based on the much better characterized FMR protein. Similar to the better characterized FMR protein, FXR1 contains two KH domains for RNA-binding and an RGG box, which is the preferred binding domain of FMRP. Using regions of TNFα 3’UTR, two different, but complementary methods were used to determine that FXR1 binds mRNA, and also validates that FXR1 tethers to mRNA. FXR1 mRNA recognition sites have not been identified. Using biotinylated cRNA pentameric probes representing various sequences of this region as cold competitors, we identified canonical AU-rich elements as a putative FXR1 binding sites, as well as a previously undescribed element comprising the sequence CUUGUGAUU, which is a fourth novel finding of this work. This corroborates experiments showing FXR1 modulation effected stability of transcripts which contained ARE in their 3’UTR. FXR1 recognition of ARE elements also suggested that FXR1 could compete with HuR for occupancy on ARE of inflammatory transcripts. FXR1 also bound the G quadruplex complex, but because this region is not present in the TNFα 3’UTR 50-mer probe, it does not exclude the possibility of competition for a common binding site with HuR on inflammatory transcripts.
RBPs such as HuR can block endonucleolytic cleavage sites to prevent mRNA degradation and therefore increase mRNA stability (Hollams et al., 2002). FXR1 reduced luciferase activity in a reporter driven by the TNFα 3’UTR, further suggesting that FXR1 may act as an mRNA de-stability protein by competing with HuR, which can also bind and stabilize the same region. Competition of FXR1 for HuR binding sites on 3’UTR is a novel function for FXR1 and a unique mechanism to dampen the inflammatory response of select transcripts. Considering FXR1’s reciprocal relationship with HuR, FXR1 could potentially compete with HuR for binding to its own 3’ UTR, reducing HuR mRNA stability, decreasing HuR mRNA abundance, and thus repressing pro-inflammatory gene protein expression. While this study does not rule out the possibility that FXR1 binds to transcripts and targets them for degradation independently of HuR, it does strongly suggest that FXR1 has the potential to oppose HuR and thus act as an mRNA de-stability factor. Many factors involved in RNA stability, as well as transcriptional and translational regulatory proteins localize in discrete cytoplasmic phase-dense stress granules. HuR, and other RBPs such as TTP and TIA-1 have been reported to localize to stress granules (Kedersha et al., 2008). The co-localization of FXR1 with HuR in cytoplasmic stress granules further points to an important role for these proteins in the post-transcriptional regulation of inflammatory mediators and resolution of the cellular inflammatory response.
IL-19 decreases atherosclerosis and vascular restenosis (Ellison et al., 2013, 2014), reduces HuR protein abundance, inflammatory mRNA stability, and abundance of proinflammatory proteins in VSMC (Cuneo et al., 2010; Tian et al., 2008). While some RBPs can be induced by pro-inflammatory stimuli, very little is known about anti-inflammatory effects of RBP as a countervailing mechanism to resolve inflammatory processes. In this report we show that stimulation of hVSMC with IL-19 increases expression of FXR1, placing FXR1 expression as part of the IL-19 anti-inflammatory pathway. We also demonstrate that FXR1 is required for HuR reduction and destabilization by IL-19, further placing FXR1 as an effector protein and part of the anti-inflammatory pathway of IL-19. Our working hypothesis is that FXR1 expression in VSMC is a counter-regulatory mechanism used by VSMC to respond to and dampen vascular inflammation.
This study is the first to describe that FXR1, a muscle-enhanced protein best known as a homologue of the neuronal protein FMR, can be induced in VSMC by anti-inflammatory stimuli. FXR1 can bind to mRNA at AREs and participate in inflammatory transcript processing by competing with HuR, resulting in a reduction in inflammatory mRNA stability. The balance of RNA-binding proteins in homeostasis and pathological conditions has not been well characterized, but based on the opposing functions of HuR and FXR1 and the shared repertoire of transcripts they regulate, we posit an equilibrium between stabilizing and destabilizing RNA-binding proteins is critical to the maintenance of inflammatory and proliferative transcripts, particularly in VSMCs, a non-immune cell-type. This work implicates FXR1 as a novel molecular mediator to resolve vascular inflammation.
Experimental Procedures
VSMC Culture.
Primary human coronary artery vascular smooth muscle cells were obtained as cryopreserved secondary culture from Lonza Corporation [Allendale, NJ, USA] and maintained as we described (Gabunia et al., 2017; Tian et al., 2008). Cells were used from passage 3–5.
Immunohistochemistry.
Ligated mouse carotid arteries, plaque from LDLR−/− mice, and human coronary arteries were collected as part of studies described previously (Ellison et al., 2013, 2014), and described in detail in the Supplemental Information section.
LC-MS/MS.
LC-MS/MS analysis was performed as described previously (Haines et al., 2012) and provided in detail in the Supplemental Information section.
Transfection, siRNA knockdown and overexpression, and luciferase.
Gene silencing was performed using ON-TARGET plus SMARTpool FXR1 siRNA, which contains a mixture of four siRNAs which target human FXR1 (10 nM) purchased from Dharmacon, Inc. (Lafayette, Co, USA) as we have described (Cuneo et al., 2010; Gabunia et al., 2016), and described in detail in Supplemental Information section.
For conditioned media transfer experiments, media was collected from hVSMCs transfected with scrambled control or FXR1 siRNA. Conditioned media from each of these groups was added to serum-starved hVSMC for four hours, RNA isolated and reverse-transcribed for qRT-PCR analysis. For proliferation, HVSMCs were seeded at 20,000 cells/well in FXR1 siRNA conditioned media or scrambled control media. Cells were counted at days 3 and 5 on a Cellometer Auto T4 Bright Field Cell Counter (Nexcelom Bioscience, Lawrence, MA, USA). P<*0.05, **0.01, or ***0.001.
RNA Extraction and Quantitative RT-PCR.
VSMCs were serum starved in 0.1% FCS for 48 hours, then stimulated with 10 ng/mL TNFα for the indicated times. RNA from cultured VSMCs was isolated and reverse transcribed into cDNA, as we have described, and target genes were amplified using an Applied Biosystems StepOne Plus Real-Time PCR System as we described (Gabunia et al., 2016, 2017) and described in detail in Supplemental Information section.
Western blotting and protein determination.
Human VSMC extracts were prepared as described (Ellison et al., 2013; England et al., 2013; Gabunia et al., 2017). Membranes were incubated with a 1:5000–9000 dilution of primary antibody, and a 1:5000 dilution of secondary antibody. Interleukin-1 beta (IL-1β), ICAM-1, FXR1, GAPDH, and HuR were from Santa Cruz Biotechnology (Dallas, TX, USA). Human antigen R (HuR), Fragile-X-mental retardation Protein 1 (FXR1) were from AbCam. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were from Cell Signaling (Danvers, MA, USA). Reactive proteins were visualized using enhanced chemiluminescence (Amersham, Piscataway, NJ, USA) according to manufacturer’s instructions. Relative intensity of bands was normalized to GAPDH, and quantitated by scanning image analysis and the Image J densitometry program.
cEMSA.
FXR1 and GST control fusion proteins were generated using a Genscript kit for GST fusion protein purification, purified from E. coli lysates, and then separated using a protease kit from GE Life Sciences PreScission Protease (Piscataway, NJ, USA). Purified proteins (2μM) were incubated with biotinylated probe (100pM) consisting of nucleotides 1333–1380 in the TNFα 3’UTR (5’CUUGUGAUUAUUUAUUAUUUAUUUAUUAUUUAUUUAUUUACAGAGAUGAA 3’) in binding buffer and glycerol. The binding reaction was run on a 5% precast polyacrylamide gel (Bio-Rad, Hercules, CA, USA). The gel is transferred to nylon membrane, cross-linked with UV light, blocked, streptavidin antibody added, and developed using chemiluminescence. Five non-biotinylated probes were used in a cold competition experiment: (UUAUUUAUU)3, (AUUUA)5 (CUUGUGAUU)4, (CAGAGAUGAA)4, and scrambled control (AUCG)5 were incubated with GST-protein and binding buffer/glycerol for 30 minutes before the addition of the biotinylated TNFα. In Supplemental Figure 3C,D the G quadruplex probe (GGGGUGGGUGGGGGGCAGUGGGGGCUGGGCGGGGGG) was used as a cold competitor to TNFα using the same experimental setup as previously described. Binding affinity was calculated as described (Bechara et al., 2007).
Co-Immunoprecipitation and RNA Immunoprecipitation (RIP).
For Co-IP, hVSMCs were washed three times in PBS and scraped off the dish into a conical tube and centrifuged to form cell pellet. HVSMC extracts were lysed in Lysis Buffer (50mM HEPES, pH 7.5, 70mM KOAc, 5mM Mg(OAc)2 in 0.1M Tris-HCl, pH 8.5 with 0.1g n-dodecyl-B-Maltoside and protease inhibitor) and incubated on a nutator at 4°C for 30 minutes. Cells were centrifuged at 16,600K for 15 minutes. Anti-FLAG M2 Affinity beads (Sigma Aldrich, St. Louis, MO, USA) were washed and added to each sample and incubated on the nutator overnight at 4°C. The samples were centrifuged at 13,000rpm and washed three times in lysis buffer. Sample buffer was added and samples were boiled and frozen or used for western blotting. For RIP, hVSMCs were transduced with AdenoFXR1 prior to serum starvation for 48 hours. Cells were treated with TNFα for 8 hours and lysed in IP buffer with RNase inhibitor. Samples were divided and half were incubated with IgG control beads or Flag-conjugated beads for 4 hours at 25 degrees C. The beads were then centrifuged and washed 5X in IP buffer. Trizol was added to the pelleted beads and RNA was extracted and reverse transcribed to cDNA. We performed qRT-PCR for the transcripts indicated.
Statistical Analysis.
Results are expressed as mean ± SEM. Differences between groups were evaluated with the use of Student’s t-test, or ANOVA, where appropriate. Differences were considered significant when p < 0.05.
Supplementary Material
Highlights:
FXR1 interacts with HuR via mRNA tethering on 3’UTR of inflammatory transcripts
FXR1 is a negative regulator of inflammatory transcript mRNA stability
FXR1 binds canonical and non-canonical sequences in the 3’UTR of TNFα
FXR1 is required for IL-19-dependent reduction of HuR mRNA stability and abundance
Acknowledgements:
This work was supported by grants HL141108 and HL117724 from the National Heart Lung, and Blood Institute of the National Institutes of Health, and grant 13GRNT1685003 from the American Heart Association to MVA. MR and AH were supported by American Heart Association pre-doctoral fellowships 16PRE31220005 and 17PRE33670798. The authors would like to acknowledge the technical expertise of Farah Kako in performing these studies.
Footnotes
Declaration of Interests: The author(s) declare no competing financial interests or any other conflict of interest.
REFERENCES
- Adinolfi S, Bagni C, Musco G, Gibson T, Mazzarella L, and Pastore A (1999). Dissecting FMR1, the protein responsible for fragile X syndrome, in its structural and functional domains. RNA N. Y. N 5, 1248–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguado A, Rodríguez C, Martínez-Revelles S, Avendaño MS, Zhenyukh O, Orriols M, Martínez-González J, Alonso MJ, Briones AM, Dixon DA, et al. (2015). HuR mediates the synergistic effects of angiotensin II and IL-1β on vascular COX-2 expression and cell migration. Br. J. Pharmacol 172, 3028–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allahverdian S, Chehroudi AC, McManus BM, Abraham T, and Francis GA (2014). Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 129, 1551–1559. [DOI] [PubMed] [Google Scholar]
- Bakheet T, Frevel M, Williams BR, Greer W, and Khabar KS (2001). ARED: human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res. 29, 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardoni B, Schenck A, and Mandel JL (2001). The Fragile X mental retardation protein. Brain Res. Bull 56, 375–382. [DOI] [PubMed] [Google Scholar]
- Barreau C, Paillard L, and Osborne HB (2005). AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 33, 7138–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara E, Davidovic L, Melko M, Bensaid M, Tremblay S, Grosgeorge J, Khandjian EW, Lalli E, and Bardoni B (2007). Fragile X related protein 1 isoforms differentially modulate the affinity of fragile X mental retardation protein for G-quartet RNA structure. Nucleic Acids Res. 35, 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballo E, Lai WS, and Blackshear PJ (1998). Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science 281, 1001–1005. [DOI] [PubMed] [Google Scholar]
- Chen Y-L, Huang Y-L, Lin N-Y, Chen H-C, Chiu W-C, and Chang C-J (2006). Differential regulation of ARE-mediated TNFalpha and IL-1beta mRNA stability by lipopolysaccharide in RAW264.7 cells. Biochem. Biophys. Res. Commun 346, 160–168. [DOI] [PubMed] [Google Scholar]
- Cuneo AA, Herrick D, and Autieri MV (2010). Il-19 reduces VSMC activation by regulation of mRNA regulatory factor HuR and reduction of mRNA stability. J. Mol. Cell. Cardiol 49, 647–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W, Zhang G, and Makeyev EV (2012). RNA-binding protein HuR autoregulates its expression by promoting alternative polyadenylation site usage. Nucleic Acids Res. 40, 787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doller A, Pfeilschifter J, and Eberhardt W (2008). Signalling pathways regulating nucleo-cytoplasmic shuttling of the mRNA-binding protein HuR. Cell. Signal 20, 2165–2173. [DOI] [PubMed] [Google Scholar]
- Doran AC, Meller N, and McNamara CA (2008). Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol 28, 812–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubé M, Huot ME, and Khandjian EW (2000). Muscle specific fragile X related protein 1 isoforms are sequestered in the nucleus of undifferentiated myoblast. BMC Genet. 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt W, Doller A, Akool E-S, and Pfeilschifter J (2007). Modulation of mRNA stability as a novel therapeutic approach. Pharmacol. Ther 114, 56–73. [DOI] [PubMed] [Google Scholar]
- Ellison S, Gabunia K, Kelemen SE, England RN, Scalia R, Richards JM, Orr AW, Orr W, Traylor JG, Rogers T, et al. (2013). Attenuation of experimental atherosclerosis by interleukin-19. Arterioscler. Thromb. Vasc. Biol 33, 2316–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison S, Gabunia K, Richards JM, Kelemen SE, England RN, Rudic D, Azuma Y-T, Munroy MA, Eguchi S, and Autieri MV (2014). IL-19 Reduces Ligation-Mediated Neointimal Hyperplasia by Reducing Vascular Smooth Muscle Cell Activation. Am. J. Pathol 184, 2134–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England RN, Preston KJ, Scalia R, and Autieri MV (2013). Interleukin-19 decreases leukocyte-endothelial cell interactions by reduction in endothelial cell adhesion molecule mRNA stability. AJP Cell Physiol. 305, C255–C265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredman G, and Tabas I (2017). Boosting Inflammation Resolution in Atherosclerosis: The Next Frontier for Therapy. Am. J. Pathol 187, 1211–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabunia K, Ellison S, Kelemen S, Kako F, Cornwell WD, Rogers TJ, Datta PK, Ouimet M, Moore KJ, and Autieri MV (2016). IL-19 Halts Progression of Atherosclerotic Plaque, Polarizes, and Increases Cholesterol Uptake and Efflux in Macrophages. Am. J. Pathol 186, 1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabunia K, Herman AB, Ray M, Kelemen SE, England RN, DeLa Cadena R, Foster WJ, Elliott KJ, Eguchi S, and Autieri MV (2017). Induction of MiR133a expression by IL-19 targets LDLRAP1 and reduces oxLDL uptake in VSMC. J. Mol. Cell. Cardiol 105, 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallouzi IE, and Steitz JA (2001). Delineation of mRNA export pathways by the use of cell-permeable peptides. Science 294, 1895–1901. [DOI] [PubMed] [Google Scholar]
- Garnon J, Lachance C, Di Marco S, Hel Z, Marion D, Ruiz MC, Newkirk MM, Khandjian EW, and Radzioch D (2005). Fragile X-related protein FXR1P regulates proinflammatory cytokine tumor necrosis factor expression at the post-transcriptional level. J. Biol. Chem 280, 5750–5763. [DOI] [PubMed] [Google Scholar]
- Hansson GK, and Libby P (2006). The immune response in atherosclerosis: a double-edged sword. Nat. Rev. Immunol 6, 508–519. [DOI] [PubMed] [Google Scholar]
- Hollams EM, Giles KM, Thomson AM, and Leedman PJ (2002). MRNA stability and the control of gene expression: implications for human disease. Neurochem. Res 27, 957–980. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Tisdale S, Hickman T, and Anderson P (2008). Real-time and quantitative imaging of mammalian stress granules and processing bodies. Methods Enzymol. 448, 521–552. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy P, Lambers E, Verma S, Thorne T, Qin G, Losordo DW, and Kishore R (2010). Myocardial knockdown of mRNA-stabilizing protein HuR attenuates post-MI inflammatory response and left ventricular dysfunction in IL-10-null mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 24, 2484–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P (2002). Inflammation in atherosclerosis. Nature 420, 868–874. [DOI] [PubMed] [Google Scholar]
- Libby P, Geng YJ, Sukhova GK, Simon DI, and Lee RT (1997). Molecular determinants of atherosclerotic plaque vulnerability. Ann. N. Y. Acad. Sci 811, 134–142; discussion 142–145. [DOI] [PubMed] [Google Scholar]
- Libby P, Tabas I, Fredman G, and Fisher EA (2014). Inflammation and its resolution as determinants of acute coronary syndromes. Circ. Res 114, 1867–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D, Wright Z, Couzens AL, Lambert J-P, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, et al. (2013). The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mientjes EJ, Willemsen R, Kirkpatrick LL, Nieuwenhuizen IM, Hoogeveen-Westerveld M, Verweij M, Reis S, Bardoni B, Hoogeveen AT, Oostra BA, et al. (2004). Fxr1 knockout mice show a striated muscle phenotype: implications for Fxr1p function in vivo. Hum. Mol. Genet 13, 1291–1302. [DOI] [PubMed] [Google Scholar]
- Musunuru K (2003). Cell-specific RNA-binding proteins in human disease. Trends Cardiovasc. Med 13, 188–195. [DOI] [PubMed] [Google Scholar]
- Palanisamy V, Jakymiw A, Van Tubergen EA, D’Silva NJ, and Kirkwood KL (2012). Control of Cytokine mRNA Expression by RNA-binding Proteins and microRNAs. J. Dent. Res 91, 651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng SS, Chen CY, and Shyu AB (1996). Functional characterization of a non-AUUUA AU-rich element from the c-jun proto-oncogene mRNA: evidence for a novel class of AU-rich elements. Mol. Cell. Biol 16, 1490–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullmann R, Juhaszova M, de Silanes IL, Kawai T, Mazan-Mamczarz K, Halushka MK, and Gorospe M (2005). Enhanced Proliferation of Cultured Human Vascular Smooth Muscle Cells Linked to Increased Function of RNA-binding Protein HuR. J. Biol. Chem 280, 22819–22826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross R (1999). Atherosclerosis--an inflammatory disease. N. Engl. J. Med 340, 115–126. [DOI] [PubMed] [Google Scholar]
- Schoenberg DR, and Maquat LE (2012). Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet 13, 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer CA, Salinthone S, Baker KJ, and Gerthoffer WT (2004). Synthesis of immune modulators by smooth muscles. BioEssays News Rev. Mol. Cell. Dev. Biol 26, 646–655. [DOI] [PubMed] [Google Scholar]
- Tian Y, Sommerville LJ, Cuneo A, Kelemen SE, and Autieri MV (2008). Expression and suppressive effects of interleukin-19 on vascular smooth muscle cell pathophysiology and development of intimal hyperplasia. Am. J. Pathol 173, 901–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber C, and von Hundelshausen P (2017). CANTOS Trial Validates the Inflammatory Pathogenesis of Atherosclerosis: Setting the Stage for a New Chapter in Therapeutic Targeting. Circ. Res 121, 1119–1121. [DOI] [PubMed] [Google Scholar]
- Wu T, Shi J-X, Geng S, Zhou W, Shi Y, and Su X (2016). The MK2/HuR signaling pathway regulates TNF-α-induced ICAM-1 expression by promoting the stabilization of ICAM-1 mRNA. BMC Pulm. Med 16, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.