

Abstract
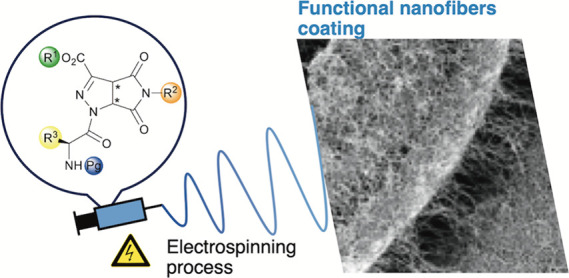
We prepared a small library of short peptidomimetics based on 3-pyrrolo-pyrazole carboxylate, a non-coded γ-amino acid, and glycine or alanine. The robust and eco-friendly synthetic approach adopted allows to obtain the dipeptides in two steps from commercial starting materials. This gives the possibility to shape these materials by electrospinning into micro- and nanofibers, in amounts required to be useful for coating surfaces of biomedical relevance. To promote high quality of electrospun fibers, different substitution patterns were evaluated, all for pure peptide fibers, free of any polymer or additive. The best candidate, which affords a homogeneous fibrous matrix, was prepared in larger amounts, and its biocompatibility was verified. This successful work is the first step to develop a new biomaterial able to produce pristine peptide-based nanofibers to be used as helpful component or stand-alone scaffolds for tissue engineering or for the surface modification of medical devices.
Introduction
Nanofibers produced by electrospinning provide a promising platform for a huge variety of applications, from energy generation and storage, water treatment and environmental remediation, to healthcare and biomedical engineering.1−4 This technique allows to produce, also on a large scale, fibers with a tunable diameter, ranging from nano- to several micrometers.5 The fibers are usually arranged in a nonwoven mat; hence, the fibers themselves exhibit a high surface-to-volume ratio and, in many cases, also a well-defined surface structure, but the mat is sponge-like, that is, has a very high porosity, which was very useful in the first documented application of aerosol filters in gas masks.3 The fiber materials reported in the large majority of the literature and, in almost all applications, are synthetic polymers. Less well documented are fibers that are composed of supramolecular assemblies, such as surfactants or host–guest complexes, for example, based on cyclodextrin.6
Considering that the development of biomaterials to improve human life is nowadays one of the hot topic in material science, a promising approach in this area involves the use of biocompatible and biodegradable polymers, some of which can be used to direct tissue repair and regeneration while providing temporary structural support for cells.2,7 However, polymer fibers often require extensive additional research to develop a biocompatible functional coating. Such a coating modifies surface charge, wettability, chemical affinity, and hydrophilicity, thus enhancing the performance in biological environment.8,9 A potentially simpler strategy combines biomolecules with synthetic polymers.10 Thus, generated fibers for tissue engineering can promote the exchange of nutrients and metabolites, while assuring structural integration into the newly formed tissue.
Obviously, ideal would be using only one material that provides all functions. In this regard, proteins2,11 and poly/oligo-peptides10,12,13 should be the perfect starting materials, based on their multiple role as a catalyst, scaffold, surfactant, and forth. Drawbacks in using pure proteins are the need of high voltages to produce nanofibers and the requirement of organic solvents, which often induce denaturation.14 A valid alternative to overcome these limits has been found in using synthetic or recombinant peptides that can be produced on a largest scale. Peptides are often characterized by specific secondary structures, which, together with the rather small size, can allow the natural self-assembly into fibers (or tubes) through noncovalent interactions.15−18 Although this process, usually achieved by adding a poor solvent, differs from the forced assembly by electrical fields, self-assembling peptides are usually also good candidates to generate high-quality electrospun fibers, since it might be due to the high anisotropy of oligomers, which should be present in both processes.
Only few papers report the electrospinning of very short peptide sequences. Bittner et al.19,20 first focused on electrospinning of diphenylalanine where the π,π-interactions possibly play a vital role in the supramolecular assembly to fibers. Other short peptides with different length are now being investigated for a range of biomedical applications.13
Applications such as tissue engineering require high proteolytic stability, which is typically inherent in most polymers but not at all in peptides. On the other hand, non-coded amino acids in the peptide sequence afford peptidomimetics, which are in general characterized by a well-defined stable conformation,20−24 possessing the features of the target peptide and granting stability against proteases. Recently, we demonstrated that short peptidomimetics containing a pyrazole-isothiazole scaffold can be electrospun into solid, continuous, homogeneous, and bead-free quasi-endless micro- and nanofibers.25 A limitation in using this scaffold is related to its long, multistep synthesis (7 steps; 22–23% overall yield) in noneco-friendly conditions.
In order to continue our researches in finding noncoded amino acids by using cycloaddition reactions,26−28 we here present a suitable alternative that avoids complex combinations (polymer and coating) and cumbersome syntheses, that is, the use of a pristine peptide for electrospinning of biocompatible nanofibers. The non-coded amino acids are synthesized by a single step, scalable, and straightforward cycloaddition reaction by commercially available starting materials, that are diazoalkanes and maleimids,29−31 which create pyrrolo-pyrazole building blocks 3 simulating γ-amino acids (Scheme 1).
Scheme 1. Synthesis of Pyrrolo-pyrazole Derivatives 3 and Dipeptides 4.
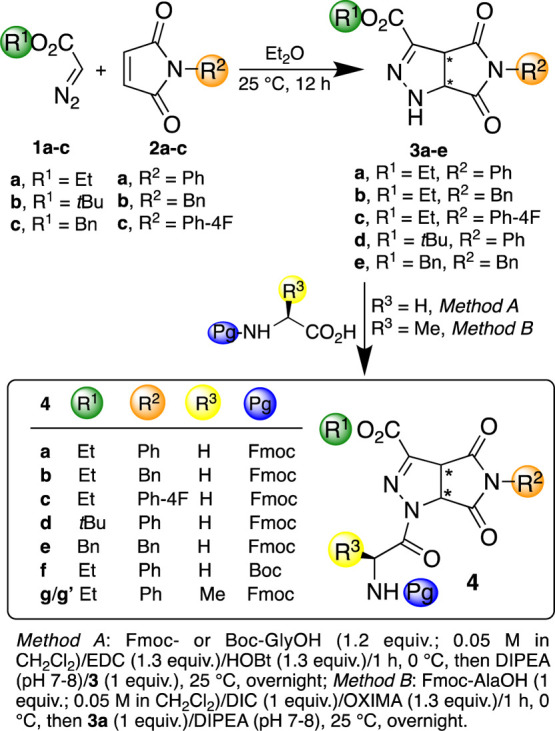
We prepared a small library of peptidomimetics 4 by varying the substitution pattern of the scaffold and of the natural amino acid. The best candidate 4a (Scheme 1), which was prepared in large amounts and electrospun from solution, affords a homogeneous nanofibrous matrix, which deposited on glass, silicon, and aluminum. We evaluated the biocompatibility of 4a-based fibers in order to elucidate potential biomedical applications.
Experimental Section
Chemistry General
NMR spectroscopic experiments were carried out either on Varian MERCURY 300 MHz (300 and 75 MHz for 1H and 13C, respectively) or Bruker Avance I 400 MHz spectrometers (400 and 101 MHz for 1H and 13C, respectively). Chemical shifts (δ) are given in ppm relative to the CHCl3 internal standard, and the coupling constants J are reported in Hertz (Hz). Mass spectra were recorded on an LCQESI MS and on a LCQ Advantage spectrometer from Thermo Finningan and a LCQ Fleet spectrometer from Thermo Scientific. Optical rotations were measured on a PerkinElmer 343 polarimeter (concentration in g/100 mL). Diazoalkanes 1a–c and maleimides 2a–c are commercially available compounds.
General Procedure for the Cycloaddition Reaction: Synthesis of Compounds 3
Maleimide 2 (2.89 mmol) was dissolved in Et2O (50 mL), and diazoacetate 1 (3.18 mmol, 15% in toluene) was added. The reaction mixture was stirred at room temperature overnight and monitored by TLC (AcOEt/hexane, 4:6). A solid was formed in the case of 3b,e that was filtered and washed with a few drops of Et2O. In the case of 3a,c,d, the solvent was evaporated at reduced pressure and the pure products were isolated after purification with a Biotage Isolera One Flash Chromatography System (hexane/AcOEt, from 100% to 40:60).
Ethyl 4,6-Dioxo-5-phenyl-1,3a,4,5,6,6a-hexahydropyrrolo[3,4-c]pyrazole-3-carboxylate (3a)
White solid. Yield: 80%; mp 193–194 °C, dec (CHCl3/Et2O; 193–194 °C29); 1H NMR (300 MHz, CDCl3): δ 7.51–7.25 (m, 5H), 7.05 (brs, 1H), 5.07, 4.72 (AX system, 2H, J = 10.8 Hz), 4.45–4.29 (m, 2H), 1.38 (t, 3H, J = 6.8 Hz).
Ethyl 4,6-Dioxo-5-benzyl-1,3a,4,5,6,6a-hexahydropyrrolo[3,4-c]pyrazole-3-carboxylate (3b)
White solid. Yield: 75%; mp 146–148 °C, dec (Et2O; 147–148 °C30); 1H NMR (300 MHz, CDCl3): δ 7.36–7.26 (m, 5H), 6.96 (s, 1H), 4.88, (dd, 1H, J = 10.8, 1.6 Hz), 4.65 (s, 2H), 4.54 (d, 1H, J = 10.8 Hz), 4.35 (q, 2H, J = 7.1 Hz), 1.36 (t, 3H, J = 7.1 Hz).
Ethyl 4,6-Dioxo-5-(4-F-phenyl)-1,3a,4,5,6,6a-hexahydropyrrolo[3,4-c]pyrazole-3-carboxylate (3c)
White solid. Yield: 65%; mp 125–126 °C, dec.; 1H NMR (300 MHz, DMSO-d6): δ 9.67 (s, 1H), 7.34–7.27 (m, 4H), 5.07, 4.63 (AX system, 2H, J = 11.3 Hz), 4.28–4.11 (m, 2H), 1.22 (t, 3H, J = 7.1 Hz); 13C NMR (75 MHz, DMSO-d6): δ 174.1, 172.4, 162.0 (JC–F = 249.5 Hz), 161.4, 134.1, 129.7 (JC–F = 9.0 Hz), 128.7 (JC–F = 2.8 Hz), 116.4 (JC–F = 22.9 Hz), 65.7, 60.7, 52.2, 14.6; MS (ESI+) m/z calcd for C14H12FN3O4 [M+] 305.27; found, 328.37 [M + Na]+. Anal. calcd for C14H12FN3O4: C, 55.08; H, 3.96; N, 13.77. Found: C, 54.98; H, 4.04; N, 13.603.
ter-Butyl 4,6-Dioxo-5-phenyl-1,3a,4,5,6,6a-hexahydropyrrolo[3,4-c]pyrazole-3-carboxylate (3d)
White solid. Yield: 89%; mp 190–193 °C, dec (CHCl3/Et2O); 1H NMR (300 MHz, CDCl3): δ 7.52–7.35 (m, 3H), 7.30–7.20 (m, 2H), 6.95 (s, 1H), 5.02, 4.66 (AX system, 2H, J = 10.8 Hz), 1.57 (s, 9H); 13C NMR (75 MHz, CDCl3): δ 173.4, 170.4, 159.6, 138.7, 131.1, 129.3, 129.1, 126.2, 83.3, 63.6, 51.9, 28.1. MS (ESI-) m/z calcd for C16H17N3O4 315.33; found, 314.68 [M-1]. Anal. calcd for C16H17N3O4: C, 60.94; H, 5.43; N, 13.33. Found: C, 60.83; H, 5.51; N, 13.22.
Benzyl 4,6-Dioxo-5-benzyl-1,3a,4,5,6,6a-hexahydropyrrolo[3,4-c]pyrazole-3-carboxylate (3e)
White solid. Yield: 78%; mp 123–125 °C, dec (Et2O); 1H NMR (400 MHz, CDCl3): δ 7.48–7.26 (m, 10H), 7.02 (s, 1H), 5.34, 5.30 (AB system, 2H, J = 11.8 Hz), 4.86, 4.54 (AX system, 2H, J = 10.4 Hz), 4.65 (s, 2H); 13C NMR (100.7 MHz, CDCl3): δ 173.6, 171.2, 160.5, 136.4, 135.2, 134.8, 128.8, 128.62, 128.60, 128.5, 128.3, 67.4, 63.8, 51.6, 43.2; MS (ESI) m/z calcd for C20H17N3O4 363.37; found, 386.51 [M + Na]+. Anal. calcd for C20H17N3O4: C, 66.11; H, 4.72; N, 11.56. Found: C, 65.96; H, 4.79; N, 11.44
General Procedure for the Synthesis of Compounds 4
Fmoc-glycine-OH (342 mg, 1.2 mmol) or Boc-glycine-OH (210 mg, 1.2 mmol) was dissolved in CH2Cl2 (0.05 M solution) at 0 °C under magnetic stirring. EDC (202 mg, 1.3 mmol) and HOBt (176 mg, 1.3 mmol) were added, and the solution was stirred for 1 h at 0 °C. Then, compound 3 (1 equiv) was added and the pH was corrected to 7–8 with DIPEA (364 μL). The reaction was stirred at 25 °C overnight. The mixture was washed with a solution of KHSO4 (5% w/v), then with a saturated solution of NaHCO3 (20 mL) and finally with a saturated solution of NaCl (20 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure. Pure products 4a–f were purified by a Biotage Isolera One Flash Chromatography System affording white solids.
Ethyl 3aR*,6aR*-1-(Fmoc-glycyl)-4,6-dioxo-5-phenyl-1,3a,4,5,6,6a-hexahydropyrrolo[3,4-c]pyrazole-3-carboxylate (4a)
TLC: AcOEt/hexane, 3:7; column chromatography: hexane/AcOEt, from 100% hexane to 50:50. Yield: 74%; mp 151–152 °C, dec.; 1H NMR (300 MHz, CDCl3): δ 7.76 (d, 2H, J = 7.4 Hz), 7.61 (d, 2H, J = 7.2 Hz), 7.50–7.20 (m, 9H), 5.81, 4.88 (AX system, 2H, J = 10.3 Hz), 5.69–5.49 (m 1H), 4.60 (dd, 1H, J = 18.2, 5.7 Hz), 4.50–4.30 (m, 5H), 4.27–4.19 (m, 1 H), 1.39 (t, 3H, J = 7.5 Hz); 13C NMR (100.7 MHz, CDCl3): δ 169.8, 169.0, 168.8, 159.7, 156.6, 143.8, 143.5, 141.28, 141.26, 131.0, 129.3, 129.1, 127.74, 127.72, 127.1, 126.3, 125.25, 125.22, 120.0, 119.9, 67.3, 62.9, 61.6, 53.0, 47.1, 43.8, 14.1; MS (ESI+) m/z calcd for C31H26N4O7: 566.57, found: 589.62 [M + Na]+. Anal. calcd for C31H26N4O7: C, 65.72; H, 4.63; N, 9.89. Found: C, 65.51; H, 4.77; N, 9.64.
Ethyl 3aR*,6aR*-1-(Fmoc-glycyl)-4,6-dioxo-5-benzyl-1,3a,4,5,6,6a-hexahydropyrrolo[3,4-c]pyrazole-3-carboxylate (4b)
TLC: AcOEt/hexane, 4:6; column chromatography: hexane/AcOEt, from 100% hexane to 60:40. Yield: 50%; mp 148–149 °C, dec (CHCl3/iPr2O); 1H NMR (400 MHz, CDCl3): δ 7.78 (d, 2H, J = 7.4 Hz), 7.67–7.58 (m, 2H), 7.45–7.23 (m, 9H), 5.62, 4.72 (AX system, 2H, J = 9.8 Hz), 5.60–5.52 (m,1H), 4.64 (s, 2H), 4.56 (dd, 1H, J = 19.1, 5.5 Hz), 4.78–4.35 (m, 4H), 4.35 (dd, 1H, J = 18.4, 4.0), 4.28–4.23 (m, 1H), 1.42 (t, 3H, J = 7.9 Hz); 13C NMR (100.7 MHz, CDCl3): δ 169.7, 168.8 168.6, 159.4, 156.4, 143.86, 143.81, 143.1, 141.3, 134.4, 129.2, 128.8, 128.5, 127.7, 127.1, 125.2, 120.0, 67.3, 62.9, 61.1, 52.6, 47.1, 43.67, 43.61, 14.1; MS (ESI-) m/z calcd for C32H28N4O7: 580.60, found: 579.49 [M – 1], 580.49 [M]. Anal. calcd for C32H28N4O7: C, 66.20; H, 4.86; N, 9.65. Found: C, 65.58; H, 4.95; N, 9.55.
Ethyl 3aR*,6aR*-1-(Fmoc-glycyl)-4,6-dioxo-5(4-F-phenyl-)1,3a,4,5,6,6a-hexahydropyrrolo[3,4-c]pyrazole-3-carboxylate (4c)
TLC: AcOEt/hexane, 3:7; column chromatography: hexane/AcOEt, from 100% hexane to 50:50. Yield: 73%; mp 144–145 °C, dec (CHCl3/iPr2O); 1H (300 MHz, CDCl3): δ 7.76, 7.61 (AA′XX′ system, 4H, J = 7.4 Hz), 7.43–7.35 (m, 2H), 7.34–7.27 (m, 2H), 7.27–7.08 (m, 4H), 5.81, 4.87 (AX system, 2H, J = 9.9 Hz), 5.54 (t, 1H, J = 5.5 Hz), 4.59 (dd, 1H, J = 18.2, 6.1), 4.48–4.31 (m, 5H), 4.26–4.19 (m, 1H), 1.39 (t, 2H, J = 6.9 Hz); 13C NMR (75 MHz, CDCl3): δ 168.9, 168.6, 168.0, 162.4 (JC–F,= 249.5 Hz), 159.3, 156.4, 143.79, 143.77, 143.2, 141.29, 141.27, 128.0 (JC–F,= 8.8 Hz), 127.7, 127.1, 126.7 (JC–F,= 3.0 Hz), 125.1, 120.0, 116.3 (JC–F,= 21.8 Hz), 67.3, 63.0, 61.1, 52.6, 47.1, 43.7, 14.0; MS (ESI+) m/z calcd for C31H25FN4O7: 584.56, found: 585.05 [M+1]. Anal. calcd for C31H25FN4O7: C, 63.70; H, 4.31; N, 9.58. Found: C, 63.49; H, 4.48; N, 9.40.
tButyl 3aR*,6aR*-1-(Fmoc-glycyl)-4,6-dioxo-5-pheny1,3a,4,5,6,6a-hexahydropyrrolo[3,4-c]pyrazole-3-carboxylate (4d)
TLC: AcOEt/hexane, 1:1; column chromatography: hexane, 100% to hexane/AcOEt, 50:50. Yield: 72%; mp 163–164 °C, dec (CHCl3/iPr2O); 1H NMR (400 MHz, CDCl3): δ 7.76 (d, 2H, J = 7.5 Hz), 7.64–7.57 (m, 2H), 7.50–7.20 (m, 9H), 5.77, 4.83 (AX system, 2H, J = 10.2 Hz), 5.54 (t, 1H), 4.61 (dd, 1H, J = 19.0, 5.6 Hz), 4.36–4.33 (m, 4H), 4.27–4.20 (m, 1H), 1.59 (s, 9H); 13C NMR (100.7 MHz, CDCl3): δ 169.1, 168.7, 168.2, 158.2, 156.4, 144.7, 143.80, 143.77, 141.3, 130.8, 129.3, 129.2, 127.7, 127.1, 126.2, 125.2, 120.0, 85.1, 67.3, 61.0, 52.8, 47.1, 43.7, 27.9; MS (ESI+) m/z calcd for C33H30N4O7: 594.62, found: 617.47 [M + Na]+. Anal. calcd for C33H30N4O7: C, 66.66; H, 5.09; N, 9.42. Found: C, 66.43; H, 5.23; N, 9.28.
Benzyl 3aR*,6aR*-1-(Fmoc-glycyl)-4,6-dioxo-5-benzyl-1,3a,4,5,6,6a-hexahydropyrrolo[3,4-c]pyrazole-3-carboxylate (4e)
Method A; column chromatography: hexane 100% to hexane/AcOEt, 6:4. Yield: 73%; mp 119–120 °C, dec (CH2Cl2/tBuOMe); 1H NMR (300 MHz, CDCl3): δ 7.75 (d, 2H, J = 7.4 Hz), 7.64–7.56 (m, 2H), 7.48–7.22 (m, 14H), 5.57, 4.69 (AX system, 2H, J = 10.4 Hz), 5.50, 4.50, 4.28 (ABX system, 3H, J = 18.4, 5.8, 4.5 Hz), 5.36 (s, 2H), 4.62 (s, 2H), 4.43–4.32 (m, 2H), 4.56–4.19 (m, 1H); 13C NMR (100.7 MHz, CDCl3): δ 169.7, 168.8 168.6, 159.4, 156.4, 143.82, 143.78, 142.8, 141.28, 141.25, 134.4, 129.2, 128.8, 128.78, 128.73, 128.5, 127.7, 127.1, 125.2, 120.0, 68.4, 67.3, 61.2, 52.6, 47.1, 43.6 (x 2); MS (ESI+) m/z calcd for C37H30N4O7: 642.67, found: 665.34 [M + Na]+. Anal. calcd for C37H30N4O7: C, 69.15; H, 4.71; N, 8.72. Found: C, 68.90; H, 4.84; N, 8.51.
Ethyl 3aR*,6aR*-1-(Boc-glycyl)-4,6-dioxo-5-phenyl-1,3a,4,5,6,6a-hexahydropyrrolo[3,4-c]pyrazole-3-carboxylate (4f)
TLC: AcOEt/hexane, 4:6; column chromatography: hexane/AcOEt, from 100% to 60:40. Yield: 70%; mp 147–148 °C, dec (CHCl3/iPr2O); 1H NMR (300 MHz, CDCl3): δ 7.50–7.20 (m, 5H), 5.80, 4.88 (AX system, 2H, J = 10.6 Hz), 5.23 (dd, 1H, J = 5.7, 4.3 Hz), 4.60 (dd, 1H, J = 18.2, 5.7 Hz), 4.46–4.35 (m, 2H), 4.28 (dd, 1H, J = 18.2, 4.3 Hz), 1.46 (s, 9H), 1.39 (t, 3H, J = 7.1 Hz); 13C NMR (75 MHz, CDCl3): δ 169.0, 168.2, 159.5, 143.0, 130.9, 129.24, 129.17, 126.2, 80.1, 62.8, 61.1, 52.6, 43.4, 28.3, 14.1; MS (ESI+) m/z calcd for C21H24N4O7: 444.44, found: 467.32 [M + Na]+. Anal. calcd for C21H24N4O7: C, 56.75; H, 5.44; N, 12.61. Found: C, 56.52; H, 5.57; N, 12.49.
Ethyl 3aR*,6aR*-1-(Fmoc-alanyl)-4,6-dioxo-5-phenyl-1,3a,4,5,6,6a-hexahydropyrrolo[3,4-c]pyrazole-3-carboxylate (4g/4′g)
Fmoc-alanine-OH (218 mg, 0.7 mmol) was dissolved in CH2Cl2 (0.05 M solution) at 0 °C under magnetic stirring. DIC (88.3 mg, 110 μL, 0.7 mmol) and OXIMA (156 mg, 1.1 mmol) were added to the solution, and the stirring was continued for 1 h at 0 °C. Then, compound 3a (201 mg, 0.7 mmol) was added and the pH was corrected with DIPEA (215 μL). The reaction was monitored by TLC (AcOEt/hexane, 1:1). The mixture was stirred at 25 °C overnight. The solution was washed with a solution of KHSO4 (5% w/v), then with a saturated solution of NaHCO3 (20 mL) and a saturated solution of NaCl (52 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure. Pure diastereoisomers 4g/4′g were separated by a Biotage Isolera One Flash Chromatography System (from hexane 100% to hexane/AcOEt 50:50) as white solids.
3aR,6aR-4g
Stereochemistry arbitrary assigned. Yield: 35%; mp 208–209 °C, dec (CHCl3/iPr2O); [α]D29.3 −2.3 (c 1.00, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 7.79 (d, 2H, J = 7.4 Hz), 7.64–7.56 (m, 2H), 7.48–7.22 (m, 9H), 5.85, 4.88 (AX system, 2H, J = 10.2 Hz), 5.54 (d, 1H, J = 8.4 Hz), 5.23–5.11 (m, 1H), 4.47–4.29 (m, 4H), 4.26–4.18 (m, 1H), 1.52 (d, 3H, J = 6.8 Hz), 1.39 (t, 3H, J = 7.1 Hz); 13C NMR (75 MHz, CDCl3): δ 172.6, 169.4, 168.3, 159.5, 155.8, 143.8, 143.7, 143.3, 141.29, 141.26, 130.8, 129.3, 129.2, 127.7, 127.1, 126.1, 125.2, 125.1, 120.0, 67.2, 62.9, 61.0, 52.6, 48.8, 47.1, 18.6, 14.1; MS (ESI+) m/z calcd for C32H28N4O7: 580.60, found: 603.93.32 [M + Na]+. Anal. calcd for C32H28N4O7: C, 66.20; H, 4.86; N, 9.65. Found: C, 69.93; H, 4.98; N, 9.46.
3aS,6aS-4′g
Stereochemistry arbitrary assigned. Yield: 34%; mp 159–160 °C, dec (CHCl3/iPr2O); [α]D29.7 + 2.08 (c 1.00, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.77 (d, 2H, J = 7.2 Hz), 7.65–7.59 (m, 2H), 7.50–7.24 (m, 9H), 5.77, 4.92 (AX system, 2H, J = 9.5 Hz), 5.67 (d, 1H, J = 6.7 Hz), 5.29–5.18 (m, 1H), 4.49–4.35 (m, 4H), 4.25–4.18 (m, 1H), 1.46 (d, 3H, J = 6.6); 1.42 (t, 3H, J = 7.0 Hz); 13C NMR (75 MHz, CDCl3): δ 172.1, 168.6, 168.0, 159.5, 155.3, 143.9, 143.8, 142.9, 141.28, 141.26, 130.8, 129.3, 129.2, 127.7, 127.1, 126.2, 125.1, 120.0, 67.1, 63.0, 61.1, 52.7, 49.2, 47.1, 19.0, 14.1; MS (ESI+) m/z calcd for C32H28N4O7: 580.60; found: 581.67 [M+1], 604.37 [M + Na]+. Anal. calcd for C32H28N4O7: C, 66.20; H, 4.86; N, 9.65. Found: C, 65.91; H, 5.01; N, 9.40.
Nanofiber Preparation
Nanofibers were prepared by using Nanon-01A (MEEC Instruments, Ltd., Ogori-shi, Fukuoka, Japan) equipped with a dehumidificator. Dipeptides 4 were dissolved in hexafluoro-2-propanol (HFIP) at the concentration of 30 wt %, as previously optimized. HFIP was an optimal solvent since it solubilizes all the compound tested and it has a boiling temperature of 58 °C suitable for the electrospinning process. Dipeptide solutions were injected through a 20G needle (Nordson EFD, Norson Italia SpA, Segrate, Italy); the nanofibers were collected on plane glass slides (22 × 22 mm, thickness 0.15 mm). The distance between needle and collector was set at 15 cm, and the environmental conditions (temperature and humidity) were monitored. To investigate the optimal process parameters, different voltages (20, 25, and 28 kV) and feed rates (0.2, 0.5, and 0.8 mL/h) were tested. After electrospinning, fabrics were kept under vacuum for 2 h and then stored in a desiccator until the following tests.
Nanofiber Characterization
Nanofibers were preliminary observed by a microscope Leica DM IL LED equipped with a digital C-mount camera TP 5200 (Sony color CDD) with 20× and 40× magnification. Further morphological characterization was performed by scanning electron microscopy (SEM, TESCAN MIRA 3, Tescan, Brno, Czech Republic) after sample coating with gold in an argon atmosphere. Images were obtained at 5 kV by using magnifications of 500× and 100 and 200 k×, and they were elaborated to measure mean sizes and size distributions. Atomic force microscopy (Horiba SmartSPM, Kyoto, Japan) was used to assess the roughness of the fiber surfaces.
Evaluation of 4a Wettability by Contact Angle Measurement
4a underwent the electrospinning process at the optimized condition. The wettability of 4a nanofibers mat was evaluated by a contact angle meter DME-211 Plus with FAMAS software. Wettability was expressed as contact angle (θ) measured by dropping 1 μL of distilled water and after a contact time of 9 s. The results were expressed as mean ± standard deviation (n = independent experiments).
In Vitro Degradation Study
The nanofibers obtained by 4a were tested to evaluate the degradation behavior in a simulated physiological condition. 4a solution (30% w/w, in HFIP) was loaded into a syringe (1 mL) and electrospun through a 20 G needle (Nordson EFD) on circular glass coverslips (1 cm diameter) placed on the flat collector. The electrospinning process was carried out at a voltage of 25 kV and a flow rate of 0.5 mL/h. The distance to the collector was set at 15 cm. The process was performed in controlled environmental conditions: internal temperature of 26.7 °C and relative humidity of 23%. The solution of 4a was electrospun for 1, 3, 10, and 15 min. Glass coverslips (n = 3 for each timing) were weighed before and after the spinning process to determine the average amount of deposited fibers (100, 300, 700, and 900 μg, respectively). Nanofibers were incubated at 37 °C enbibed in filtered Dulbecco’s Modified Eagle Medium (DMEM) at pH 8, 7.4, and 7.2 with FBS 10% v/v. At scheduled time points (10, 30, 60, 120, 240, and 360 min), nanofibers were recovered and observed at the microscope; images were elaborated by ImageJ software.
At the same incubation time points, the related incubation media were withdrawn and stored for the biocompatibility test (see below).
Cell Culture
Normal human dermal fibroblasts (NHDFs) were used for the biocompatibility study (see below). Cells were cultured in DMEM culture medium and added with FBS (10% v/v) and penicillin/streptomycin (1% v/v) at 37 °C, in a humidified atmosphere, containing 5% CO2.
Evaluation of 4a-Based Electrospun Matrix Biocompatibility
Ten thousand NHDFs were seeded in a 96-well plate and incubated at 37 °C, 5% CO2 for 24 h to reach the 70% of confluence. After that, cell media were withdrawn and NHDFs were treated for 24 h with 200 μL of the incubation media from in vitro degradation study. Cells treated with fresh media were used as control (CTRL). Cell viability was evaluated by the 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazole bromide (MTT) assay.32 The results are expressed as a percentage of cell viability calculated as
| 1 |
Results and Discussion
Synthetic Procedures
A small library of pyrrolo-pyrazole compounds 3, differing for the ester function and the residue at nitrogen of maleimide, was prepared by using a 1,3-dipolar cycloaddition reaction29−31 from commercially available diazoalkanes 1a–c (toluene solution) and maleimides 2a–c operating in Et2O at 25 °C (12 h). As expected, compounds 3a–e were obtained in good to excellent yields (68–89%) as single regioisomer (Δ2-pyrazoline) and as a mixture of enantiomers (Scheme 1).
Compounds 3 were then made to react with glycine derivatives (Scheme 1). Several attempts were tested to find the best reaction conditions. Accordingly, Fmoc- or Boc-GlyOH (1.2 equiv) was first activated with 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC; 1.3 equiv) and 1-hydroxybenzotriazole (HOBt; 1.3 equiv) operating in CH2Cl2 (0.05 M, 1 h, 0 °C). Then, compound 3 (1 equiv) was added, and the pH was adjusted with diisopropylethylamine (DIPEA; pH 7–8). After stirring at 25 °C overnight, peptides 4a–f were isolated in good yields (50–74%) after purification.
Since we succeeded in the formation of good quality of electrospun fibers starting from 4a (see below), scaffold 3a was made to react also with Fmoc-Ala (Scheme 1). Very low yields of the corresponding diastereoisomers 4g/4′g were achieved by using the above conditions. The use of 1,3-disopropylcarbodiimide (DIC, 1 equiv)/ethyl cyanohydroxyiminoacetate (OXIMA, 1.3 equiv), as activating agents, before the addition of the scaffold 3a (1 equiv) and DIPEA gave a mixture of diastereoisomers 4g/4′g (69%), then successfully separated by silica gel chromatography.
All compounds were characterized by NMR spectroscopy, and details for 4g/4′g (Figures S1–S5) are reported in the Supporting Information. Noesy/Roesy experiments did not help to find significant spatial proximities to define the absolute stereochemistry of each dipeptide. On the other hand, information on the secondary structure of 4g is given. Any attempt to obtain crystals of the single diastereoisomer 4g or 4′g for X-ray analysis failed. As a result, the stereochemistry of 4g and 4′g was arbitrary assigned.
Electrospinning Studies and Fiber Characterization
HFIP is selected for electrospinning-based manufacturing studies because it is highly polar, of high vapor pressure and low surface tension. As a result, it easily evaporates from thin liquid jets. Further HFIP is a suitable solvent for all the compounds of this work. However, the resulting solution has low viscosity that may cause beads or droplets in fibers (stemming from jet instabilities). This problem could be overcome by increasing the concentration of the substance as much as possible. Electrospinning was performed on a NANON-01A nanofiber electrospinning system for lab use with a B Braun Injekt syringe. The temperature and the humidity were controlled, and different voltages and flow rates were tested to find the correct combination to produce fibers of good quality. A first analysis of the electrospun nanofibers is performed acquiring images with optical microscopy with a magnification up to 20×.
Furthermore, in our previous study on peptides containing pyrazolo-isothiazole core,25 the use of a 30 % wt concentration (compound wt/compound + solvent %wt) of HFIP was proved to be the best one and was selected also in this study.
Accordingly, scaffolds 3a,b,e, Fmoc-GlyOH, Fmoc-AlaOH, and dipeptides 4 were tested for their electrospinnability (30 % wt in HFIP), in the first three cases to evaluate the potential capability to generate fibers “per se” of the single component of the dipeptide, that is, the heterocyclic scaffold and the Fmoc-protected amino acid.
All compounds 4 appeared to be able to generate nanofibers through forced assembly but with different tendency (Figure 1).
Figure 1.

Microscope images of (A) 4a, (D) 4d, (F) 4f: 25 kV, 0.5 mL/h; (B) 4b: 20 kV, 0.5 mL/h; (C) 4c: 25 kV; 0.8 mL/h; (E) 4e: 20 kV; 0.2 mL/h; (G) 4g: 28 kV, 0.5 mL/h. Magnification is 20×, and scale bars are 20 μm.
Compounds 4a–e, characterized by Fmoc-glycine moiety linked to pyrrolo-pyrazole scaffold, showed different fibers with different structure according to the various electrospinning parameters tested. Dipeptide 4a lead to produce nanofibers in a wider range of process parameters compared to the other compounds. The optimal process parameters were identified as 25 kV voltage and a feed rate from 0.2 to 0.8 mL/h (Table TS1) for which homogenous and continuous fibers were obtained (Figure 1A; 0.8 mL/h) with a diameter of around 1 μm (n = 100). By increasing the voltage up to 28 kV, fibers showed smaller diameter (below to 1 μm) when combined with a low feed rate (0.2 mL/h) and discontinuous fibers when combined with a high feed rate (0.5–0.8 mL/h). 4b-based fibers were obtained at 20 kV with a feed rate of 0.5–0.8 mL/h. Figure 1B shows fibers collected at 20 kV voltage and 0.5 mL/h flow rate. An increase of the voltage (25–28 kV) triggered a fluid jet destabilization and the formation of discontinuous fibers and beads (Table TS1). The addition of fluorine, increasing lipophilicity, on the phenyl ring in compound 4c did not improve the self-assembly of the moieties during the electrospinning process since only fiber fragments were collected regardless the manufacturing parameters (Figure 1C and Table S1). The manufacturing of 4d fibers was unsteady (Table S1). However, as shown in Figure 1D, few continuous fibers can be collected by setting the voltage at 25 kV and varying the feed rate from 0.2 to 0.8 mL/h. Similar behavior was observed for sample 4e, and the electrospinning allowed to obtain fibers intermittently. The best 4e-based fibers were produced by working at 20 kV with a feed rate of 0.2 mL/h (Figure 1E). Changing the protective group of the glycine from Fmoc to Boc (compound 4f), a decrease of the quality of the fibers and spherical beads was gathered for all the parameters tested (Figure 1F and Table S1).
Finally, by 4g, functionalized with Fmoc-Ala instead of Fmoc-Gly, long, endless fibers mainly at high voltage (28 kV, Figure 1G) were obtained. Comparing the behavior of 4g with 4′g, fibers of different quality were detected, being the first ones of good quality (compare Figure 1G with Figure S6). This difference can be ascribed to the different orientation of the substitutes due to the different stereochemistry of the scaffold, as confirmed also by our previous work.25
The effect of the voltage on the fibers diameter is shown in Figure 2, derived from the elaboration of the optical microscope images. The decrease in mean diameter from 1.67 ± 0.47 to 0.98 ± 0.47 μm (n = 100) was detected by varying the voltage from 20 to 28 kV (Table TS1). No significant effect was revealed about the feed rate.
Figure 2.
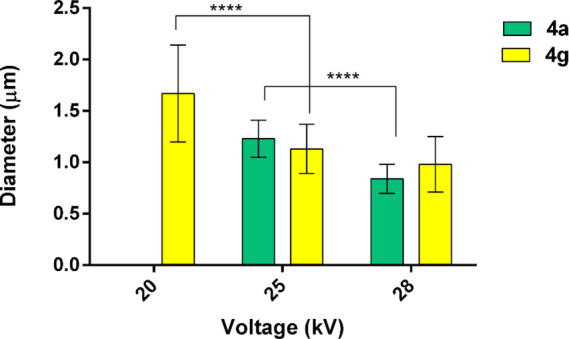
Effect of the voltage on the diameter of the electrospun fibers. Fibers (n = 100) were measured, the mean and standard deviation was calculated, and then, two-way ANOVA (Tukey’s multiple comparison test) was used to assess the statistical difference. Tukey’s multiple comparison test revealed differences for ****, p value <0.0001. 4a at 20 kV did not lead to suitable fibers.
Focusing on the single component of dipeptides, neither Fmoc-Gly and Fmoc-Ala nor 3a,b,e gave good results when electrospun in HFIP (30 wt %/wt solution). Images obtained with compound 3a are shown Figure S7, demonstrating the lack of spinnability of this compound that produced only droplets.
The above results demonstrated that the pyrrolo-pyrazole core alone lacks the requisites for the assembly needed for fiber formation in the applied conditions and that the synergic presence of both the heterocyclic ring and Fmoc-amino acid allows to succeed in obtaining electrospun fibers. The substituents on the heterocyclic ring influence the quality of the fibers. The N-phenyl ring is the best substituent able to stabilize π,π-interactions, thus optimizing intermolecular interactions, probably due to its coplanarity with the heterocyclic ring. Worse results were achieved with the N-benzyl group (compare 4a,c with 4b,e), characterized by a larger rotational freedom. Furthermore, π,π-interactions derive from the Fmoc group of the amino acid. In fact, the variation of the protective group from Fmoc to Boc significantly affects the quality of the fibers: 4f did not allow the formation of a continuous jet of peptide solution and only beads can be collected. The contribution of the amino acid seems to be very important to obtain good fibers. The use of Ala moiety disfavors the formation of good fibers, as demonstrated also in our previous work,25 probably due to the presence of the methyl group that, increasing the distances between the peptidomimetic moieties, decreases π,π-interactions (compare 4a with 4g). This hypothesis agrees also with the bulkiness of ester function, stabilizing the lipophilic interactions: the smallest is the alcoholic moiety, the highest is the quality of the fiber (compare 4a with 4d).
A morphological analysis of the fibers of the best candidate 4a was carried out by SEM (Figure 3; gold coating of dried samples under vacuum).
Figure 3.

SEM images of 4a-based electrospun fibers obtained at (A) 500×, (B) 10 k×, and (C) 20 k× and (D) diameter distribution obtained by the elaboration of SEM images by ImageJ (n = 100).
As shown in Figure 3B,C, fibers displayed an alternation of thinner portions with diameter in a rank of 0.15–0.3 μm and thicker portion with diameter ranging from 0.7 to 1.5 μm. SEM images were processed by Image software to assess the fiber diameter distribution: 4a-based fibers showed a bimodal diameter distribution (Figure 3D) since the highest frequency of diameters was reached at 0.5 and 0.85 μm indicating that the majority of the sample is made of nanofibers.
AFM (Figure 4; Horiba SmartSPM, semicontact mode) was used to investigate fiber surfaces.
Figure 4.

AFM images of 4a-based electrospun fibers on silica wafer, dried overnight.
As shown from Figure 4A,B, fibers were not porous. However, they presented longitudinal grooves of the surface (Figure 4C,D). It is reported in the literature that this feature may be due to three different phenomena: void-based elongation, wrinkle-based elongation, and collapsed jet-based elongation.33 The first one is based on the formation of voids on the jet surface at the early stage of electrospinning caused by the fast evaporation of highly volatile solvent and the subsequent elongation and solidification of tiny voids into grooved fibers. In the second mechanism (wrinkle-based elongation), the grooved surface is formed due to buckling of a cylindrical fluid jet under compressive radial stresses, triggered by the removal of solvent from the core of the jet. Finally, the third mechanism consists in the collapse of the jet because of the low evaporation rate and high viscosity of the solution followed by the stretching into smooth grooved structures. Considering the experimental conditions used in this work, the third mechanism can be excluded.
Deeper studies with 4a were also undertaken with a home-built setup in horizontal geometry, with either on the <5 μL scale with the microliter electrospinning technique or on the 0.5 mL scale with a conventional setup (flow rate: 0.5 mL/h). In the first case, the high voltage is applied to a vertically oriented flat tip syringe steel needle (Terumo 18G 38 mm, inner diameter 0.84 mm, outer diameter 1.27 mm), which placed 15 cm above the collector. The solution was directly placed from a micropipette on the tip syringe steel needle. In this way, it is possible to test the electrospinnability of microgram amounts. In the second case, the high voltage is applied to a steel needle connected to a standard syringe pump to control the flow rate of the solution, which placed at various distances above the collector.
Creating stable and biocompatible surface coatings for interfaces in contact with biological fluids is critical to prevent device-associated complications such as clot formation, fouling, and device failure.
To verify the ability to coat different surfaces, 4a-based fibers were collected on a precleaned silicon wafer or an aluminum substrate. A positive voltage of 15 kV (HP030R, Applied Kilovolts) was applied at the tip; other values gave less satisfactory results. The setup is situated in a large chamber (>500 L), which is fitted with a low suction vacuum in order to remove toxic gases. SEM images are reported in Figure 5A,B showing the deposition on a silica wafer: forked but continuous fibers were observed, with a diameter of 3.5 ± 0.6 μm. As shown in Figure 5C, 4a fibers successfully coated the aluminum substrates and they appeared long and straight with a mean diameter of 3.1 ± 0.9 μm (Figure 5D). No significant differences in fiber size were revealed among the different collectors. However, the conductivity of the substrates affected the fiber morphology and orientation of the fibers. Therefore, the last collector tested was duly amended to harvest highly oriented fibers of pristine 4a compound. Figure 5F displays the aligned fibers deposited between the two side of the conductive collectors showing that the compound has the necessary strength resistant to remain suspended in a gap of 0.5 cm. Instead, Figure 5G shows the ability of the compound to cover a pit in the aluminum collector. This is an important achievement for the elettrospinning technique applied to small molecules.
Figure 5.

SEM images of 4a based on electrospun fibers obtained on silica wafer at (A) 1345× and (B) 53× and on aluminum substrate at (C) 156× (flat), (D) 800× (flat); (E) diameter distribution obtained by the elaboration of SEM images by ImageJ (n = 100). (F) Optical photo of aligned free-standing fibers on an aluminum window, scale bar = 5 mm; (G) 101× SEM images of fibers covering a pit of the aluminum collector.
Based on this good results, 4a-based electrospun matrix biocompatibility was evaluated for cytocompatibility, cell proliferation, and cell arrangement on the matrices.
Since hydrophobicity and hydrophilicity play a major role in biomedical materials, understanding how the coated surfaces interact with water is important. Glass surfaces were coated with nanofibers of 4a through an electrospinning manufacturing of 10 min to obtain dense and uniform coating. The water contact angle of the surfaces exhibited an increase from 36° for bare glass to 113 ± 1.9° (Figure 6), indicating an increase of the hydrophobicity that likely stems from the features of the peptide. The hydrophobicity degree is comparable to the most frequently used synthetic polymers in tissue engineering (around 120°).34
Figure 6.

Wettability of 4a-based electrospun fibers evaluated as contact angle.
4a-Based Electrospun Matrix Degradation in Biological Medium
Another important features for materials with biomedical purposes include the biodegradation kinetic and the biocompatibility of degradation products.
To gain insight into these features, glass surfaces fully covered with 4a-based nanofibers were incubated at 37 °C in DMEM with the addition of FBS (10% v/v; final pH = 8). At schedule time points (10, 30, 60, 120, 240, 360, and 420 min), they were observed by a Leica DM IL LED inverted optical microscope to assess the fiber morphology (Figure 7).
Figure 7.

Optical microscope images (20× magnification, scale bar of 20 μm) of 4a-based electrospun fibers incubated at 37 °C in DMEM with FBS at pH 8 for several timing.
The fibrous matrices were unchanged up to 60 min of incubation. At 120 min of incubation, some structural modifications were observed. However, the glass coverslips were homogeneously covered by fiber-like structures. Prolonging the incubation to 240 min, the first degradation phenomena were revealed: fibers partially covered the glass coverslips showing important structural changes and the degradation process was almost completed at 420 min when only few fiber residues were observed. Image elaboration (Figure S8) showed that a fiber swelling arises in combination with their degradation since the mean diameter increase from 1.33 ± 0.22 to 2.21 ± 0.49 μm prolonging the incubation time from 10 to 420 min.
4a-Based Electrospun Matrix Degradation at Different pH
We studied the degradation of 4a fibers incubated in DMEM and FBS at different pH (7.0, 7.2, and 7.4) for different times (4 and 6 h; Figure 8).
Figure 8.

Optical microscope images (20× magnification, scale bar = 40 μm) of 4a electrospun fibers incubated in DMEM and FBS at different pH 7.0, 7.2, and 7.4 for 240 (4 h) and 360 (6 h) min.
As at pH 8 (Figure 7), 4a fibers incubated at pH 7.4 underwent a fast degradation within 6 h of incubation (Figure 8). The degradation rate is slowed down at pH 7.2 where fiber-like structure covered the glass slide surface showing some structural modifications both after 4 and 6 h. Finally, at pH 7, the effect of the matrix degradation was enhanced: no fibers were detected at 240 min of incubation. From these results, the degradation of the fibers seems to be sensitive to the pH. We could hypothesize a chemical degradation due to the deprotection of Fmoc-Gly moiety in basic environment. However, the results obtained at pH 7 seem to rule out this hypothesis. On the other hand, we can speculate a physical degradation of the nanofibers due to the dipeptide reassembly into nanoparticles after dilution in water-based medium. This behavior can be helpful in surface modification of polymer-based biomedical devices to get materials with anticoagulant and antifouling to prevent microbial growth and/or adsorption of bio-organisms.
In Vitro Biological Studies
Aiming to assess potential side effects of the degradation products released from electrospun fiber degradation, incubation media recovered by each time point of the in vitro degradation test were incubated on NHDFs for 24 h at 37 °C in controlled environmental conditions, and then, cell viability was evaluated by the MTT assay. Cells treated with fresh medium at the same pH of the sample [pH 8.0 (Figure 9A) and 7.4, 7.2, 7.0 (Figure 9B)] were used as control (Figure 9).
Figure 9.
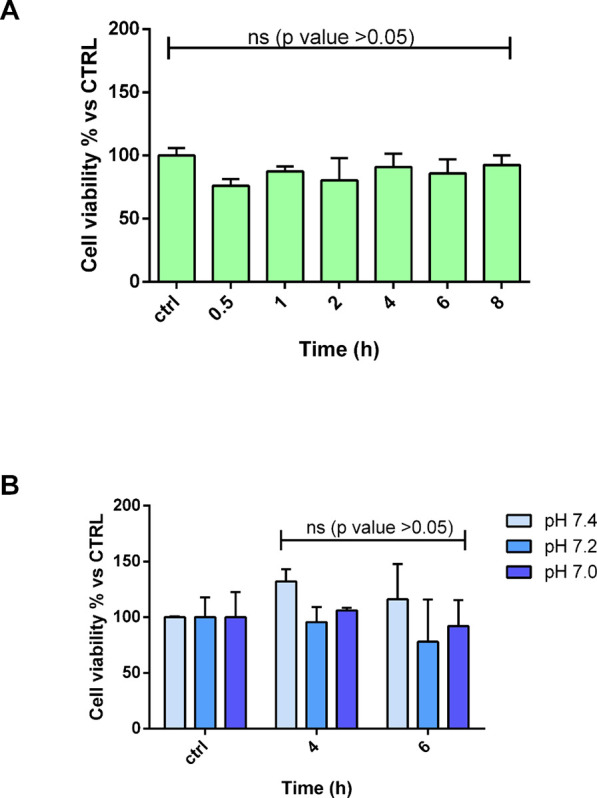
NHDF viability (A) after treatment with degradation media at pH 8.0 collected at several time points and (B) after treatment with degradation media at pH 7.4, 7.2, and 7.0. Tukey’s comparison test did not reveal statistical differences (ns, p value <0.05).
As shown, no evident toxicity was highlighted since there are not significant differences between the degradation media collected at several time points and the control (ctrl). Moreover, the viability of NHDFs is always higher than 60% proving the biocompatibility of the 4a-based electrospun nanofibers. This evidence is confirmed by the morphological examination of cells after the treatment if compared by the control cells (Figure S9 in the Supporting Information).
Conclusions
A very straightforward synthesis of dipeptides 4, obtained in large amount and containing the pyrrolo-pyrazole scaffold linked to a natural amino acid, was performed in two steps and excellent yields starting from commercially available starting materials, that are, diazoalkanes, maleimides, and a natural amino acid.
Electrospinning of the small library of dipeptides 4, characterized by a different substitution pattern and amino acid, evidenced the importance of both π–π and hydrophobic interactions between the substituents that are at the base of the stabilization of the aggregates. On the other hand, steric hindrances disfavor the formation of good quality of fibers (compare 4a vs 4d, 4f). This is also demonstrated by the use of Fmoc-Gly vs the most hindered Fmoc-Ala (compare 4a vs 4g).
Best results in terms of electrospinning manufacturing were obtained by compound 4a: a continuous and homogeneous process of bead-less fiber formation with diameters in the range of nanometers was observed. Furthermore, 4a-based fibers are able to adhere and coat substrates as glass slides, silica wafer, and aluminum substrate, thus increasing the hydrophobicity of the surface. Moreover, the optimized electrospinning process allows to produce aligned fibers.
The fast degradation of the fibers in a biological environment (6 h of incubation) is the main limitation revealed in this study. However, the released degradation products were biocompatible, and they did not alter the cell growth.
In conclusion, the achievement of electrospun fibers from small molecules without the use of any polymers is an actual bottleneck of this technique and represents a very important not trivial achievement. The properties of 4a-based fibers revealed that it can be potentially useful for the surface modification of biomedical devices adding anticoagulant and antifouling properties, opening the way of its possible future biomedical applications.
Acknowledgments
We thank the European Union for funding received in the Marie Skłodowska-Curie (grant agreement no. 101072645).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.3c01310.
NMR characterization of compounds 4; electrospinning data and images; 1H and 13C NMR spectra for compounds 3a–d and 4a–g (PDF)
Author Contributions
E.C., F.C., I.G., and M.L.G. conceptualized the research; F.A., R.B., and M.B. synthesized all compounds and characterized them by NMR; E.C., M.B., I.G., and A.M.B. generated and characterized the electrospun fibers; E.C. and I.G. studied the stability of the fibers in biological media; M.P. performed the AFM analysis of the fibers; E.C., F.C., I.G., A.M.B., and M.L.G. interpreted the data; E.C., M.L.G., and A.M.B. wrote the manuscript. All authors revised and approved the last version of the article.
This project has received funding from the European Union’s research and innovation program under the Marie Skłodowska-Curie grant agreement No 101072645.
The authors declare no competing financial interest.
Special Issue
Published as part of Biomacromoleculesvirtual special issue “Peptide Materials”.
Supplementary Material
References
- Fadil F.; Affandi N. D. N.; Misnon M. I.; Bonnia N. N.; Harun A. M.; Alam M. K. Review on Electrospun Nanofiber-Applied Products. Polymers (Basel) 2021, 13, 2087. 10.3390/polym13132087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yıldız A.; Kara A. A.; Acartürk F. Peptide-Protein Based Nanofibers in Pharmaceutical and Biomedical Applications. Int. J. Biol. Macromol. 2020, 148, 1084. 10.1016/j.ijbiomac.2019.12.275. [DOI] [PubMed] [Google Scholar]
- Xue J.; Wu T.; Dai Y.; Xia Y. Electrospinning and Electrospun Nanofibers: Methods, Materials, and Applications. Chem. Rev. 2019, 119 (8), 5298. 10.1021/acs.chemrev.8b00593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenry; Lim C. T. Nanofiber Technology: Current Status and Emerging Developments. Prog. Polym. Sci. 2017, 70, 1. 10.1016/j.progpolymsci.2017.03.002. [DOI] [Google Scholar]
- Reneker D. H.; Yarin A. L. Electrospinning Jets and Polymer Nanofibers. Polymer (Guildf) 2008, 49 (10), 2387. 10.1016/j.polymer.2008.02.002. [DOI] [Google Scholar]
- Singer J. C.; Ringk A.; Giesa R.; Schmidt H.-W. Melt Electrospinning of Small Molecules. Macromol. Mater. Eng. 2015, 300 (3), 259. 10.1002/mame.201400296. [DOI] [Google Scholar]
- Taskin M. B.; Ahmad T.; Wistlich L.; Meinel L.; Schmitz M.; Rossi A.; Groll J. Bioactive Electrospun Fibers: Fabrication Strategies and a Critical Review of Surface-Sensitive Characterization and Quantification. Chem. Rev. 2021, 121 (18), 11194. 10.1021/acs.chemrev.0c00816. [DOI] [PubMed] [Google Scholar]
- Song J.; Winkeljann B.; Lieleg O. Biopolymer-Based Coatings: Promising Strategies to Improve the Biocompatibility and Functionality of Materials Used in Biomedical Engineering. Adv. Mater. Interfaces 2020, 7, 2000850 10.1002/admi.202000850. [DOI] [Google Scholar]
- Badv M.; Bayat F.; Weitz J. I.; Didar T. F. Single and Multi-Functional Coating Strategies for Enhancing the Biocompatibility and Tissue Integration of Blood-Contacting Medical Implants. Biomaterials 2020, 258, 120291 10.1016/j.biomaterials.2020.120291. [DOI] [PubMed] [Google Scholar]
- Bucci R.; Vaghi F.; Erba E.; Romanelli A.; Gelmi M. L.; Clerici F. Peptide Grafting Strategies before and after Electrospinning of Nanofibers. Acta Biomater. 2021, 122, 82. 10.1016/j.actbio.2020.11.051. [DOI] [PubMed] [Google Scholar]
- DeFrates K. G.; Moore R.; Borgesi J.; Lin G.; Mulderig T.; Beachley V.; Hu X. Protein-Based Fiber Materials in Medicine: A Review. Nanomaterials 2018, 8 (7), 457. 10.3390/nano8070457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuansing W.; Georgilis E.; de Oliveira T. V. A. G.; Charalambidis G.; Eleta A.; Coutsolelos A. G.; Mitraki A.; Bittner A. M. Electrospinning of Tetraphenylporphyrin Compounds into Wires. Part. Part. Sys. Charact. 2014, 31 (1), 88. 10.1002/ppsc.201300293. [DOI] [Google Scholar]
- Bucci R.; Georgilis E.; Bittner A. M.; Gelmi M. L.; Clerici F. Peptide-Based Electrospun Fibers: Current Status and Emerging Developments. Nanomaterials 2021, 11 (5), 1262. 10.3390/nano11051262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuansing W.; Frauchiger D.; Huth F.; Rebollo A.; Hillenbrand R.; Bittner A. M. Electrospinning of Peptide and Protein Fibres: Approaching the Molecular Scale. Faraday Discuss. 2013, 166 (0), 209. 10.1039/c3fd00069a. [DOI] [PubMed] [Google Scholar]
- Bonetti A.; Pellegrino S.; Das P.; Yuran S.; Bucci R.; Ferri N.; Meneghetti F.; Castellano C.; Reches M.; Gelmi M. L. Dipeptide Nanotubes Containing Unnatural Fluorine-Substituted Β2,3-Diarylamino Acid and l-Alanine as Candidates for Biomedical Applications. Org. Lett. 2015, 17 (18), 4468. 10.1021/acs.orglett.5b02132. [DOI] [PubMed] [Google Scholar]
- Reches M.; Gazit E. Casting Metal Nanowires Within Discrete Self-Assembled Peptide Nanotubes. Science (1979 2003, 300 (5619), 625. 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- Levin A.; Hakala T. A.; Schnaider L.; Bernardes G. J. L.; Gazit E.; Knowles T. P. J. Biomimetic Peptide Self-Assembly for Functional Materials. Nat. Rev. Chem. 2020, 4 (11), 615–634. 10.1038/s41570-020-0215-y. [DOI] [Google Scholar]
- Firdous S. O.; Sagor Md. M. H.; Arafat M. T.. Advances in Transdermal Delivery of Antimicrobial Peptides for Wound Management: Biomaterial-Based Approaches and Future Perspectives. ACS Appl. Bio. Mater. 2023. 10.1021/acsabm.3c00731. [DOI] [PubMed] [Google Scholar]
- Nuansing W.; Rebollo A.; Mercero J. M.; Zuñiga J.; Bittner A. M. Vibrational Spectroscopy of Self-Assembling Aromatic Peptide Derivates. J. Raman Spectrosc. 2012, 43 (10), 1397. 10.1002/jrs.4063. [DOI] [Google Scholar]
- Singh G.; Bittner A. M.; Loscher S.; Malinowski N.; Kern K. Electrospinning of Diphenylalanine Nanotubes. Adv. Mater. 2008, 20 (12), 2332. 10.1002/adma.200702802. [DOI] [Google Scholar]
- Bucci R.; Giofré S.; Clerici F.; Contini A.; Pinto A.; Erba E.; Soave R.; Pellegrino S.; Gelmi M. L. Tetrahydro-4H-(Pyrrolo[3,4-d]Isoxazol-3-Yl)Methanamine: A Bicyclic Diamino Scaffold Stabilizing Parallel Turn Conformations. J. Org. Chem. 2018, 83 (19), 11493. 10.1021/acs.joc.8b01299. [DOI] [PubMed] [Google Scholar]
- Contini A.; Ferri N.; Bucci R.; Lupo M. G.; Erba E.; Gelmi M. L.; Pellegrino S. Peptide Modulators of Rac1/Tiam1 Protein-Protein Interaction: An Alternative Approach for Cardiovascular Diseases. Peptide Science 2018, 110 (5), e23089 10.1002/bip.23089. [DOI] [PubMed] [Google Scholar]
- Bucci R.; Bonetti A.; Clerici F.; Contini A.; Nava D.; Pellegrino S.; Tessaro D.; Gelmi M. L. Tandem Tetrahydroisoquinoline-4-Carboxylic Acid/β-Alanine as a New Construct Able To Induce a Flexible Turn. Chem.—Eur. J. 2017, 23 (45), 10822. 10.1002/chem.201701045. [DOI] [PubMed] [Google Scholar]
- Bucci R.; Contini A.; Clerici F.; Pellegrino S.; Gelmi M. L. From Glucose to Enantiopure Morpholino β-Amino Acid: A New Tool for Stabilizing γ-Turns in Peptides. Org. Chem. Front. 2019, 6 (7), 972. 10.1039/C8QO01116H. [DOI] [Google Scholar]
- Locarno S.; Eleta-Lopez A.; Lupo M. G.; Gelmi M. L.; Clerici F.; Bittner A. M. Electrospinning of Pyrazole-Isothiazole Derivatives: Nanofibers from Small Molecules. RSC Adv. 2019, 9 (36), 20565. 10.1039/C9RA02486G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci R.; Dapiaggi F.; Macut H.; Pieraccini S.; Sironi M.; Gelmi M. L.; Erba E.; Pellegrino S. On-Resin Multicomponent 1,3-Dipolar Cycloaddition of Cyclopentanone–Proline Enamines and Sulfonylazides as an Efficient Tool for the Synthesis of Amidino Depsipeptide Mimics. Amino Acids 2020, 52 (1), 15. 10.1007/s00726-019-02805-3. [DOI] [PubMed] [Google Scholar]
- Bucci R.; Vaghi F.; Di Lorenzo D.; Anastasi F.; Broggini G.; Lo Presti L.; Contini A.; Gelmi M. L. A Non-Coded Β2,2-Amino Acid with Isoxazoline Core Able to Stabilize Peptides Folding through an Unprecedented Hydrogen Bond. Eur. J. Org. Chem. 2022, 2022 (39), e202200601 10.1002/ejoc.202200601. [DOI] [Google Scholar]
- Oliva F.; Bucci R.; Tamborini L.; Pieraccini S.; Pinto A.; Pellegrino S. Bicyclic Pyrrolidine-Isoxazoline γ Amino Acid: A Constrained Scaffold for Stabilizing α-Turn Conformation in Isolated Peptides. Front. Chem. 2019, 7, 1. 10.3389/fchem.2019.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa A.; Zayed S. M. A. D.; Khattab S. Reactions with Diazoalkanes. V. Action of Diazoalkanes and of Aryl Azides on N-Arylmaleimides. J. Am. Chem. Soc. 1956, 78 (1), 145. 10.1021/ja01582a041. [DOI] [Google Scholar]
- Brighty K.; Castaldi M. Synthesis of (1α,5α,6α)-6-Amino-3-Azabicyclo[3.1.0]Hexane, a Novel Achiral Diamine. Synlett 1996, 11, 1097. 10.1055/s-1996-5684. [DOI] [Google Scholar]
- Chen H.-J.; Liu Y.; Wang L.-N.; Shen Q.; Li J.; Nan F.-J. Discovery and Structural Optimization of Pyrazole Derivatives as Novel Inhibitors of Cdc25B. Bioorg. Med. Chem. Lett. 2010, 20 (9), 2876. 10.1016/j.bmcl.2010.03.040. [DOI] [PubMed] [Google Scholar]
- Chiesa E.; Dorati R.; Pisani S.; Bruni G.; Rizzi L. G.; Conti B.; Modena T.; Genta I. Graphene Nanoplatelets for the Development of Reinforced PLA–PCL Electrospun Fibers as the Next-Generation of Biomedical Mats. Polymers (Basel) 2020, 12 (6), 1390. 10.3390/polym12061390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaarour B.; Zhu L.; Jin X. A Review on the Secondary Surface Morphology of Electrospun Nanofibers: Formation Mechanisms, Characterizations, and Applications. ChemistrySelect 2020, 5 (4), 1335. 10.1002/slct.201903981. [DOI] [Google Scholar]
- Maria Tottoli E.; Chiesa E.; Ceccarelli G.; Pisani S.; Bruni G.; Genta I.; Conti B.; Dorati R. BioFiber: An Advanced Fibrous Textured Dressing to Manage Exudate in Severe Wounds. Int. J. Pharm. 2022, 625, 122073 10.1016/j.ijpharm.2022.122073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.