

Abstract
Purpose
Axenfeld–Rieger syndrome (ARS) is characterized by ocular anomalies including posterior embryotoxon, iridocorneal adhesions, corectopia/iris hypoplasia, and developmental glaucoma. Although anterior segment defects and glaucoma contribute to decreased visual acuity, the role of potential posterior segment abnormalities has not been explored. We used high-resolution retinal imaging to test the hypothesis that individuals with ARS have posterior segment pathology.
Methods
Three individuals with FOXC1-ARS and 10 with PITX2-ARS completed slit-lamp and fundus photography, optical coherence tomography (OCT), OCT angiography, and adaptive optics scanning light ophthalmoscopy (AOSLO). Quantitative metrics were compared to previously published values for individuals with normal vision.
Results
All individuals demonstrated typical anterior segment phenotypes. Average ganglion cell and inner plexiform layer thickness was lower in PITX2-ARS, consistent with the glaucoma history in this group. A novel phenotype of foveal hypoplasia was noted in 40% of individuals with PITX2-ARS (but none with FOXC1-ARS). Moreover, the depth and volume of the foveal pit were significantly lower in PITX2-ARS compared to normal controls, even excluding individuals with foveal hypoplasia. Analysis of known foveal hypoplasia genes failed to identify an alternative explanation. Foveal cone density was decreased in one individual with foveal hypoplasia and normal in six without foveal hypoplasia. Two individuals (one from each group) demonstrated non-foveal retinal irregularities with regions of photoreceptor anomalies on OCT and AOSLO.
Conclusions
These findings implicate PITX2 in the development of the posterior segment, particularly the fovea, in humans. The identified posterior segment phenotypes may contribute to visual acuity deficits in individuals with PITX2-ARS.
Keywords: Axenfeld–Rieger syndrome, PITX2, FOXC1, foveal hypoplasia, posterior segment
Axenfeld–Rieger syndrome (ARS) is characterized by anterior segment ocular anomalies with variable other systemic features.1 Approximately 71% of ARS is explained by pathogenic variants in either PITX2 (41%) or FOXC1 (30%), which result in overlapping ocular phenotypes but distinct patterns of additional anomalies.1 Typical ocular anomalies include posterior embryotoxon, iridocorneal adhesions, and corectopia or iris hypoplasia, with other anterior segment diagnoses reported in a subset of affected individuals including aniridia, Peters anomaly/corneal opacities, and primary congenital glaucoma.1 Both PITX2-ARS and FOXC1-ARS are associated with an increased risk of developmental glaucoma (56%–72%), with an earlier age of onset more likely in FOXC1-ARS.1,2 In most situations, glaucoma is associated with increased intraocular pressure; however, several cases of normal-tension glaucoma have also been reported.2–4 Visual acuity is highly variable, but low vision is common; one review identified average (± SD) best-corrected visual acuity (BCVA) as 0.58 ± 0.91 logMAR and 0.95 ± 1.00 logMAR for FOXC1-ARS and PITX2-ARS, respectively.5 Although corneal opacities, early-onset glaucoma, and severe corectopia all contribute to lowered visual acuity, additional unidentified factors in the posterior segment may also play a role.
Animal models suggest a role for these genes in posterior segment development with a reduced posterior vitreous chamber noted in pitx2-deficient zebrafish6 and hyaloid vasculature anomalies seen in both pitx2- and foxc1a-deficient zebrafish.7,8 Posterior segment anomalies have been occasionally reported in humans, including persistent fetal vasculature,9 macular retinoschisis,5 and absent foveal pit10 in one individual each with PITX2-ARS; persistent fetal vasculature11 and dysplastic optic nerves with peripapillary chorioretinal atrophy12 in one individual each with FOXC1-ARS; and optic nerve/retinal colobomas in three cases with 6p25 deletions (involving other genes in addition to FOXC1).1,13,14 An overlapping phenotype of typical aniridia, caused by variants in PAX6 and characterized by a variable degree of iris hypoplasia, includes malformations in multiple additional ocular tissues, with foveal hypoplasia identified in up to 98.5% of cases and considered to be one of the differentiating features of that phenotype.15,16 Characterization of the posterior segment has not been systematically explored in individuals with ARS; yet, those additional features, if present, may contribute to reduced visual acuity in affected individuals. Moreover, this knowledge will advance our understanding of the roles of the FOXC1 and PITX2 genes in human ocular development. Here, we used high-resolution retinal imaging to test the hypothesis that individuals with PITX2-ARS and FOXC1-ARS have posterior segment pathology.
Methods
Individual Demographics
This human study was approved by the Institutional Review Boards of Children's Wisconsin and Medical College of Wisconsin, and written informed consent was obtained from every participant or legal guardian, as appropriate. Forty individuals with a genetic diagnosis of ARS met the inclusion criteria (older than 7 years of age, with sufficient corneal clearance in at least one eye), including 22 individuals with FOXC1 variants (age range, 7–61 years; mean ± SD, 27.7 ± 16.4 years) and 18 individuals with PITX2 variants (age range, 8–67 years; mean ± SD, 34.2 ± 17.8 years). We successfully imaged 13 White individuals (eight females and five males; age range, 12–52 years; mean ± SD, 27.2 ± 14.3 years) (see Supplementary Note). Imaging included fundus photography, slit-lamp photography, optical coherence tomography (OCT), OCT angiography (OCTA), and adaptive optics scanning light ophthalmoscopy (AOSLO). Axial length measurements were obtained using a partial-coherence interferometer (IOLMaster; Carl Zeiss Meditec, Dublin, CA, USA). Clinical ophthalmology records from the treating provider were reviewed for documentation of structural anomalies, diagnosis of glaucoma, and BCVA.
Genetic screening was completed by exome or genome sequencing in all participants as previously described1,17 and analyzed using VarSeq (Golden Helix, Bozeman, MT, USA). MANE transcripts were used for variant nomenclature (NM_001453.3 for FOXC1 and NM_000325.6 for PITX2; of note, most prior literature reports use NM_153427.2 for PITX2). Nine individuals had genetic and limited clinical data previously reported.1,6,17,18 The data was also examined for pathogenic variants in several genes associated with foveal hypoplasia: SLC38A8, TYR, OCA2, ATF6, and PAX6. Because abnormal foveal morphology has also been reported with homozygosity for the TYR hypomorphic alleles p.R402Q and p.S192Y,19,20 genotypes for these common alleles were reviewed for all individuals.
Qualitative Analysis of Fundus Characteristics
Fundus photographs were acquired for each individual using VISUCAM (Carl Zeiss Meditec). These images were reviewed by a pediatric ophthalmologist (DMC), and notable characteristics were described, including optic nerve pallor, atrophy in optic nerve margins, and appearance of the vessels and macula. It was noted when image quality or clarity inhibited the ability to extract notable characteristics from the images.
Optical Coherence Tomography
The CIRRUS HD-OCT (Carl Zeiss Meditec) was used to image a nominal 6 × 6-mm retinal area (128 B-scans and 512 A-scans per B-scan). Additional line scans were acquired through the foveal region to ensure that the foveal center was captured. Foveal hypoplasia was graded from the corresponding OCT images by determining whether one or more inner retinal layers (outer plexiform layer, inner nuclear layer, inner plexiform layer, or ganglion cell layer) persisted through the fovea.21 Retinal ganglion cell thinning was evaluated from CIRRUS ganglion cell analysis, where the average sum of the ganglion cell layer and inner plexiform layer (GCL/IPL) was approximated based on horizontal B-scans and compared to normative data. The analysis was used to describe the distribution of ganglion cell thinning in relationship to the location of the fovea. Because the retinal ganglion cell thickness was often asymmetric between eyes, all eyes were included in the statistical analysis.
Quantitative analysis of foveal pit characteristics was acquired from CIRRUS macular cubes to describe foveal pit depth (mm) and volume (mm3). Mathematical modeling was used to calculate foveal pit metrics for each individual by applying an algorithm based on the difference of Gaussian functions to approximate the foveal pit contour.22 As a brief overview, retinal pigment epithelium and inner limiting membrane thickness data were extracted from the CIRRUS software using the Advanced Export feature as a .dat file and imported into the algorithm. This data was segmented into radial slices spaced 1° apart through the fovea center, which was determined using the FoveaFinder algorithm for the CIRRUS software. The radial slices were fit to a difference in Gaussian functions model and used to measure foveal metrics (depth and volume). The foveal pit metrics were compared to values from a subset of previously published19 data from 137 individuals with no vision-limiting pathology (83 females and 54 males; age range, 9–70 years; mean ± SD, 31.03 ± 12.9 years). Based on previously reported ethnic differences in foveal pit morphology,23,24 we compared our data to an ethnicity-matched subset of this normative cohort.
OCT Angiography
OCTA was attempted on all 13 individuals. Imaging was not successful on one individual with PITX2-ARS and one individual with FOXC1-ARS. Single OCTA images were acquired for three individuals and multiple (two to five) OCTA images were acquired for eight individuals. The nominal scan size was 3 × 3 mm, and for each angiogram two volumes consisting of 304 B-scans and 304 A-scans per B-scan were acquired; one volume had a horizontal fast scanning axis and the other volume had a vertical fast scanning axis. The two volumes were registered (AngioVue 2018.1.0.43) to create a single angiogram volume. For each volume, the full thickness slab (inner limiting membrane to 9 µm above the outer plexiform layer) was extracted. For the eight individuals with multiple angiogram images, their images were registered in ImageJ (National Institutes of Health, Bethesda, MD, USA) using StackReg and averaged using the Z Project tool in ImageJ.
Using the ImageJ Point Tool,25 a single observer (author SZ) segmented the foveal avascular zone (FAZ) in each angiogram image. The segmentation coordinates were entered into a custom MATLAB (MathWorks, Natick, MA, USA) script, which has been previously described.26,27 The area of the FAZ (or the largest avascular area if the FAZ was fragmented) was computed in pixels using the polyarea function in MATLAB. This was converted to square millimeters using the nominal image scale (9.87 µm/pixel) and the ratio of the participant's measured axial length to the axial length assumed by the device (23.95 mm).28 The FAZ area data in our individuals were compared to values from a subset of previously published19 data from 133 individuals with no vision-limiting pathology (81 females and 52 males; age range, 9–70; mean ± SD, 30.86 ± 12.6 years). Again, based on previously reported ethnic differences in FAZ size,29,30 we compared our data to an ethnicity-matched subset of this normative cohort.
AOSLO Retinal Imaging
AOSLO imaging was attempted on one eye for all individuals using a previously described device.31 Dilation drops were applied prior to imaging, and AOSLO videos were acquired on one of two devices (using an imaging wavelength of either 790 or 775 nm). During imaging, the participant's head was stabilized using a dental impression on a bite bar. Individual videos subtended either 1° × 1° or 1.5° × 1.5° field of view. Image sequences (videos) consisting of 150 to 200 frames were recorded at different locations using an internal fixation target (consisting of a crosshair projected onto a dichroic for the individual to view) to guide fixation.32 The raw frames from each video were corrected for sinusoidal distortions as previously described.33 Following this, a reference frame was automatically selected34 for registration of 50 to 80 frames and averaged to produce an image with improved signal-to-noise ratio.33 For each eye, the processed AOSLO images were semi-automatically montaged using a multimodal montaging algorithm that rescaled images from different fields of view to a common scale.35 Alignment was checked and manual correction was performed where needed using Photoshop CS6 (Adobe Systems, San Jose, CA, USA). The location of peak cone density (referred to here as the foveal center) was manually identified by marking the locus of the globally most tightly packed cones. This location was used as the anchor to define retinal eccentricity for subsequent analyses.
Regions of interest (ROIs) were extracted from each montage using Mosaic (Translational Imaging Innovations, Cary, NC, USA). ROIs were extracted at the foveal center and at 0.3°, 0.6°, 0.9°, 1.2°, 1.5°, 1.8°, 2.1°, 3.0°, 4.0°, 5.0°, 6.0°, 7.0°, 8.0°, 9.0°, 10.0°, 11.0°, and 12.0° in the temporal and superior directions. The size of each ROI was 0.3° × 0.3° at locations less than 2.1° eccentricity from the fovea and 0.4° × 0.4° at locations 2.1° and greater eccentricity from the fovea. If a montage was incomplete or did not extend sufficiently to include all intended ROI locations, only ROIs with underlying AOSLO images were extracted. ROIs were then assessed by a single grader (author JC) in a masked fashion. If the image quality was deemed insufficient to reliably identify cone structures, the ROI was not used for the analysis. In the end, we used between four and 28 ROIs (average, 11.4 ROIs) per individual. Cones were semi-automatically identified using Mosaic, and metrics (density and intercone distance) were computed as previously described.36 The lateral scale of the AOSLO images for a given individual (S′R(x); µm/pixel) was estimated by using the following equation:
where T represents the periodicity of a Ronchi ruling (µm/cycles), fl represents the focal length of the model eye in our system (µm), Ts represents the sampling period of the lines in the model eye image of the Ronchi ruling (pixels/cycle), RMF represents the assumed retinal magnification factor (291 µm/°) of an eye with a 24.0-mm axial length (represented by lA,0), and lA represents the actual axial length of the individual eye in millimeters. This scale was used to convert raw metrics in pixels to either millimeters or micrometers. Data were compared to values from 19 individuals with no known vision pathology, taken from a previously published study.36
Statistical Analyses
All statistical analyses were performed in Prism 10 (GraphPad, Boston, MA, USA). A Shapiro–Wilk test was used to assess the normality of each dataset and guide the choice of parametric or non-parametric test.
Results
Genotype and Systemic Phenotypes
Twenty-two individuals diagnosed with FOXC1-ARS and 18 individuals with PITX2-ARS qualified for the study; three of the 22 individuals with FOXC1-ARS (14%) and 10 of the 18 individuals with PITX2-ARS (55%) completed the imaging procedures. The higher rate of participation by individuals with PITX2-ARS may be due to the more complex ocular presentation and syndromic features observed in individuals with FOXC1-ARS (or their children) that made travel and overall participation more challenging; additionally, the average age of individuals with FOXC1 was younger, so more individuals approached had young children at home, making travel difficult. The majority of individuals had FOXC1 or PITX2 variants identified via their prior participation in our genetic studies with all FOXC1 variants1,18 and seven out of 10 PITX2 alleles1,6,17 previously reported. Three PITX2 alleles were novel (Supplementary Table S1).
The FOXC1-ARS group consisted of individual 1 with a deletion of FOXC1 and individuals 2 and 3 (siblings) with a frameshift allele leading to premature truncation of FOXC1, removing a part of the DNA-binding domain and C-terminal region. Both variants are predicted to result in loss-of-function alleles and are typical for the FOXC1-ARS–associated spectrum (Supplementary Table S1). Two individuals had a clinical diagnosis of glaucoma with elevated intraocular pressure in both. Systemic anomalies were present in all three individuals and included commonly observed features such as skeletal, cardiac, dental, and hearing defects.1
The PITX2-ARS group consisted of 10 individuals from nine families. The variants included premature truncations, missense variants within the homeodomain region of PITX2, and a complex rearrangement affecting the coding and regulatory regions of the gene; all variants are predicted to result in loss-of-function alleles and are typical for the PITX2-ARS–associated spectrum (Supplementary Table S1). All individuals had a clinical diagnosis of glaucoma with elevated intraocular pressure noted in all with records available. Systemic anomalies were present in all individuals and consisted of characteristic features for PITX2-ARS1 including microdontia/hypodontia/oligodontia, redundant periumbilical skin, umbilical hernia, and Meckel diverticulum.
Ocular Phenotype
Slit-lamp evaluation in individuals with FOXC1-ARS revealed the typical ocular features with posterior embryotoxon in all three, iridocorneal adhesions in two, and subtle iris hypoplasia in the other (Table, Figs. 1A–1C). Corneal phenotypes included mild corneal haze and Haab's striae in one individual each. Fundus photos were reviewed for all participants, and the optic disc was grossly normal in all three (Table). Additional anomalies identified in the retina included an unusual pattern of diffuse, linear streaks across the retina (lacquer cracks) and peripapillary atrophy in individual 2 (Fig. 2A); OCT evaluation showed the linear deformities occurring at the inner and outer segment junction in the OCT B-scan (Fig. 2B). The fundus image for individual 1 was blurred and unable to be assessed outside the optic disc.
Table.
Summary of Anterior and Posterior Segment Features in Individuals With ARS
| Fundus | Foveal Pit | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Individual* | Age (y); Sex | Glaucoma† | Slit Lamp‡ | Optic Disc | Other | Average GCL/IPL (OD; OS) (µm)‡ | Depth (mm) | Volume (mm3) | FAZ Metrics (mm2) | Axial Length (OD; OS) (mm)‡ | BCVA (OD; OS)‡ |
| FOXC1-ARS | |||||||||||
| 11 | 12; F | N | PE, ICA, mild CO, mild IH | WNL (OU) | Limited | 82; 89 | 0.1142 | 0.11085 | NA | 22.71; 21.33 | 20/50; 20/70 |
| 218 | 17; M | Y, 13d, SX | PE, mild ICA | WNL (OD), PPA | Lacquer cracks (OD) | 58§; 66§ | 0.0683 | 0.03475 | 0.091# | 23.39; 23.36 | 20/25; 20/40 |
| 318 | 14; F | Y, 12d, DO | PE, mild IH, Haab's striae | WNL (OD) | NA | 74; 75 | 0.10455 | 0.06955 | 0.283 | 24.17; 24.34 | 20/30; 20/70 |
| PITX2-ARS | |||||||||||
| 46 | 27; M | Y, 7/14y, SX | PE, ICA, CT, IH, corneal neovascularization (OS) | Large C/D (0.8) (OS) | Limited | NA; 60§ | 0.0538# | 0.0251# | NA | NA; 26.40 | NLP; 20/40 |
| 51 | 38; M | Y, 20s, DO/SX | PE, ICA, severe IH/PC, CT | WNL (OS) | Subretinal deposits | NA; 41§ | 0.0656 | 0.0478 | 0.413 | NA; 25.18 | 20/200; 20/63 |
| 6 | 14; M | Y, 13y, SX/DO | PE, CT, severe IH (OS) | WNL (OS) | NA | 40;§ 58§ | 0.0323# | 0.02095# | 0.05# | 28.69; 27.15 | 20/150; 20/40 |
| 71 | 52; F | Y, 45y, DO | PE, ICA, mild IH (OD); severe CT (OS) | WNL (OD) | NA | 76; 80 | 0.10285 | 0.058 | 0.321 | 24.24; 23.41 | 20/20; CF |
| 81 | 18; F | Y, 10y, DO/SX | PE, mild IH (OD); AN (OS) | WNL (OD) | Limited | 64§; NA | 0.0046# | 0.0189# | 0.098# | 26.56; NA | 20/60; 20/600 |
| 9 | 45; F | Y, 8y | PE, severe IH/PC, CT (OD); mild IH (OS) | WNL (OS) | NA | 57§; 79 | 0.0602 | 0.03035 | 0.168 | 25.93; 26.42 | 20/250; 20/25 |
| 10 | 44; F | Y, 30s, DO | PE, ICA, severe IH/CT | WNL (OS) | Darkly pigmented fundus | 79; 72 | 0.0951 | 0.06325 | 0.364 | Failed | NA |
| 111 | 22; F | Y, 4–5y, SX | PE, severe CT, mild IH (OD); severe IH, lens opacity (OS) | WNL (OD) | Limited | 72; NA | 0.1112 | 0.0902 | 0.578 | Failed; failed | 20/200; 20/200 |
| 12 | 13; M | Y, 11y, DO | PE, severe CT, IH/PC (OD); mild IH, CT (OS) | WNL (OS) | NA | 86; 85 | 0.0322 | 0.04705 | 0.168 | Failed; 25.48 | 20/80; 20/40 |
| 1317 | 38; F | Y, unknown, SX/DO | PE, mild IH; PC (OD); CT (OS) | Cupping (OD > OS), PPA | Pigmented fundus/WNL vasculature | 64§; 81 | 0.05495# | 0.04645# | 0.099# | 27.10; failed | 20/25; 20/40 |
AN, aniridia; C/D, cup-to-disc ratio; CF, counting fingers; CO, corneal opacity; CT, corectopia; DO, drops only; F, female; ICA, iridocorneal adhesions; IH, iris hypoplasia; M, male; N, no; NA, not available; NLP, no light perception; OD, right eye; OS, left eye; OU, both eyes; PC, polycoria; PE, posterior embryotoxon; PPA, peripapillary atrophy; SX, surgery; WNL, within normal limits; Y, yes.
Individual number; previous papers reporting corresponding individuals are indicated by reference number in superscript.
Glaucoma column shows the age of diagnosis and treatment, if it differed between eyes (OD/OS).
OU unless specified.
Indicates eyes with abnormal GCL/IPL thickness as assessed using a Z-score analysis.
Grade 1b foveal hypoplasia and/or fragmented FAZ.
Figure 1.

Anterior segment images illustrating features of ARS in imaged individuals. (A–M) Slit-lamp images demonstrating typical ARS features in all individuals, including iridocorneal adhesions (arrow in A), posterior embryotoxon (arrow in D), iris hypoplasia (asterisk in E), and corectopia (arrow in K). Pigment deposits in the cornea (arrow in G1) and lens opacity (asterisk in K1) were also noted in some individuals. Individual number is indicated in the upper right corner of each image. Both eyes are shown for individuals with asymmetric iris phenotypes. Surgical history includes glaucoma surgery in the eyes pictured in B, D, E, F1, H1, K, and K1 and cataract surgery as well as corneal transplant in H1 and I.
Figure 2.

Posterior segment images of selected individuals. (A–F) In individual 2, fundus photography (A) demonstrated peripapillary atrophy and linear streaks across the retina with corresponding focal disruptions of the ellipsoid zone band on OCT (B, arrow) and linear regions of abnormal photoreceptor pattern by AOSLO (C, dashed line). In individual 5, fundus photography (D) revealed subretinal deposits along the fovea with irregularities in the inner and outer segment junction on OCT (E, bracket) and patchy regions of abnormal photoreceptor pattern on AOSLO with several outlined with dashed lines (F). Scale bars: 250 µm.
For individuals with PITX2-ARS, slit-lamp evaluation revealed the typical ocular features with posterior embryotoxon and variable degrees of iridocorneal adhesions and iris hypoplasia (Table, Figs. 1D–1M) in all individuals. The majority of individuals had severe iris phenotypes in at least one eye, including significant corectopia in eight, severe iris hypoplasia with polycoria in six, and absent iris in one. Striking asymmetry of the iris phenotype between the two eyes with a relatively intact iris in one eye was noted in six of the nine individuals with both eyes imaged (one individual had prior enucleation of one eye) (Figs. 1F–1I, 1K, 1L) and has been reported previously for PITX2.37 Pigment deposits in the cornea were noted in many individuals. Fundus photographs were reviewed for all participants, and the optic disc was grossly normal in all except individual 4 who had a large cup-to-disc ratio (0.8) (Table). Additional anomalies included peripapillary atrophy in individual 13 and subretinal deposits of unknown origin along the fovea in individual 5; OCT analysis showed irregularities in the inner and outer segment junction in these regions (Figs. 2D–2F). Three images (from individuals 4, 8, and 11) were blurred and unable to be assessed outside the optic disc.
Altered Foveal Morphology in PITX2-ARS but not FOXC1-ARS
None of the three individuals with FOXC1-ARS had foveal hypoplasia, whereas four of the 10 individuals with PITX2-ARS (individuals 4, 6, 8, and 13) had grade 1b foveal hypoplasia (as defined by the Leicester Grading Scale21) (Fig. 3). A history of premature birth was reported for only one individual in the cohort, who had an estimated gestational age of 25 weeks in addition to foveal hypoplasia (individual 8).
Figure 3.
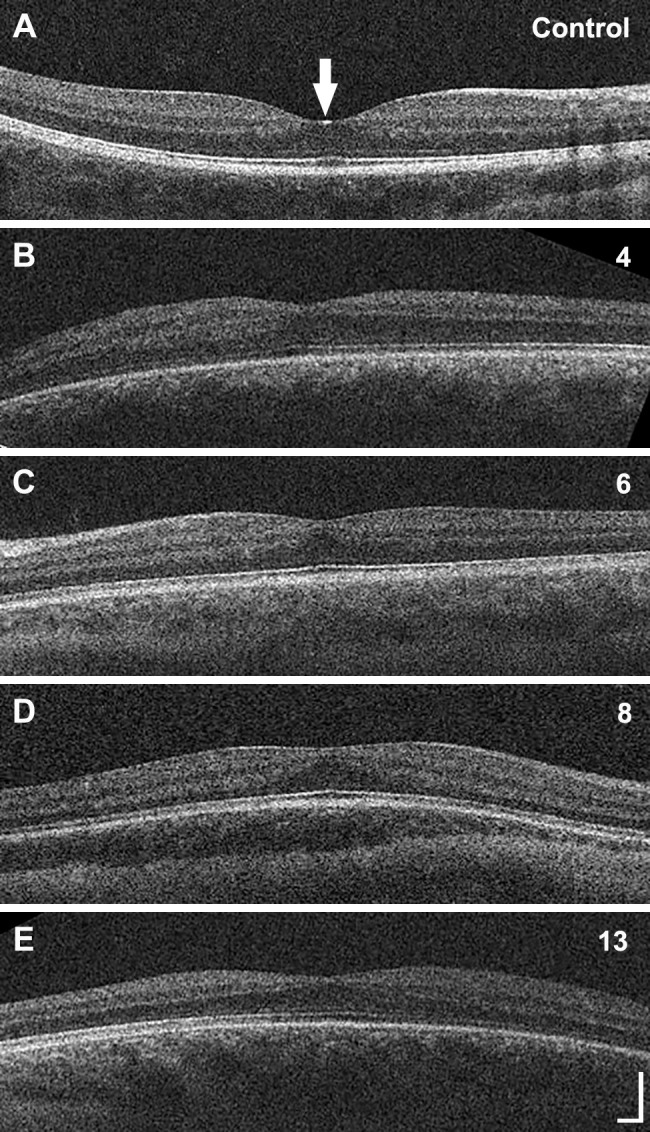
Foveal hypoplasia in PITX2-ARS. (A–E) OCT images from 15-year-old female with no vision-limiting pathology (A, control) and individuals 4 (B), 6 (C), 8 (D), and 13 (E) displaying grade 1b foveal hypoplasia. The arrow in A indicates the location of the fovea. Scale bar: 100 µm in each direction for all images.
Because foveal hypoplasia has been associated with variants in multiple genes (e.g., TYR, OCA2, SLC38A8, ATF6, PAX6), we analyzed relevant genetic data in our cohort. No pathogenic or likely pathogenic variants were detected in any genes. Heterozygous rare variants in OCA2 were identified in two of the individuals with foveal hypoplasia: NM_000275.3:c.1465A>G p.Asn489Asp and NM_000275.2:c.1966C>G p.Leu656Val, both classified as variants of uncertain significance with no second allele detected (Supplementary Table S1). No rare coding variants were identified in TYR, SLC38A8, ATF6, or PAX6. Further analyses of the common TYR hypomorphic alleles (p.S192Y and p.R402Q) previously associated with abnormal foveal morphology19,20 identified seven individuals homozygous for one of the two alleles but no individuals homozygous for both variants in our cohort (Supplementary Table S1). Comparison of the rate of homozygotes for both hypomorphic alleles (p.S192Y and p.R402Q) in the PITX2 cohort versus the control cohort did not identify a significant difference between the two populations (P = 0.24 and P = 0.14, respectively, Fisher's exact test). Comparison of the TYR genotypes of individuals with and without foveal hypoplasia within our PITX2 cohort did not identify any significant enrichment for homozygotes for either hypomorphic allele (P = 0.13 and P > 0.99, respectively, Fisher's exact test). Comparison of the TYR genotypes between the two genetic groups (FOXC1 vs. PITX2) similarly did not identify any significant enrichment for homozygotes for either variant (P > 0.99 and P = 0.20, respectively, Fisher's exact test).
Foveal pit depth and volume were calculated using OCT data on total retinal thickness (internal limiting membrane to the retinal pigment epithelium). In comparing foveal morphology data between eyes of the same individual, there was no significant interocular difference in either depth (P = 0.25, Wilcoxon matched-pairs signed rank test) or volume (P = 0.26, paired t-test) for the three individuals with FOXC1-ARS. Similar symmetry was observed in the individuals with PITX2-ARS for both depth (P = 0.20, paired t-test) and volume (P = 0.14, paired t-test). We thus averaged the values between eyes to generate a single per-individual value for comparison with normal controls.
Average (± SD) foveal pit depths were 0.096 ± 0.024 mm in the individuals with FOXC1-ARS, 0.061 ± 0.034 mm in the individuals with PITX2-ARS, and 0.115 ± 0.022 mm in the 137 normal controls. Testing with a one-way ANOVA and Dunnett's multiple comparisons post hoc testing revealed that the PITX2-ARS foveal pits were significantly shallower than normal (P < 0.0001), but the FOXC1-ARS foveal pits were not significantly different from normal (P = 0.26). This difference remained significant even after excluding the four individuals with foveal hypoplasia (P = 0.0001).
Average (± SD) foveal pit volumes were 0.072 ± 0.038 mm3 in the individuals with FOXC1-ARS, 0.045 ± 0.022 mm3 in the individuals with PITX2-ARS, and 0.088 ± 0.034 mm3 in the 137 normal controls. Kruskal–Wallis and Dunn's multiple comparisons post hoc testing revealed that the PITX2-ARS foveal pits were significantly smaller than normal (P = 0.0002), but the FOXC1-ARS foveal pits were not significantly different from normal (P = 0.85). Again, this difference remained significant (P = 0.024) even after excluding the four individuals with foveal hypoplasia (Fig. 4).
Figure 4.

Foveal comparisons. (A). Foveal pit images showing variability among non-hypoplastic foveal pits. Largest (individual 1), intermediate (individual 7), and smallest (individual 9) non-hypoplastic pit images were chosen. (B, C) Plotting of foveal pit depth and volume demonstrates a statistically significant decrease in both metrics compared to normal controls for PITX2-ARS (blue symbols) but not FOXC1-ARS (yellow symbols). The normal range from control individuals is indicated by black lines in B and C. The symbol assigned to each individual is indicated in the lower left corner.
OCTA was successfully completed on one eye each from two individuals with FOXC1-ARS and nine individuals with PITX2-ARS , including three of the individuals with foveal hypoplasia (Fig. 5, Supplementary Fig. S1). The two individuals with FOXC1-ARS had FAZ areas of 0.09 mm2 and 0.28 mm2, and the PITX2-ARS group had an average (± SD) FAZ area of 0.25 ± 0.17 mm2. The overall ARS group average (± SD) FAZ area was 0.24 ± 0.17 mm2. Although the mean FAZ area was lower in the individuals with ARS compared to the normative controls (0.26 ± 0.10 mm2), this difference was not significant (P = 0.53, unpaired t-test). However, the FAZ area was positively correlated with foveal pit volume (Spearman r = 0.85; P = 0.0015), as well as depth (Spearman r = 0.65; P = 0.034), in our ARS cohort, which has been reported in individuals with normal vision.38 Consistent with this, all individuals with foveal hypoplasia and OCTA data had a fragmented FAZ; one individual without foveal hypoplasia also displayed a fragmented FAZ (individual 2). Fragmented FAZs have been reported in individuals with and without foveal hypoplasia,27 suggesting some independence in the mechanisms establishing these two features of the human fovea.39
Figure 5.

OCT Angiography. (A–D) OCTA images demonstrate normal FAZ in individual 7 and a fragmented FAZ in individuals 6, 8, and 13, all of whom showed a foveal hypoplasia phenotype (Figure 3). Scale bar: 500 µm (all images).
Variably Altered GCL/IPL Thickness
Across the three individuals with FOXC1-ARS, the mean (± SD) average GCL/IPL thickness in all six eyes was 74.00 ± 11.04 µm. Across 137 control eyes, the mean (± SD) average GCL/IPL thickness was 83.25 ± 7.40 µm. With the control data, we employed a Z-score analysis to determine which eyes had abnormal GCL/IPL thickness values (α < 0.05). Diffuse retinal ganglion cell thinning was noted in both eyes of individual 2 with a clinical diagnosis of glaucoma requiring surgery at 13 days of age (Table). Two individuals had normal GCL/IPL thickness (individual 1 without glaucoma and individual 3 with glaucoma controlled with drops only).
Macular cube analysis on OCT was able to be completed on 16 eyes from the 10 individuals with PITX2-ARS and showed a mean (± SD) average GCL/IPL thickness of 68.38 ± 14.36 µm. Again, we employed a Z-score analysis to determine which eyes had abnormal GCL/IPL thickness values (α < 0.05). Significant retinal ganglion cell thinning was noted in at least one eye in six of 10 individuals (Table); all of these individuals had a clinical diagnosis of glaucoma. Four individuals did not show a significant deviation from normal (Table); although these individuals also had a clinical diagnosis of glaucoma, three of the four were considered controlled with drops only.
Cone Mosaic Results From AOSLO
AOSLO imaging was attempted on all three individuals with FOXC1-ARS, with full/partial success in two, followed by cone mosaic analysis on the montage that appeared normal (Fig. 6). Resolution of the central-most foveal cones was successful only in individual 3, who had a peak foveal cone density of 92,304 cones/mm2, which is within previously published normative values from multiple studies.40–43 AOSLO evaluation of the retinal anomalies in individual 2 showed linear patterns of absent photoreceptors on the confocal modality (Fig. 2C). It was not possible to resolve cells on split-detection AOSLO in this location, so whether these cones were missing, degenerated, or simply had impaired waveguiding is not known.
Figure 6.

Cone mosaic analysis. Cone density (A, B) and spacing (C, D) were determined from AOSLO images from individuals with PITX2-ARS (blue symbols) and FOXC1-ARS (yellow symbols) along both the superior (A, C) and temporal (B, D) eccentricity. Only individual 6 (with foveal hypoplasia) had values consistently outside the normal range. The symbol assigned to each individual is indicated and consistent with Figure 4 (but AOLSO data are only available for a subset of participants).
AOSLO imaging was attempted on all 10 individuals with PITX2-ARS, with full/partial success in five. Although some individuals occasionally had cone density values falling outside 2 SD from the normal mean, individual 6 (Fig. 6, blue-filled diamonds) consistently had values outside the expected normal range. Interestingly, this individual also has foveal hypoplasia; altered cone topography has been reported in some individuals with foveal hypoplasia due to other conditions (e.g., albinism).22 Resolution of the central-most foveal cones was only successful in individual 7, who had a peak foveal cone density of 131,459 cones/mm2, which is consistent with previously published normative values.40–43 AOSLO evaluation of the retinal anomalies in individual 5 showed multiple dark patches. Again, it was not possible to resolve cells on split-detection AOSLO in this location, so whether these cones were missing, degenerated, or simply had impaired waveguiding is not known.
Discussion
Slit-lamp evaluation revealed the typical spectrum of ocular anomalies associated with ARS; iris phenotypes were more severe and more asymmetric in individuals with PITX2-ARS compared to FOXC1-ARS. Interestingly, foveal phenotypes were symmetric, even in eyes with asymmetric anterior segment features. Systematic evaluation of the posterior segment revealed several anomalies. As expected, most of the individuals with a clinical diagnosis of glaucoma, especially those requiring surgical intervention, showed evidence of optic nerve thinning. Average GCL/IPL thickness was lower in the group with PITX2-ARS, consistent with the older ages and higher prevalence of glaucoma requiring surgery in this cohort.
Foveal hypoplasia was noted by OCT in 40% (4/10) of the individuals with PITX2-ARS but in none of the three with FOXC1-ARS in this study. Moreover, the foveal pit was significantly smaller in individuals with PITX2-ARS compared to normal controls, even after removing the four individuals with foveal hypoplasia. These findings implicate PITX2 in the development of the fovea in humans. Foveal hypoplasia is frequently seen in association with PAX6 variants, another common genetic cause of anterior segment phenotypes such as aniridia, Peters anomaly, and iris hypoplasia,16 as well as several other genetic factors (TYR, OCA2, SLC38A8, and ATF6). No likely pathogenic variants sufficient to provide an alternative explanation for the foveal hypoplasia phenotype were identified in the affected individuals, but some possible risk factors included OCA2 variants of uncertain significance,44 premature birth,45 and homozygosity for TYR hypomorphic alleles. However, there was no significant difference in the frequency of TYR homozygotes between the PITX2 and FOXC1 groups nor between the individuals with foveal hypoplasia and those without, suggesting that the phenotype was specific to PITX2. The foveal hypoplasia observed in PITX2-ARS is relatively mild (grade 1b) but nonetheless known to be associated with reduced visual acuity46–48 and thus likely to affect vision in younger children with ARS, with glaucoma becoming a more significant factor later in life. Further evaluation is necessary to confirm this association. Due to the small number of individuals imaged with FOXC1-ARS, it is not clear at this time whether this finding is specific to PITX2 or if our FOXC1-ARS cohort was too small to detect a foveal phenotype.
AOSLO imaging was successfully completed in about half of the attempted individuals with ARS (7/13; 54%), but foveal cone imaging was only successful in two (2/13; 23%), which is not unexpected given that the resolution of foveal cones is challenging even in eyes with excellent optical quality.49 Unsurprisingly, individuals with significant corectopia and those with significant corneal or lens opacities were more likely to be unsuccessful in AOSLO imaging. Axial length was also unable to be measured in participants with significant corectopia using the IOLMaster. Outside of individual 6 (PITX2-ARS) with consistently decreased cone density, the AOSLO imaging did not show any significant deviation from the normal optical signature of cone photoreceptors within the retina. In terms of other anomalies, one person with FOXC1-ARS showed an unusual pattern of diffuse, linear streaks across the retina with deformities at the inner and outer segment junction in the retinal cross-section, and one individual with PITX2-ARS displayed subretinal deposits along the fovea and irregularities in the inner and outer segment junction in these regions. Confocal AOSLO imaging revealed a discontiguous cone mosaic in both of these regions. Several human conditions are associated with abnormalities at the junction between the inner and outer segments of the photoreceptors.50 At this time, it is not clear whether the observed phenotypes are associated with FOXC1 and/or PITX2 deficiencies or represent coincidental findings, although our data would suggest that further examination of additional individuals with ARS is warranted.
Overall, these data suggest a role for PITX2 in the development of the posterior segment of the eye and specifically in the human fovea, in addition to the anterior segment structures. In humans, foveal development begins early in gestation with the creation of a central rod-free zone and continues throughout gestation with further maturation occurring through the first decade of life.51,52 The foveal pit specifically initiates prior to 30 weeks’ gestation and is thought to require the presence of the FAZ. The size of the foveal pit correlates with the size of the FAZ,38 so the OCTA results in this cohort are consistent with the foveal phenotypes. Although the fovea is important in visual acuity in primates and some other vertebrates, this structure is not present in commonly used animal models such as mice and zebrafish, which may explain why a role for PITX2 in foveal development was not identified previously.51,52 Both ARS genes, PITX2/pitx2 and FOXC1/foxc1, are known to be expressed in the neural-crest– and mesoderm-derived periocular mesenchymal cell populations that contribute to multiple ocular lineages such as the corneal endothelium and stroma, trabecular meshwork, Schlemm's canal, ciliary body muscles, iris stroma, extraocular muscles, sclera, choroid, and hyaloid vasculature (the transient embryonic blood system that maintains the growth and development of the posterior lens, vitreous, and retina).53,54 Interactions between the retina and other tissues in the posterior region, including the developing choroid and sclera, are believed to be responsible for foveal morphogenesis.55 Deficiencies in pitx2 or foxc1 lead to anterior segment defects as well as highly anomalous hyaloid vasculature with aberrant branching, thinner vessels, and increased vascular permeability in zebrafish.7,8,56 It is possible that PITX2 deficiency in the developing hyaloid vascular system, choroid, and/or sclera due to PITX2 deficiency affect various aspects of human posterior segment development, particularly the foveal avascular zone and foveal pit, thus requiring additional studies into the underlying mechanisms.
Supplementary Material
Acknowledgments
The authors thank the participants for their willingness to contribute to what is known about Axenfeld–Rieger syndrome.
Supported in part by a grant from the National Eye Institute of the National Institutes of Health (R01EY015518). This investigation was conducted in a facility constructed with support from a grant from the Research Facilities Improvement Program (C06RR016511) of the National Center for Research Resources at the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional support was provided by the Children's Research Institute Foundation at Children's Wisconsin (EVS).
Disclosure: A. Untaroiu, None; L.M. Reis, None; B.P. Higgins, None; A. Walesa, None; S. Zacharias, None; D. Nikezic, None; D.M. Costakos, None; J. Carroll, Translational Imaging Innovations (P, I), MeiraGTx (F), AGTC (F); E.V. Semina, None
References
- 1. Reis LM, Maheshwari M, Capasso J, et al.. Axenfeld-Rieger syndrome: more than meets the eye. J Med Genet. 2023; 60: 368–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Souzeau E, Siggs OM, Zhou T, et al.. Glaucoma spectrum and age-related prevalence of individuals with FOXC1 and PITX2 variants. Eur J Hum Genet. 2017; 25: 839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Or L, Barkana Y, Hecht I, Weiner C, Einan-Lifshitz A, Pras E. FOXC1 variant in a family with anterior segment dysgenesis and normal-tension glaucoma. Exp Eye Res. 2020; 200: 108220. [DOI] [PubMed] [Google Scholar]
- 4. Weisschuh N, Dressler P, Schuettauf F, Wolf C, Wissinger B, Gramer E. Novel mutations of FOXC1 and PITX2 in patients with Axenfeld-Rieger malformations. Invest Ophthalmol Vis Sci. 2006; 47: 3846–3852. [DOI] [PubMed] [Google Scholar]
- 5. Zhou L, Wang X, An J, Zhang Y, He M, Tang L. Genotype-phenotype association of PITX2 and FOXC1 in Axenfeld-Rieger syndrome. Exp Eye Res. 2023; 226: 109307. [DOI] [PubMed] [Google Scholar]
- 6. Hendee KE, Sorokina EA, Muheisen SS, et al.. PITX2 deficiency and associated human disease: insights from the zebrafish model. Hum Mol Genet. 2018; 27: 1675–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ferre-Fernández JJ, Sorokina EA, Thompson S, et al.. Disruption of foxc1 genes in zebrafish results in dosage-dependent phenotypes overlapping Axenfeld-Rieger syndrome. Hum Mol Genet. 2020; 29: 2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu Y, Semina EV.. pitx2 Deficiency results in abnormal ocular and craniofacial development in zebrafish. PLoS One. 2012; 7: e30896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arikawa A, Yoshida S, Yoshikawa H, et al.. Case of novel PITX2 gene mutation associated with Peters' anomaly and persistent hyperplastic primary vitreous. Eye (Lond). 2010; 24: 391–393. [DOI] [PubMed] [Google Scholar]
- 10. Golaszewska K, Dub N, Saeed E, Mariak Z, Konopinska J.. Axenfeld-Rieger syndrome combined with a foveal anomaly in a three-generation family: a case report. BMC Ophthalmol. 2021; 21: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suzuki K, Nakamura M, Amano E, Mokuno K, Shirai S, Terasaki H. Case of chromosome 6p25 terminal deletion associated with Axenfeld-Rieger syndrome and persistent hyperplastic primary vitreous. Am J Med Genet A. 2006; 140: 503–508. [DOI] [PubMed] [Google Scholar]
- 12. Jacobson A, Bohnsack BL.. Posterior segment findings in Axenfeld-Rieger syndrome. J AAPOS. 2022; 26: 320–322. [DOI] [PubMed] [Google Scholar]
- 13. Beby F, Des Portes V, Till M, Mottolese C, Denis P.. Chromosome 6p25 deletion syndrome: report of a case with optic disc coloboma and review of published ophthalmic findings. Ophthalmic Genet. 2012; 33: 240–248. [DOI] [PubMed] [Google Scholar]
- 14. Walsh LM, Lynch SA, Clarke MP.. Ocular abnormalities in a patient with partial deletion of chromosome 6p. A case report. Ophthalmic Genet. 1997; 18: 151–156. [DOI] [PubMed] [Google Scholar]
- 15. Daruich A, Robert MP, Leroy C, et al.. Foveal hypoplasia grading in 95 cases of congenital aniridia: correlation to phenotype and PAX6 genotype. Am J Ophthalmol. 2022; 237: 122–129. [DOI] [PubMed] [Google Scholar]
- 16. Daruich A, Duncan M, Robert MP, et al.. Congenital aniridia beyond black eyes: from phenotype and novel genetic mechanisms to innovative therapeutic approaches. Prog Retin Eye Res. 2022; 95: 101133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Farris J, Khanna C, Smadbeck JB, et al.. Complex balanced intrachromosomal rearrangement involving PITX2 identified as a cause of Axenfeld-Rieger syndrome [published online ahead of print January 17, 2024]. Am J Med Genet A, 10.1002/ajmg.a.63542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kumar M, Chambers C, Dhamija R.. Axenfeld-Rieger syndrome and leukoencephalopathy caused by a mutation in FOXC1. Pediatr Neurol. 2017; 66: 113–114. [DOI] [PubMed] [Google Scholar]
- 19. Ayala GD, Linderman RE, Valenzuela RK, et al.. Assessing foveal structure in individuals with TYR R402Q and S192Y hypomorphic alleles. Ophthalmol Sci. 2021; 1: 100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Currant H, Hysi P, Fitzgerald TW, et al.. Genetic variation affects morphological retinal phenotypes extracted from UK Biobank optical coherence tomography images. PLoS Genet. 2021; 17: e1009497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thomas MG, Kumar A, Mohammad S, et al.. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography a predictor of visual acuity? Ophthalmology. 2011; 118: 1653–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wilk MA, McAllister JT, Cooper RF, et al.. Relationship between foveal cone specialization and pit morphology in albinism. Invest Ophthalmol Vis Sci. 2014; 55: 4186–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wagner-Schuman M, Dubis AM, Nordgren RN, et al.. Race- and sex-related differences in retinal thickness and foveal pit morphology. Invest Ophthalmol Vis Sci. 2011; 52: 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zouache MA, Silvestri G, Amoaku WM, et al.. Comparison of the morphology of the foveal pit between African and caucasian populations. Transl Vis Sci Technol. 2020; 9: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schneider CA, Rasband WS, Eliceiri KW.. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012; 9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kraker JA, Omoba BS, Cava JA, et al.. Assessing the influence of OCT-A device and scan size on retinal vascular metrics. Transl Vis Sci Technol. 2020; 9: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Linderman RE, Cava JA, Salmon AE, et al.. Visual acuity and foveal structure in eyes with fragmented foveal avascular zones. Ophthalmol Retina. 2020; 4: 535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Linderman R, Salmon AE, Strampe M, Russillo M, Khan J, Carroll J. Assessing the accuracy of foveal avascular zone measurements using optical coherence tomography angiography: segmentation and scaling. Transl Vis Sci Technol. 2017; 6: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moir J, Rodriguez SH, Chun LY, Massamba N, Skondra D.. Racial differences in quantitative optical coherence tomography angiography findings between older non-diabetics with co-morbidities. PLoS One. 2023; 18: e0285360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chun LY, Silas MR, Dimitroyannis RC, Ho K, Skondra D.. Differences in macular capillary parameters between healthy black and white subjects with optical coherence tomography angiography (OCTA). PLoS One. 2019; 14: e0223142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dubra A, Sulai Y.. Reflective afocal broadband adaptive optics scanning ophthalmoscope. Biomed Opt Express. 2011; 2: 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Steven S, Sulai YN, Cheong SK, Bentley J, Dubra A.. Long eye relief fundus camera and fixation target with partial correction of ocular longitudinal chromatic aberration. Biomed Opt Express. 2018; 9: 6017–6037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dubra A, Harvey Z.. Registration of 2D Images from Fast Scanning Ophthalmic Instruments. Berlin: Springer; 2010: 60–71. [Google Scholar]
- 34. Salmon AE, Cooper RF, Langlo CS, Baghaie A, Dubra A, Carroll J. An automated reference frame selection (ARFS) algorithm for cone imaging with adaptive optics scanning light ophthalmoscopy. Transl Vis Sci Technol. 2017; 6: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen M, Cooper RF, Han GK, Gee J, Brainard DH, Morgan JIW. Multi-modal automatic montaging of adaptive optics retinal images. Biomed Opt Express. 2016; 7: 4899–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cooper RF, Wilk MA, Tarima S, Carroll J.. Evaluating descriptive metrics of the human cone mosaic. Invest Ophthalmol Vis Sci. 2016; 57: 2992–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perveen R, Lloyd IC, Clayton-Smith J, et al.. Phenotypic variability and asymmetry of Rieger syndrome associated with PITX2 mutations. Invest Ophthalmol Vis Sci. 2000; 41: 2456–2460. [PubMed] [Google Scholar]
- 38. Dubis AM, Hansen BR, Cooper RF, Beringer J, Dubra A, Carroll J. Relationship between the foveal avascular zone and foveal pit morphology. Invest Ophthalmol Vis Sci. 2012; 53: 1628–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Linderman RE, Georgiou M, Woertz EN, et al.. Preservation of the foveal avascular zone in achromatopsia despite the absence of a fully formed pit. Invest Ophthalmol Vis Sci. 2020; 61: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang T, Godara P, Blanco ER, et al.. Variability in human cone topography assessed by adaptive optics scanning laser ophthalmoscopy. Am J Ophthalmol. 2015; 160: 290–300.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Y, Bensaid N, Tiruveedhula P, Ma J, Ravikumar S, Roorda A. Human foveal cone photoreceptor topography and its dependence on eye length. eLife. 2019; 8: e47148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cava JA, Allphin MT, Mastey RR, et al.. Assessing interocular symmetry of the foveal cone mosaic. Invest Ophthalmol Vis Sci. 2020; 61: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Domdei N, Reiniger JL, Holz FG, Harmening WM.. The relationship between visual sensitivity and eccentricity, cone density and outer segment length in the human foveola. Invest Ophthalmol Vis Sci. 2021; 62: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gronskov K, Ek J, Brondum-Nielsen K. Oculocutaneous albinism. Orphanet J Rare Dis. 2007; 2: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fieß A, Pfisterer A, Gissler S, et al.. Retinal thickness and foveal hypoplasia in adults born preterm with and without retinopathy of prematurity: the gutenberg prematurity eye study. Retina. 2022; 42: 1716–1728. [DOI] [PubMed] [Google Scholar]
- 46. Rufai SR, Thomas MG, Purohit R, et al.. Can structural grading of foveal hypoplasia predict future vision in infantile nystagmus?: a longitudinal study. Ophthalmology. 2020; 127: 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kuht HJ, Maconachie GDE, Han J, et al.. Genotypic and phenotypic spectrum of foveal hypoplasia: a multicenter study. Ophthalmology. 2022; 129: 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kuht HJ, Thomas MG, McLean RJ, Sheth V, Proudlock FA, Gottlob I. Abnormal foveal morphology in carriers of oculocutaneous albinism. Br J Ophthalmol. 2023; 107: 1202–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Putnam NM, Hammer DX, Zhang Y, Merino D, Roorda A.. Modeling the foveal cone mosaic imaged with adaptive optics scanning laser ophthalmoscopy. Opt Express. 2010; 18: 24902–24916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tao LW, Wu Z, Guymer RH, Luu CD.. Ellipsoid zone on optical coherence tomography: a review. Clin Exp Ophthalmol. 2016; 44: 422–430. [DOI] [PubMed] [Google Scholar]
- 51. He Y, Chen X, Tsui I, Vajzovic L, Sadda SR.. Insights into the developing fovea revealed by imaging. Prog Retin Eye Res. 2022; 90: 101067. [DOI] [PubMed] [Google Scholar]
- 52. Bringmann A, Syrbe S, Gorner K, et al.. The primate fovea: structure, function and development. Prog Retin Eye Res. 2018; 66: 49–84. [DOI] [PubMed] [Google Scholar]
- 53. Gage PJ, Rhoades W, Prucka SK, Hjalt T.. Fate maps of neural crest and mesoderm in the mammalian eye. Invest Ophthalmol Vis Sci. 2005; 46: 4200–4208. [DOI] [PubMed] [Google Scholar]
- 54. Lutty GA, McLeod DS.. Development of the hyaloid, choroidal and retinal vasculatures in the fetal human eye. Prog Retin Eye Res. 2018; 62: 58–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rasys AM, Wegerski A, Trainor PA, Hufnagel RB, Menke DB, Lauderdale JD. Dynamic changes in ocular shape during human development and its implications for retina fovea formation. Bioessays. 2023; 46: e2300054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Skarie JM, Link BA.. FoxC1 is essential for vascular basement membrane integrity and hyaloid vessel morphogenesis. Invest Ophthalmol Vis Sci. 2009; 50: 5026–5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.