

Abstract
Modern glycoproteomics experiments require the use of search engines due to the generation of countless spectra. While these tools are valuable, manual validation of search engine results is often required for detailed analysis of glycopeptides as false-discovery rates are often not reliable for glycopeptide data. Near-isobaric mismatches are a common source of misidentifications for the popular glycopeptide-focused search engine pGlyco3.0, and in this technical note we share a strategy and script that improves the accuracy of the search utilizing two manually validated datasets of the glycoproteins CD16a and HIV-1 Env as proof-of-principle.
Keywords: Glycopeptide, glycoprotein, Glycoproteomics, mass spectrometry
Introduction
As mass spectrometry instrumentation matures, more in-depth data is being produced than ever before. The complexity of glycoproteomic data necessitates the use of specialized search engines (e.g. Byonic (Bern et al. 2012), pGlyco (Zeng et al. 2021), MSFragger (Polasky et al. 2022)) to assist the automated and high-throughput data analysis of intact glycopeptides (Polasky and Nesvizhskii 2023). However, one of the biggest challenges for glycoproteomic data analysis is the quality control of the search output, where a false discovery rate (FDR) evaluation is often underestimated leading to inaccurate glycopeptide spectral matches (GPSMs). Manual validation can increase the accuracy of GPSMs but requires large amounts of analyst time, effort, and expertise to complete as the searches often scale to thousands of GPSMs (Hoffmann et al. 2018). Manually validated results can differ significantly from the FDR-filtered search engine output. The pGlyco search engine is of particular interest because it computes respective FDR values on peptide and glycan moieties (N-linked or O-linked) as well as an integrated FDR value on glycopeptide entities therefore providing a comprehensive FDR estimation, whereas most of the other search engines only consider FDRs on the peptides (Zeng et al. 2021). To streamline the process of validating pGlyco search outputs, in this technical note, we looked to identify common errors in glycopeptide spectral matching and build automated solutions.
Results and discussion
When examining large sets of spectra that failed manual inspection, common themes emerge. A major issue is the monoisotopic mass being incorrectly assigned to the 1 or 2 13C-isotope peak that shifts the predicted mass of the glycopeptide by ~1–2 daltons. Glycopeptides, in general, are more likely to have large masses and a higher number of Carbon atoms than a typical non-glycosylated peptide resulting in the base peak often not being the monoisotopic peak. This incorrect calculation of the monoisotopic mass can result in common errors given that various combinations of sugars result in mass shifts of ~1 or ~2 daltons. One of the most common errors found in glycopeptide spectral matching search engines is confusion between a pair of fucoses (Fuc) and a single N-acetylneuraminic acid (Neu5Ac). For reference, the m/z values for the oxonium ions of monosaccharides are: Fuc at m/z 147.0652; hexose (Hex) at m/z 163.0601; N-acetylhexosamine (HexNAc) at m/z 204.0866; Neu5Ac at m/z 292.1027. Two fucoses are roughly 1 Dalton (1.0204) greater than a single Neu5Ac. This problem can even be doubled, where 2 sialic acids are mistaken for 4 fucoses, which leads to erroneous GPSMs with multiple fucoses when the peak with two-13C isotopes is misidentified as the monoisotopic peak. Note the small difference in weight between two fucose for 1 Neu5Ac (1.0204) and a single proton (1.0073).
In some cases, this problem can be easily identified with the presence of Neu5Ac oxonium ions in the MS/MS spectra (m/z 274 and m/z 292). An example of an assigned non-sialylated, triply-fucosylated glycopeptide is shown that is in fact a mono-fucosylated, mono-sialyated glycopeptide by manual examination (Fig. 1a, denoted using symbol nomenclature for glycans (Neelamegham et al. 2019)). However, spotting this error becomes more difficult in glycopeptides with both and multiple Neu5Acs and fucoses, as the presence of Neu5Ac oxonium ions can be explained by the presence of a single or multiple Neu5Acs. Additionally, the Y-ions (defined as the peptide backbone with glycan attached, Fig. 1a) rarely contain terminal monosaccharides, such as Neu5Acs and fucoses, due to non-reducing end fragmentation as well as limited mass range in the mass spectrometer. The neutral losses of the terminal Neu5Acs and fucoses are seldom observed for the aforementioned reasons as well. The latest version of pGlyco at the time of writing (pGlyco3.0_build20210615) attempts to find Neu5Ac oxonium ions (m/z 274 and m/z 292) in multiply fucosylated GPSMs and corrects them to assignment with Neu5Acs where the Neu5Ac oxonium ions are present. This results in a few additional output columns from the program (e.g. “CorrectedGlycan(x,x,x,x)”) that reflect where corrections have been made. While this solution seems to fix some of the erroneous GPSMs, it does not capture all of them. This motivated us to find additional solutions to the problem.
Fig. 1.
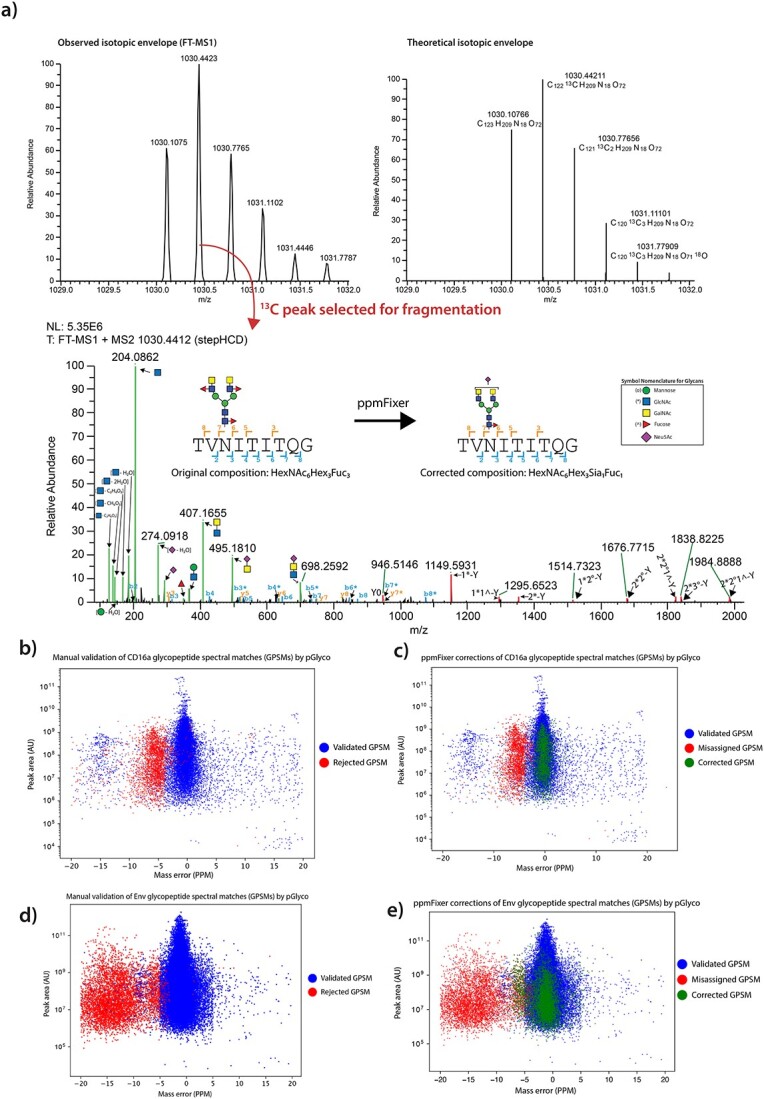
pGlyco misidentification is associated with a mass error shift. a) Example spectra of a misidentified CD16a glycopeptide being corrected using ppmFixer. Upper—MS1 precursor ion selected (left) and theoretical isotopic envelope (right). Lower—MS2 from precursor ion stepHCD fragmentation. * indicates an GlcNAc attached to a peptide b- or y-ion. Glycan Y-ions annotated with */ °/^ for clarity. (Color guide—blue: peptide backbone b-ion, orange: peptide backbone y-ion, red: peptide backbone with glycan–attached Y-ion, green: oxonium ion). b) Mass error vs. monoisotopic peak area of identified CD16a GPSMs (glycopeptide spectral matches), colored by agreement with manual validation. c) Demonstration of the shift of revised CD16a GPSMs (green) from original mass error (red). GPSMs rejected in both sets excluded from plot. d) Demonstration of the shift of revised Env GPSMs (green) from original mass error (red). GPSMs rejected in both sets excluded from plot. e) Demonstration of the shift of revised Env GPSMs (green) from original mass error (red). GPSMs rejected in both sets excluded from plot.
Another common error in pGlyco search output is the confusion between four N-acetylhexosamines (HexNAc) and five hexoses (Hex) where the mass difference is approximately 2 Daltons (2.0534). For reasons discussed earlier, such as multiply-charged species reducing mass difference as well as mis-identified monoisotopic peaks, this confusion often leads to the mis-assignment of high mannose (Man) and N-acetylgalactosamine β1–4 N-acetylglucosamine (GalNAcβ1–4GlcNAc, or LacDiNAc) glycans. For example, the program often mis-identifies Man8GlcNAc2 as HexNAc4Man3GlcNAc2 with the two–13C peak being misidentified as the monoisotopic peak (note that two–13C peak is a mass shift of 2.0146 daltons).
An additional abundant error is the misidentification of certain high-mannose glycans as complex or bizarre (and unlikely) hybrid N-glycans containing a single hexose. The misidentified glycans have two GlcNAcs, three fucoses, and a sialic acid assigned which is ~1 dalton (1.0581) heavier than simply being 7 hexoses.
An alternative approach to full manual validation (and the one being proposed in this manuscript) involves a comparison of mass errors to the average of the population. In order to test our approach, we utilized a highly detailed dataset of the human glycoprotein CD16a that we had previous characterized (Adams et al. 2022). CD16a has 5 sites of N-linked glycosylation and our analyses generated 18,586 GPSMs (see Supplementary Methods). When individual GPSMs from this dataset are plotted against their mass error, there is a distribution of GPSMs that are both offset from the overall distribution of mass errors and are rejected during manual validation (Fig. 1b). During manual validation, it was noted that a large proportion of these corrections were due to the near-isobaric conundrums detailed previously. This is caused by these glycan species not being exactly isobaric; there is a small offset caused by nuclear binding energy that is reflected in the mass error of the resulting GPSMs (for example proton weighing 1.0073 daltons but 2 fucoses weighting 1.0204 daltons more than a Neu5Ac). Therefore, likely candidates for near-isobaric mismatches can be found by identifying outliers in the mass error distribution, which can easily be done by identifying those that have a mass error greater than the standard deviation of the entire population. To correct this issue, we developed a Python script titled “ppmFixer” that does the following:
1) Identifies GPSMs with a mass error > 1 standard deviation from the mean mass error.
-
2) Recalculates mass error for these GPSMs if they fit the following cases:
Where #Fucose
 2, two fucoses are replaced by a Neu5Ac, move down an isotope.
2, two fucoses are replaced by a Neu5Ac, move down an isotope.Where #Fucose
 4, two fucoses are replaced by a Neu5Ac, move down two isotopes.
4, two fucoses are replaced by a Neu5Ac, move down two isotopes.Where #HexNAc >5, 5 hexoses are added and 4 HexNAcs removed, move down two isotopes.
Where #HexNAc = 4, 7 hexoses are added and 2 HexNAcs removed and all sialics/fucoses removed, move down two isotopes.
3) If |new mass error| < |original mass error|, adjusts the composition. Outputs this adjusted composition as a new column in the pGlyco output file (CorrectedGlycan_ppmFixer). Note: If multiple cases apply, accept the revision with the lowest mass error. Additionally, the native pGlyco correction utilizing the sialic acid oxonium ion takes precedence over all other corrections due to spectral evidence.
4) Recalculates mass error average and stdDev with new corrections, cycles through script again.
5) Once a round adds no new changes, stop execution.
6) Outputs the adjusted compositions and mass errors as new columns in the pGlyco output file (CorrectedGlycan_ppmFixer, Glycan(H,N,A,F)_ppmFixer).
This script identifies a significant proportion of the misassigned and mass offset GPSMs resulting in mass error shifts more in line with the rest of the distribution (Fig. 1c). We also tested ppmFixer on a larger glycoproteomics dataset from the HIV−1 envelope glycoprotein (Env), that we have previously characterized with sequence perturbations many times (Zhou et al. 2017; Duan et al. 2018; Yu et al. 2018; Escolano et al. 2019; Wang et al. 2020), where we observed similar patterns of mass error shifts (Fig. 1d and e).
We probed what corrections were being made in both the CD16a and Env datasets. The two datasets showed diverging revision profiles (Fig. 2a), with the most common revision in the CD16a dataset being the conversion of two fucoses two a sialic acid (F2− > A1) and the most common revision in the Env dataset being the conversion of a complex glycan to high-mannose (N×Hy− > N(×−4)H(y + 5)). This is consistent with the fact that the N-glycans from CD16a are dominated by highly sialylated, complex glycans while the HIV−1 Env has a large degree of high mannose N-linked glycans. This demonstrates that the revisions needed can vary from dataset to dataset and justifies the inclusion of all revision types, even if they may not greatly impact a specific experiment.
Fig. 2.

ppmFixer adjusts pGlyco results to more closely match manual validation for multiple datasets. a) Types of revisions made by ppmFixer in CD16a and Env datasets. b) Comparison of agreement with manual validation between default pGlyco output and ppmFixer (revised) output. c) Depiction of ppmFixer accuracy. d) Demonstration of the effect of the adjustment on the most abundant glycoforms (for CD16a: Filtered for GPSM count >250, for Env: Filtered for GPSM count >500). e) Effects of ppmFixer revisions on levels of CD16a Neu5Ac:Fucose ratios. f) Effects of ppmFixer on site-specific Env N-glycan classes.
The results of the ppmFixer output were compared to fully manually validated data, which can be viewed as a positive control that automated results should seek to emulate. Utilizing the CD16a dataset, ppmFixer improved the default pGlyco output from a 76.5% match with manual validation to an 88.0% match with manual validation, a large improvement in GPSM accuracy (Fig. 2b). Additionally, ppmFixer erroneously adjusted only 0.37% of total GPSMs, which is a marginal error compared to the 11.7% of total GPSMs that were corrected (Fig. 2c). This effectively means that where ppmFixer made a change, 97% of the time it agreed with manual validation for CD16a. ppmFixer effectively cuts overall GPSM misidentifications to half of their volume when compared to the default pGlyco output for this CD16a dataset. ppmFixer performed similarly well with the Env dataset, with agreement with manual validation increasing from 87.2% to 95.5% (Fig. 2b).
Next, we compared the most abundant GPSMs identified in the default pGlyco output to those found in the revised ppmFixer output (Fig. 2d). Substantial changes were observed, with many of the top glycan compositions being affected. In particular, in the Env dataset, 3 of the top 10 most abundant glycoforms were eliminated entirely. We then compared the default and revised outputs with regards to fucosylation and sialylation for the CD16a dataset (Fig. 2e). Overall, ppmFixer output contained a greater proportion of sialic acids in comparison to fucoses when compared to the default pGlyco output. Identification of this error explains the previous published finding that pGlyco output contains more fucosylated glycans than other search engines (Riley et al. 2019). Finally, we examined how ppmFixer altered site-specific glycan distributions by comparing relative amounts of glycan types using the Env dataset (Fig. 2f). The resulting data illustrates a shift away from hybrid/complex N-glycans and towards high-mannose N-glycans. This can be explained by the most common correction in this dataset being the conversion of a complex N-glycan to high-mannose (Fig. 2a). This data demonstrates that ppmFixer’s revisions can have a dramatic effect on site-specific glycopeptide analysis.
Overall, ppmFixer is a simple, yet effective, improvement to the current pGlyco3.0 workflow. It is important to note that ppmFixer is only effective when using high-accuracy, high-resolution instrumentation that is able to generate a mass error distribution with a standard deviation less than the small mass errors generated by the described near-isobaric mismatches, as this strategy relies on identifying distinct populations of mass error distributions. While manual validation remains the gold standard of glycopeptide analysis, corrections to search engine results help reduce the burden of the process while also providing more accurate results especially for high-throughput analyses where manual validation isn’t feasible.
Methods
Methods can be found in the Supplementary Methods.
Supplementary Material
Contributor Information
Trevor M Adams, Department of Biochemistry and Molecular Biology, Complex Carbohydrate Research Center, University of Georgia, 315 Riverbend Road, Athens 30602, Georgia.
Peng Zhao, Department of Biochemistry and Molecular Biology, Complex Carbohydrate Research Center, University of Georgia, 315 Riverbend Road, Athens 30602, Georgia.
Rui Kong, Department of Pathology and Laboratory Medicine, Emory Vaccine Center, Emory University, 7 Frist Ave, Atlanta 30317, Georgia.
Lance Wells, Department of Biochemistry and Molecular Biology, Complex Carbohydrate Research Center, University of Georgia, 315 Riverbend Road, Athens 30602, Georgia.
Author contributions
Trevor Adams (Conceptualization [Equal], Data curation [Lead], Formal analysis [Lead], Investigation [Lead], Methodology [Lead], Resources [Supporting], Software [Lead], Validation [Lead], Writing—original draft [Lead], Writing—review & editing [Supporting], Peng Zhao (Data curation [Supporting], Formal analysis [Supporting], Investigation [Supporting], Methodology [Supporting], Resources [Supporting], Validation [Supporting], Writing—review & editing [Supporting], Rui Kong (Funding acquisition [Equal], Methodology [Supporting], Resources [Supporting]), Lance Wells (Conceptualization [Equal], Funding acquisition [Equal], Methodology [Supporting], Project administration [Lead], Resources [Equal], Supervision [Lead], Validation [Supporting], Writing—review & editing [Lead]).
Funding
Support for this work was provided by the NIH (R01AI162267 to RK, R01GM130915 to LW, and R01GM111939 to LW), Emory Vaccine Center (RK), Georgia Research Alliance (RK and LW), and Glycoscience Training Program (5T32GM107004) from NIGMS (TMA).
Conflict of interest statement: None declared.
Data availability
ppmFixer can be downloaded from https://github.com/trvadams/ppmFixer.
References
- Adams TM, Zhao P, Chapla D, Moremen KW, Wells L. Sequential in vitro enzymatic N-glycoprotein modification reveals site-specific rates of glycoenzyme processing. J Biol Chem. 2022:298(10):102474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bern M, Kil YJ, Becker C. Byonic: advanced peptide and protein identification software. Curr Protoc Bioinformatics. 2012:Chapter 13(20):13.20.1–13.20.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan H, Chen X, Boyington JC, Cheng C, Zhang Y, Jafari AJ, Stephens T, Tsybovsky Y, Kalyuzhniy O, Zhao P, et al. Glycan masking focuses immune responses to the HIV-1 CD4-binding site and enhances elicitation of VRC01-class precursor antibodies. Immunity. 2018:49(2):301–311.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escolano A, Gristick HB, Abernathy ME, Merkenschlager J, Gautam R, Oliveira TY, Pai J, West AP, Barnes CO, Cohen AA, et al. Immunization expands B cells specific to HIV-1 V3 glycan in mice and macaques. Nature. 2019:570(7762):468–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Pioch M, Pralow A, Hennig R, Kottler R, Reichl U, Rapp E. The fine art of destruction: a guide to in-depth Glycoproteomic analyses-exploiting the diagnostic potential of fragment ions. Proteomics. 2018:18(24):1800282. [DOI] [PubMed] [Google Scholar]
- Neelamegham S, Aoki-Kinoshita K, Bolton E, Frank M, Lisacek F, Lütteke T, O’Boyle N, Packer NH, Stanley P, Toukach P, et al. Updates to the symbol nomenclature for glycans guidelines. Glycobiology. 2019:29(9):620–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polasky DA, Nesvizhskii AI. Recent advances in computational algorithms and software for large-scale glycoproteomics. Curr Opin Chem Biol. 2023:72:102238. [DOI] [PubMed] [Google Scholar]
- Polasky DA, Geiszler DJ, Yu F, Nesvizhskii AI. Multiattribute glycan identification and FDR control for Glycoproteomics. Mol Cell Proteomics. 2022:21(3):100205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley NM, Hebert AS, Westphall MS, Coon JJ. Capturing site-specific heterogeneity with large-scale N-glycoproteome analysis. Nat Commun. 2019:10(1):1311. [accessed 2019 Mar 25]. http://www.nature.com/articles/s41467-019-09222-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Voronin Y, Zhao P, Ishihara M, Mehta N, Porterfield M, Chen Y, Bartley C, Hu G, Han D, et al. Glycan profiles of gp120 protein vaccines from four major HIV-1 subtypes produced from different host cell lines under non-GMP or GMP conditions. J Virol. 2020:94(7):e01968–e01919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W-H, Zhao P, Draghi M, Arevalo C, Karsten CB, Suscovich TJ, Gunn B, Streeck H, Brass AL, Tiemeyer M, et al. Exploiting glycan topography for computational design of Env glycoprotein antigenicity. PLoS Comput Biol. 2018:14(4):e1006093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W-F, Cao W-Q, Liu M-Q, He S-M, Yang P-Y. Precise, fast and comprehensive analysis of intact glycopeptides and modified glycans with pGlyco3. Nat Methods. 2021:18(12):1515–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Doria-Rose NA, Cheng C, Stewart-Jones GBE, Chuang G-Y, Chambers M, Druz A, Geng H, McKee K, Kwon YD, et al. Quantification of the impact of the HIV-1-glycan shield on antibody elicitation. Cell Rep. 2017:19(4):719–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ppmFixer can be downloaded from https://github.com/trvadams/ppmFixer.