

Abstract
Increased chromatin accessibility is a feature of cell-type-specific cis-regulatory elements; therefore, mapping of DNase I hypersensitive sites (DHSs) enables the detection of active regulatory elements of transcription, including promoters, enhancers, insulators and locus-control regions. Single-cell DNase sequencing (scDNase-seq) is a method of detecting genome-wide DHSs when starting with either single cells or <1,000 cells from primary cell sources. This technique enables genome-wide mapping of hypersensitive sites in a wide range of cell populations that cannot be analyzed using conventional DNase I sequencing because of the requirement for millions of starting cells. Fresh cells, formaldehyde-cross-linked cells or cells recovered from formalin-fixed paraffin-embedded (FFPE) tissue slides are suitable for scDNase-seq assays. To generate scDNase-seq libraries, cells are lysed and then digested with DNase I. Circular carrier plasmid DNA is included during subsequent DNA purification and library preparation steps to prevent loss of the small quantity of DHS DNA. Libraries are generated for high-throughput sequencing on the Illumina platform using standard methods. Preparation of scDNase-seq libraries requires only 2 d. The materials and molecular biology techniques described in this protocol should be accessible to any general molecular biology laboratory. Processing of high-throughput sequencing data requires basic bioinformatics skills and uses publicly available bioinformatics software.
INTRODUCTION
Regulatory DNA regions of the genome coordinate cell-type-specific gene expression1. In these regions, nucleosomes can be dynamically removed or added to control accessibility of chromatin-binding proteins to the DNA during transcription and other nuclear processes. Genomic regions with increased chromatin accessibility serve as markers for a wide range of regulatory features, including enhancers, silencers, promoters, locus-control regions and insulators. Therefore, measurement of chromatin accessibility provides a means to identify regulatory DNA elements.
DNase sequencing (DNase-seq) and assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) are the most common methods used to identify DNA accessibility on a genome-wide scale2–5. In this protocol, we describe single-cell sequencing (scDNase-seq)—a modified DNase-seq protocol for single-cell or limited-cell inputs6. Our recent work highlights the use of scDNase-seq for mapping DHSs at the single-cell level in embryonic stem cells and NIH3T3 cells, as well as in a small number of tumor and normal cells recovered from FFPE tissue slides. At the single-cell level, scDNase-seq can reproducibly detect the same DHSs from a bulk population while enabling the study of cell-to-cell variation within the population. A limitation of conventional DNase-seq on a bulk population of cells is that it cannot identify cell-to-cell variations in DHSs. The differences in DHSs between individual cells in an apparently ‘homogeneous’ population of cells may be biologically meaningful7. ScDNase-seq offers a method of identifying cell-to-cell variation in DHSs within a bulk population of cells.
Development and overview of scDNase-seq
The original DNase-seq method requires millions of cells, and, therefore, its application is limited to cell lines or abundant primary cell types3. The scDNase-seq method adapts DNase-seq for use with single cells or limited cells by including a bacterial circular carrier DNA to minimize loss of sample DNA during processing. Additional changes to conventional DNase-seq include the omission of nuclei isolation, optimization of the DNase-I digestion steps for limited cell numbers and omission of fractionation of DNA fragments by agarose gel to minimize loss of DNA, as well as optimization of PCR conditions to preferentially amplify the desired small DNA fragments.
Otherwise, the stages of the scDNase-seq procedure are similar to those of conventional DNase-seq. First, the cellular and nuclear membranes are permeabilized so that the DNase I enzyme can enter the nucleus. DNase I enzyme is added to the cells to create single- and double-stranded nicks in the DNA. Genomic regions unbound by histones, transcription factors or other regulatory proteins will be relatively more accessible to the DNase I enzyme. This relative increased DNA accessibility to the DNase I enzyme generates short DNA fragments from DNase I hypersensitive regions. The digested DNA is purified, next-generation sequencing libraries are prepared and the smallest DNA fragments are sequenced by high-throughput sequencing.
Data processing in scDNase-seq and conventional DNase-seq is also comparable. Short reads generated by high-throughput sequencing are aligned to the appropriate genome, and unique, nonredundant reads are identified. The accumulation of multiple reads at a particular genomic location indicates the presence of a DHS peak and can be identified with existing peak-calling software. However, peak calling is not possible when starting with fewer than ten cells, because of the limited number of reads at each DHS. Necessary modifications to the analysis process in the scDNase-seq procedure are discussed further in the Experimental design section.
Comparison with other methods
The original ATAC-seq method is optimized for exactly 50,000 cells, to ensure the proper ratio of cellular DNA to the transposase that tags accessible regions. Sensitivity and specificity for ATAC-seq drops considerably when starting with 500 or 5,000 cells2. Use of the commercially available transposase for ATAC-seq increases the cost per sample in comparison with scDNase-seq, although it is now possible to independently produce the enzyme8.
Two recent publications report methods for performing single-cell ATAC-seq (scATAC-seq)9,10. In these protocols, single cells are isolated for scATAC-seq, either by using a programmable microfluidics platform (Fluidigm) or by combinatorial indexing of nuclei. The advantage of these approaches is that they enable higher throughput to efficiently assay a larger number of single cells than does scDNase-seq. However, expertise with a microfluidics machine is mandatory for using scATAC-seq, making this a more technically challenging approach compared with scDNase-seq. With scDNase-seq, each single cell on average has >300,000 unique reads when the library is sequenced to saturation. By contrast, individual cells passing quality filters for scATAC-seq have ~5,000 unique reads. Owing to the lack of library complexity, <10% of promoters identified in bulk ATAC-seq are detected in any individual cell—in contrast to scDNase-seq, in which ~50% of bulk DHS promoter sites can be detected. Therefore, scATAC-seq captures only a small percentage of DHSs for an individual cell, making it necessary to combine the results from hundreds of single cells to draw meaningful conclusions. scDNase-seq also works well with both native and formaldehyde-cross- linked cells, which allows flexibility in sample preparation and storage. ATAC-seq on formaldehyde-fixed cells has recently been described using 50,000 starting cells11.
Formaldehyde-assisted isolation of regulatory elements sequencing (FAIRE-seq) is another assay of DNA accessibility and is more sensitive for the detection of distal regulatory elements than is DNase-seq12,13. However, similar to the standard DNase-seq protocol, FAIRE-seq requires 1 million to 10 million cells, thus limiting its applicability to abundant cell types.
Applications
ScDNase-seq enables the detection of genome-wide DHSs in a wide range of cell types and tissues without limitations owing to the abundance of the starting material. Our group and others have used scDNase-seq to map regulatory regions in embryonic stem cells, hematopoietic stem cells and developing embryos6,14. We expect that the method will be broadly applicable in areas such as stem-cell biology, development and cancer biology7,15.
Limitations
Automation.
At present, the scDNase-seq method cannot be performed in an automated or semiautomated manner because of the phenol extraction/ethanol precipitation steps. Thus, it is not capable of processing hundreds or thousands of single cells at a time as is possible with a microfluidics approach.
Sequencing efficiency.
Although library complexity is substantially higher with scDNase-seq as compared with scATAC-seq, the percentage of unique, nonredundant reads mapped to the genome for single-cell libraries is low. For scDNase-seq, on average, 1–3% of the total sequenced reads uniquely map to the reference genome. Generation of a library with >300,000 unique reads requires 10–40 × 106 total sequencing reads. Sequencing each single-cell library to saturation requires a large number of reads, which increases the cost of sequencing. However, the mappability and library complexity can be substantially improved by increasing the cell number. Increasing the input to ten cells results in a log increase in the number of unique reads per library.
DNase I enzyme variability.
DNase I has a relatively narrow optimal enzymatic range to achieve properly digested DNA. In addition, the enzymatic activity of DNase I varies from lot to lot and can decline even when properly stored. The relative chromatin accessibility also varies from cell type to cell type. For this reason, it is necessary to perform a titration of DNase I concentrations when using a new cell type or adjusting the starting cell number.
Experimental design
DNase I enzyme titration.
Detection of chromatin accessibility by DNase-seq is based on the principle that the nucleosome-free genomic regions are hypersensitive to DNase I digestion as compared with the regions associated with nucleosomes. An ideal experiment would thus detect only the nucleosome-free genomic regions that are associated with regulatory elements. However, if the degree of digestion is not well-controlled, the regions associated with nucleosomes can also be completely digested by DNase I, which leads to increased background and decreased specificity. On the other hand, underdigestion reduces the number of detectable hypersensitive sites.
In our experience, for single-cell or limited cell numbers (<10,000 cells), the 30×, 45× and 60× DNase I dilutions (0.33 U/μl, 0.22 U/μl and 0.16 U/μl, respectively) cited in the protocol have worked for a number of cell types, including NIH3T3 cells, mouse embryonic stem (ES) cells, mouse hematopoietic stem cells, and human and mouse T cells. These concentrations worked over a range of starting cell numbers and generated the data from our labs in the recent scDNase-seq paper6. When possible, the use of two to three enzyme concentrations for each sample mitigates some of the variability in DNase I digestion efficiency. Performing an enzyme titration across a broader range of enzyme concentrations may be necessary when adapting the method to new cell types, in order to identify the optimal two or three enzyme concentrations for a particular cell type (Fig. 1).
Figure 1 |.

Results of pilot experiment to determine optimal DNase I enzyme concentration. 5,000 human CD8+ T cells per lane digested with decreasing concentrations (indicated in red (U/μl)) of DNase I. All samples were PCR-amplified for 15 cycles. Increasing the enzyme concentration yields more short fragments. However, the optimal DNase I concentration cannot be determined solely from the gel image. In this example, the optimal concentration range for future experiments was determined to be 0.2–0.3 U/μl by limited sequencing of each library (Fig. 2).
The first step in assessing the quality of a DNase I library is to look at the range of fragments generated after PCR amplification. The majority of DNase I-digested fragments should be between 150 and 400 bp. However, gel images alone are insufficient to determine the quality of a DNase I library. From each enzyme concentration, we therefore generate a small number of sequencing reads (0.5–1 million) to predict the signal-to-noise ratio for each concentration. By plotting the read tag density surrounding genome-wide transcription start sites (TSSs) for each concentration, it is possible to identify which concentration generates the highest relative enrichment (Fig. 2). As highlighted by the pilot DNase I titration experiments here, at higher concentrations of DNase I, more small fragments are recovered during library preparation. However, these additional small reads come from non-DHSs, as evidenced by the decreasing TSS enrichment at higher enzyme concentrations.
Figure 2 |.

Use of proximal and distal DHS read enrichment to determine optimal DNase I enzyme concentration. (a) Plot of the relative enrichment of normalized read density near transcription start sites (TSSs) that represent reads from proximal DHSs. 5,000 human CD8+ cells were collected by FACS sorting. DNase I was diluted as indicated to generate an enzyme concentration titration. Suboptimal concentrations (0.4 U/μl and above) have a broader distribution of reads near the TSS. The highest concentrations (0.5–1 U/μl) result in overdigestion of chromatin and show a depletion of reads proximal to the TSS. (b) Enrichment of reads near H3K27ac peaks, which serve as a marker of active distal enhancers, indicates that DNase I enzyme concentrations of 0.2–0.4 U/μl generate the highest enrichment at distal DHSs. On the basis of enrichment at proximal and distal DHSs, a DNase I enzyme concentration of 0.2–0.3 U/μl is optimal.
Starting cell number.
At the single-cell level, scDNase-seq does not detect every DHS seen in a bulk cell population. This is due to both technical limitations of detecting all fragments from a single cell and biological variability of ‘homogeneous’ cell populations. As bulk cell populations represent an average of accessibility across the entire population, it is more likely that DHSs with lower peaks from ensemble data will display more variability at the single-cell level. Conversely, regulatory elements for highly expressed genes are the most readily detected DHSs at the single-cell level.
When starting with limited cell numbers, the ability to detect DHSs improves with increasing cell numbers. The most notable improvement comes from increasing the starting cell numbers into the 100–1,000 cell range. At this cutoff, the signal-to-noise ratio improves markedly, and the detected DHSs have high concordance with ENCODE data generated from millions of starting cells. In addition, the efficiency of sequencing increases markedly when starting with 1,000 cells rather than 100 cells.
When the cell source is FFPE tissue slides, it is usually sufficient to scrape off cells from an area of 5 × 5 mm, although the exact cell number has not been determined.
The number of individual single cells to assay to fully capture all possible DHSs from a bulk population will vary depending on the underlying characteristics of the bulk population, making it difficult to generate universal recommendations16. Generation of DHS profiles from the bulk population of interest can help to estimate the total number of DHSs that can possibly be detected by scDNase-seq. Comparison of DHSs from the bulk population with those from pooled single cells will provide an estimate of how well a collection of single cells captures the range of variation within a bulk population.
Starting -cell quality.
It is critical to ensure that the cells used for scDNase-seq do not include apoptotic cells. Apoptotic cells degrade genomic DNA non-specifically into nucleosomal fragments (~147 bp in length). As DNase sequencing detects small DNA fragments in the size range of nucleosomal DNA, any nonspecific DNA fragments from apoptotic cells will be sequenced and will contribute to a high background. Use of a control sample without the addition of any DNase I enzyme is essential to confirm that the sample does not contain any fragmented DNA (Fig. 3). In our experiments, apoptotic cells are excluded during fluorescence-activated cell sorting (FACS). Removal of dead/dying cells can effectively decrease background noise and increase the signal-to-noise ratio (Fig. 4).
Figure 3 |.

Gel image demonstrating the importance of excluding dead or apoptotic cells before DNase I digestion. Mouse T cells were cultured in Th1-differentiating medium for 2 d. FACS-sorted lanes excluded 7-AAD+/annexin+ apoptotic cells. Column exclusion of apoptotic cells was performed using a Dead Cell Removal Kit (Miltenyi Biotec) per the manufacturer’s instructions before scDNase-seq (6.6% residual 7-AAD+/annexin+ cells). PBS-washed lanes were washed twice in PBS before DNase I digestion (29% 7-AAD+/annexin+ apoptotic cells determined by FACS). 500 cells per sample were used, and DNase I was either omitted or added at a concentration of 0.3 U/μl, as indicated. Bands seen in lanes 2 and 3 slightly below 300 bp indicate the presence of nucleosomal DNA in the cells that will create nonspecific background if sequenced; note that nucelosomal DNA is absent from lane 1 (FACS sorted). Although lanes 4–6 appear similar, only lane 4 represents a successful library for sequencing, as lanes 5 and 6 will generate more background due to the presence of nucleosomal DNA, which can be seen in the corresponding control lanes (lanes 2 and 3; no DNase I).
Figure 4 |.

Representative IGV genome browser images of scDNase-seq experiments using cells of differing viabilities22. Both scDNase-seq tracks (red and orange) are derived from 5,000 mouse naive CD4+ T cells using the 0.3 U/μl DNase I enzyme concentration, with 95 and 80% viable cells, respectively. The top track is from the ENCODE (accession no. GSM1014192) project and shows the highest signal-to-noise ratio23. The lower background seen in the red track is the result of starting with cells with higher viability, and this track is similar to conventional DNase-seq results. The presence of apoptotic cells in a library (orange track, 80% viable cells) increases the background and substantially reduces the sensitivity and specificity of scDNase-seq.
It is possible to use cells directly from cell culture, but care should be taken to ensure that the viability is greater than 95%. As described in our original paper, FFPE slides can serve as the starting material, but they will have a lower signal-to-noise ratio than fresh cells because of DNA fragmentation caused by the FFPE sample preparation procedure. It is also possible to start with fresh or previously frozen cells that are fixed with 1% (vol/vol) formaldehyde, so long as dead cells were excluded from the sample before fixation.
Controls.
As mentioned above, including a no-DNase I control for each sample allows the detection of any contaminating short fragments of DNA. If there is visible DNA on the gel in the 150–350 bp range in the no-DNase I control lane, then there will probably be an unacceptably high background in the DNase I-digested samples.
Library size selection.
The size of recovered fragments after DNase I digestion has a critical role in ensuring the highest signal-to-noise ratio. Fragment inserts between 50 and 100 bp (library DNA length = 172–222 bp) are more likely to contain meaningful reads from within hypersensitive sites, as compared with reads containing fragment inserts of 100–200 bp or longer17. As gel-based size selection is not precise, we include a broader band (150–300 bp, including linkers). Including higher-molecular-weight DNA generally only decreases the signal-to-noise ratio. Although not an absolute requirement, paired-end sequencing provides the precise fragment length of each read in a library, which is useful for obtaining a narrower size selection. Our laboratory has obtained good results with a gel-based size-selection strategy. Our collaborators have subsequently reported the use of solid-phase reversible immobilization (SPRI) beads as an alternative strategy for size selection14.
Determining sequencing depth.
When using single cells as the input for scDNase-seq, the efficiency of sequencing is low. To generate >300,000 unique, nonredundant reads per single-cell library, we typically sequence each library to >30 million reads in 50-bp single-end mode. Paired-end sequencing is desirable for FFPE samples because of chromatin fragmentation before DNase I digestion. An initial round of sequencing to generate a few million reads is helpful in estimating library complexity and quality. The final sequencing depth is dependent on the library complexity and read redundancy. Libraries predicted to have adequate complexity (>200,000 estimated unique DNA fragments) can be selected for deeper sequencing.
Even from a partially sequenced library, it should be possible to identify the relative enrichment of unique reads within 2,000 bp of genome-wide TSSs. Libraries with minimal enrichment near the TSSs are likely to have a high level of background and a lower sensitivity for detecting DHSs when sequenced to saturation. As the cell number per library increases, the efficiency of detecting unique sequencing reads improves rapidly. For libraries with >100 cells, scDNase-seq usually needs only 10 to 20 million total raw reads in order to obtain several million unique reads, which are sufficient for genome-wide detection of DHSs.
PCR amplification of libraries.
The total number of PCR cycles for each library depends on the starting cell number and the efficiency of the DNase I digestion. For single-cell libraries, 23 cycles are generally required to generate a sufficient amount of DNA for sequencing. As the input number of cells increases, the number of PCR cycles should be reduced to minimize overamplification, which reduces library complexity. Starting with 100 cells or 1,000 cells, we perform 16 or 13 cycles of PCR, respectively. Gel extraction and additional amplification for 3–4 additional cycles can be performed if there is insufficient DNA after the initial PCR steps.
Level of expertise.
scDNase-seq does not require advanced technical skills and is accessible to any molecular biology laboratory. Processing of high-throughput sequencing data uses existing software packages for sequencing alignment, marking of duplicate reads, generation of genome browser visualizations and peak calling. Complex analysis of genome-wide chromatin accessibility requires the additional technical expertise of a bioinformatics expert.
Data analysis.
When starting with more than a single cell, processing of sequencing reads and peak calling from scDNase-seq is the same as for standard DNase-seq protocols1,18,19. Our approach is to align reads to the appropriate reference genome with Bowtie2 and use nonredundant reads with a mapping quality (MAPQ) ≥ 10, followed by peak calling using model-based analysis of ChIP sequencing (MACS)18,20. BED graph files can be generated using hypergeometric optimization of motif enrichment (HOMER) and can be visualized on the University of California, Santa Cruz (UCSC) Genome Browser19,21. If the background is high, selection of smaller fragments (50–100 bp) for analysis may increase the specificity of detecting DHSs (Fig. 5).
Figure 5 |.
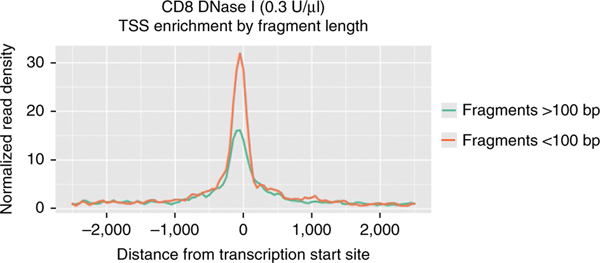
Use of shorter fragments improves enrichment at the TSS. The fragment length of each read can be determined with paired-end (PE) sequencing. Use of PE sequencing instead of single-end sequencing is optional, but it provides an additional means of size selection beyond gel- or bead-based size selection. In this example, read length was determined by PE sequencing. Reads <100 bp have a higher TSS signal-to-noise ratio.
It is not possible to use peak-calling algorithms on single-cell libraries because of the limited numbers of reads at each DHS. Instead, we consider a read from a single-cell library to represent a DHS if it falls within a DHS peak from an ensemble data set such as ENCODE, as described in the original scDNase-seq paper6. The false-discovery rate when using this approach is estimated to be between 11 and 13%.
MATERIALS
REAGENTS
▲ CRITICAL All reagents must be kept DNase-free.
-
NIH/3T3 cells (ATCC, cat. no. CRL-1658)
! CAUTION The cell lines used in your research should be regularly checked to ensure they are authentic and are not infected with mycoplasma.
Nuclease-free water (Life Technologies, cat. no. AM9930)
1 M Tris-HCl (pH 7.5; Quality Biological, cat. no. 351–006-101)
Triton X-100 (Quality Biological, cat. no. A611-M143–13)
1 M Magnesium chloride (Quality Biological, cat. no. A611-E525–06)
10% Sodium dodecyl sulfate (Quality Biological, cat. no. 351–032-101)
3 M Sodium acetate (pH 4.5; Quality Biological, cat. no. 351–309-101)
0.5 M EDTA (Quality Biological, cat. no. 351–027-721)
5 M Sodium chloride, molecular-biology-grade (Promega, cat. no. V4221)
Glycogen, molecular-biology-grade (Sigma-Aldrich, cat. no. 10901393001)
Proteinase K, recombinant, PCR-grade (Sigma-Aldrich, cat. no. 3115836001)
DNase I, recombinant, RNase-free (Sigma-Aldrich, cat. no. 4716728001)
dATP (10 mM; Thermo Fisher Scientific, cat. no. 18252015)
EB buffer (Qiagen, cat. no. 19086)
TE buffer (Invitrogen, cat. no. 12090015)
16% formaldehyde solution (Thermo Scientific, cat. no. 28908)
Deparaffinization solution (Qiagen, cat. no. 19093)
End-It DNA End-Repair Kit (Epicentre, cat. no. ER0720)
Klenow fragment (3′→5′ exo-; 5,000 U/ml; New England BioLabs, cat. no. M0212S)
T4 DNA ligase (400,000 U/ml; New England BioLabs, cat. no. M0202S)
Phusion High-Fidelity PCR Master Mix with HF Buffer (New England BioLabs, cat. no. M0531S)
NEBuffer 2 (New England BioLabs, cat. no. B7002S)
pUC19 vector (New England BioLabs, cat. no. N3041S)
APC Annexin V Apoptosis Detection Kit with 7-AAD (BioLegend, cat. no. 640930)
Dead Cell Removal Kit (Miltenyi Biotec, cat. no. 130–090-101)
Ethyl alcohol (Warner-Graham, cat. no. 64–17-5) ! CAUTION Ethanol is highly flammable.
-
Phenol–chloroform, pH 6.7/8.0 (Amresco, cat. no. 0883)
! CAUTION Phenol–chloroform can burn the eyes and the skin. Use in a chemical hood.
Illumina paired end adaptor oligo mix (Illumina, cat. no. 1001782)
Illumina multiplex PCR primer 1.0 (see Supplementary Table 1)
Illumina multiplex PCR primer 2 index (see Supplementary Table 1)
E-Gel EX Agarose Gels Starter Kit, 2% (Invitrogen, cat. no. G6512ST)
E-Gel EX Agarose Gels, 2% (Invitrogen, cat. no. G401002)
E-Gel 1 Kb Plus DNA Ladder (Invitrogen, cat. no. 10488090)
MinElute Gel Extraction Kit (Qiagen, cat. no. 28604)
EQUIPMENT
0.2-ml PCR tubes (Molecular BioProducts, cat. no. 3418)
Microtube, 1.5-ml, DNA LowBind (Sarstedt, cat. no. 72.706.700)
Tabletop microcentrifuge (model no. MicroStar 17R; VWR, cat. no. 521–1647)
Precision general-purpose water baths (Thermo Fisher Scientific, cat. no. 2824)
Thermal cycler (model no. PTC-200; MJ Research, cat. no. 8252–30-0001)
Freezers, −20 °C and −80 °C (Liebherr, cat. no. GPESF1476)
Qubit fluorometer (Life Technologies, cat. no. Q32866)
Cell sorter (BD Biosciences, model no. FACSAria II)
Sequencing system (Illumina, model no. HiSeq 2500)
Software
Any Linux/UNIX environment
HOMER (v4.8.2; http://homer.salk.edu)
Bowtie 2 (v2.2.9; http://bowtie-bio.sourceforge.net/bowtie2)
Samtools (v1.3.1; http://htslib.org)
MACS (v1.4.2; http://liulab.dfci.harvard.edu/MACS/)
REAGENT SETUP
Lysis buffer
Prepare lysis buffer by mixing 10 mM Tris-HCl (pH 7.4), 0.1% (vol/vol) Triton X-100, 10 mM NaCl and 3 mM MgCl2. Place the buffer on ice. Freshly prepare the buffer before each experiment.
DNase buffer
We divide DNase I (10 U/μl) into 5-μl aliquots and store them at −20 °C for single use (store for up to 1 year).
DNase buffer/enzyme mix
Immediately before DNase digestion, add 145 μl of lysis buffer to a 5-μl aliquot of DNase I (10 U/μl) and gently mix the solution with a pipette (this is the 30× dilution). Prepare three dilutions (30×, 45× and 60×) with lysis buffer. ▲ CRITICAL It may be necessary to optimize the DNase I concentration when using a new cell type. Do not refreeze or reuse diluted DNase I.
Stop buffer
Prepare stop buffer by mixing 10 mM Tris-HCl (pH 7.4), 10 mM NaCl and 10 mM EDTA (can be mixed and left at room temperature (20–25 °C) before cell sorting). Add 0.15% (vol/vol) SDS and 0.25 μg/μl proteinase K immediately before DNase digestion. We freshly prepare stop buffer before each experiment.
Illumina adaptor
The Illumina paired-end adaptor oligo mix can be purchased premade from Illumina, or it can be prepared by purchasing single-stranded DNA and annealing the Illumina top and bottom DNA (Supplementary Table 1) to make a double-stranded adaptor. To anneal the adaptors, resuspend both top and bottom DNA sequences in EB buffer (50 μM). Combine the top and bottom adaptor DNA in equal molar amounts. Heat the mixture in a PCR machine to 95 °C for 3 min, then unplug the PCR machine and allow the mixture to cool for 30 min or until the PCR heating block has returned to room temperature. Dilute a double-stranded Illumina adaptor mixture to 15 μl in EB buffer. The mixture can be stored at −20 °C for 1 year.
PROCEDURE
Cell collection, lysis and DNase digestion (day 1) ● TIMING 3–20 h
-
1To prepare cultured or primary cells, follow option A. For FFPE tissue, follow option B.
- Facs collection of cells or cultured cells with viability >95% ● TIMING 3–20 h
-
For single-cell collection, prepare one 32-μl aliquot of ice-cold lysis buffer per tube in 0.2-ml PCR tubes on ice. For non-single-cell collection, prepare 32-μl aliquots of ice-cold lysis buffer in 1.5-ml microtubes on ice.▲ CRITICAL STEP For single-cell sorting, we use the smallest tubes possible to minimize the chance of the cell adhering to the wall of the tube.
-
FACS-sort viable cells directly into the lysis buffer; briefly vortex the tubes, briefly centrifuge them and return the tubes to ice for 10–30 min. If processing from cell culture, pellet the desired number of cells, remove the supernatant, resuspend it in lysis buffer and return the tubes to ice for 10–30 min.▲ CRITICAL STEP The cell pellet will not be visible. Leave 2–3 μl of liquid at the bottom of the tube to avoid aspiration of the cell pellet.
- Add 0.15% (vol/vol) SDS and 0.25 μg/μl proteinase K to the stop buffer (see Reagent Setup), which will be used in Step 1A(vii).
-
Add 8 μl of each dilution of the DNase buffer/enzyme mix (see Reagent Setup) to the appropriate tube. Gently pipette the mixture up and down several times.▲ CRITICAL STEP Avoid vortexing the DNase I buffer/enzyme mix, as this will reduce the enzymatic activity of the DNase I.
- Add 8 μl of DNase buffer without any added enzyme to the no-DNase control sample. The no-DNase control should be included in all subsequent steps.
-
Incubate the samples at 37 °C in a water bath for 5 min.▲ CRITICAL STEP If processing more than 6 samples, consider processing them in batches to ensure that digestion occurs for exactly 5 min.
- Add 80 μl of stop buffer (from Step 1A(iii)) to each tube, mix well and spin the tube briefly.
-
Pipette 5 μl of 6 ng/μl circular carrier DNA (pUC19 vector) into each tube.▲ CRITICAL STEP Carrier DNA will be degraded if it is added before DNase I has been inactivated by the SDS and EDTA in the stop buffer.
-
Digestion of proteins. If you are using fresh cells, incubate them at 55 °C for at least 1 h to digest the protein. If you are using formaldehyde-fixed cells, incubate them at 65 °C overnight to digest the protein and reverse formaldehyde cross-linking.■ pause poInt Incubation of fresh cells can also be performed overnight.
- If you are using 0.2-ml PCR tubes, transfer them to 1.5-ml microtubes and proceed to step 2 to perform DNA extraction.
-
- FFPE tissue cell collection ● TIMING 19 h
- Mark the regions of a paraffin-embedded slide containing the cells of interest.
- Using a razor blade, carefully scrape the cells from the slide, and transfer them to a 1.5- ml microtube.
- Immediately add 150 μl of deparaffinization solution to the tube, and vortex the mixture vigorously for 10 s.
- Incubate the tube in a 56 °C heating block for 3 min.
- Allow the sample to cool to room temperature.
- Add 150 μl of lysis buffer to the sample.
- Incubate the tube at 37 °C in a water bath for 1 h.
- Aspirate and discard the top layer of deparaffinization solution.
- Centrifuge the sample at 400g for 2 min at 25 °C.
-
Carefully aspirate and discard the supernatant while leaving the pelleted nuclei intact.▲ CRITICAL STEP Nuclei may not be visible, depending on the starting number of cells. Leave a residual 10 μl at the bottom of the tube to avoid disturbing the pellet.
- Wash the pellet by adding 150 μl of room-temperature lysis buffer, and proceed to the next step immediately.
- Centrifuge the sample at 400g for 2 min at 25 °C.
-
Carefully remove the supernatant, and resuspend the pellet in 32 μl of lysis buffer.▲ CRITICAL STEP Nuclei may not be visible, depending on the starting number of cells. Leave a residual 5 μl at the bottom of the tube to avoid disturbing the pellet.
- Add 0.15% (vol/vol) SDS and 0.25 μg/μl proteinase K to the stop buffer (see Reagent Setup), which will be used in Step 1B(xviii).
-
Add 8 μl of each dilution of the DNase buffer/enzyme mix (see Reagent Setup) to the appropriate tube. Gently pipette the mixture up and down several times.▲ CRITICAL STEP Avoid vortexing the DNase I buffer/enzyme mix, as this will reduce the enzymatic activity of DNase I.
- Add 8 μl of DNase buffer without any added enzyme to the no-DNase control sample. The no-DNase control should be included in all subsequent steps.
-
Incubate the tube at 37 °C in a water bath for exactly 5 min.▲ CRITICAL STEP If processing more than 6 samples, consider processing them in batches to ensure that digestion occurs for exactly 5 min.
- Add 80 μl of stop buffer (from Step 1B(xiv)) to each tube, mix the well and centrifuge them brieflys.
-
Pipette 5 μl of 6 ng/μl circular carrier DNA (pUC19 vector) into each tube.▲ CRITICAL STEP The carrier DNA will be degraded if it is added before DNase I has been inactivated by the SDS and EDTA in the stop buffer.
- Incubate the tubes at 65 °C overnight to digest the protein and reverse the formaldehyde cross-links.
Extraction of digested DNA (day 1) ● TIMING 1 h
-
2
Pipette 125 μl of phenol–chloroform into each tube and vortex the tubes vigorously for 10 s.
! CAUTION Phenol–chloroform can cause serious burns. Perform all the extraction steps in a chemical hood.
-
3
Spin the tubes at 16,000g at 4 °C in a microcentrifuge for 3 min.
-
4
Transfer the top aqueous layer to a new 1.5-ml microtube.
▲ CRITICAL STEP Pipette slowly to ensure that the entire aqueous layer is transferred without transferring the phenol layer.
-
5
Add 1 μl of 20 mg/ml glycogen, 12.5 μl of 3 M sodium acetate and 345 μl of 100% (vol/vol) ethanol.
-
6
Incubate the tubes at −80 °C for at least 15 min to precipitate the DNA.
■ PAUSE POINT The DNA can be stored at −80 °C indefinitely.
-
7
Spin the tubes in a microcentrifuge at maximum speed at 4 °C for 15 min.
-
8
Remove the supernatant without disturbing the pellet.
-
9
Add 400 μl of ice-cold 70% (vol/vol) ethanol to wash the pellet.
-
10
Spin the tube in a microcentrifuge at maximum speed at 4 °C for 10 min.
-
11
Remove the supernatant without disturbing the pellet.
▲ CRITICAL STEP The pellet is easily disturbed after the 70% (vol/vol) ethanol wash.
-
12
Air-dry the pellet briefly until the ethanol has evaporated and the DNA pellet becomes colorless (5–10 min).
▲ CRITICAL STEP Avoid overdrying the pellet, as this can make the DNA more difficult to resuspend.
Library preparation ● TIMING 2–4 d
-
13
Resuspend the pellet from Step 12 with 10 μl of EB buffer.
-
14Prepare DNA end-repair master mix on ice, using the End-It DNA End-Repair Kit as shown below:
Component Amount (μl) Final concentration 10× dNTP mix 2.5 1× 10× ATP mix 2.5 1× 10× end-repair buffer 2.5 1× End-repair enzyme mix 0.5 Molecular-biology-grade water 7 -
15
Add 15 μl of end-repair master mix to the DNA from Step 13 and gently mix by pipetting.
-
16
Incubate the mixture at 37 °C for 20 min.
-
17
Add 100 μl of TE buffer to the reaction mixture.
-
18
Repeat Steps 2–12 to phenol-extract and ethanol-precipitate the samples.
-
19
Resuspend the pellet in 10 μl of EB buffer.
-
20Prepare A-tailing master mix on ice as shown below:
Component Amount (μl) Final concentration 10× dATP mix 0.5 1× 10× NEBuffer 2 2.5 1× 5,000 U/ml Klenow fragment (3′→5′ exo-) 1 Molecular-biology-grade water 11 -
21
Add 15 μl of A-tailing master mix to the samples from Step 19.
-
22
Incubate the samples at 37 °C for 20 min.
-
23
Add 100 μl of TE buffer to the reaction mixture.
-
24
Repeat Steps 2–12 to phenol-extract and ethanol-precipitate the samples.
-
25
Resuspend the pellet in 7 μl of EB buffer.
-
26Prepare the ligation master mix as shown below:
Component A+mount (μl) Final concentration 10× T4 DNA ligase buffer 2 1× 15-mM Illumina adaptor oligo mix 1.2 0.9 M Molecular-biology-grade water 8.2 -
27
Add 11.4 μl of ligation master mix to each sample from Step 25.
-
28
Add 1.6 μl of 400 U/μl T4 DNA ligase to each sample.
▲ CRITICAL STEP Do not add T4 DNA ligase to the master mix, in order to avoid ligation of Illumina adaptors without DNA insert.
-
29
Incubate the mixture at room temperature for 2 h. If you are using <1,000 cells, incubate the mixture overnight.
■ PAUSE POINT Incubation can be extended at room temperature overnight.
-
30Prepare the PCR master mix as shown below:
Component Amount (μl) Final concentration 2× NEB Phusion master mix 21.5 1× 10 mM Illumina multiplexing PCR 1.0 primer 0.8 0.9 M -
31
Barcode each unique sample by adding 0.8 μl of 10 M Illumina PCR multiplexing primer 2 with a unique index (supplementary table 1) to each sample from Step 29.
▲ CRITICAL STEP Each tube from Step 29 requires a unique index in order to identify each library after sequencing.
-
32
Add 21.5 μl of PCR master mix from Step 30 to each sample and mix well.
-
33PCR-amplify the library using the program and the number of cycles outlined in the tables below. The number of cycles to use will depend on the number of starting cells:
Cycle no. Denature Anneal Extend Hold 1 98 °C, 30 s — — — Varies with the starting cell number (see below) 98 °C, 10 s 68 °C, 30 s 72 °C, 30 s — Final — — — 4 °C Starting cell no. Total PCR cycles 1 23 100 18 1,000 15 10,000 13 -
34
Run the PCR reaction directly on a 2% E-gel for 26 min.
▲ CRITICAL STEP A prominent DNA smear should be present, with the strongest intensity between 150 and 280 bp. If faint bands are present, proceed with gel purification and then perform three additional cycles of PCR.
? TROUBLESHOOTING
-
35
Cut the band from the gel, isolating the size range of 160–260 bp.
▲ CRITICAL STEP Smallest fragments are enriched for DNA from hypersensitive sites. Isolating larger fragments will increase the background signal-to-noise ratio.
-
36
Process the gel bands per the Qiagen instructions for the MinElute Gel Extraction Kit.
-
37
Sequence the libraries on an Illumina next-generation-sequencing platform.
▲ CRITICAL STEP The no-DNase I control samples are not sequenced.
Data analysis ● TIMING 1–3 d
-
38
Align the reads to the reference genome, remove low-mapping-quality reads and coordinate the sort-resulting BAM file.
Example command:export BOWTIE2_INDEXES=/path_to_reference_genome gunzip -c sample.fastq.gz | bowtie2 −4 -x genome -U - --no-unal | samtools view -q 10 -uSh - | samtools sort -O bam - > sample.sorted.bam
-
39
Mark duplicate reads using Picard
Example command:java -Xmx8g -jar picard.jar MarkDuplicates \ INPUT= sample.sorted.bam \ OUTPUT= sample.dedup.bam \
-
40
Generate UCSC Genome Browser bedGraph files for visualization of DHS sites using HOMER.
Example command:makeTagDirectory sample_Tag_dir/ -unique sample.dedup.bam makeUSCSfile sample_Tag_dir/ -o auto
? TROUBLESHOOTING
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
TABLE 1 |.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 34 | No DNA is present after PCR | Inadequate amplification | Extract DNA in the 150–400 bp range and repeat PCR for three additional cycles |
| Overdigestion or underdigestion | Perform DNase I enzyme titration | ||
| DNA was lost during processing | Exercise caution during phenol extraction to avoid losing the DNA | ||
| DNA is not present in the 160–330 bp range with a high-molecular-weight smear | Underdigestion | Perform DNase I enzyme titration | |
| Excessive adaptor dimers | Ratio of free adaptor to fragmented DNA is too high | Reduce the concentration of Illumina adaptor (Step 26) | |
| Perform a two-step PCR. Amplify the sample for six PCR cycles, and perform gel extraction of fragments (the fragments will not be visible on the gel) between 150 and 400 bp. Perform second- stage PCR for a number of remaining cycles | |||
| DNA is present in the no-DNase I control lane | Excessive dead cells are present in samples | FACS-sort the cells to exclude apoptotic cells | |
| 40 | High background | Nucleosomal DNA from apoptotic/dead cells is present Suboptimal DNase I concentration Excessive high- molecular-weight DNase fragments |
Ensure that the no-DNase control does not amplify. Perform DNase I enzyme titration. Size-select libraries between 150 and 300 bp. Use paired-end sequencing to exclude reads with fragment inserts >150 bp |
| Variability between replicates and samples | DNase I digestion time was not exactly 5 min | Process samples in smaller batches to ensure the correct digestion time |
● TIMING
Step 1A(i and ii), collection of cells: 1–3 h
Step 1A(iii–vii), DNase I digestion reaction: 1 h
Step 1A(viii–x), proteinase K digestion of proteins: 1 h or overnight
Step 1B(i–xi), extraction of cells from FFPE tissue slide: 2 h
Step 1B(xii–xvii), DNase I digestion reaction: 1 h
Step 1B(xviii–xx), reversal of formaldehyde cross-links: overnight
Steps 2–12, extraction of DNA: 1 h
Steps 13–19, library preparation (end repair): 1.5 h
Steps 20–25, library preparation (A-tailing): 1.5 h
Steps 26–29, library preparation (adaptor ligation): 2.5 h to overnight
Steps 30–33, library preparation (PCR amplification): 2 h
Steps 33–36, gel-size selection of PCR products: 1 h
Step 37: sequencing: variable, based on sequencing platform (1–3 d)
Steps 38–40, data analysis: variable
ANTICIPATED RESULTS
After PCR amplification of the scDNase-seq library, a smear of DNA should be present, with the highest intensity between ~150 and 300 bp (Fig. 1). There should not be any DNA smear in the no-DNase I control lanes. If there is DNA within the no-DNase I control lanes, this indicates the presence of apoptotic and/or dead cells (see Troubleshooting section). Determination of the optimal range of concentrations for the DNase I enzyme cannot be made solely based on the library gel image. By generating plots of the relative enrichment of reads near the TSSs for all enzyme concentrations, it is possible to identify the optimal concentration range for the DNase I enzyme (Fig. 2).
The efficiency of sequencing depends on the starting-cell input (see Supplementary Tables S2 and S6 from ref. 6). A successful library from a single cell should have >200,000 unique, high-quality reads when sequenced to saturation. A single-cell library sequenced to saturation will have <1% nonredundant reads. For larger cellular inputs (>100 cells), the percentage of unique reads in the sequencing library increases to 30–40% of the total reads. Starting with ≥1,000 cells, we obtain highly reproducible DHSs (R2 > 0.97). At the single-cell level, the R2 values for different cells are variable because of both biological and technical variability.
When starting with 100 cells, genome browser images from a successful scDNase-seq experiment should contain discrete peaks with low background similar to ENCODE tracks (Figs. 4 and 6a). Genome browser images from single-cell libraries will not have typical peaks because of the limited number of reads per DHS. To assess the quality of single-cell libraries, we generate plots of the relative enrichment of reads normalized to total library reads near the TSSs. Successful single-cell libraries should have at least fivefold enrichment at the TSS as compared with background (Fig. 6b).
Figure 6 |.

scDNase-seq library quality can be estimated by TSS enrichment profiles. (a) Genome browser images from scDNase-seq experiments using 5,000 human CD8+ T cells and a 0.3 U/μl enzyme concentration show that they correlate with previously reported ENCODE bulk CD8 DNase I data from conventional DNase-seq experiments starting with 107 cells (track accession no. GSM701499, https://www.encodeproject.org). (b) TSS enrichment seen from scDNA-seq experiments in NIH-3T3 cells with various starting cell numbers using a 0.3 U/μl enzyme concentration. ENCODE tracks start with 107 cells (track accession no. ENCSR000CNS; https://www.encodeproject.org). Single-cell libraries should have enrichment near the TSS, but the signal-to-noise ratio will be lower than those of non-single-cell libraries.
Supplementary Material
ACKNOWLEDGMENTS
We thank W. Jin for his insights into the bioinformatics analysis of the data, the National Heart, Lung, and Blood Institute (NHLBI) DNA Sequencing Core facility for sequencing the libraries and the NHLBI Flow Cytometry Core facility for sorting the cells. The work was supported by the Division of Intramural Research, NHLBI (K.Z.).
Footnotes
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html. Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyle AP et al. High-resolution mapping and characterization of open chromatin across the genome. Cell 132, 311–322 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crawford GE et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res. 16, 123–131 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crawford GE et al. DNase-chip: a high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat. Methods 3, 503–509 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin W. et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 528, 142–146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wen L. & Tang F. Single-cell sequencing in stem cell biology. Genome Biol. 17, 71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Picelli S. et al. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 24, 2033–2040 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buenrostro JD et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cusanovich DA et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen X. et al. ATAC-see reveals the accessible genome by transposase-mediated imaging and sequencing. Nat. Methods 13, 1013–1020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song L. et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 21, 1757–1767 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simon JM, Giresi PG, Davis IJ & Lieb JD Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat. Protoc. 7, 256–267 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu F. et al. Establishing chromatin regulatory landscape during mouse preimplantation development. Cell 165, 1375–1388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X. et al. Single-cell sequencing for precise cancer research: progress and prospects. Cancer Res. 76, 1305–1312 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Shapiro E., Biezuner T. & Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 14, 618–630 (2013). [DOI] [PubMed] [Google Scholar]
- 17.He HH et al. Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat. Methods 11, 73–78 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heinz S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langmead B. & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kent WJ et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson JT et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yue F. et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.