

Abstract
Background:
Cyclin and MAPK/MEK-related gene alterations are implicated in cell-cycle progression and cancer growth. Yet, monotherapy to target the cyclin (CDK4/6) or the MEK pathway has often yielded disappointing results. Because co-alterations in cyclin and MEK pathway genes frequently co-occur, we hypothesized that resistance to CDK4/6 or MEK inhibitor monotherapy might be mediated via activation of oncogenic co-drivers, and that combination therapy might be useful.
Patients and methods:
Herein, we describe nine patients with advanced malignancies harboring concomitant CDKN2A and/or CDKN2B alterations (up-regulate CDK4/6) along with KRAS or BRAF alterations (activate the MEK pathway) who were treated with palbociclib (CDK4/6 inhibitor) and trametinib (MEK inhibitor) combination-based regimens.
Results:
Two patients (with pancreatic cancer) achieved a partial remission (PR) and, overall, five patients (56%) had clinical benefit (stable disease ≥6 months/PR) with progression-free survival of ~7, 9, 9, 11, and 17.5+ months. Interestingly, one of these patients whose cancer (gastrointestinal stromal tumor) had progressed on MEK targeting did well for about one year after palbociclib was added.
Conclusions:
These observations suggest that co-targeting cyclin and MEK signaling can be successful when tumors bear genomic co-alterations that activate both of these pathways. Further prospective studies using this matching precision strategy in order to overcome resistance are warranted.
Keywords: Cell cycle, next-generation sequencing, targeted therapy, precision oncology, MAP kinase
Brief summary:
Molecularly matched targeted therapy approach to co-target MEK and cyclin gene alterations for advanced solid tumors
STATEMENT OF TRANSLATIONAL RELEVANCE
The advent of next-generation sequencing has identified potentially actionable genomic alterations for patients that previously were not characterized. However, targeting MAP kinase pathway alterations with MEK inhibitors or cyclin alterations with CDK4/6 inhibitors as monotherapy has not been effective. In this study, advanced cancer patients with co-alterations in both MAP kinase and cell cycle pathways were treated with trametinib (MEK inhibitor) and palbociclib (CDK4/6 inhibitor) based therapy. Co-targeting of MAP kinase and cyclin pathway demonstrated durable clinical benefit, including in patients with pancreatic cancer. Further investigation with co-targeting of cyclin and MEK pathway aberrations is warranted.
INTRODUCTION
The cell cycle is highly regulated by various proteins that are required for proliferation. These proteins include cyclins, cyclin-dependent kinases, as well as growth factors and retinoblastoma proteins.(1,2) The cyclin complexes allow cell division to proceed and propagate the replication process.(3) In certain cancers, any part of this feedback cascade may be altered leading to dysregulation of proliferation.(4) The most well-described pathogenesis models suggest that pathway changes include increased production of cyclin D1 (CCND1), upregulation in expression/amplification of CDK4 and CDK6, and/or alterations in the Rb, CCNE1, CDKN2A, or CDKN2B genes.(5–9) Theoretically, amplification of cyclin D(10) or CDK4/6,(11) as well as alterations in CDKN2A/B,(12) all lead to increased CDK4/6, which can be targeted with CDK4/6 inhibitors; however, sufficient clinical data is lacking or suggests that the presence of pathway alterations do not influence responsiveness. Indeed, in the Targeted Agent and Profiling Utilization Registry (TAPUR) Study, when participants with pancreatic and biliary cancers and CDKN2A loss or mutation were treated with palbociclib monotherapy, meaningful response rates were absent; all patients with biliary malignancies showed tumor progression at or before 10 weeks and all pancreatic cancers progressed by week 16.(13)
One of the potential reasons that CDK4/6 inhibitor monotherapy has not demonstrated significant impact on clinical outcomes, even when given to patients with genomic alterations that upregulate CDK4/6, may be due to molecular co-alterations as well as tumor heterogeneity. Notably, for instance, cell cycle-associated genes are altered in ~31% of RAS-altered malignancies.(14) RAS alterations lead to the activation of the canonical mitogen-activated protein kinase (MAPK) pathway, which invokes a downstream cascade involving RAF, MEK, and ERK.(15) Activation of the MEK pathway at any step along the cascade leads to up-regulation of cell division and further cellular proliferation.(16) RAS and RAF alterations can potentially be targeted with drugs that inhibit one of the later steps, including MEK and ERK inhibitors, as well as specific inhibitors of KRAS G12C (for cancers that harbor a specific KRAS G12C mutation).(17,18) Importantly in this regard, we have recently demonstrated that survival was negatively impacted when patients had malignancies that demonstrated alterations in both RAS and cell cycle-associated genes as compared to patients with only one of these pathways altered,(14) and some authors hypothesize that co-targeting MEK and cyclin pathways might be important.(19,20)
In an effort to evaluate the clinical impact of a genomic matching combination approach in patients whose advanced tumors harbor both MEK and cell cycle pathway abnormalities, we analyzed individuals who received concomitant MEK inhibitors and CDK inhibitors. Herein we report that the combination was well tolerated and that patients can achieve objective responses and/or durable disease stability.
METHODS
Patients and therapy:
Electronic medical records were reviewed for patient characteristics and outcome for all individuals treated with a matched MEK inhibitor (for a RAS or BRAF alteration) combined with a matched CDK4/6 inhibitor (for a CDKN2A and/or CDKN2B alteration) and included in the PREDICT database. Patients who received immunotherapy or chemotherapy as part of their matched regimen were excluded from this analysis. Patients were presented at a Molecular Tumor Board, which occurred either in person (weekly) or electronically on demand, and included medical oncologists, surgeons, radiation oncologists, gynecologic oncologists, clinical trial coordinators/navigators, medication acquisition specialists, geneticists, pathologists, radiologists, basic/translational scientists, and bioinformatics specialists.(21–23) Patient-specific molecular diagnostic data in concert with patient characteristics, prior treatment, and review of pathology and imaging were discussed. Dialogues focused on the role of the various alterations in signaling cascades (somatic or germline) and whether there were drugs, either Food and Drug Administration (FDA) approved or in active clinical trials, to target the specific alteration(s).
Study approval:
This study followed the guidelines of the Internal Review Board (IRB) approved University of California San Diego (UCSD) Profile-Related Evidence Determining Individualized Cancer Therapy (PREDICT) study (NCT02478931) and any investigational studies for which the patients gave consent.
Next generation sequencing (NGS) of tissue DNA:
Next-generation sequencing (236–405 genes) was performed on formalin-fixed, paraffin-embedded tissue submitted to a Clinical Laboratory Improvement Amendments (CLIA) certified lab for genomic sequencing (Foundation Medicine). The details of sample requirement, DNA extraction and NGS were described previously.(24) Average depth of sequencing was greater than 250x, with 100x at >99% of exons. This method of sequencing enabled detection of copy number changes, gene rearrangement, and single nucleotide variants with 99% sensitivity and 99% specificity for base substitution, and 95% sensitivity for copy number changes. Amplification was defined as copy number increase of ≥ 8 copies (equivocal, 6 to 7 copies).
Statistical methods and clinical endpoints:
Patient and molecular characteristics were summarized in a descriptive manner. We evaluated PFS, which was defined as time between therapy initiation and the date of progression, determined by imaging or clinical findings. Patients whose tumor had not progressed at last evaluation were censored at that point. Responses were evaluated by RECIST 1.1 criteria.(25)
RESULTS
We report a group of nine patients with metastatic malignancies who had both cyclin and MEK pathway activating alterations and received therapy targeting both pathways after presentation at our Molecular Tumor Board (either face-to-face or electronic) (Table 1, Figure 1, Supplemental Table 1 and Supplemental Figure 1). The median age of patients was 65 years old (range, 24–88 years). Five of the 9 patients were women. The median treatment line was 2 (range, 1–10). Patient diagnoses included pancreatic adenocarcinoma (n = 6), colorectal adenocarcinoma (n = 1), gastrointestinal stromal tumor (GIST) (n = 1), and rhabdomyosarcoma (n = 1). Genomic alterations activating the cyclin pathway (hence up-regulating CDK4/6) included CDKN2A, CDKN2B or both in all patients. Genomic alterations activating the MEK pathway included BRAF (n = 2) and KRAS (n = 7). The most common additional pathogenic molecular alterations were in the TP53 gene (n = 5). The median number of pathogenic alterations per patient was 6 (range, 3–7).
Table 1:
Patient characteristics and tumor molecular findings in nine patients with metastatic malignancies treated with matched CDK4/6 and MEK inhibitors
| Median Age (range) | 65 years (24–88 years) | |
| Sex (N (%)) | Men | 4 (44.4%) |
| Women | 5 (55.6%) | |
| Ethnicity (N (%)) | White | 5 (55.6%) |
| Asian | 2 (22.2%) | |
| Hispanic | 2 (22.2%) | |
| Type of Cancer, N (%) | Pancreatic adenocarcinoma | 6 (66.7%) |
| Colorectal adenocarcinoma | 1 (11.1%) | |
| Gastrointestinal stromal tumor | 1 (11.1%) | |
| Rhabdomyosarcoma | 1 (11.1%) | |
| Median treatment line (range) | 2 (1–10) | |
| Median number of pathogenic genomic alterations per patient | 6 (range, 3–7) | |
| Genomic alterations in RAS or RAF genes |
BRAF alteration KRAS alteration |
2 (22.2%) 7 (77.7%) |
| Selected other genomic alterations | CDKN2A and/or 2B | 9 (100%) |
| TP53 | 5 (55.5%) | |
| SMAD4 | 2 (22.2% | |
| GNAS | 2 (22.2%) | |
| ERBB2 (amplification) | 1 (11.1%) | |
| APC | 1 (11.1%) | |
| AKT1 | 1 (11.1%) | |
| PIK3CA | 1 (11.1%) |
Figure 1.
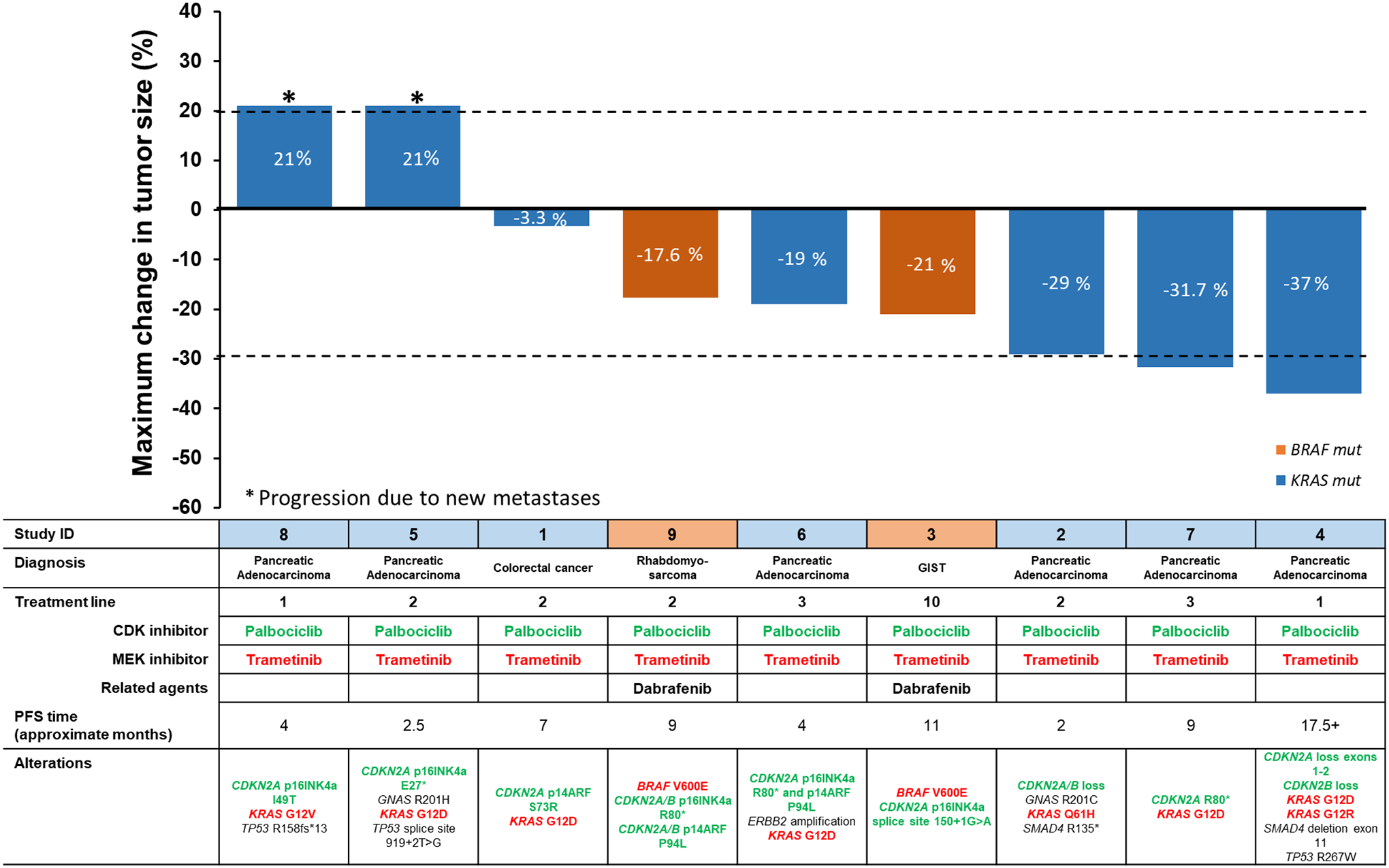
Waterfall plot showing patient and tumor molecular characteristics, treatment and percent change in tumor size. (New metastases were graphed as 21% progression. Treatment line refers to therapies in the metastatic setting.
Patients ID 4,5 and 8 also received bevacizumab. ID 6 also received lapatinib and trastuzumab.
Overall, 2 of the 9 patients (22%) achieved a partial response; progression-free survival (PFS) in these patients was 9.2 and 17.5+ months. Three additional patients achieved stable disease (SD) that lasted ≥6 months; therefore, the clinical benefit rate was 56% (5 of 9 patients) (Figure 1). Two of the six patients with pancreatic cancer (33%) achieved a PR. In all patients, the matched drugs used were palbociclib (CDK4/6 inhibitor) together with trametinib (MEK inhibitor). The most commonly used doses were 75 mg orally once a day (3 weeks on, 1 week off) for palbociclib together with 1 mg orally daily of trametinib. (Approved doses of the drugs as monotherapy are as follows: palbociclib 125 mg by mouth daily (three weeks on and one week off); trametinib 2 mg by mouth daily). Therapy was well tolerated and patients did not experience severe adverse events (grade 3 or 4) that were considered possibly drug related. There were no eye toxicities in these patients or significant changes in QTc interval or in ejection fraction. In three of the five patients with clinical benefit, there were additional drugs given. One patient with pancreatic cancer received bevacizumab along with trametinib and palbociclib (see Figure 1, ID 4); the two patients with BRAF V600E alterations received the BRAF inhibitor dabrafenib (in addition to palbociclib and trametinib); notably, one of the patients (Figure 1, GIST: ID 3) had already failed the combination of dabrafenib and trametinib, after which palbociclib was added with a prolonged PFS of about one year.
As mentioned, the most common MEK pathway alterations in these patients were in KRAS (n = 7 patients) and BRAF V600E (n = 2 patients). KRAS mutations have, until recently, been considered undruggable; however, there are now compounds that can specifically impact the protein produced as a result of the KRAS G12C mutation.(17,26,27) None of our patients had the latter aberration. It should also be noted that three of our patients had GNAS and/or SMAD4 alterations, in addition to KRAS alterations (Figure 1); both GNAS and SMAD4 alterations can activate the MEK pathway.(28–31)
Some patients are of special interest. For example, a patient with GIST and multiple prior lines of therapy (10th treatment line) (Figure 1, ID: 3), including imatinib and sunitinib, was treated with combined dabrafenib, trametinib and palbociclib. His immediate prior regimen consisted of dabrafenib and trametinib, given because his tumor harbored a BRAF V600E mutation. He was on the dabrafenib and trametinib regimen for six months, followed by disease progression, some of which was resected. He was continued on dabrafenib and trametinib; however, he developed new lung metastases and progressive disease in the rectum. Since the tumor also showed a CDKN2A alteration, he was continued on the dabrafenib and trametinib, and palbociclib was added; his PFS on this triplet was ~11 months. Another notable 63-year-old woman had pancreatic cancer (Figure 1, ID:4) with CDKN2A/B, KRAS, SMAD4 and TP53 alterations; the patient achieved a PR lasting 17.5+ months. Additionally, an 86 year-old woman with pancreatic cancer (Figure 1, ID: 2) who had alterations in CDKN2A/B loss, FAM123B E370*, GATA6 amplification, GNAS R201C, KRAS Q61H and SMAD4 R135* demonstrated 29% tumor regression, albeit short lived (Figure 2). Finally, patient ID:7 (Figure 1) achieved a PR (with PFS of ~9 months) on the matched trametinib and palbociclib despite her tumor showing previous progression on a regimen of nab-paclitaxel combined with palbociclib. These examples highlight the importance of co-targeting of the MEK and cyclin pathways and the potential for co-targeting to achieve benefit even when targeting one of the pathways fails.
Figure 2.
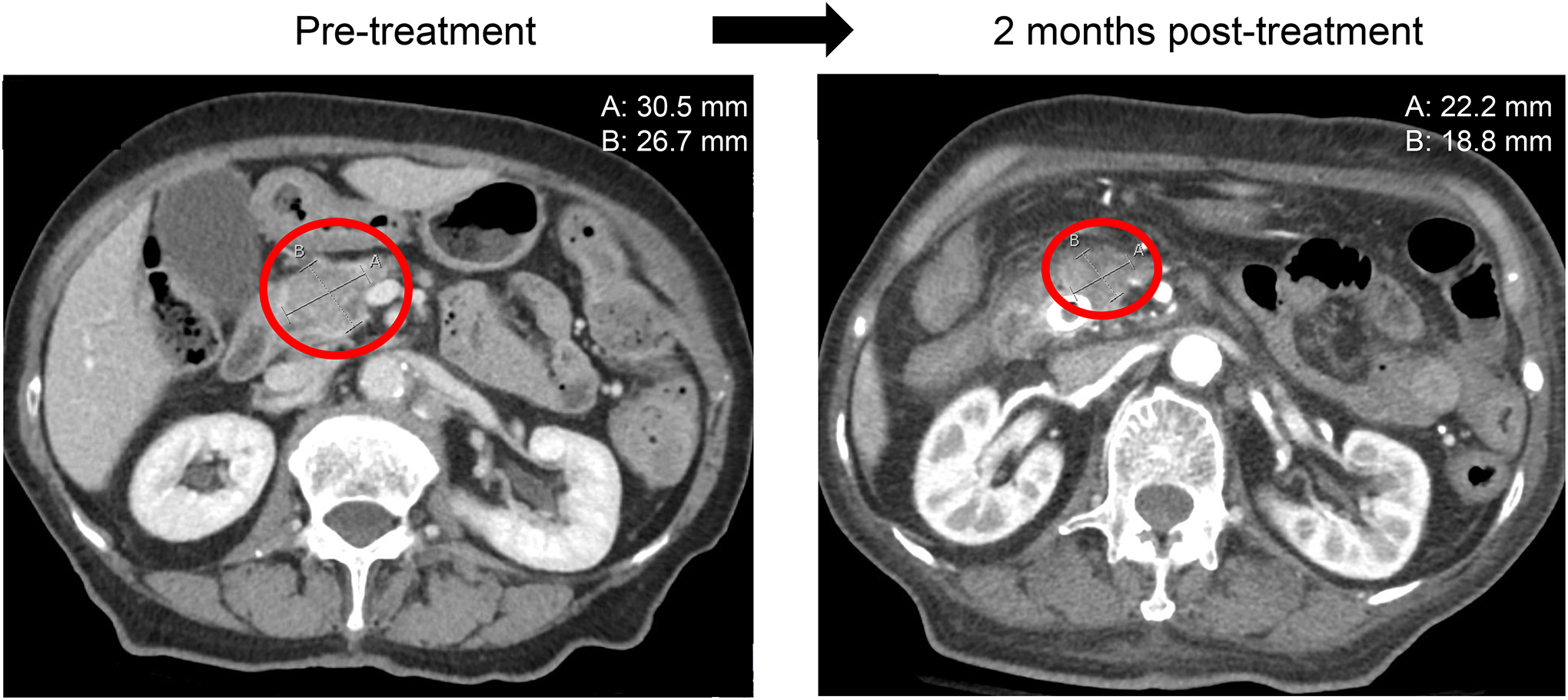
Regression on computerized tomographic scan of the abdomen of pancreatic cancer in a patient with KRAS and CDKN2A/B alterations managed with trametinib and palbocicib
This is an 86-year-old woman (study ID: 2; Figure 1) with metastatic adenocarcinoma of pancreas. Patient was initially treated with gemcitabine and albumin-bound paclitaxel. Upon progression, under the I-PREDICT study, patient was started on trametinib (for KRAS Q61H) and palbociclib (for CDKN2A/B loss). Imaging showed decrease in pancreatic mass (29% reduction) (measurements in upper right hand corner); images with largest diameter of tumor selected in each case.
DISCUSSION
Targeting one gene at a time for both cell cycle genes and RAS genes (other than KRAS G12C for which specific effective inhibitors are now in clinical trials (17,26,27,32)) has shown limited clinical efficacy.(13,33) Moreover, about 30% of tumors with RAS mutations are reported to have co-alterations in cell cycle genes, and about 5.5% of patients with diverse cancer harbor alterations in both BRAF/KRAS and cell cycle genes (136/2457) patients with Foundation Medicine tissue testing (Supplemental Figure 1) which may also explain why targeting only a single pathway can be challenging(14). As reported previously, targeting as many genomic alterations as possible can yield better clinical outcomes.(23,34) Other mechanisms may also be operative. For instance, prior studies show that, in the presence of KRAS pathway inhibition, cancers become dependent on autophagy for survival, and that removing this protective mechanism, in part via MEK inhibition, may be effective.(35)
We were able to find three ongoing trials (on clinicaltrials.gov) that include the use of a cell cycle inhibitor (CDK4/6 inhibitor) in combination with a MAPK pathway/MEK inhibitor (NCT02703571, NCT03434262, NCT02065063). In these trials, having cell cycle alterations (i.e. CDK4/6 amplification, CDKN2A, CDKN2B alterations) are not requisites for inclusion. One trial has KRAS mutations (but not cyclin gene alterations) as a requisite to receive ribociclib (CDK4/6 inhibitor) with trametinib (MEK inhibitor) in colorectal carcinoma (NCT02703571). Another trial (NCT03434262) employing the same regimen--ribociclib with trametinib--for various brain tumors has no mention of cell cycle or MAPK pathway alterations in the inclusion criteria. The last trial (NCT02065063) using palbociclib (CDK4/6 inhibitor) with trametinib in solid tumors requires patients’ tumors to be BRAF V600-wild type with or without NRAS mutations. These trials all use a combination strategy aimed at cell cycle and MEK pathways, but none requires aberrations within both of these pathways for entry into their trials.
The study presented herein has limitations. The sample size is small with various histologic subtypes and allowed combination therapies beyond CDK4/6 and MEK inhibitors. Further, this study is a retrospective analysis of data derived from a master observational trial and implicit bias may be present. Further validation with a larger cohort of patients studied prospectively is needed.
In summation, patients with cell cycle pathway alterations have historically derived minimal benefit from CDK4/6 inhibition monotherapy.(13) Similarly, patients with MAPK pathway alterations who receive single-agent MEK inhibitors mostly do not show salutary effects.(36) However, these pathway alterations often co-exist, and patients who have concomitant cell cycle and MAPK alterations have a poor prognosis compared to those who have neither or either alteration.(14) The logic behind dual inhibition is further strengthened by preclinical work that suggests that the MEK pathway drives cyclin activation and that there is strong interplay between these pathways.(37,38) Our study suggests that a significant subset of patients with metastatic neoplasms who have both cyclin and MEK genomic co-alterations can benefit from palbociclib and trametinib combinations, even if they have very heavily pretreated disease or difficult-to-manage malignancies such as pancreatic cancer. Larger prospective trials are needed in order to confirm the benefit of dual MEK and CDK4/6 inhibitors in patients with advanced malignancies that bear molecular abnormalities affecting both signaling cascades (Figure 3).
Figure 3.
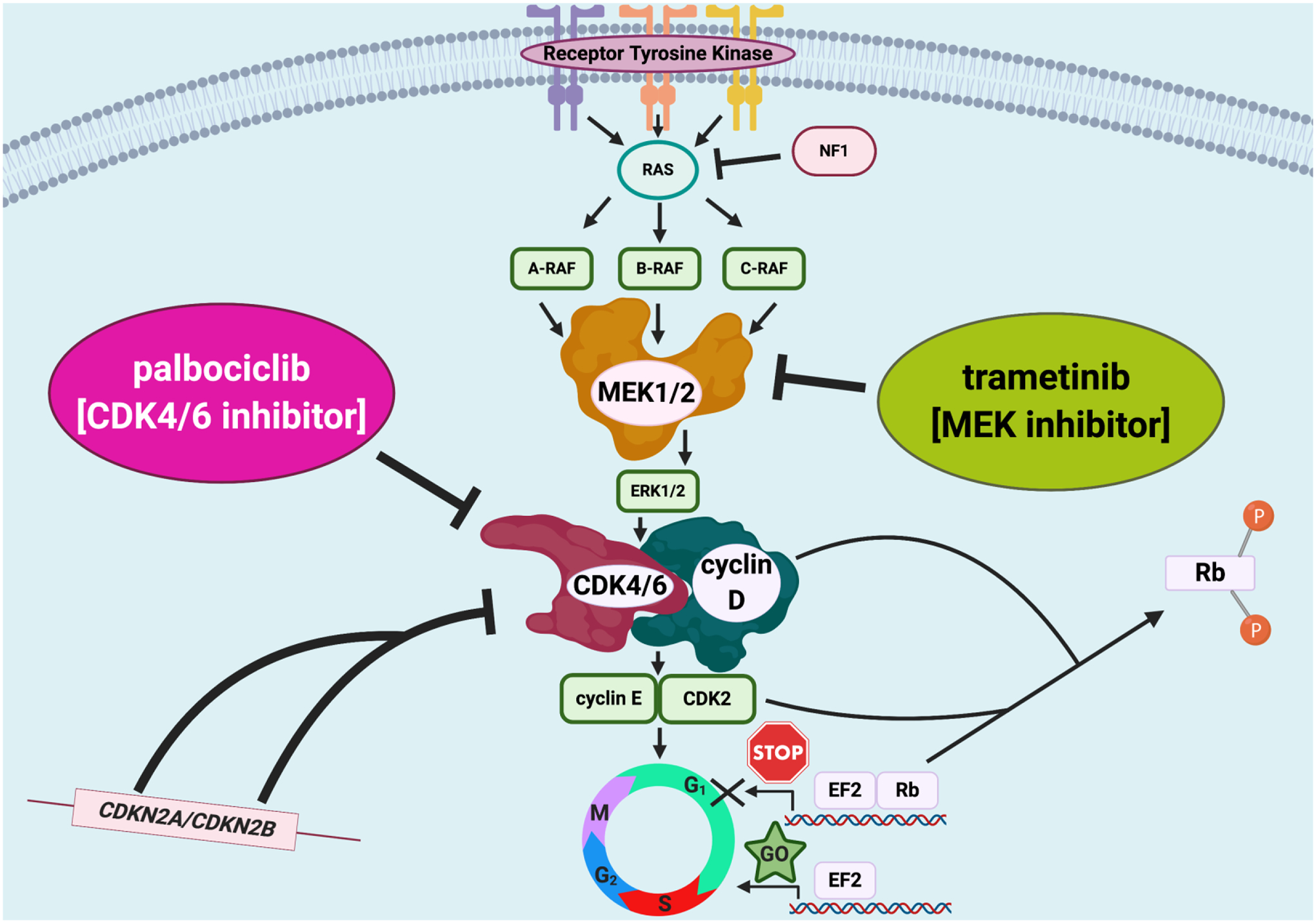
Dual MEK and CDK4/6 inhibitor approach in patients with advanced malignancies that harbor molecular abnormalities affecting both signaling cascades.
Supplementary Material
Acknowledgment:
Funded in part by the Joan and Irwin Jacobs Fund, and by National Cancer Institute grants P30 CA023100 (RK, JKS). The authors also acknowledge the support of NIH K08CA168999 and R21CA192072, as well as Pedal the Cause, David Foundation, and Kristen Ann Carr Fund (JKS).
Disclosures:
Shumei Kato serves as a consultant for Foundation Medicine. Speaker’s fee: Roche.
Research grant: ACT Genomics, Sysmex, Konica Minolta, OmniSeq. Jacob J Adashek has no disclosures. Jason Sicklick receives research funds from Foundation Medicine Inc., Novartis Pharmaceuticals, Blueprint Medicines, and Amgen, as well as consultant fees from Loxo, Biotheranostics, and Grand Rounds.
Razelle Kurzrock receives research funding from Genentech, Incyte, Merck, Serono, Pfizer, Sequenom, Foundation Medicine, Grifols, and Guardant, as well as consultant fees from Loxo, X Biotech, NeoMed, and Actuate Therapeutics, speaker fees from Roche, and an ownership interest in IDbyDNA and Curematch Inc and is a Board member of CureMatch and CureMetrix Inc.
REFERENCES
- 1.Bates S, Parry D, Bonetta L, Vousden K, Dickson C, Peters G. Absence of cyclin D/cdk complexes in cells lacking functional retinoblastoma protein. Oncogene 1994;9(6):1633–40. [PubMed] [Google Scholar]
- 2.Evans T, Rosenthal ET, Youngblom J, Distel D, Hunt T. Cyclin: a protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 1983;33(2):389–96 doi 10.1016/0092-8674(83)90420-8. [DOI] [PubMed] [Google Scholar]
- 3.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993;75(4):805–16 doi 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 4.Hamel PA, Hanley-Hyde J. G1 cyclins and control of the cell division cycle in normal and transformed cells. Cancer Invest 1997;15(2):143–52 doi 10.3109/07357909709115767. [DOI] [PubMed] [Google Scholar]
- 5.Schwaederle M, Daniels GA, Piccioni DE, Fanta PT, Schwab RB, Shimabukuro KA, et al. Cyclin alterations in diverse cancers: Outcome and co-amplification network. Oncotarget 2015;6(5):3033–42 doi 10.18632/oncotarget.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol 2006;24(11):1770–83 doi 10.1200/JCO.2005.03.7689. [DOI] [PubMed] [Google Scholar]
- 7.Sheppard KE, McArthur GA. The cell-cycle regulator CDK4: an emerging therapeutic target in melanoma. Clin Cancer Res 2013;19(19):5320–8 doi 10.1158/1078-0432.CCR-13-0259. [DOI] [PubMed] [Google Scholar]
- 8.Helsten T, Kato S, Schwaederle M, Tomson BN, Buys TP, Elkin SK, et al. Cell-Cycle Gene Alterations in 4,864 Tumors Analyzed by Next-Generation Sequencing: Implications for Targeted Therapeutics. Mol Cancer Ther 2016;15(7):1682–90 doi 10.1158/1535-7163.MCT-16-0071. [DOI] [PubMed] [Google Scholar]
- 9.Kato S, Schwaederle M, Daniels GA, Piccioni D, Kesari S, Bazhenova L, et al. Cyclin-dependent kinase pathway aberrations in diverse malignancies: clinical and molecular characteristics. Cell Cycle 2015;14(8):1252–9 doi 10.1080/15384101.2015.1014149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 2011;11(8):558–72 doi 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 11.Hamilton E, Infante JR. Targeting CDK4/6 in patients with cancer. Cancer Treat Rev 2016;45:129–38 doi 10.1016/j.ctrv.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 12.Peguero JA, O’Neil BH, Sohal D, Bauer TM, Subbiah V, Kelly K, et al. Genomic mutation profiling (GMP) and clinical outcome in patients (pts) treated with ribociclib (CDK4/6 inhibitor) in the Signature program. Journal of Clinical Oncology 2016;34(15_suppl):2528- doi 10.1200/JCO.2016.34.15_suppl.2528. [DOI] [Google Scholar]
- 13.Baghdadi TA, Halabi S, Garrett-Mayer E, Mangat PK, Ahn ER, Sahai V, et al. Palbociclib in Patients With Pancreatic and Biliary Cancer With CDKN2A Alterations: Results From the Targeted Agent and Profiling Utilization Registry Study. JCO Precision Oncology 2019(3):1–8 doi 10.1200/po.19.00124. [DOI] [PubMed] [Google Scholar]
- 14.Kato S, Okamura R, Sicklick JK, Daniels GA, Hong DS, Goodman A, et al. Prognostic Implications of RAS Alterations in Diverse Malignancies and Impact of Targeted Therapies. Int J Cancer 2019. doi 10.1002/ijc.32813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gardner AM, Vaillancourt RR, Johnson GL. Activation of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase by G protein and tyrosine kinase oncoproteins. J Biol Chem 1993;268(24):17896–901. [PubMed] [Google Scholar]
- 16.Macdonald SG, Crews CM, Wu L, Driller J, Clark R, Erikson RL, et al. Reconstitution of the Raf-1-MEK-ERK signal transduction pathway in vitro. Mol Cell Biol 1993;13(11):6615–20 doi 10.1128/mcb.13.11.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. Discovery of a Covalent Inhibitor of KRAS(G12C) (AMG 510) for the Treatment of Solid Tumors. J Med Chem 2020;63(1):52–65 doi 10.1021/acs.jmedchem.9b01180. [DOI] [PubMed] [Google Scholar]
- 18.Bhagwat SV, McMillen WT, Cai S, Zhao B, Whitesell M, Shen W, et al. ERK Inhibitor LY3214996 Targets ERK Pathway-Driven Cancers: A Therapeutic Approach Toward Precision Medicine. Mol Cancer Ther 2020;19(2):325–36 doi 10.1158/1535-7163.MCT-19-0183. [DOI] [PubMed] [Google Scholar]
- 19.Yadav V, Chen SH, Yue YG, Buchanan S, Beckmann RP, Peng SB. Co-targeting BRAF and cyclin dependent kinases 4/6 for BRAF mutant cancers. Pharmacol Ther 2015;149:139–49 doi 10.1016/j.pharmthera.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 20.Pek M, Yatim S, Chen Y, Li J, Gong M, Jiang X, et al. Oncogenic KRAS-associated gene signature defines co-targeting of CDK4/6 and MEK as a viable therapeutic strategy in colorectal cancer. Oncogene 2017;36(35):4975–86 doi 10.1038/onc.2017.120. [DOI] [PubMed] [Google Scholar]
- 21.Patel M, Kato SM, Kurzrock R. Molecular Tumor Boards: Realizing Precision Oncology Therapy. Clin Pharmacol Ther 2018;103(2):206–9 doi 10.1002/cpt.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwaederle M, Parker BA, Schwab RB, Fanta PT, Boles SG, Daniels GA, et al. Molecular tumor board: the University of California-San Diego Moores Cancer Center experience. Oncologist 2014;19(6):631–6 doi 10.1634/theoncologist.2013-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sicklick JK, Kato S, Okamura R, Schwaederle M, Hahn ME, Williams CB, et al. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med 2019;25(5):744–50 doi 10.1038/s41591-019-0407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31(11):1023–31 doi 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwartz LH, Litiere S, de Vries E, Ford R, Gwyther S, Mandrekar S, et al. RECIST 1.1-Update and clarification: From the RECIST committee. Eur J Cancer 2016;62:132–7 doi 10.1016/j.ejca.2016.03.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.OA01.06 Safety E, and Pharmacokinetics of AMG 510, a Novel KRASG12C Inhibitor, in Patients with Non-Small Cell Lung Cancer, Govindan Rea, Journal of Thoracic Oncology V, Issue 11, S1125 – S1126. [Google Scholar]
- 27.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov 2020;10(1):54–71 doi 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parish AJ, Nguyen V, Goodman AM, Murugesan K, Frampton GM, Kurzrock R. GNAS, GNAQ, and GNA11 alterations in patients with diverse cancers. Cancer 2018;124(20):4080–9 doi 10.1002/cncr.31724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iglesias M, Frontelo P, Gamallo C, Quintanilla M. Blockade of Smad4 in transformed keratinocytes containing a Ras oncogene leads to hyperactivation of the Ras-dependent Erk signalling pathway associated with progression to undifferentiated carcinomas. Oncogene 2000;19(36):4134–45 doi 10.1038/sj.onc.1203764. [DOI] [PubMed] [Google Scholar]
- 30.Ozawa H, Ranaweera RS, Izumchenko E, Makarev E, Zhavoronkov A, Fertig EJ, et al. SMAD4 Loss Is Associated with Cetuximab Resistance and Induction of MAPK/JNK Activation in Head and Neck Cancer Cells. Clin Cancer Res 2017;23(17):5162–75 doi 10.1158/1078-0432.CCR-16-1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ai X, Wu Y, Zhang W, Zhang Z, Jin G, Zhao J, et al. Targeting the ERK pathway reduces liver metastasis of Smad4-inactivated colorectal cancer. Cancer Biol Ther 2013;14(11):1059–67 doi 10.4161/cbt.26427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fell JB, Fischer JP, Baer BR, Blake JF, Bouhana K, Briere DM, et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRAS(G12C) Inhibitor for the Treatment of Cancer. J Med Chem 2020. doi 10.1021/acs.jmedchem.9b02052. [DOI] [PubMed] [Google Scholar]
- 33.Blumenschein GR Jr., Smit EF, Planchard D, Kim DW, Cadranel J, De Pas T, et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)dagger. Ann Oncol 2015;26(5):894–901 doi 10.1093/annonc/mdv072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodon J, Soria JC, Berger R, Miller WH, Rubin E, Kugel A, et al. Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat Med 2019;25(5):751–8 doi 10.1038/s41591-019-0424-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seton-Rogers S Eliminating protective autophagy in KRAS-mutant cancers. Nat Rev Cancer 2019;19(5):247 doi 10.1038/s41568-019-0137-5. [DOI] [PubMed] [Google Scholar]
- 36.Kim RD, McDonough S, El-Khoueiry AB, Bekaii-Saab TS, Stein SM, Sahai V, et al. Randomised phase II trial (SWOG S1310) of single agent MEK inhibitor trametinib Versus 5-fluorouracil or capecitabine in refractory advanced biliary cancer. Eur J Cancer 2020;130:219–27 doi 10.1016/j.ejca.2020.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Modi PK, Komaravelli N, Singh N, Sharma P. Interplay between MEK-ERK signaling, cyclin D1, and cyclin-dependent kinase 5 regulates cell cycle reentry and apoptosis of neurons. Mol Biol Cell 2012;23(18):3722–30 doi 10.1091/mbc.E12-02-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leontieva OV, Demidenko ZN, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Differ 2013;20(9):1241–9 doi 10.1038/cdd.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.