

Abstract
Background and Aims
Allopolyploidy is an important driver of diversification and a key contributor to genetic novelty across the tree of life. However, many studies have questioned the importance of extant polyploid lineages, suggesting that the vast majority may constitute evolutionary ‘dead ends’. This has important implications for conservation efforts where polyploids and diploid progenitors often compete for wildlife management resources. Isoetes appalachiana is an allotetraploid that is broadly distributed throughout the eastern USA alongside its diploid progenitors, I. valida and I. engelmannii. As such, this species complex provides an excellent opportunity to investigate the processes that underpin the formation and survival of allopolyploid lineages.
Methods
Here we utilized RADseq and whole-chloroplast sequencing to unravel the demographic and evolutionary history of hybridization in this widespread species complex. We developed a modified protocol for phasing RADseq loci from an allopolyploid in order to examine each progenitor’s genetic contribution independently in a phylogenetic context. Additionally, we conducted population-level analyses to examine genetic diversity and evidence of gene flow within species.
Key Results
Isoetes appalachiana is the product of multiple phylogenetic origins, suggesting that formation and establishment of allopolyploids are common in this group. Hybridization appears to be unidirectional, with I. engelmannii consistently being the maternal progenitor. Additionally, we find that polyploid lineages are genetically isolated, rarely if ever experiencing gene flow between geographically distinct populations.
Conclusions
Allopolyploid lineages of I. appalachiana appear to form frequently and experience a high degree of genetic isolation following formation. Thus, our results appear to corroborate the hypothesis that the vast majority of recently formed polyploids may represent evolutionary dead ends. However, this does not necessarily lessen the evolutionary importance or ecological impact of polyploidy per se. Accordingly, we propose a conservation strategy that prioritizes diploid taxa, thus preserving downstream processes that recurrently generate allopolyploid diversity.
Keywords: Isoetes, population genomics, RADseq, conservation, polyploidy, hybridization, genome skimming, admixture, phylogenetics, Appalachian, gene flow
INTRODUCTION
The significance of allopolyploidy has long been recognized as a key driver of plant evolution and diversification since Stebbins’ treatise on the subject over 80 years ago (Stebbins, 1940). Polyploidization results in a sudden reproductive isolation of parents and offspring while potentially providing new material for evolution of novel genotypes and phenotypes (Levin, 1983). Due to its prominent evolutionary role, many studies have sought to characterize the effects of ancient polyploidization in plants both with and without hybridization (Vanneste et al., 2014; Van de Peer et al., 2017; Landis et al., 2018; Barrett et al., 2019). However, the evolutionary significance of neopolyploidy is less clear. While neopolyploidy is common among extant plants, instances of ancient whole-genome duplication (WGD) are infrequent, suggesting that polyploids are rarely successful (Mayrose et al., 2011; Li and Barker, 2020). This, combined with their putative genetic isolation from other lineages, has led many authors to declare most neopolyploids as evolutionary ‘dead ends’ (Mayrose et al., 2011; Arrigo and Barker, 2012; Soltis et al., 2014; Van de Peer et al., 2017). This has important implications not only for the study of plant evolution but also for conservation biology. If polyploids form frequently but rarely survive, conservation efforts should perhaps primarily focus on diploid lineages, thus maintaining the processes that generate genomic complexity in hybrid and polyploid species complexes (Ennos et al., 2005, 2012; Hamston et al., 2018).
Nowhere is the disparity between neoploidy and ancient polyploidy so apparent as in Isoetes (Isoetaceae). Commonly known as quillworts or merlin’s grass, Isoetes is a globally distributed genus of lycophyte inhabiting a wide range of terrestrial and freshwater environments. While they are morphologically simple, quillworts are genomically complex. The lineage exhibits frequent hybridization and possesses one of the highest rates of polyploidy among vascular plants (Hoot et al., 2004). Though diploid (homoploid) hybrids of Isoetes are typically sterile, they can restore fertility by duplicating their entire genome (Taylor and Hickey, 1992). As a result, Isoetes are remarkable for their high incidence of both polyploidy and hybridization. Nearly 50 % of the 192 named species are polyploids, the majority of these presumably of hybrid origin (Troia et al., 2016) and many are listed as threatened or endangered around the world. A notable example is the federally listed allopolyploid I. louisianensis, found in the southern USA. In spite of the extraordinarily high abundance of extant polyploids, a recent study of the I. taiwanensis DeVol genome found conclusive evidence for just one ancient WGD in its evolutionary history (Wickell et al., 2021). While this suggests that polyploids are evolutionarily ephemeral, the degree to which they are genetically isolated from other polyploid lineages, as well as their diploid progenitors, is not well understood in this group. Though the prevalence of hybridization, polyploidy and cryptic species complexes in Isoetes presents a formidable challenge to modern genomic analysis, it also provides a rare opportunity to gain deeper insight into the formation and evolutionary trajectory of nascent polyploids.
Isoetes appalachiana (Fig. 1A) is one such polyploid. A mostly aquatic species found along the margins of lakes and streams throughout the eastern USA, it is an allotetraploid whose range broadly overlaps with that of its diploid progenitors, I. engelmannii (Fig. 1B) and I. valida (Fig. 1C) (Brunton and Britton, 1997). In addition, prior research utilizing a low-copy nuclear marker has indicated that I. appalachiana is the product of at least two separate hybridization events involving distinct genotypes of I. engelmannii (Schafran, 2019). The large, overlapping ranges of this species and its diploid progenitors, combined with existing evidence of recurrent formation, make I. appalachiana an excellent test case to investigate how polyploid lineages become established and persist alongside their diploid progenitors.
Fig. 1.
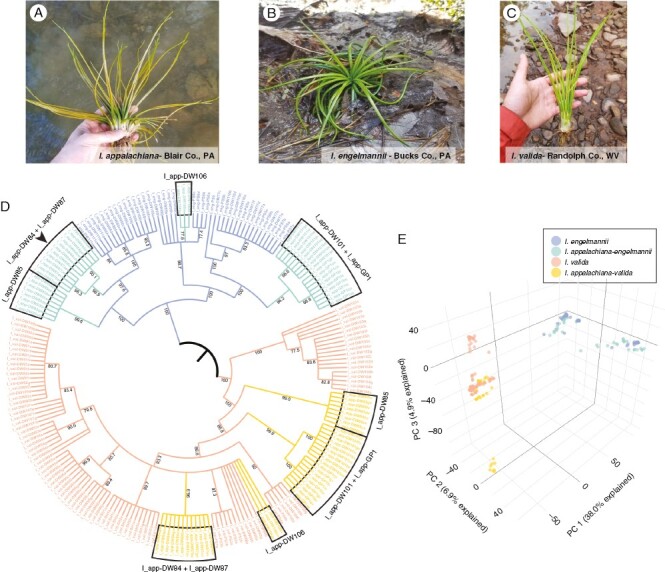
Phylogeny and PCA of phased RADseq data place homoeologous I. appalachiana loci among diploid progenitors. (A–C) Photographs of focal species I. appalachiana (A), I. engelmannii (B) and I. valida (C) in the field. (D) SVDquartets phylogeny of phased homoeologous RADseq loci from I. appalachiana with diploid progenitors I. valida and I. engelmannii. Multiple origins of I. appalachiana are marked by boxes. (E) PCA of RADseq data for all three species following separation of homoeologous loci.
With this study, we combine next-generation sequencing data with a read-phasing approach, similar to that used by Sherman-Broyles et al. (2017) and Chase et al. (2023), to identify and compare homoeologous RADseq loci from multiple allopolyploid populations of I. appalachiana with their diploid counterparts in I. valida and I. engelmannii. In doing so we manage to gain valuable insight into the history of hybridization and polyploidization in this widespread species complex. Specifically, we find phylogenetic evidence for the recurrent but not reciprocal formation of polyploid lineages of I. appalachiana. Following establishment, our admixture analyses suggest that populations rarely come into secondary contact. This appears to be true for both polyploids and diploids – only a handful of localities in our dataset show any evidence of admixture between individuals at different sites. We believe that the methods and insights described here represent an important stepping stone on the path to understanding processes of hybridization, polyploidization and speciation in Isoetes and vascular plants in general. Furthermore, our research has important implications for the conservation of rare and imperilled polyploid species of Isoetes and other plants.
MATERIALS AND METHODS
Sample collection
Vouchers and samples of leaf tissue were collected from Isoetes appalachiana Brunt. & Britton and its diploid progenitors I. valida and I. engelmannii from across their range in the eastern USA (Fig. 2, Supplementary Data Table 1). We took leaf samples at random from up to ten plants at each site, with a special focus on sampling individuals at varying distances from one another across their local distribution. Sampling methods necessarily varied based on habitat, population size and density. Detailed notes are provided on sampling methods for each site in Supplementary Data Table S1. Leaf tissue was immediately placed on silica for rapid desiccation. Individuals were chosen from each site for RADseq analysis in a manner that maximized the total area covered by samples at each site. A subset of the individuals used in RADseq analyses was used for genome skimming and whole-chloroplast genome assembly. Vouchers for each population, excluding those from Shenandoah and Great Smoky Mountain National Parks, where no vouchers were taken, are deposited at the L. H. Bailey Hortorium at Cornell University.
Fig. 2.
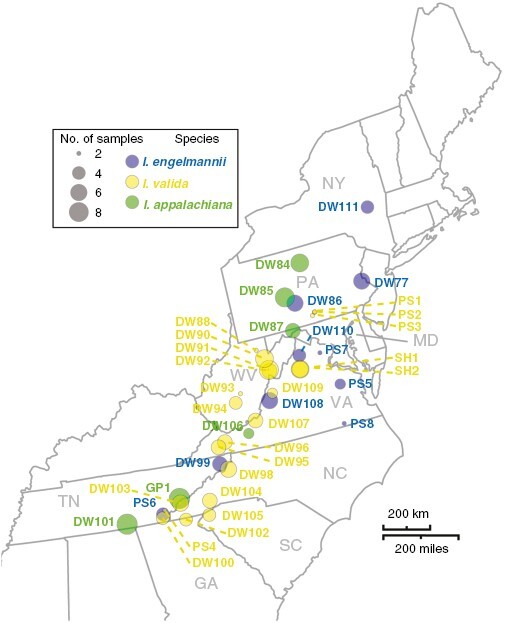
Map of localities sampled for this study. Circles are coloured by species, with I. engelmannii shown in blue, I. valida shown in yellow and I. appalachiana shown in green. Circle diameter indicates the number of samples taken from each locality.
In addition to samples collected specifically for this study, we utilized silica-dried leaf material to achieve more robust sampling for our RADseq dataset. These samples comprised 8 specimens of I. valida from southern Pennsylvania and eastern Tennessee as well as 11 specimens of I. engelmannii from Tennessee and Virginia (Fig. 2, Supplementary Data Table S1). Any associated vouchers are deposited at the Old Dominion Herbarium (ODU).
Species’ identities were confirmed based on megaspore morphology. In addition, individuals assigned to I. appalachiana based on morphology were assumed to be tetraploid if they did not produce the small misshapen spores that are characteristic of diploid hybrids.
RADseq
A total of 192 samples of silica-dried leaf tissue were sent to University of Wisconsin–Madison Biotechnology Center for extraction, library preparation and sequencing. DNA extraction was conducted using the Qiagen DNeasy mericon 96 QIAcube HT Kit and total DNA was quantified with the Quant-iT™ PicoGreen dsDNA kit (Life Technologies, Grand Island, N, USA). Library preparation followed the procedure of Elshire et al. (2011). Genomic DNA was digested using PstI and MspI restriction enzymes (New England Biolabs, Ipswich, MA, USA) and barcoded Illumina adapters were attached using T4 ligase (New England Biolabs, Ipswich, MA, USA). All 192 samples were pooled and amplified prior to removal of adapter dimer contamination by SPRI bead purification. Finally, the completed libraries were assessed for quantity using the Qubit dsDNA HS Assay Kit (Life Technologies, Grand Island, NY, USA) and quality using the Agilent Bioanalyzer High Sensitivity Chip (Agilent Technologies, Santa Clara, CA, USA) prior to paired-end (2 × 150) sequencing on an Illumina NovaSeq 6000.
Phasing of homoeologous RADseq reads
To phase homoeologous reads from RADseq data, we produced a polyploid pseudo-reference using reads from diploid progenitor species, I. engelmannii and I. valida. First, raw reads were demultiplexed and clustered separately for each diploid species using the process_radtags program and denovo_map.pl wrapper provided with Stacks v2.61 (Rochette et al., 2019). Consensus sequences were generated for each diploid species by running the populations command with the ‘--fasta-loci’ option. Next, a draft genome assembly of I. engelmannii was downloaded from NCBI (GenBank assembly accession GCA_011763485.2) and the concatenated diploid consensus sequences were mapped using BWA-MEM version 0.7.17 (Li and Durbin, 2009). Following alignment to the reference, loci were selected only if one contig from each diploid species successfully aligned only once (i.e. an alignment depth of 2) using a custom python script (get_2read_loci_fromSAM.py). Contigs aligning to these regions were placed into a single fasta file to create a polyploid pseudo-reference. Reads from the polyploid samples were then aligned to the pseudo-reference using BWA-MEM version 0.7.17 (Li and Durbin, 2009). Finally, reads that preferentially aligned to one diploid or the other were retained and assigned to a diploid progenitor using a custom python script (parse_bam_4homoeologs.py). Reads were retained if they aligned only once to either I. valida or I. engelmannii, or if they aligned twice (once to each diploid species) and the mapping quality scores differed by more than ten points, in which case they were assigned to the species with the higher score. Reads that did not map, mapped multiple times (more than once to the same species) or mapped ambiguously to both species with similar alignment scores were excluded from further analysis. While this reduced the number of reads available by roughly 80 % for polyploid taxa, it allowed us to perform subsequent analyses on homoeologous loci as though they were diploid. Python scripts used for phasing are available at https://github.com/dawickell/public_scripts. Raw RADseq data were deposited in the NCBI SRA database (PRJNA1005833).
Chloroplast phylogenomics
Genomic DNA was extracted from 50 samples using the Omega E.Z.N.A.® Plant DNA Kit according to the manufacturer’s instructions. Whole genomic dsDNA was then quantified using the BR Assay kit on a Qubit 3.0 fluorometer (ThermoFisher Scientific, Waltham, MA, USA). Quality was assessed by 260:280 nm absorption ratio using a NanoDrop One spectrophotometer (ThermoFisher Scientific, Waltham, MA, USA). Finally, gel electrophoresis was conducted and highly fragmented samples (those where the majority of fragments were shorter than our intended insert length of 300 bp) were excluded.
A total of 20 µL of genomic DNA was diluted to a concentration of ~100 ng/µL and sheared on a Q800R2 sonicator (Qsonica, Newtown, CT, USA) at 20° amplitude for 3 min and 30 s in 10-s pulses separated by 10-s pauses, yielding fragments ~500 bp in length. Library preparation was conducted using the KAPA HyperPrep kit (Roche Diagnostics, Wilmington, MA, USA) according to the manufacturer’s instructions. Libraries were then pooled for paired-end (2 × 150) sequencing on an Illumina NovaSeq 6000 by Novogene.
Demultiplexed reads were checked for quality using FastQC (Andrews, 2010). The resulting reads were trimmed using fastp v0.23.2 (Chen et al., 2018) to automatically detect and remove adapters and poly-G sequences. Chloroplast genomes were assembled in NOVOPlasty 4.3.1 (Dierckxsens et al., 2017) under the default settings using the I. engelmannii chloroplast genome (refseq accession NC_038080.1) as a seed. Taxa that did not produce a circularized assembly from NOVOPlasty were reassembled using Geneious Prime 2022.2.2 (www.geneious.com) by aligning to a reference genome. The reference genome used for alignment was from the closest relative, selected based on the RADseq phylogeny.
Following assembly of chloroplast genomes, sequence alignment was conducted using full plastome sequences in MAFFT v7.123b (Katoh and Standley, 2013) using the iterative refinement method with up to 1000 iterations. Aligned sequences were used to construct a maximum likelihood phylogeny in IQ-TREE2 v2.2.0 (Minh et al., 2020) with 10 000 ultrafast bootstrap replicates. Raw WGS data were deposited in the NCBI SRA database (PRJNA1005833).
Phylogenetic analysis of RADseq data
VCF file outputs from Stacks were filtered in vcftools v0.1.15 (Danecek et al., 2011) to remove sites with >40 % missing data and singleton loci with a minor allele count of <2. Individual samples were removed if they had >40 % total missing data after filtering by site. After filtering, an alignment of concatenated SNPs was obtained from Stacks v2.61 (Rochette et al., 2019) using the populations command with --phylip_var output flag. In this way, alignments were obtained for individual samples as well as consensus sequences of all the samples in a site. Coalescent trees were then generated from these alignments with 1000 bootstrap replicates using SVDquartets in PAUP* v4.0 (Swofford, 2003).
Admixture analyses
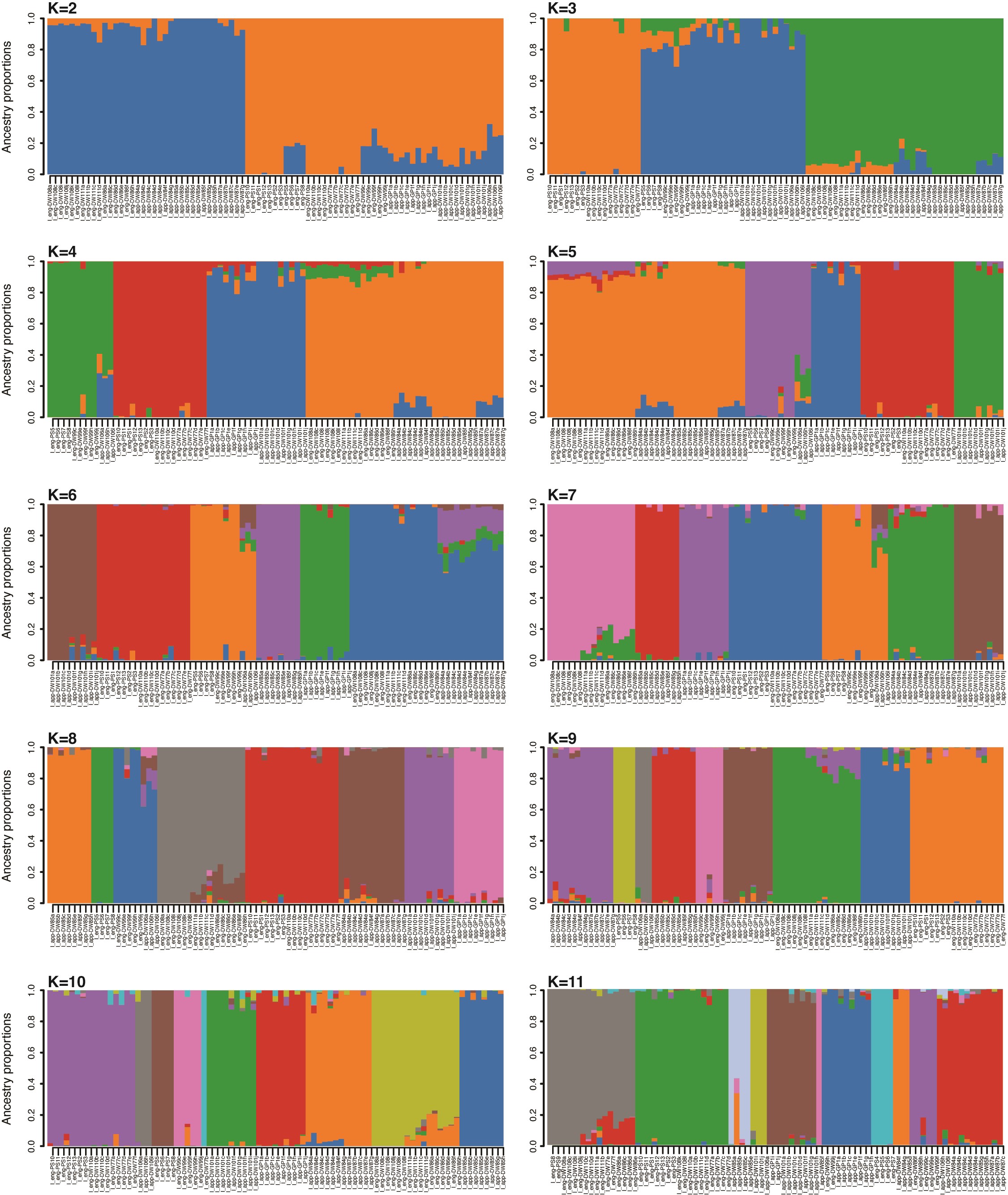
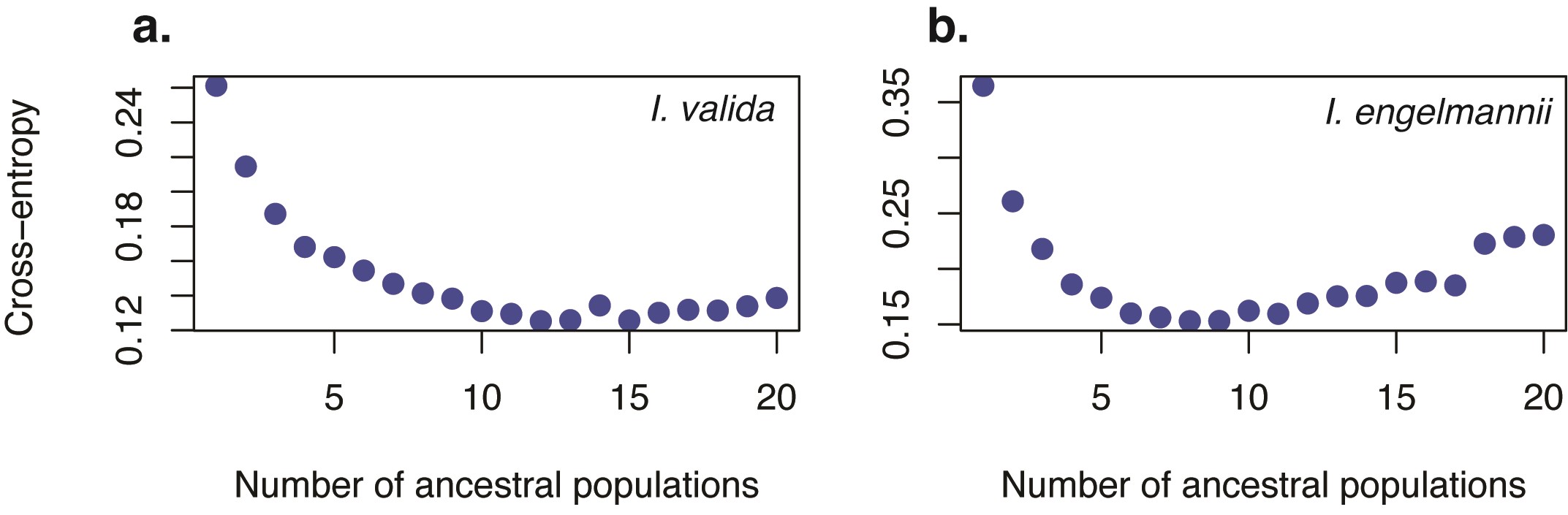
Prior to population genomics analysis, RADseq data were initially filtered using vcftools v0.1.15 (Danecek et al., 2011) to exclude individuals with >40 % missing data and singleton loci with a minor allele count of <2. RADseq loci were then further filtered at 30, 20 and 10 % missing data to investigate the effects of our missingness threshold on population structure analyses. Generally, lower levels of missingness were preferable despite comprising fewer loci as they produced similar groupings with less ‘noise’ in both admixture plots and PCA. However, at and above a missingness threshold of 10 % we recovered far fewer loci (<1000 SNPs) for I. engelmannii. Therefore, to strike a balance between the total number of sites and proportion of missing data, all population analyses were ultimately conducted on data filtered for <20 % missingness and a minor allele count of >2. After filtering, PCA plots were constructed with the R package adegenet v2.1.10 (Jombart, 2008). Barplots of shared ancestry proportions were produced using the LEA v3.10.2 package (Frichot and François, 2015) in R. Optimal values for K were selected based on evaluation of cross-entropy plots by selecting K with the lowest cross-entropy (Supplementary Data Fig. 1). However, a range of values for K from K = 2 to K = optimal K + 2 were also plotted (Supplementary Data Figs S2 and S3) for comparison with each other as well as comparison with PCA, phylogenetic trees and associated f-branch statistics.
Analyses were done without filtering for linkage disequilibrium to retain as many markers as possible. However, a separate analysis was conducted on far fewer markers where only one site was selected from each contig using the radiator v1.2.5 package (Gosselin et al., 2020) in R. We obtained nearly identical grouping and optimal K based on cross-entropy plots, but poorer resolution in homoeologous samples (Supplementary Data Fig. S4).
To obtain a quantitative measure of gene flow among localities, f-branch statistics were calculated in Dsuite v0.5 (Malinsky et al., 2021). Initially, D-statistics were calculated for each diploid species and associated homoeologous loci using the dtrios command with the -tree flag. Phylogenetic trees were generated for each species by pruning branches from the SVDquartets tree generated from consensus sequences of individuals from each locality using the R packages ape v5.7-1 (Paradis and Schliep, 2019) and phytools v1.5-1 (Revell, 2012). Four outgroup taxa were chosen from different localities for each analysis on the basis of which individuals had the least missing data. All outgroup taxa had <10 % missing data. Following the generation of D-statistics, f-branch statistics were generated using the fbranch command in Dsuite v0.5 (Malinsky et al., 2021) with a P-value cut-off of 0.001.
To avoid misinterpreting noise in admixture plots as evidence of admixture we took a conservative approach to assessing gene flow between populations. Two localities were only considered to have experienced admixture if: (1) they showed contributions of >20 % from multiple ancestral populations in admixture plots; (2) they also showed evidence of gene flow based on f-branch statistics; and (3) individuals from that locality had variable or uncertain placement in our SVDquartets phylogeny.
Phylogenetic network construction
VCF file outputs by Stacks were divided by species and filtered to remove sites with >40 % missing data and singleton loci with a minor allele count of <2. Samples were removed if they had >40 % missing data after filtering. After filtering each dataset, one for I. valida + phased I. appalachiana and another for I. engelmannii + phased I. appalachiana, was used to construct a phylogenetic network in SplitsTree v4.19.0 (Huson et al., 2008) using the default settings.
Isolation by distance analysis
To investigate isolation by distance, samples were first sorted according to species and each dataset was filtered for <40 % missingness. Nei’s FST was then calculated between localities for each species using the R package hierfstat v0.5-11 (Goudet, 2005). Next, the R package fields v14.1 (Nychka et al., 2017) was used to calculate the great-circle distance between individual localities based on native GPS coordinates. Finally, isolation by distance was estimated using the adegenet v2.1.10 package (Jombart, 2008) in R and a Mantel test with 999 replicates was used to assess the significance of our result.
RESULTS
Homoeologue-phased RADseq data resolves phylogenetic relationships of polyploid lineages
Based on our phylogenetic reconstruction incorporating phased allopolyploid RADseq loci, we were able to infer at least four independent hybridization events leading to the extant distribution of I. appalachiana. The grouping of polyploid lineages appears to be largely similar between I. engelmannii and I. valida clades (Fig. 1D). In both clades, individuals from Tom Pack Lake (I_eng-DW101, Franklin County, TN) and Great Smoky Mountain National Park (GRSMNP) form a well-supported, monophyletic group. In the I. engelmannii clade, these samples are sister to a poorly resolved but well supported lineage composed of diploid individuals from Virginia and a single population from eastern Pennsylvania. In the I. valida clade, they are sister to a large, poorly resolved clade containing I. appalachiana samples from Rattlesnake Rock (I_app-DW84, Lycoming Co., PA) and Sideling Hill Creek (I_app-DW87, Allegheny Co., MD) along with all the diploids collected north of GRSMNP. Similarly, individuals from Rattlesnake Rock (I_app-DW84, Lycoming Co., PA) and Sideling Hill Creek (I_app-DW87, Allegheny Co., MD) consistently form a monophyletic clade in both lineages. In the I. engelmannii clade they are sister to a lineage containing geographically distant samples from Lake Myosotis (I_eng-DW111, Albany Co., NY), the Juniata River (I_eng-DW86, Huntingdon Co., PA) and Clifton Forge reservoir (I_eng-DW108, Allegheny Co., VA). In the I. valida clade samples from Rattlesnake Rock (I_app-DW84, Lycoming Co., PA) and Sideling Hill Creek (I_app-DW87, Allegheny Co., MD) are nested within a large polytomy composed of all the I. valida samples collected north of GRSMNP.
There are two notable examples where the placement of polyploid lineages relative to each other differed between the I. valida and I. engelmannii clades. The first is the placement of I. appalachiana individuals from Gatewood Reservoir (I_app-DW106; Pulaski Co., VA) in a clade with individuals from Rattlesnake Rock (I_app-DW84; Lycoming Co., PA) and Sideling Hill Creek (I_app-DW87; Allegheny Co., MD) in the I. valida portion of the phylogeny despite appearing to be more distantly related in the I. engelmannii clade (Fig. 1D). The second and more intriguing difference between the placement of homoeologous samples between the two diploid clades involves individuals from I. appalachiana’s type locality at Tipton Reservoir (I_app-DW85; Blair Co., PA). In the I. engelmannii portion of the phylogeny, DW85 is placed in a clade with polyploids DW84 and DW87 and diploids representing the ‘northern’ genotype (see below) of I. engelmannii (Fig. 1D). However, in the I. valida clade, DW85 is distantly related to other polyploids, with all individuals forming a monophyletic clade outside the large clade containing the other homoeologous samples (Fig. 1D). This distant phylogenetic placement contrasts with the position of DW85 in the I. engelmannii clade as well as its geographic proximity to DW84 and DW87.
No evidence for reciprocal hybridization in I. appalachiana
In the phylogenetic reconstruction generated from whole chloroplast sequences every allopolyploid population shares I. engelmannii as the maternal progenitor (Fig. 3). We also see some notable discordance between the nuclear (RADseq) and chloroplast phylogenetic analyses. In the I. valida clade of the chloroplast tree (Fig. 3), most differences in topology are associated with clades that have lower support (<90 %) in the SVDquartets phylogeny (Figs 1D and 4C). One exception to this is found in the placement of individuals from four localities south of GRSMNP (DW102, Towns Co., GA; DW103, Swain Co., NC; DW104, Transylvania Co., NC; DW105, Pickens Co., SC). While individuals from these sites form a well-supported clade in both nuclear and chloroplast phylogenies, their placement is different between them. In the nuclear phylogeny, they form a monophyletic lineage sister to all other samples of I. valida (Figs 1D and 4C). However, in the chloroplast phylogeny they appear to be closely related to individuals from the Black Fork of the Cheat River (I_val-DW90, Tucker Co., WV) and Three Fork Creek (I_val-DW88, Taylor Co., WV) (Fig. 3). In the I. engelmannii clade, the placement of I. appalachiana samples from Tom Pack Lake (I_eng-DW101, Franklin Co., TN) shifts from being sister to other polyploid individuals in GRSMNP (I_app-GP1, Blount Co., TN) in the nuclear phylogeny (Figs 1D and 4C) to being sister to diploid I. engelmannii samples collected from the Watauga River (DW99, Carter Co., TN) in the plastome phylogeny (Fig. 3). As a result, the chloroplast genome of DW101 (Franklin Co., TN) samples appear to be more closely related to those of polyploids from central Pennsylvania and Maryland (I_app-DW84, Lycoming Co., PA; I_app-DW85, Blair Co., PA; I_app-DW87, Allegheny Co., MD) than those in GRSMNP.
Fig. 3.
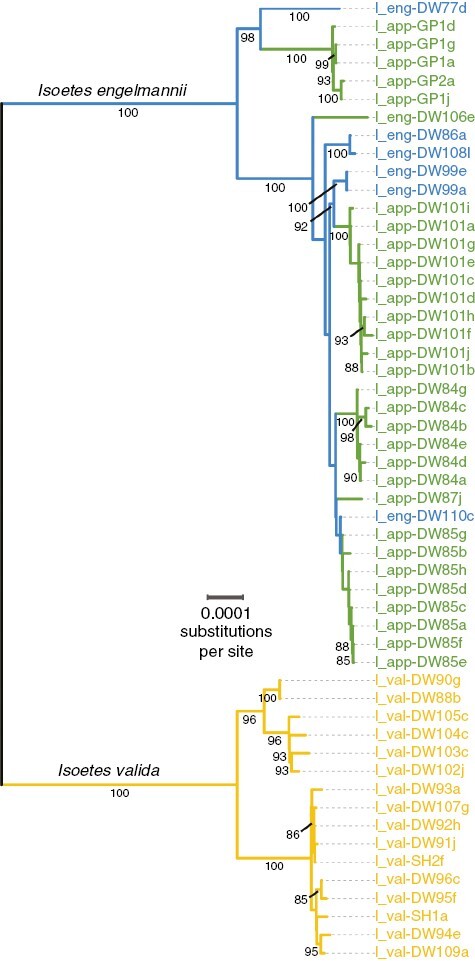
Maximum likelihood phylogeny of whole chloroplast sequences. Branches are coloured by species, with I. engelmannii shown in blue, I. valida shown in yellow and I. appalachiana shown in green. Support values are shown for branches with >75 % bootstrap support.
Fig. 4.
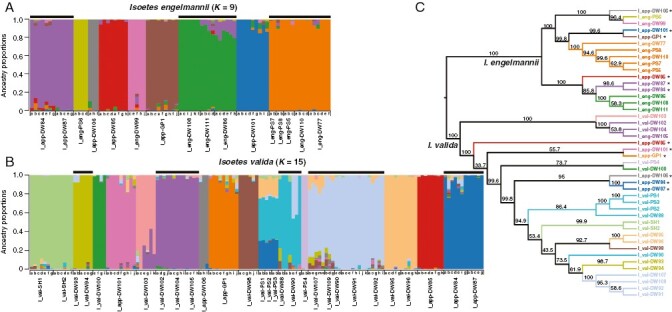
Admixture plots and phylogeny of RADseq data. (A, B) Bar plot of ancestry proportions in (A) I. engelmannii and (B) I. valida at optimal values of K. (C) Phylogeny built in SVDquartets from consensus loci of each locality with tips coloured to indicate the predominant ancestry in each locality. Phased polyploid taxa are marked with an asterisk in the phylogeny. Black bars over admixture plots indicate ancestral clusters spanning multiple watershed regions.
Analysis of RADseq loci reveals little admixture between lineages
Admixture analysis and PCA generally recapitulate phylogenetic groupings from the homoeologue-resolved phylogenetic analysis in SVDquartets. In our PCA (Fig. 1E), the first principal component (PC1) explains 38 % of variation between samples and corresponds to species-level differences, with I. engelmannii and I. valida forming distinct clusters. Similar to analysis using the LEAFY intron 2 marker, I. engelmanni can be broadly subdivided along the second principal component (PC2) (explaining 6.9 % of variation) into two distinct clusters that roughly correspond to northern and southern portions of its range. Once again, as in our phylogenetic analysis, I. valida generally exhibits relatively little structure and forms a single cluster in PC1 and PC2 in the PCA (Fig 1E). However, In PC3 (explaining 4.9 % variation) I. valida can generally be separated into two clusters, where samples from the Smoky Mountain region form a distinct group that is widely separated from other diploids as well as all the homoeologous samples of I. appalachiana. This genetic distinctness is also seen in the phylogenetic analysis of RADseq data (Figs 1A and 4C), where the same individuals comprise a highly supported clade sister to a poorly supported clade containing the phased I. appalachiana samples and all other diploid samples of I. valida.
The ‘best’ value for K was found to be 9 for I. engelmannii and 15 for I. valida using the cross-entropy method in the LEA package (Supplementary Data Fig. 1). At these values we find that many samples tend to form distinct clusters by locality with little to no admixture, suggesting that dispersal between populations and subsequent gene flow are rare (Fig. 4A, B). However, there are multiple instances where samples from two or more localities cluster together in each of the three species (indicated by black bars in Fig. 4A, B), some of which comprise geographically distant samples occupying distinct watershed regions (e.g. I_eng-DW111, Albany Co., NY; I_eng-DW86, Huntingdon Co., PA; I_eng-DW108, Allegheny Co., VA).
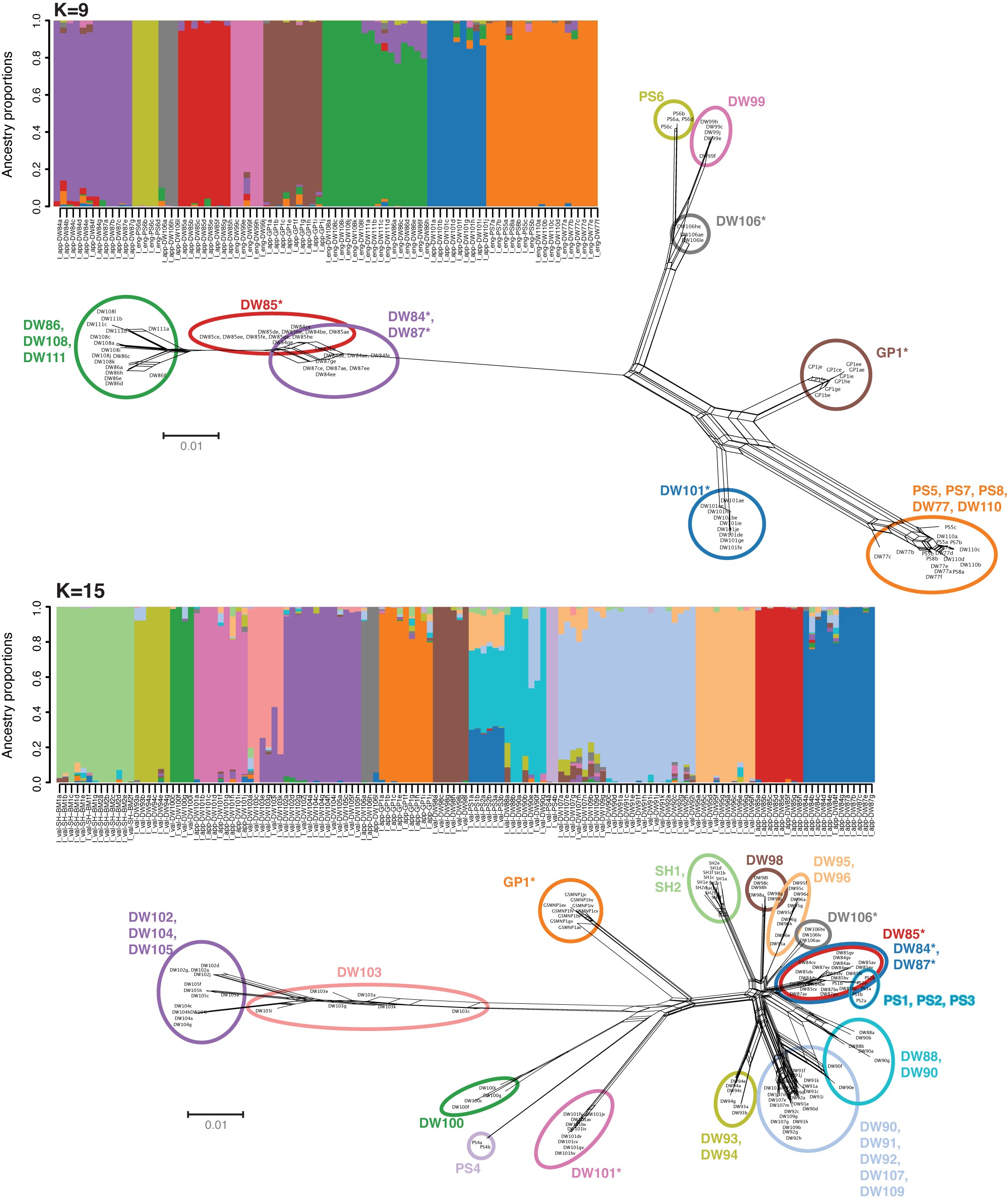
In addition to our phylogenetic analysis in SVDquartets, a phylogenetic network was constructed in SplitsTree to provide a general overview of reticulate relationships between members of each diploid species and phased allopolyploids (Supplementary Data Fig. 5). While the network indicates some evidence of historical admixture, individuals tend to form distantly separated clusters that align with the ancestral populations identified in our admixture analysis. That being said, the phylogenetic network does reveal highly reticulate relationships within some ancestral populations and appears to corroborate evidence of recent admixture between a few distinct populations. Most notably, in I. valida we find evidence of gene flow between multiple sites (I_val-DW91, Randolph Co., WV; I_val-DW92, Randolph Co., WV; I_val-DW107, Giles Co., VA; I_val-DW109, Bath Co., VA) comprising a single ancestral population in our admixture analysis with individuals from Three Fork Creek (I_val-DW88, Taylor Co., WV). In our phylogenetic network and admixture analysis, samples from the Cheat River (I_val-DW90, Tucker Co., WV) clustered with one population or the other with two individuals appearing intermediate between both. Similarly, both analyses seem to find evidence of admixture in a population from Cheoah Lake (I_val-DW103, Swain Co., NC) with three other geographically distant localities that comprise a single ancestral population in our admixture analysis (I_val-DW102, Towns Co., GA; I_val-DW104, Transylvania Co., NC; I_val-DW105, Pickens Co., SC). In our phylogenetic network Cheoah Lake individuals are scattered along several long branches that separate the large ancestral population from other members of the I. valida clade (Supplementary Data Fig. 5a). In the I. engelmannii network we find that I. appalachiana samples from Gatewood Reservoir (I_app-DW106, Pulaski Co., VA) appear to share reticulate relationships with I. engelmannii individuals from the Watauga River (I_eng-DW99, Carter Co., TN) despite showing no evidence of gene flow in our admixture plots (Supplementary Data Fig. 5b). Finally, we observe reticulate relationships between I. appalachiana samples from Tipton Reservoir and an ancestral population containing individuals from Pine Creek (I_app-DW84, Lycoming Co., PA) and Sideling Hill Creek (I_app-DW85, Blair Co., PA) in both I. valida and I. engelmannii phylogenetic networks. However, we find no evidence of gene flow in our admixture analysis at optimal values of K.
To gain additional insight into the possibility of historic or ongoing gene flow between populations we calculated f-branch statistics for individual sites within each diploid species and phased homoeologous samples of I. appalachiana. In general, results from D-suite corroborated the rarity of admixture between phylogenetically and geographically distant lineages (Fig. 5). In I. engelmannii, while we find evidence of admixture between diploids from Passage Creek (I_eng-DW110, Shenandoah Co., VA) with individuals from two distant sites in Pennsylvania and Virginia (I_eng-DW77, Bucks Co., PA; I_eng-PS7, Prince William Co., VA), the populations comprise a single ancestral population in our admixture plots and occupy a well-supported monophyletic clade in our phylogeny. Thus, it is unclear if this represents ongoing gene flow or recent radiation of a single ancestral population. Similarly, f-branch statistics indicate significant gene flow between the I. appalachiana population from Gatewood Reservoir (I_app-DW106, Pulaski Co., VA) and the I. engelmannii population from the Watauga River (I_eng-DW99, Carter Co., TN). While these populations form distinct ancestral populations in our admixture plots at optimal K, they do appear to be admixed at lower values of K.
Fig. 5.
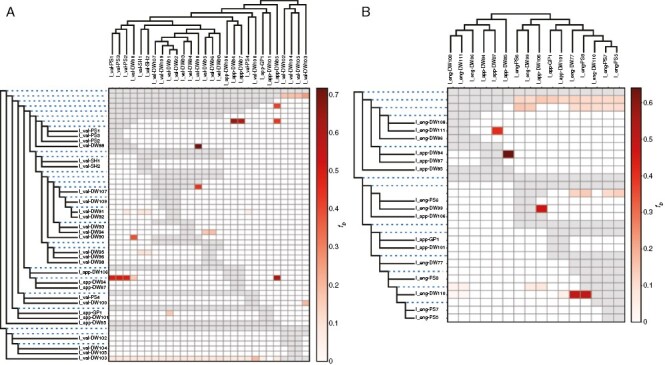
f-Branch statistics calculated from RADseq data aggregated by locality with associated phylogenies showing gene flow and phylogenetic relationships among sites of (A) I. valida and (B) I. engelmannii with associated phased I. appalachiana localities. Displayed f-branch statistics have a P-value of <0.005. Grey cells correspond to combinations for which the f-branch cannot be calculated given the topology of the tree.
In I. valida we find at least two instances where gene flow appears to be confirmed by results from f-branch statistics and admixture plots. In one of these cases, at optimal K (K = 15) in admixture plots, samples from Michaux State Forest, Pennsylvania (I_val-PS1, Cumberland Co., PA; I_val-PS2, Cumberland Co., PA; I_val-PS3, Cumberland Co., PA) exhibit shared ancestry with I. valida from the Tennessee watershed region in western Virginia (DW95, Washington Co., VA; DW96, Russell Co., VA) as well as I. appalachiana from central Pennsylvania and Maryland (I_app-DW84, Lycoming Co., PA; I_app-DW87, Allegheny Co., MD). However, f-branch statistics only appear to corroborate admixture between the most recent common ancestor of the Michaux State Forest localities and Three Fork Creek locality (I_val-DW88; Taylor Co., WV) with polyploid individuals. The second putative case of admixture in I. valida involves individuals from the Black Fork of the Cheat River (I_val-DW90, Tucker Co., WV) and Three Fork Creek (I_val-DW88, Taylor Co., WV). Here, in addition to admixture analysis and f-branch statistics, gene flow appears to be supported by our phylogeny as individuals from the Cheat River vary in their placement, with individuals that show signs of admixture in our LEA analysis being more closely related to those from Three Fork Creek. This example is particularly interesting as the localities involved originate in different watersheds.
Finally, the genomic distinctness of the Tipton Reservoir population (I_app-DW85, Blair Co., PA) seen in phylogenetic analysis of RADseq data is also captured by PCA and admixture plots. It is assigned to a unique ancestral population in admixture plots of I. engelmannii and I. valida at 6 < K < 11 and 3 < K < 17, respectively, while individuals from Rattlesnake Rock (I_app-DW84, Lycoming Co., PA) and Sideling Hill Creek (I_app-DW87, Allegheny Co., MD) tend to form a single cluster at the same values of K (Supplementary Data Figs 2 and 3). However, our analysis of f-branch scores as well as our phylogenetic network both seem to indicate an unusually high rate of admixture with individuals from Rattlesnake Rock and Sideling Hill Creek.
No evidence for isolation by distance
Comparison of genetic and geographic distance demonstrated no evidence of isolation by distance between populations of either species (Supplementary Data Fig. 6). In I. engelmannii this result is corroborated by a high degree of phylogenetic structure, with most sites forming well supported monophyletic clades in phylogenetic analyses of RADseq data. While the phylogeny of I. valida is poorly resolved, we still fail to see a strong correlation between genetic and geographic distance. Similarly, both species show relatively high degrees of isolation between populations regardless of distance based on pairwise FST (Supplementary Data Fig. 7). That being said, we do see some evidence for repeated long-range dispersal events both within and between distinct watersheds in both parental diploid species.
DISCUSSION
The utility of RADseq data in resolving an allopolyploid complex
Here we demonstrate that even in the absence of high-quality reference genomes it is possible to phase homoeologous loci in a non-model allotetraploid. Previous studies have conducted similar phasing of RADseq loci in Glycine (Sherman-Broyles et al., 2017) and Nicotiana (Chase et al., 2023) using higher quality reference assemblies. Based on a single short-read draft assembly of I. engelmannii, we were able to recover enough loci to provide insight into population structure among related species of different ploidy in a widespread allopolyploid species complex. A notable downside to our phasing approach is the large number of loci that were lost during phasing. This could be mitigated to some extent by sequencing polyploids at a higher depth according to their ploidy (i.e. sequencing tetraploids at twice the depth of diploids). However, in cases such as this where diploid progenitors are closely related, reads that map to both genomes equally well will still be substantial and will be discarded. A second factor that has likely affected the number of loci we recovered is the quality of our reference genome. A higher quality reference would likely increase the number of initial loci recovered for our pseudo-polyploid assembly, thus increasing the number of reads that map to it from polyploids. Ideally, the pseudo reference would comprise high-quality genome assemblies from each parent. However, our study demonstrates the utility of even a single low-quality draft assembly in recovering homoeologous loci from an allopolyploid arising from two shallowly diverged parental diploids.
Evidence for the recurrent but not reciprocal formation of allopolyploids
Previous research using the intron of low-copy nuclear gene LEAFY has indicated that I. appalachiana is the product of at least two independent hybridization events involving distinct genotypes of I. engelmannii (Schafran, 2019). While our PCA does seem to divide I. engelmannii and its associated hybrids into two separate groupings, we were not able to determine whether these constitute the same groupings identified by Schafran as we could not reliably recover the same LEAFY intron sequences from genome skimming data. Regardless, our results indicate that polyploids form frequently in Isoetes with the six populations sampled here, resulting from no less than four separate hybridization events. This lower bound represents a conservative estimate as the only I. appalachiana sites that reliably cluster together in nuclear and chloroplast phylogenies, as well as our population analyses, are Rattlesnake Rock and Sideling Hill Creek (I_app-DW84, Lycoming Co., PA; I_app-DW87, Allegheny Co., MD).
In addition, our results call into question whether formation can occur reciprocally between I. valida and I. engelmannii as previously reported in other lineages (Suissa et al., 2022), even when parents are of equal ploidy. A previous in vitro study of interspecific hybridization found that the number of archegonia produced by Isoetes megagametophytes can vary widely (Santos et al., 2020), which can influence which species tend to be the maternal progenitor. However, most of the variation was between species of differing ploidies, and I. valida was not included. Thus, the extent to which the rate of archegonial formation might limit reciprocal hybridization in this case remains unclear. Biased maternal contribution by I. engelmannii in this case might also be explained by cytonuclear incompatibility. Due to the coevolutionary relationship between nuclear and organellar genomes, cytonuclear incompatibility can arise as a result of hybridization between divergent taxa when organellar genomes are suddenly placed in a novel nuclear genomic background (Postel and Touzet, 2020). While the divergence between I. valida and I. engelmannii is relatively recent (Larsén and Rydin, 2016), it is possible that conflict between their nuclear and organellar genomes is contributing to the observed pattern of biased plastid inheritance in I. appalachiana.
Admixture is rare despite evidence of long-distance dispersal
We find little evidence of admixture among geographically distinct populations in both diploid and polyploid members of the I. appalachiana species complex. While there are several instances where a single ancestral population from our admixture analysis spans a large region comprising multiple watersheds, it is unclear if this is the result of ongoing gene flow between the associated localities or recent radiation of a single population. Previous studies have generally found that rates of gene flow are substantially higher among diploids than polyploid species of Isoetes (Kim et al., 2009; Chen et al., 2012; Santos et al., 2020). In addition, while our study found a relatively high degree of population structure in all three species considered here, based on phylogenetic and population genetic analyses, I. valida seemed to exhibit slightly more frequent admixture paired with lower population structure. This is similar to what was found by Wood et al. (2018) in I. lacustris. They theorized that the reduced population structure, in this case relative to an aquatic, clonal angiosperm, was a result of obligate sexual reproduction in Isoetes as opposed to frequent dispersal between ponds. As there is no notable difference in how I. engelmannii and I. valida disperse or reproduce, it is unclear what is driving this disparity between our diploid taxa. In any case, despite the apparent paucity of admixture, we do find some notable examples suggesting that limited gene flow does occur, certainly within diploids and possibly among polyploid taxa as well.
Our study found only two instances of gene flow that were supported by admixture plots, phylogenetic networks and f-branch statistics, both involving the diploid species I. valida. One of these examples appears to indicate admixture between populations of differing ploidy. Thus, we think that this more likely represents an admixture between the sampled diploid lineage and an unidentified ancestral diploid population that then gave rise to the polyploid lineages. The second example involves two I. valida localities along the Cheat River and Three Fork Creek that inhabit separate but adjacent watersheds in West Virginia. In contrast to I. valida, we find no evidence of admixture between diploid lineages of I. engelmannii. While f-branch statistics seem to indicate gene flow between individuals from Passage Creek with two geographically distant sites in Pennsylvania and Virginia, all three are members of a large, uniform ancestral population in our admixture analysis. This is corroborated by our phylogenetic network, where we see a large number of edges connecting individuals within this group. However, we are unable to determine if this represents ongoing gene flow between these sites or merely results from a combination of shared ancestry and incomplete lineage sorting.
Finally, there is the notable case of I. appalachiana at Tipton Reservoir in central Pennsylvania. While individuals from this locality form a single, well-supported clade in our SVDquartets phylogeny and comprise a unique ancestral population at most values of K, f-branch statistics and our SplitsTree network consistently show evidence of gene flow between it and two other I. appalachiana sites along Pine Creek in Pennsylvania and Sideling Hill Creek in Maryland. This is evident in our analysis of both I. engelmannii as well as I. valida loci. However, near optimal values of K we find no evidence of gene flow in our admixture plots, and only at lower values of K do we begin to see some evidence of shared ancestry. Based on the close sister relationship between individuals from Pine Creek and Three Fork Creek, we hypothesize that they are either the product of a single hybridization event or separate hybridizations involving the same ancestral diploid populations. Thus, the most parsimonious explanation for the observed admixture between these lineages is that gene flow occurred between Tipton Reservoir and the ancestral allopolyploid population that gave rise to individuals at Pine Creek and Three Fork Creek. This is the only scenario that would not require multiple independent dispersal events either between ancestral diploid populations or from Tipton Reservoir to both geographically distant populations at Pine Creek and Three Fork Creek.
The low frequency of admixture between populations is surprising in light of evidence that Isoetes is clearly capable of long-range dispersal, as indicated by shared ancestry between populations spanning multiple watershed regions. Indeed, phylogeographic evidence has suggested that the distributions of many species of Isoetes are likely explained by dispersal over great distances (Larsén and Rydin, 2016; Pereira et al., 2021). Thus our results could be explained by two separate, but possibly overlapping, scenarios. First, while we do find evidence of long-range dispersal, it may be that such events are rare. In this case, we might see radiation of a single lineage to occupy new and distant habitats resulting in multiple widespread localities sharing nearly identical genotypes due to founder effects (Novak and Mack, 1993; Ibrahim et al., 1996; Shirk et al., 2014). However, in the event that they disperse to an area that is already occupied, especially if dispersal is rare, they may fail to become established in competition with the dominant genotype. Second, it is possible that significant postzygotic barriers exist for intraspecific hybridization of genetically distinct lineages of Isoetes. While previous studies have demonstrated that different species readily hybridize, those hybrids subsequently tend to produce non-viable, malformed spores and thus suffer from high levels of infertility (Kang et al., 2005; Kim et al., 2008; Santos et al., 2020). To date, no in vitro studies have been conducted to test the viability of intraspecific crosses of genetically or geographically distinct populations of Isoetes. Thus we are currently unable to ascertain to what extent one or both of these factors might contribute to the observed patterns of genetic diversity in this enigmatic group.
Recurrent formation of allopolyploids has important implications for conservation
In addition to having implications for the evolutionary importance of neopolyploid lineages, our study may have significant ramifications for conservation efforts in Isoetes as well as other lineages with high rates of genetic isolation, hybridization and polyploidization. The IUCN Red List currently lists 26 out of 68 assessed species of Isoetes as vulnerable, endangered or critically endangered. Many of these species are allopolyploids, some of uncertain parentage. In the USA, I. louisianensis, one of the three federally listed endangered species of Isoetes, is an allotetraploid of unknown parentage. Similarly in China, I. sinensis is an imperilled allotetraploid formed from two diploids (I. yunguiensis in China and I. taiwanensis in Taiwan) that are considered critically endangered in their own right.
The high degree of differentiation between various populations in our study suggests that much of the species diversity within Isoetes exists between populations rather than within them. This result generally agrees with other studies that have found similar patterns in some species of Isoetes in Asia (Kim et al., 2008; Chen et al., 2010, 2012) and New Zealand (Hofstra and de Winton, 2016). In our study, population structure was loosely associated with watershed regions where there is potential for flowing water and flood events to promote frequent dispersal and possibly gene flow between localities. With this in mind, conservation should broadly focus on preserving individuals from as many distinct watershed regions as possible to avoid irreversible loss of genetic diversity. In addition, the apparent isolation of polyploid lineages in our study combined with the scarcity of ancient WGD relative to the number of polyploid species in Isoetes (Wickell et al., 2021) and other groups (Mayrose et al., 2011) indicates that young polyploid lineages may rarely survive over macro-evolutionary timescales. Consequently, we propose a diploids-first approach to conservation that seeks to maintain polyploid diversity by protecting their progenitors. This will in turn preserve the processes that lead to the repeated formation of polyploid lineages. Indeed, there is substantial evidence that, however rare, polyploidy has played an important role in the evolutionary success of vascular plants.
Future directions
While our study provides novel insight into the formation and proliferation of polyploid lineages in Isoetes, we have barely scratched the surface of this diverse and understudied group. It is our hope that this research will provide a starting point for future studies investigating the processes of hybridization and polyploid speciation. Our results indicate that it is possible to recover meaningful species- and population-level relationships with even a relatively small number of markers given sufficient sampling and read depth. Even so, future investigation into ongoing gene flow would benefit from deeper sampling of individual localities in order to more accurately estimate population-level statistics and thus increase the likelihood of detecting ancient or infrequent periods of introgression. In addition, alternative sequencing methods such as RAD capture (Ali et al., 2016) or genomic resequencing in the presence of a high-quality reference may improve phylogenetic resolution and aid in the characterization of gene flow within and between species of Isoetes. Furthermore, the continued development of genomic resources, in particular whole-genome assemblies, will serve to generally increase the utility of RADseq data and facilitate phasing of homoeologous loci. Finally, subsequent studies should incorporate niche modelling to assess whether polyploid lineages are expanding into new habitats or merely occupying unfilled niches that overlap with their progenitors.
Conclusions
In conclusion, allopolyploid lineages of I. appalachiana appear to form frequently and rarely if ever experience gene flow between geographically isolated populations. Our study further corroborates mounting evidence that many diploid and polyploid species of Isoetes experience a high degree of genetic isolation despite the apparent ability to disperse over long distances. Though we are able to identify three separate origins with a high degree of certainty, it is possible that nearly every polyploid locality is the product of its own hybridization event. In light of the single ancient duplication reported for this genus, our results appear to corroborate the hypothesis that the vast majority of recently formed polyploids may represent evolutionary dead ends. However, the fact that polyploid lineages rarely survive does not diminish the evolutionary significance or ecological importance of polyploidy per se. In fact, our research suggests that it is not enough to merely preserve polyploids and may even inadvertently divert precious resources from preservation of parental diploid species. Instead, conservation efforts should focus on preserving diploid progenitors along with suitable habitat to facilitate the recurrent formation of polyploids, some of which may eventually survive, diploidize and diversify.
SUPPLEMENTARY DATA
Supplementary data are available at Annals of Botany online and consist of the following. Figure S1: plots of cross-entropy scores calculated by LEA for ascending values of K from 1 to 20 in I. valida with phased I. appalachiana loci and I. engelmannii with phased I. appalachiana loci. Figure S2: admixture plots of I. valida and phased I. appalachiana samples for values of K = 2 to K = 17. Figure S3: admixture plots of I. engelmannii and phased I. appalachiana samples for values of K = 2 to K = 11. Figure S4: plots of cross-entropy scores calculated by LEA for values of K = 1 to K = 20 for the linkage disequilibrium-pruned dataset in I. valida with phased I. appalachiana loci and I. engelmannii with phased I. appalachiana loci. Figure S5: phylogenetic networks and admixture plots for I. engelmannii and I. valida with phased I. appalachiana loci. Clades are circled with colours corresponding to principal ancestry proportions for each clade. Figure S6: isolation by distance plots and histograms showing results of Mantel test for (a) I. engelmannii and (b) I. valida with phased I. appalachiana loci. Figure S7: pairwise FST of RADseq data between localities of (a) I. engelmannii and (b) I. valida with phased I. appalachiana loci. Table S1: sample information with collection notes.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
This work was supported in part by a Smithsonian Institution 10-week graduate student fellowship. We would like to acknowledge the L. H. Bailey Hortorium and Old Dominion Herbarium, where vouchers are deposited, and The National Parks Service and The United States Forest Service for providing permits and support in the field. We would like to thank PhD committee members Drs Jeff Doyle and Kelly Zamudio for their help with experimental design and suggestions on the manuscript, Drs Peter Schafran, Jacob Suissa and W. Carl Taylor for their help locating and identifying Isoetes in the field, Dr Peter Schafran for providing leaf material, Gabriel Johnson for his invaluable experience and assistance with preparing RADseq samples for sequencing, and Dr Michael Windham for discussion on the diploid-first approach for conservation.
Contributor Information
David Wickell, Plant Biology Section, School of Integrative Plant Science, Cornell University, Ithaca, NY 14853, USA; Boyce Thompson Institute, Ithaca, NY 14853, USA.
Jacob Landis, Plant Biology Section, School of Integrative Plant Science, Cornell University, Ithaca, NY 14853, USA; Boyce Thompson Institute, Ithaca, NY 14853, USA.
Elizabeth Zimmer, National Museum of Natural History, Smithsonian Institution, Washington D.C., USA.
Fay-Wei Li, Plant Biology Section, School of Integrative Plant Science, Cornell University, Ithaca, NY 14853, USA; Boyce Thompson Institute, Ithaca, NY 14853, USA.
LITERATURE CITED
- Ali OA, O’Rourke SM, Amish SJ, et al. 2016. RAD capture (rapture): flexible and efficient sequence-based genotyping. Genetics 202: 389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (25 November 2023, date last accessed). [Google Scholar]
- Arrigo N, Barker MS.. 2012. Rarely successful polyploids and their legacy in plant genomes. Current Opinion in Plant Biology 15: 140–146. [DOI] [PubMed] [Google Scholar]
- Barrett CF, McKain MR, Sinn BT, et al. 2019. Ancient polyploidy and genome evolution in palms. Genome Biology and Evolution 11: 1501–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunton DF, Britto DM.. 1997. Appalachian quillwort (Isoetes appalachiana, sp. nov.; Isoetaceae), a new pteridophyte from the eastern United States. Rhodora 99:118–133. [Google Scholar]
- Chase MW, Samuel R, Leitch AR, et al. 2023. Down, then up: non-parallel genome size changes and a descending chromosome series in a recent radiation of the Australian allotetraploid plant species, Nicotiana section Suaveolentes (Solanaceae). Annals of Botany 131: 123–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-Y, Liao L, Li W, Li Z-Z.. 2010. Genetic diversity and population structure of the endangered alpine quillwort Isoetes hypsophila Hand.-Mazz. revealed by AFLP markers. Plant Systematics and Evolution 290: 127–139. [Google Scholar]
- Chen Y-Y, Kong D-R, Huang C-H, Xu Y-X, Li Z-Z.. 2012. Microsatellite analysis reveals the genetic structure and gene flow of the aquatic quillwort Isoetes sinensis, a critically endangered species in China. Aquatic Botany 96: 52–57. [Google Scholar]
- Chen S, Zhou Y, Chen Y, Gu J.. 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34: i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, et al. ; 1000 Genomes Project Analysis Group. 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierckxsens N, Mardulyn P, Smits G.. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Research 45: e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennos RA, French GC, Hollingsworth PM.. 2005. Conserving taxonomic complexity. Trends in Ecology & Evolution 20: 164–168. [DOI] [PubMed] [Google Scholar]
- Ennos RA, Whitlock R, Fay MF, et al. 2012. Process-Based Species Action Plans: an approach to conserve contemporary evolutionary processes that sustain diversity in taxonomically complex groups. Botanical Journal of the Linnean Society 168: 194–203. [Google Scholar]
- Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE.. 2011. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PloS one 6: 19379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frichot E, François O.. 2015. LEA: an R package for landscape and ecological association studies. Methods in Ecology and Evolution 6: 925–929. [Google Scholar]
- Gosselin T, Lamothe M, Devloo-Delva F, Grewe P.. 2020. radiator: RADseq data exploration, manipulation and visualization using R. R packageversion. https://rdrr.io/github/thierrygosselin/radiator/ (25 November 2023, date last accessed). [Google Scholar]
- Goudet J. 2005. hierfstat, a package for R to compute and test hierarchical F-statistics. Molecular Ecology Notes 5: 184–186. [Google Scholar]
- Hamston TJ, de Vere N, King RA, et al. 2018. Apomixis and hybridization drives reticulate evolution and phyletic differentiation in Sorbus L.: implications for conservation. Frontiers in Plant Science 9: 1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofstra D, de Winton M.. 2016. Geographic scales of genetic variation amongst Isoëtes in New Zealand. Aquatic Botany 131: 28–37. [Google Scholar]
- Hoot SB, Napier NS, Taylor WC.. 2004. Revealing unknown or extinct lineages within Isoetes (Isoetaceae) using DNA sequences from hybrids. American Journal of Botany 91: 899–904. [DOI] [PubMed] [Google Scholar]
- Huson DH, Kloepper T, Bryant D.. 2008. SplitsTree 40 – computation of phylogenetic trees and networks. Bioinformatics 14: 68–73. [Google Scholar]
- Ibrahim KM, Nichols RA, Hewitt GM.. 1996. Spatial patterns of genetic variation generated by different forms of dispersal during range expansion. Heredity 77: 282–291. [Google Scholar]
- Jombart T. 2008. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24: 1403–1405. [DOI] [PubMed] [Google Scholar]
- Kang M, Ye Q, Huang H.. 2005. Genetic consequence of restricted habitat and population decline in endangered Isoetes sinensis (Isoetaceae). Annals of Botany 96: 1265–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Na HR, Choi H-K.. 2008. Genetic diversity and population structure of endangered Isoetes coreana in South Korea based on RAPD analysis. Aquatic Botany 89: 43–49. [Google Scholar]
- Kim C, Shin H, Choi H.. 2009. Genetic diversity and population structure of diploid and polyploid species of Isoëtes in East Asia based on amplified fragment length polymorphism markers. International Journal of Plant Sciences 170: 496–504. [Google Scholar]
- Landis JB, Soltis DE, Li Z, et al. 2018. Impact of whole-genome duplication events on diversification rates in angiosperms. American Journal of Botany 105: 348–363. [DOI] [PubMed] [Google Scholar]
- Larsén E, Rydin C.. 2016. Disentangling the phylogeny of Isoetes (Isoetales), using nuclear and plastid data. International Journal of Plant Sciences 177: 157–174. [Google Scholar]
- Levin, D.A., 1983. Polyploidy and novelty in flowering plants. The American Naturalist 122: 1-25. [Google Scholar]
- Li Z, Barker MS.. 2020. Inferring putative ancient whole-genome duplications in the 1000 plants (1KP) initiative: access to gene family phylogenies and age distributions. GigaScience 9: giaa004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinsky M, Matschiner M, Svardal H.. 2021. Dsuite – fast D-statistics and related admixture evidence from VCF files. Molecular Ecology Resources 21: 584–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayrose I, Zhan SH, Rothfels CJ, et al. 2011. Recently formed polyploid plants diversify at lower rates. Science 333: 1257. [DOI] [PubMed] [Google Scholar]
- Minh BQ, Schmidt HA, Chernomor O, et al. 2020. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution 37: 1530–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak SJ, Mack RN.. 1993. Genetic variation in Bromus tectorum (Poaceae): comparison between native and introduced populations. Heredity 71: 167–176. [Google Scholar]
- Nychka D, Furrer R, Paige J, Sain S.. 2017. fields: tools for spatial data. R package version. https://cran.r-project.org/web/packages/fields/index.html (25 November 2023, date last accessed). [Google Scholar]
- Paradis E, Schliep K.. 2019. ape 50: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35: 526–528. [DOI] [PubMed] [Google Scholar]
- Van de Peer Y, Mizrachi E, Marchal K.. 2017. The evolutionary significance of polyploidy. Nature Reviews. Genetics 18: 411–424. [DOI] [PubMed] [Google Scholar]
- Pereira JBS, Giulietti AM, Prado J, et al. 2021. Plastome-based phylogenomics elucidate relationships in rare Isoëtes species groups from the Neotropics. Molecular Phylogenetics and Evolution 161: 107177. [DOI] [PubMed] [Google Scholar]
- Postel Z, Touzet P.. 2020. Cytonuclear genetic incompatibilities in plant speciation. Plants (Basel) 9: 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revell LJ. 2012. phytools: an R package for phylogenetic comparative biology (and other things). Methods in Ecology and Evolution 3: 217–223. [Google Scholar]
- Rochette NC, Rivera-Colón AG, Catchen JM.. 2019. Stacks 2: analytical methods for paired-end sequencing improve RADseq-based population genomics. Molecular Ecology 28: 4737–4754. [DOI] [PubMed] [Google Scholar]
- Santos MP, Araujo JVSR, Lopes AVS, et al. 2020. The genetic diversity and population structure of two endemic Amazonian quillwort (Isoetes L.) species. PeerJ 8: e10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafran PW. 2019. Molecular Systematics of Isoëtes (Isoëtaceae) in Eastern North America. PhD Thesis, Old Dominion University. USA. [Google Scholar]
- Sherman-Broyles S, Bombarely A, Doyle J.. 2017. Characterizing the allopolyploid species among the wild relatives of soybean: utility of reduced representation genotyping methodologies: genotyping Glycine allopolyploids. Journal of Systematics and Evolution 55: 365–376. [Google Scholar]
- Shirk RY, Hamrick JL, Zhang C, Qiang S.. 2014. Patterns of genetic diversity reveal multiple introductions and recurrent founder effects during range expansion in invasive populations of Geranium carolinianum (Geraniaceae). Heredity 112: 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis DE, Segovia-Salcedo MC, Jordon-Thaden I, et al. 2014. Are polyploids really evolutionary dead-ends (again)? A critical reappraisal of Mayrose et al. (2011). New Phytologist 202: 1105–1117. [DOI] [PubMed] [Google Scholar]
- Stebbins GL. 1940. The significance of polyploidy in plant evolution. American Naturalist 74: 54–66. [Google Scholar]
- Suissa JS, Kinosian SP, Schafran PW, Bolin JF, Taylor WC, Zimmer EA.. 2022. Homoploid hybrids, allopolyploids, and high ploidy levels characterize the evolutionary history of a western North American quillwort (Isoëtes) complex. Molecular Phylogenetics and Evolution 166: 107332. [DOI] [PubMed] [Google Scholar]
- Swofford DL. 2003. PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4. Sunderland, MA: Sinauer Associates. [Google Scholar]
- 622. Taylor WC, Hickey RJ.. 1992. Habitat, evolution, and speciation in Isoetes. Annals of the Missouri Botanical Garden 79: 613–. [Google Scholar]
- Troia A, Pereira JBS, Kim C, Taylor WC.. 2016. The genus Isoetes (Isoetaceae): a provisional checklist of the accepted and unresolved taxa. Phytotaxa 277: 140–145. [Google Scholar]
- Vanneste K, Baele G, Maere S, Van de Peer Y.. 2014. Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the Cretaceous–Paleogene boundary. Genome Research 24: 1334–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickell D, Kuo L-Y, Yang H-P, et al. 2021. Underwater CAM photosynthesis elucidated by Isoetes genome. Nature Communications 12: 6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood DP, Olofsson JK, McKenzie SW, Dunning LT.. 2018. Contrasting phylogeographic structures between freshwater lycopods and angiosperms in the British Isles. Botany Letters 165: 476–486. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.