

Abstract
Objectives:
Osteoarthritis (OA) features aging-related defects in cellular homeostasis mechanisms in articular cartilage. These defects are associated with suppression of Forkhead Box O (FoxO) transcription factors. FoxO1 or FoxO3 deficient mice show early onset OA while FoxO1 protects against oxidative stress in chondrocytes and promotes expression of autophagy genes and the essential joint lubricant proteoglycan 4 (PRG4). The objective of this study was to identify small molecules that can increase FoxO1 expression.
Methods:
We constructed a reporter cell line with FoxO1 promoter sequences and performed high-throughput screening (HTS) of the Repurposing, Focused Rescue, and Accelerated Medchem (ReFRAME) library. Hits from the HTS were validated and function was assessed in human chondrocytes, meniscus cells and synoviocytes and following administration to mice. The most promising hit, the histone deacetylase inhibitor (HDACI) Panobinostat was tested in a murine OA model.
Results:
Among the top hits were HDACI and testing in human chondrocytes, meniscus cells and synoviocytes showed that Panobinostat was the most promising compound as it increased the expression of autophagy genes and PRG4 while suppressing the basal and IL-1β induced expression of inflammatory mediators and extracellular matrix degrading enzymes.
Intraperitoneal administration of Panobinostat also suppressed the expression of mediators of OA pathogenesis induced by intraarticular injection of IL-1β. In a murine OA model, Panobinostat reduced the severity of histological changes in cartilage, synovium and subchondral bone and improved pain behaviors.
Conclusion:
Panobinostat has a clinically relevant activity profile and is a candidate for OA symptom and structure modification.
Keywords: Osteoarthritis, FoxO, Histone deacetylase inhibitor, Panobinostat
INTRODUCTION
Osteoarthritis (OA) is the most prevalent joint disease with aging1, abnormal mechanical loading2, metabolic syndrome3, female sex4, and potentially alterations of the gut microbiota5 as its major risk factors. Despite considerable efforts in OA drug discovery and develpment there are no approved disease modifying OA drugs (DMOAD) that would halt or slow disease progression which ultimately leads to the need for joint replacement surgery6. Potential reasons for the failures in DMOAD development are persisting challenges in OA clinical trials design7,8, heterogeneity of OA patient populations and a diverse set of OA pathogenesis pathways and molecular therapeutic targets9.
Our focus in the discovery of mechanisms and molecules was on aging-related changes in joint cartilage, and our more recent findings point to deficient cellular homeostasis mechanisms, including autophagy10,11 and proteasome function12. In our search for dysregulated transcription factors that might contribute to the abnormal cellular homeostasis mechanisms we investigated Forkhead Box O (FoxO) transcription factors which are known to regulate fundamental mechanisms of cellular aging13,14. We found that the expression of FoxO1 and FoxO3 is suppressed in human OA cartilage and in mouse models where it precedes cartilage and meniscus damage15,16. In chondrocytes, FoxO increase cellular resistance to oxidative stress17,18. Cartilage-specific deletion of FoxO leads to abnormal postnatal cartilage maturation and more severe spontaneous OA and experimental OA induced by destabilization of the medial meniscus (DMM)19. The early onset of OA in these mice appears first as a disruption of the superficial zone with suppression of PRG4 and autophagy genes, considered as drivers of the increased disease severity19. We also found that viral ectopic FoxO1 expression increased PRG4 and synergized with transforming growth factor-β stimulation. In OA chondrocytes, overexpression of FoxO1 reduced inflammatory mediators and cartilage-degrading enzymes, increased protective genes, and antagonized interleukin-1β (IL-1β) effects18.
Collectively, these findings of FoxO suppression in OA and its protective functions in chondrocytes and cartilage suggest that it is a promising DMOAD target. The goal of the present study was to discover small molecules that can induce FoxO1 and test hit compounds in vitro and in vivo.
METHODS
Detailed experimental procedures are described in the online Supplementary Materials and Methods and Supplemental Tables.
RESULTS
Screening of ReFrame library
To find compounds that upregulate FoxO1 expression for OA treatment, we prepared a monoclonal human SW1353 chondrosarcoma cell line containing a FoxO1 0.8 kb promoter-luciferase construct (see methods for details) and screened the Repurposing, Focused Rescue, and Accelerated Medchem (ReFRAME) Library. The ReFRAME library is composed of 11,948 small molecules that have reached clinical development or undergone significant preclinical profiling22. The primary screen was performed at 10 μM drug concentrations. Hit selection cutoffs for the primary screen were selected at 100% increase in luciferase activity compared to DMSO control. To validate the hits obtained from our primary screen, compounds were re-spotted in duplicate in a 10-point, 3-fold dilution dose response. There was a dose-dependent increase in luciferase activity for 59 compounds (46% hit confirmation rate), with EC50 values as low as 0.33 μM. Among the top 59 hits were 24 histone deacetylase inhibitors (HDACI) including Panobinostat with the lowest EC50 of 0.42 μM (Table 1; Supplementary Figure 1). The ReFame library contains an additional 24 compounds that are classified as HDACI which did not significantly increase luciferase levels (Supplementary Table 1).
Table 1.
HDAC inhibitors with significant activity in the FoxO1 drug screen.
| Panobinostat (LBH589) | 4.15329E-08 |
| Vorinostat (SAHA) | 8.62446E-08 |
| Dacinostat (LAQ824) | 1.43602E-07 |
| Givinostat | 1.52729E-07 |
| QUISINOSTAT | 1.55606E-07 |
| AR-42 | 2.78339E-07 |
| Pracinostat (SB939) | 3.15689E-07 |
| Abexinostat (PCI-24781) | 3.67361E-07 |
| Ivaltiniostat (CG-200745) | 4.13034E-07 |
| JNJ-16241199 | 4.30184E-07 |
| EVP-0334 | 4.8753E-07 |
| CUDC-101 | 5.14518E-07 |
| M344 | 9.07171E-07 |
| Belinostat (PXD101) | 1.41954E-06 |
| CAY10603 | 1.67958E-06 |
| CHR-3996 | 1.79217E-06 |
| Resminostat (RAS2410) | 2.20604E-06 |
| Scriptaid | 2.29618E-06 |
| Rocilinostat (ACY-1215) | 2.51576E-06 |
| Pyroxamide | 3.47248E-06 |
| CY-190602 | 5.18846E-06 |
| Oxamflatin | 6.39766E-06 |
| CXD101 | 6.60552E-06 |
| Tacedinaline | 5.14518E-07 |
Hit confirmation
We selected the top four HDACI (Panobinostat, Vorinostat/SAHA, Givinostat/ITF2357 and Dacinostat/LAQ824) for further testing. In SW1353 chondrosarcoma cells, Panobinostat induced the largest increases in the expression of FoxO1 and its target gene Sesn3 (Figure 1A) compared to the 3 other HDACI. In human OA chondrocytes, Panobinostat was similar to Dacinostat and Givinostat and superior to Vorinostat in inducing the expression of FoxO1, Sesn3, MAP1LC3 and PRG4, and it was superior to the other HDACI in suppressing IL-6 and MMP-13 (Figure 1B). In normal human chondrocytes, Panobinostat Dacinostat and Givinostat also increased FoxO1, Sesn3, MAP1LC3 and PRG4 (Figure 1C). The drugs did not change IL-6 and MMP-13 as the basal levels of these genes in normal cells were very low.
Figure 1. Comparison of top four HDACI.
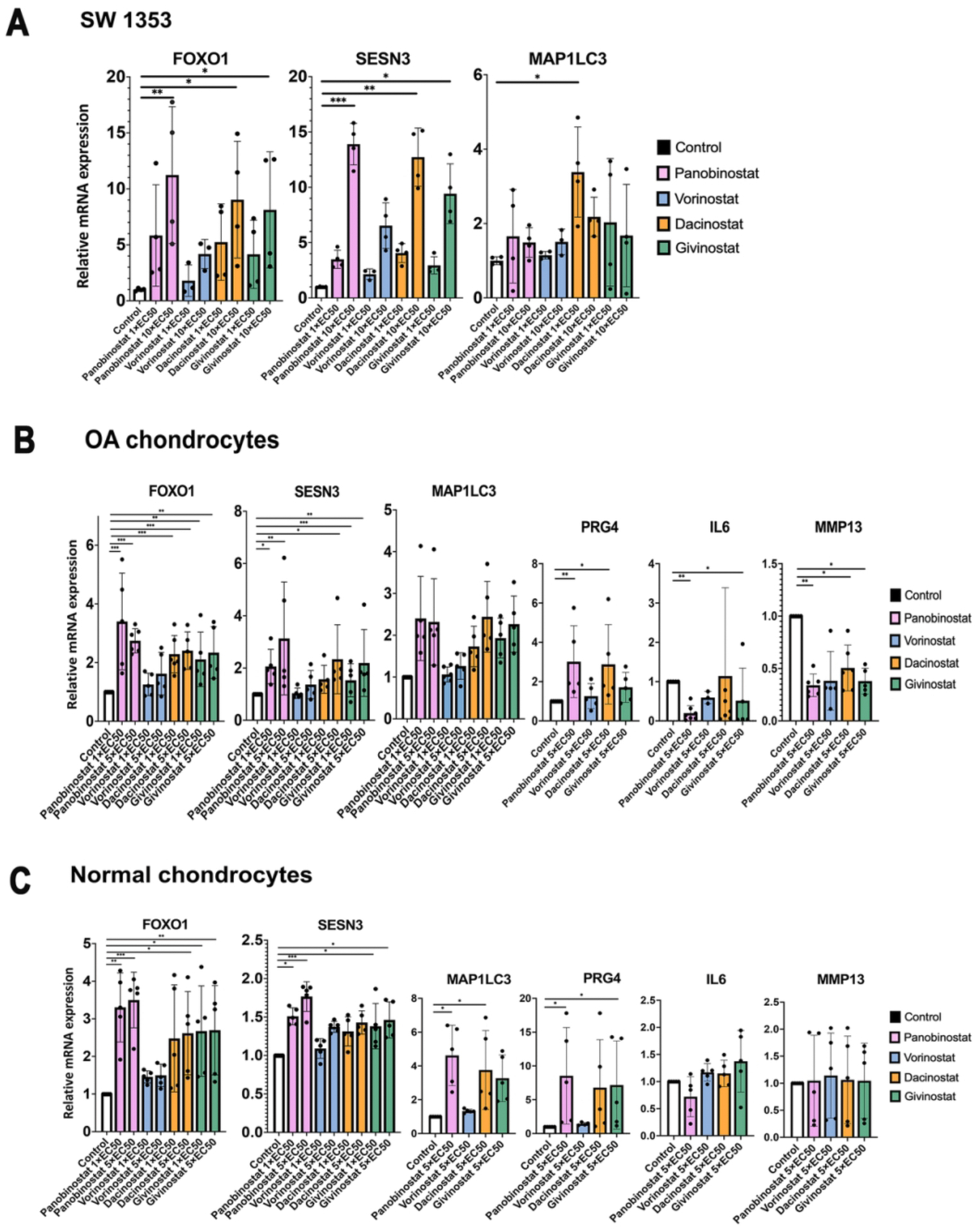
(A) SW1353 human chondrosarcoma cells were cultured in the presence of the indicated EC50 values of Panobinostat, Vorinostat, Givinostat and Dacinostat for 24 h, and RNA was isolated for qRT-PCR analysis. Independent experiments (N=3) were performed where each condition was tested in duplicate.
(B) Human OA chondrocytes (passage 1) from 5 donors were incubated with the indicated doses of Panobinostat, Vorinostat, Givinostat and Dacinostat for 24 hours and RNA was isolated for qRT-PCR analysis.
(B) Normal human chondrocytes (passage 1) from 5 donors were incubated with the indicated doses of Panobinostat, Vorinostat, Givinostat and Dacinostat for 24 hours and RNA was isolated for qRT-PCR analysis.
Panobinostat effects on chondrocytes in vitro
The effects of Panobinostat were profiled in more detail in human OA chondrocytes, and genes related to cellular homeostasis and cartilage ECM were analyzed. Panobinostat did not affect cell viability at 20 nM but caused a small reduction at 40 nM with the higher doses up to 320 nM not causing additional changes in viability (Supplementary Figure 2). Panobinostat increased expression of FoxO1 and the homeostasis genes Sesn3 and MAP1LC3. Among the cartilage ECM genes, it increased PRG4 at all concentrations tested, while increasing ACAN at 5–10 nM but decreasing it at 40–160 nM (Figure 2A). Col2A1 was not increased at any dose but decreased at 10–160 nM. Dio2, an OA risk gene31, was increased at lower doses but suppressed at higher doses of Panobinostat (Figure 2A). In SW1353 human chondrosarcoma cells, Panobinostat increased FoxO1, Sesn3, PRG4 and Col2A1, while LC3 and ACAN were not significantly changed (Figure 2B).
Figure 2. Panobinostat and homeostasis and cartilage ECM genes.
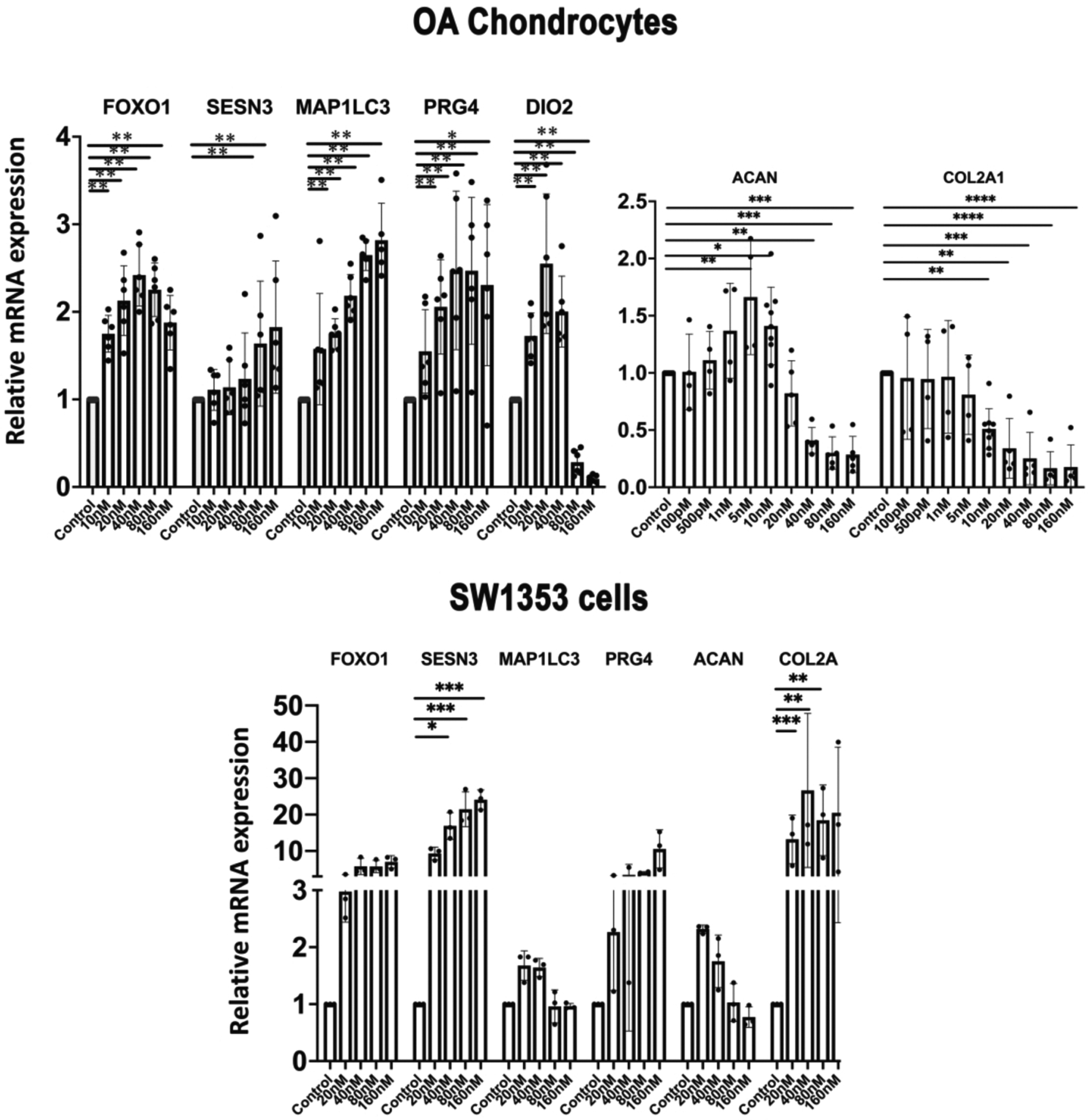
(A). Chondrocytes
Human OA chondrocytes (passage 1) from 5 donors were incubated with the indicated doses of Panobinostat for 24 h and RNA was isolated for qRT-PCR analysis.
(B). SW1353 human chondrosarcoma cells
SW1353 cells were treated with the indicated doses of Panobinostat for 24 h and RNA was isolated for qRT-PCR analysis. Independent experiments (N=3) were performed where each condition was tested in duplicate for each condition.
****= p<0.0001; ***= p<0.001; **= p<0.01; *= p<0.05
Next, we analyzed genes related to inflammation and cartilage ECM degradation. Panobinostat suppressed the basal levels of IL-6, MMP13, PTGES2 and NOS2 in OA chondrocytes. In response to IL-1β treatment, these genes were further increased compared to vehicle control, and Panobinostat dose-dependently suppressed the IL-1β effect on above four genes (Figure 3).
Figure 3. Panobinostat and catabolic genes.
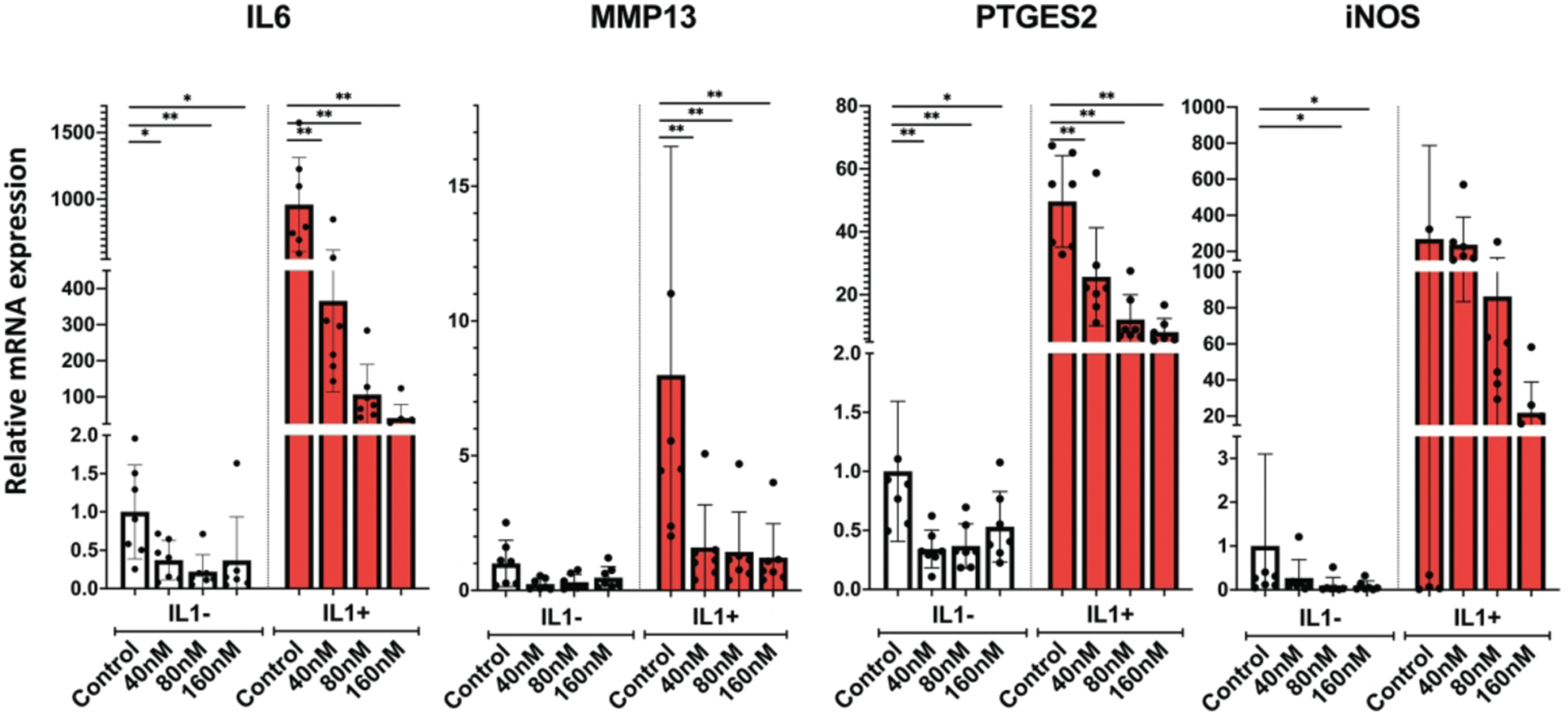
Human OA chondrocytes (passage 1 from 6 donors) were pre-incubated with the indicated doses of Panobinostat for 24 hours. IL-1β (1 ng/ml) was added during the last 6 h and RNA was isolated for qRT-PCR analysis
**= p<0.01; *= p<0.05
As HDAC inhibitors are known to not only change histone acetylation but also affect other signaling mechanisms, we determined which of the Panobinostat effects were dependent on FoxO1. Treatment of chondrocytes with the FoxO1 inhibitor AS1842856 significantly suppressed the effect of Panobinostat on MAP1LC3 but not on the other genes that were analyzed, including PRG4, SESN3, COL2A1 and ACAN (Supplementary Figure 3).
Panobinostat in synoviocytes and meniscus cells
To extend the analysis of Panobinostat on other joint tissue cells, we included meniscus cells and synoviocytes. In meniscus cells (Figure 4A), Panobinostat dose-dependently increased FoxO1, Sesn3 and MAP1LC3. PRG4 was increased significantly at 10 nM but not at the higher doses. Similar effects were observed in synoviocytes (Figure 4B). The basal levels of IL-6 were not significantly changed by Panobinostat in either cell type. The IL-1β effect on MMP13 in synoviocytes was significantly suppressed at all doses of Panobinostat, and IL-1β-induced IL-6 was suppressed by 160 nM Panobinostat in both cell types (Figure 4).
Figure 4. Panobinostat effects on human meniscus cells and synoviocytes.
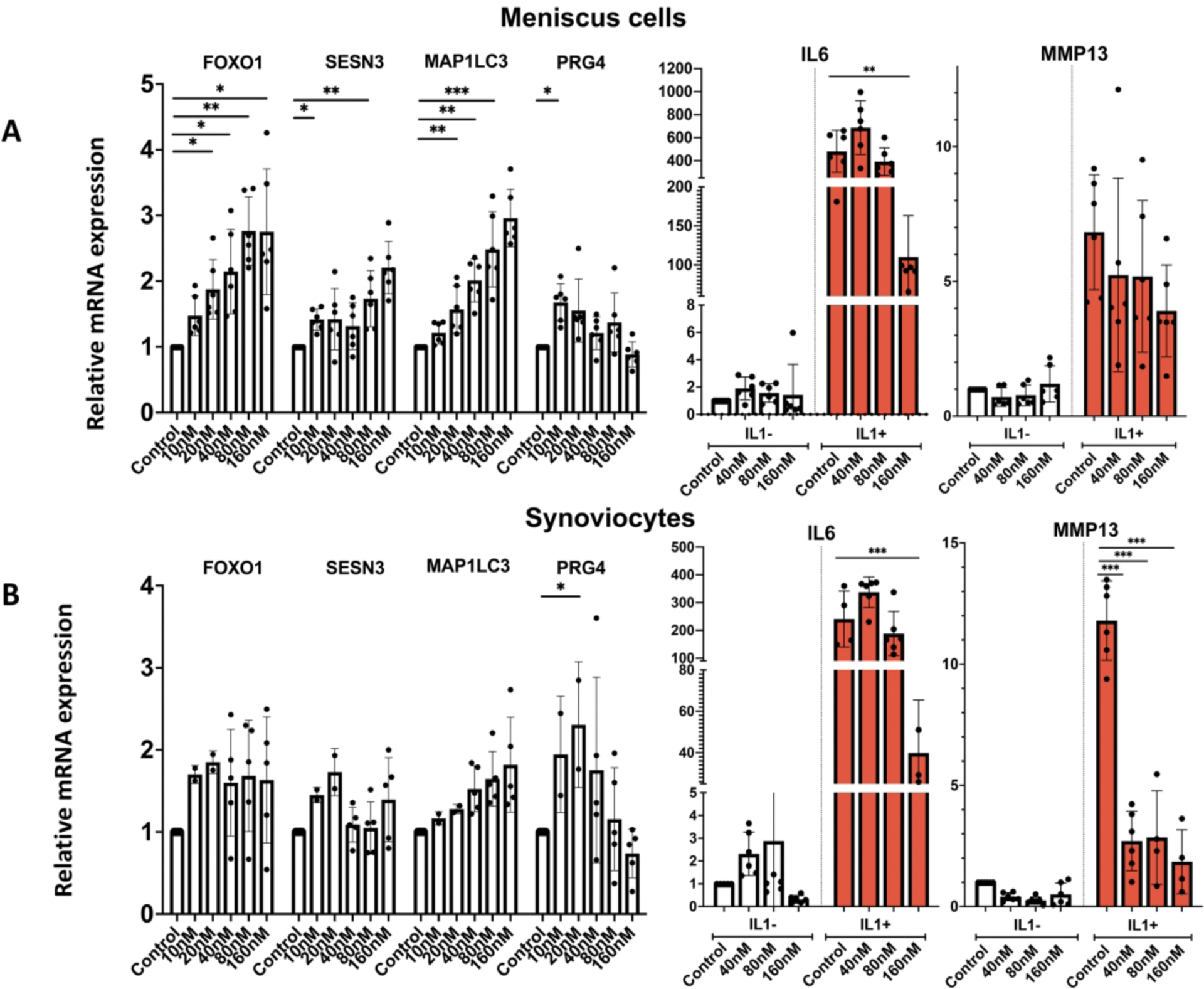
Human OA meniscus cells at passage 1 from 6 donors (Panel A) and OA synoviocytes at passage 1 from 6 donors (Panel B) were treated with Panobinostat for 24 h when IL-1β (1 ng/ml) was added for additional 6 h and RNA was isolated for RT-qPCR analysis.
***= p<0.001; **= p<0.01; *= p<0.05
Effects of Panobinostat on gene expression in normal mouse joints
To explore Panobinostat effects on joint tissues in vivo, we injected the drug intraperitoneally into normal C57BL/6J mice (n=6). Following injection on day 0, 2 and 4, Panobinostat at 100 μ g/kg increased FoxO1, Prg4 and Acan while suppressing Nos2 at 2.5 mg/kg (Figure 5A). To test whether the drug can suppress OA pathogenic genes, normal C57BL/6J mice (n=8) were injected intraperitoneally with Panobinostat at 2.5 mg/kg and 2 hours later with IL-1β (5 ng/joint) in the right knee and with saline in the left knee. Six hours after IL-1β injection, cartilage and synovium were collected for RNA isolation and PCR analysis. Panobinostat significantly suppressed the IL-1β effect on Mmp3, Mmp13 and Nos2 in cartilage (Figure 5B) and synovium (Figure 5C).
Figure 5. Panobinostat effects on joint tissues in normal mice.
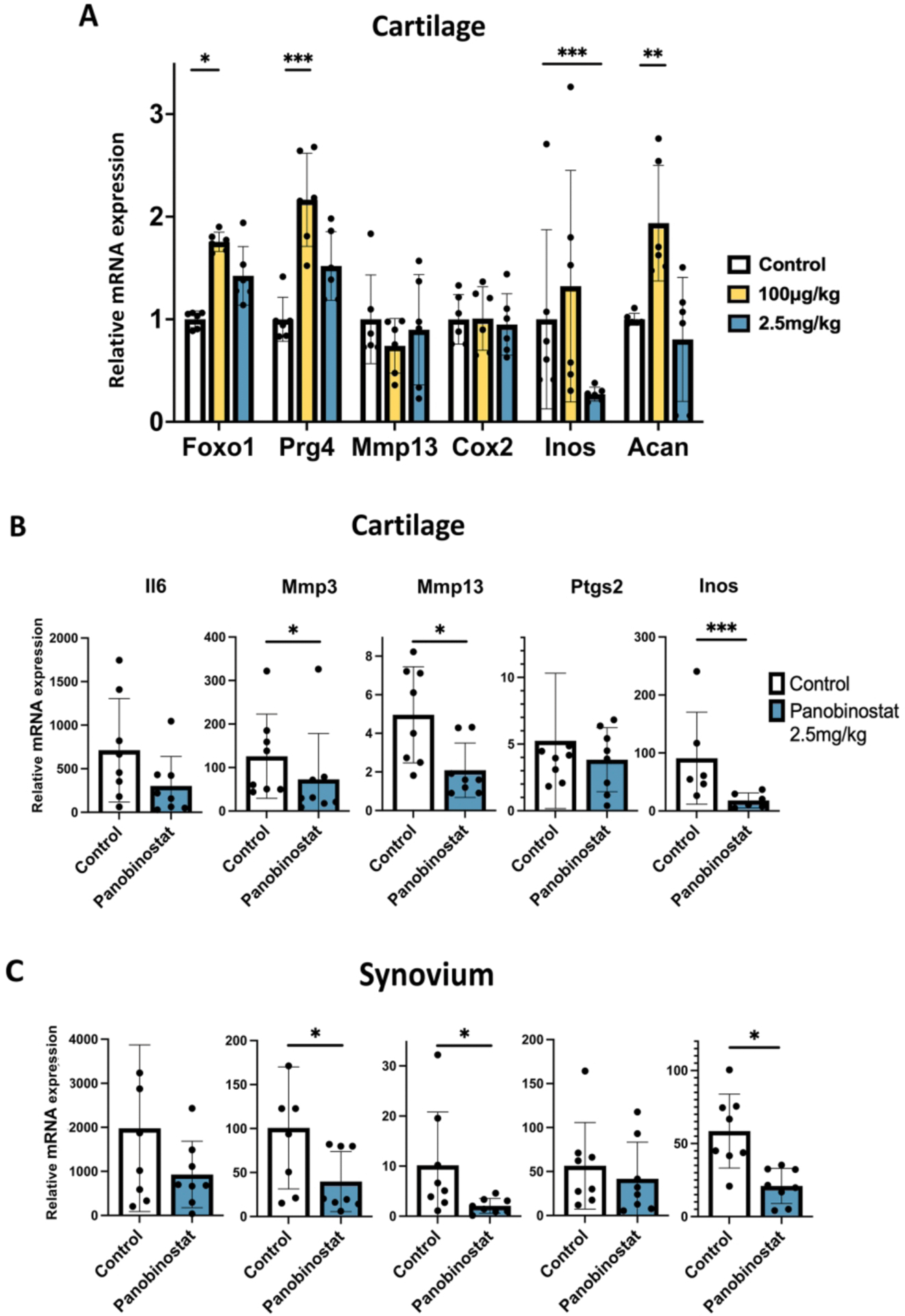
(A). C57BL/6J mice (n=6) received intraperitoneal injections of Panobinostat on Day 0, day 2 and Day 4 and were euthanized 1–3 h after the last injection.
(B). C57BL/6J mice (n=8 each) received one intraperitoneal injection of vehicle and Panobinostat (2.5 mg/kg) and 2 h later IL-1β (50 ng in 10 μl of 5% dextrose) was injected into the right knee and 10 μl of 5% dextrose injected into the left knee. Six h after the IL-1β injection the mice were euthanized. Cartilage and synovium were resected for RNA isolation and RT-qPCR analysis. Data are shown as fold increase in response to IL-1β in Control animals and in Panobinostat treated animals.
***= p<0.001; **= p<0.01; *= p<0.05
Analysis of structural changes and pain behaviors in mice with experimental OA
The DMM model of experimental OA25,26 was used to test whether intraperitoneal injections of Panobinostat (100 μg/kg or 2.5 mg/kg) can improve joint pathology and pain behaviors. First, we measured knee swelling following the early post-surgical period after DMM surgery as an indicator of acute anti-inflammatory effects of Panobinostat. Mice developed detectable swelling at the knee with DMM surgery, and this swelling decreased to basal levels by day 9. The Panobinostat 2.5 mg/kg group (n=14) showed significantly less swelling compared to the control group on days 1, 3 and 5 (Supplementary Figure 4).
Eight weeks following DMM surgery, the OARSI scores for cartilage pathology were significantly and dose-dependently reduced by Panobinostat (Figure 6A). In a separate experiment, mice were treated with Panobinostat (1 mg/kg) for 12 weeks and this also significantly improved OARSI scores (Figure 6B). Scoring of subchondral bone showed that both concentrations of Panobinostat significantly reduced the thickness of the subchondral bone plate and the area of cancellous bone (Figure 6C). Synovitis scores were significantly reduced at 1 mg/kg (Figure 6D).
Figure 6. Analysis of structural changes and symptoms in mice with experimental OA.
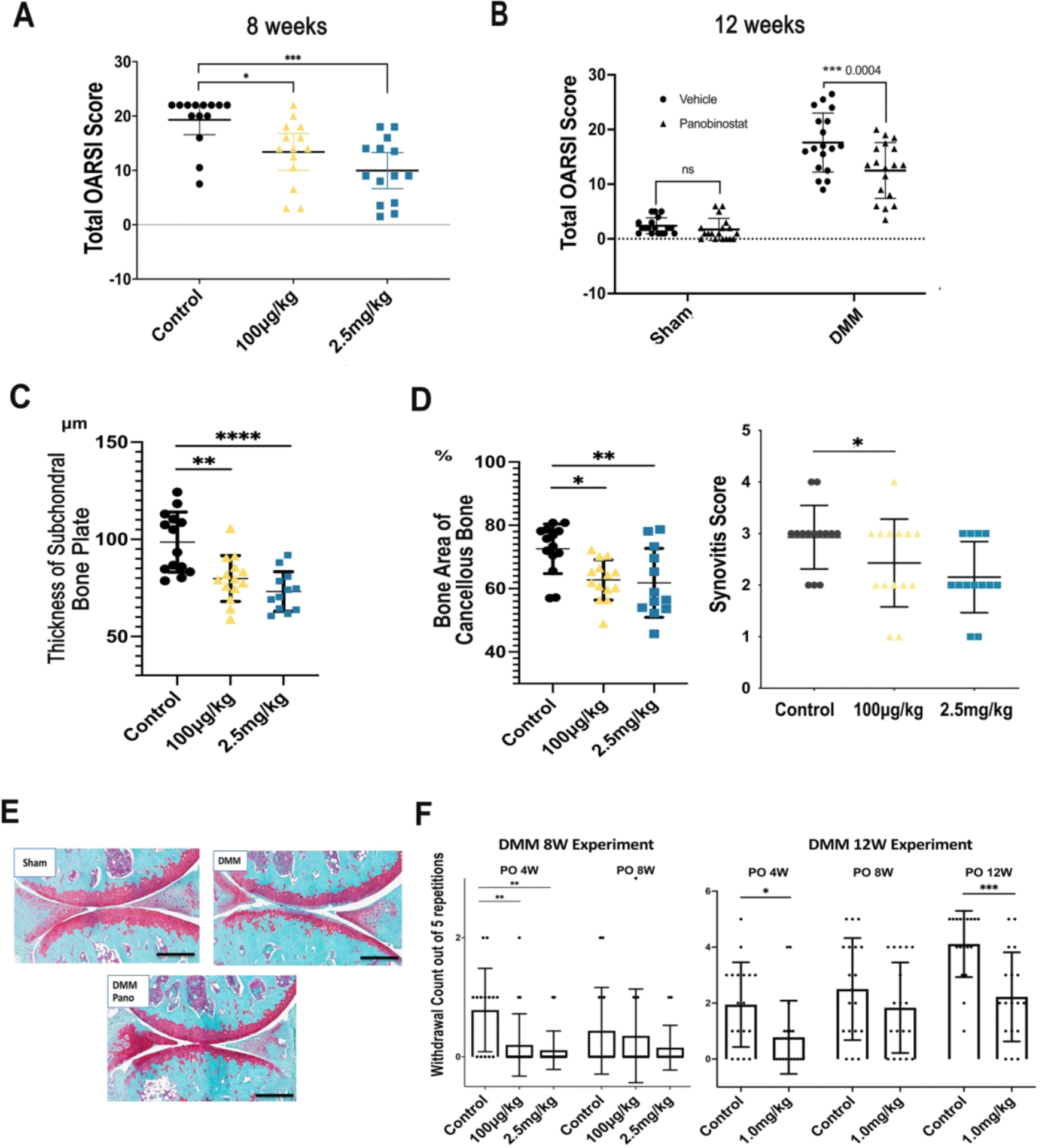
Male C57BL/6J mice (n=70), 16-weeks old, were subjected to DMM surgery on the right knee and sham surgery on the left knee. Fourteen mice were randomly assigned to one of three groups, Control (IP injection of 10 μl/g body weight) of 5% dextrose with 0.72% DMSO, Panobinostat 100 μg/kg, and Panobinostat 2.5 mg/kg. IP injections were given every other day starting from one-week post-surgery. Two separate experiments were performed with treatment duration for 8 or 12 weeks after DMM.
(A). Knee cartilage changes were scored according to the OARSI system following 8-weeks of Panobinostat treatment (100 μg/kg or 2.5 mg/kg).
(B). Knee cartilage changes were scored according to the OARSI system following 12-weeks of Panobinostat treatment (2.5 mg/kg).
(C). The area of subchondral bone plate and subchondral cancellous bone were measured by Image J at 8 weeks after DMM.
(D). Synovitis score was measured at 8 weeks after DMM.
(E). Representative images of Safranin-O stained knee sections from sham surgery control mice, mice with DMM and mice with DMM and Panobinostat (Pano) treatment (2.5 mg/kg).
Scale bars, 200 μm
(F). Von Frey testing for evaluating mechanical allodynia was performed with 1 g filaments at the indicated time points after DMM in the 8-week and 12-week Panobinostat treatment experiments.
****= p<0.0001; ***= p<0.001; **= p<0.01; *= p<0.05
Von Frey testing for evaluating mechanical allodynia30,32 showed a significant improvement at 4 weeks in response to both dosages of Panobinostat when probing was done with the 1g filament in the 8-week experiment (Figure 6E).
In the separate 12-week DMM experiment, improvements in von Frey testing were seen with the 1g filament at 4 and 12 weeks (Figure 6F). Testing with the 2g filament did not show significant differences in the two experiments (Supplementary Figure 5).
There were no apparent systemic adverse reactions to the Panobinostat injections. All animals completed the study without animal deaths or signs of distress.
DISCUSSION
The motivation to discover inducers of FoxO expression was prior findings that FoxO1 expression is reduced in OA-affected human and mouse cartilage15,16. In addition, the FoxO deletion in mice leads to spontaneous OA and more severe surgical OA, and FoxO1 overexpression of FoxO has protective effects in joint cells in vitro and in OA animal models19. Here, we performed drug screening with the ReFRAME library22 using SW1353 human chondrosarcoma clones with a reporter construct containing a region from the human FoxO1 promoter. This collection of known drugs or advanced compounds that have optimized pharmacokinetics, safety, known mechanisms and that have been tested in humans leverages this information for more rapid application in new indications. We found that HDACI represented the largest class of hit compounds. In vitro hit validation was first performed with SW1353 cells and subsequently with normal and OA human chondrocytes. We tested Panobinostat, the most potent HDACI, in human chondrocytes, synoviocytes and meniscus cells and in an OA animal model and performed mechanistic analyses in vitro and in vivo.
The ReFRAME library contains 11,948 compounds and 48 compounds that have HDACI activity22. Among the HDACI, 24 had significant activity in the primary and secondary screens, while the other 24 compounds showed no significant effects (Table S1).
HDACs fall into four classes: class I HDACs (HDACs 1, 2, 3, and 8), class II HDACs (HDACs 4, 5, 6, 7, 9, and 10), class IV (HDAC 11) and class III (sirtuin family). Class II HDACs are further divided into two subgroups: class IIa, which has a large C-terminus, and class IIb, which has two deacetylase domains33.
HDACI are classified based on the presence of a metal chelating group, and further subdivided by the chemical architecture of the cap and linker region. All HDACI, except for sirtuin inhibitors, have a group that can chelate with Zn2+ in HDACs34. There are two main HDACI classes, hydroxamates and non-hydroxamates. Non-hydroxamates include short-chain fatty acids, benzamides, cyclic tetrapeptides and sirtuin inhibitors35,36. Benzamides demonstrate relative isoform selectivity targeting primarily Class I HDACs, with weaker activity against Class II HDACs37,38. The hydroxamic acid group includes the potent non-selective HDAC inhibitors Panobinostat, Givinostat, Trichostatin A (TSA) and Vorinostat/SAHA.
We found that most of the hit HDACI are hydroxamates (Table S2), including the top 4 hits, Panobinostat, Vorinostat/SAHA, Givinostat/ITF2357 and Dacinostat/LAQ824 which were analyzed in more detail in the present study. The main difference between hit and non-hit compounds in the FoxO1 screen is the inhibition of Class IIa HDACs (Table S3) by hydroxamates. Non-hydroxamates exhibit very weak or no activity against the Class IIa HDACs35,36. By contrast, Panobinostat and the other top hits are pan HDACI with potent activity against Class IIa HDACs at low nanomolar concentrations39.
Among the HDACI in the ReFRAME library, Panobinostat had not only the lowest EC50 of all HDACI in the primary screen and also a more favorable in vitro profile compared to the other three top inhibitors (Vorinostat/SAHA, Givinostat/ITF2357 and Dacinostat/LAQ824) from the same class when tested on human chondrocytes where it increased the expression of autophagy and cellular homeostasis genes (sestrin3, LC3), PRG4 and Acan which are known FoxO target genes40, while suppressing genes encoding mediators of inflammation (IL-6, iNOS, PTGES2), and ECM degradation (MMP3, MMP13). The anti-inflammatory activities have been observed for several HDACI, especially for broad spectrum HDACI. HDACI interfere with expression and/or secretion of IL-1β, IL-6, and TNF-β41. Importantly, Panobinostat inhibited the effects of IL-1β, a potent inducer of OA mediators in chondrocytes.
The Panobinostat effects in vitro showed linear dose-responses for the induction of Sesn3 and MAPLC3 and linear dose responses for suppression of inflammatory mediators (IL-6, iNOS, PTGES2) and ECM degrading enzymes (MMP3, MMP13). The ECM genes (PRG4, ACAN, COL2A1) were induced at low Panobinostat concentrations (<40 nM) but suppressed dose-dependently at higher doses. In vivo analyses also showed that Panobinostat had more favorable effects in stimulating PRG4 at 100 μg/kg as compared to 1.5 mg/kg. These findings are consistent with the notion that global HDACI have a tendency of a dual response with divergent effects at low versus high concentrations42. It is assumed that the role of diverse HDACs associated with single pathophysiology are quite different and the overall inhibition effect of HDACI also varies according to concentration.
The HDAC enzymes appear to be the major target of the HDACI43. HDACs have an important role both in transcription regulation and in protein modification. Histone deacetylation leads to chromatin compaction and repression of mRNA synthesis, which are likely to be involved in at least some of the effects observed in chondrocytes. Besides the role in transcription repression, HDACs also function as regulators in posttranslational modification and deacetylate non-histone proteins including both, transcription factors, such as E2F, p53, c-Myc, NF-κB and FoxO, and signal mediators, such as Stat3, Smad7, mitogen-activated protein kinases (MAPK) and β-catenin homeostasis33,44. HDAC4 is known to be a major regulator of chondrocyte hypertrophy and abnormal expression of HDAC4 in OA cartilage suggests its involvement in promoting the catabolc activity of chondrocytes associated with OA pathogenesis45. HDAC4 is also an upstream mediator of MAPK and promotes ADAMTS4, ADAMTS5, and COX2 expression in rat articular chondrocytes and stimulates IL-1β46,47. HDAC7 evokes cartilage damage and ECM degradation through the over-expression of MMP3 and MMP13, which is consistent with the inhibition of HDAC7 in vitro leading to suppression of inflammatory induced MMP13 gene expression48,49.
Prior studies tested HDAC and HDACI in the context of OA50. HDACI suppressed mediators of ECM degradation in human OA chondrocytes45,51,52. The pan HDACI (Trichostatin A) and specific Class I HDACI (Valproic acid and MS275) prevented the expression of MMPs in human OA chondrocytes53,54. TSA and MS275 suppressed RUNX2, ADAMTS5 and MMP3 through the inhibition of p38 MAPK, ERK1/2 and JNK activation in human chondrocytes55. TSA is the only HDACI that was previously tested in OA animal models. Systemic administration of TSA prevented cartilage destruction in a mouse model of surgically induced OA54 and in a rabbit experimental OA model56.
The present results show that Panobinostat induced several FoxO target genes, including PRG4, autophagy genes, and sestrin3 which functions as an antioxidant to reduce oxidative damage in cells57. FoxO and Panobinostat also share the ability to reduce the basal or IL-1β induced expression of inflammatory mediators and ECM degrading enzymes. Thus, Panobinostat replicates some but not all protective effects of FoxO1 in joint tissues which is consistent with the Panobinostat effects beyond HDAC inhibition.
The present study has used DMM which is a model of posttraumatic OA. To determine utility of Panobinostat in primary OA, testing in spontaneous aging-related animal models would be required.
In summary, the present in vitro and in vivo results suggest that Panobinostat has desirable activities in protecting against OA. Concerns have been raised about using systemic administration of broad spectrum HDACI to treat OA, which are related to their adverse effects. Panobinostat has been approved for the treatment of patients with multiple myeloma where it induces hematological adverse events58. However, the present study used much lower concentrations (2.5 mg/kg and 100 μg/kg) than what is typically used in animal model studies of tumors (10 mg/kg). For application in OA, an alternative and feasible route would be the development of sustained release formulations of Panobinostat for intraarticular administration.
Supplementary Material
Funding:
This study was supported by NIH grants AG049617 and AG059418.
Competing interests:
No financial support or other benefits have been obtained from any commercial sources for this study and the authors declare that they have no competing financial interests.
Footnotes
Patient and public involvement: Patient and/or the public were not involved in the design, or conduct, or reporting or dissemination plans of this research.
Patient consent for publication: Obtained.
Ethics approval: All experimental procedures involving mice were conducted with the approval of the Scripps Animal Care and Use Committee. Human joint tissues were collected following informed written patient consent with approval from Scripps IRB Committee.
Data and materials availability:
All data are included in the manuscript and supplemental material.
REFERENCES
- 1.Collins JA, Diekman BO, Loeser RF. Targeting aging for disease modification in osteoarthritis. Curr Opin Rheumatol 2018;30:101–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, Perry TA, Arden N, Chen L, Parsons CM, Cooper C, et al. Occupational Risk in Knee Osteoarthritis: A Systematic Review and Meta-Analysis of Observational Studies. Arthritis Care Res (Hoboken) 2020;72:1213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Courties A, Berenbaum F, Sellam J. The Phenotypic Approach to Osteoarthritis: A Look at Metabolic Syndrome-Associated Osteoarthritis. Joint Bone Spine 2019;86:725–30. [DOI] [PubMed] [Google Scholar]
- 4.Hawker GA. Osteoarthritis is a serious disease. Clin Exp Rheumatol 2019;37 Suppl 120:3–6. [PubMed] [Google Scholar]
- 5.Biver E, Berenbaum F, Valdes AM, Araujo de Carvalho I, Bindels LB, Brandi ML, et al. Gut microbiota and osteoarthritis management: An expert consensus of the European society for clinical and economic aspects of osteoporosis, osteoarthritis and musculoskeletal diseases (ESCEO). Ageing Res Rev 2019;55:100946. [DOI] [PubMed] [Google Scholar]
- 6.Katz JN, Arant KR, Loeser RF. Diagnosis and Treatment of Hip and Knee Osteoarthritis: A Review. JAMA 2021;325:568–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kraus VB, Simon LS, Katz JN, Neogi T, Hunter D, Guermazi A, et al. Proposed study designs for approval based on a surrogate endpoint and a post-marketing confirmatory study under FDA’s accelerated approval regulations for disease modifying osteoarthritis drugs. Osteoarthritis Cartilage 2019;27:571–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emery CA, Whittaker JL, Mahmoudian A, Lohmander LS, Roos EM, Bennell KL, et al. Establishing outcome measures in early knee osteoarthritis. Nat Rev Rheumatol 2019;15:438–48. [DOI] [PubMed] [Google Scholar]
- 9.McClurg O, Tinson R, Troeberg L. Targeting Cartilage Degradation in Osteoarthritis. Pharmaceuticals (Basel) 2021;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carames B, Olmer M, Kiosses WB, Lotz MK. The relationship of autophagy defects to cartilage damage during joint aging in a mouse model. Arthritis Rheumatol 2015;67:1568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meckes JK, Carames B, Olmer M, Kiosses WB, Grogan SP, Lotz MK, et al. Compromised autophagy precedes meniscus degeneration and cartilage damage in mice. Osteoarthritis Cartilage 2017;25:1880–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serrano RL, Chen LY, Lotz MK, Liu-Bryan R, Terkeltaub R. Impaired Proteasomal Function in Human Osteoarthritic Chondrocytes Can Contribute to Decreased Levels of SOX9 and Aggrecan. Arthritis Rheumatol 2018;70:1030–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martins R, Lithgow GJ, Link W. Long live FOXO: unraveling the role of FOXO proteins in aging and longevity. Aging Cell 2016;15:196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci 2014;39:159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akasaki Y, Hasegawa A, Saito M, Asahara H, Iwamoto Y, Lotz MK. Dysregulated FOXO transcription factors in articular cartilage in aging and osteoarthritis. Osteoarthritis Cartilage 2014;22:162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fisch KM, Gamini R, Alvarez-Garcia O, Akagi R, Saito M, Muramatsu Y, et al. Identification of transcription factors responsible for dysregulated networks in human osteoarthritis cartilage by global gene expression analysis. Osteoarthritis Cartilage 2018;26:1531–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.[The present status of schools for public health nursing, midwifery and nursing]. Kango Kyoiku 1968;9:61–3. [PubMed] [Google Scholar]
- 18.Akasaki Y, Alvarez-Garcia O, Saito M, Carames B, Iwamoto Y, Lotz MK. FoxO transcription factors support oxidative stress resistance in human chondrocytes. Arthritis Rheumatol 2014;66:3349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuzaki T, Alvarez-Garcia O, Mokuda S, Nagira K, Olmer M, Gamini R, et al. FoxO transcription factors modulate autophagy and proteoglycan 4 in cartilage homeostasis and osteoarthritis. Sci Transl Med 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Repa JJ, Turley SD, Lobaccaro JA, Medina J, Li L, Lustig K, et al. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science 2000;289:1524–9. [DOI] [PubMed] [Google Scholar]
- 21.Deng X, Zhang W, I OS, Williams JB, Dong Q, Park EA, et al. FoxO1 inhibits sterol regulatory element-binding protein-1c (SREBP-1c) gene expression via transcription factors Sp1 and SREBP-1c. J Biol Chem 2012;287:20132–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janes J, Young ME, Chen E, Rogers NH, Burgstaller-Muehlbacher S, Hughes LD, et al. The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis. Proc Natl Acad Sci U S A 2018;115:10750–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kjaer SK, de Villiers EM, Haugaard BJ, Christensen RB, Teisen C, Moller KA, et al. Human papillomavirus, herpes simplex virus and cervical cancer incidence in Greenland and Denmark. A population-based cross-sectional study. Int J Cancer 1988;41:518–24. [DOI] [PubMed] [Google Scholar]
- 24.Alvarez-Garcia O, Matsuzaki T, Olmer M, Plate L, Kelly JW, Lotz MK. Regulated in Development and DNA Damage Response 1 Deficiency Impairs Autophagy and Mitochondrial Biogenesis in Articular Cartilage and Increases the Severity of Experimental Osteoarthritis. Arthritis Rheumatol 2017;69:1418–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carames B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis 2012;71:575–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage 2007;15:1061–9. [DOI] [PubMed] [Google Scholar]
- 27.Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 2010;18 Suppl 3:S17–23. [DOI] [PubMed] [Google Scholar]
- 28.Krenn V, Morawietz L, Haupl T, Neidel J, Petersen I, Konig A. Grading of chronic synovitis--a histopathological grading system for molecular and diagnostic pathology. Pathol Res Pract 2002;198:317–25. [DOI] [PubMed] [Google Scholar]
- 29.Nagira K, Ikuta Y, Shinohara M, Sanada Y, Omoto T, Kanaya H, et al. Histological scoring system for subchondral bone changes in murine models of joint aging and osteoarthritis. Sci Rep 2020;10:10077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994;53:55–63. [DOI] [PubMed] [Google Scholar]
- 31.Meulenbelt I, Min JL, Bos S, Riyazi N, Houwing-Duistermaat JJ, van der Wijk HJ, et al. Identification of DIO2 as a new susceptibility locus for symptomatic osteoarthritis. Hum Mol Genet 2008;17:1867–75. [DOI] [PubMed] [Google Scholar]
- 32.Miller RE, Belmadani A, Ishihara S, Tran PB, Ren D, Miller RJ, et al. Damage-associated molecular patterns generated in osteoarthritis directly excite murine nociceptive neurons through Toll-like receptor 4. Arthritis Rheumatol 2015;67:2933–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoon S, Eom GH. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med J 2016;52:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bieliauskas AV, Pflum MK. Isoform-selective histone deacetylase inhibitors. Chem Soc Rev 2008;37:1402–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ediriweera MK, Tennekoon KH, Samarakoon SR. Emerging role of histone deacetylase inhibitors as anti-breast-cancer agents. Drug Discov Today 2019;24:685–702. [DOI] [PubMed] [Google Scholar]
- 36.Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int J Mol Sci 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moradei OM, Mallais TC, Frechette S, Paquin I, Tessier PE, Leit SM, et al. Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J Med Chem 2007;50:5543–6. [DOI] [PubMed] [Google Scholar]
- 38.Garcia Olmos L [Morbidity in primary health care: patients versus visits]. Gac Sanit 1991;5:34–8. [DOI] [PubMed] [Google Scholar]
- 39.Neckers LM, Nordan RP. Regulation of murine plasmacytoma transferrin receptor expression and G1 traversal by plasmacytoma cell growth factor. J Cell Physiol 1988;135:495–501. [DOI] [PubMed] [Google Scholar]
- 40.Duffy T, Bekki H, Lotz MK. Genome-Wide Occupancy Profiling Reveals Critical Rol of FoxO1 in Regulating Extracellular Matrix and Circadian Rhythm Genes in Human Chondrocytes. Arthritis Rheumatol 2020;72:1514–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoon S, Kang G, Eom GH. HDAC Inhibitors: Therapeutic Potential in Fibrosis-Associated Human Diseases. Int J Mol Sci 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Halili MA, Andrews MR, Labzin LI, Schroder K, Matthias G, Cao C, et al. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J Leukoc Biol 2010;87:1103–14. [DOI] [PubMed] [Google Scholar]
- 43.Lakshmaiah KC, Jacob LA, Aparna S, Lokanatha D, Saldanha SC. Epigenetic therapy cancer with histone deacetylase inhibitors. J Cancer Res Ther 2014;10:469–78. [DOI] [PubMed] [Google Scholar]
- 44.Zhang H, Ji L, Yang Y, Zhang X, Gang Y, Bai L. The Role of HDACs and HDACi in Cartilage and Osteoarthritis. Front Cell Dev Biol 2020;8:560117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu J, Sun Y, Ge Q, Teng H, Jiang Q. Histone deacetylase 4 alters cartilage homeostasis in human osteoarthritis. BMC Musculoskelet Disord 2014;15:438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song J, Jin EH, Kim D, Kim KY, Chun CH, Jin EJ. MicroRNA-222 regulates MMP-13 via targeting HDAC-4 during osteoarthritis pathogenesis. BBA Clin 2015;3:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang P, Mao Z, Pan Q, Lu R, Huang X, Shang X, et al. Histone deacetylase-4 and histone deacetylase-8 regulate interleukin-1beta-induced cartilage catabolic degradation through MAPK/JNK and ERK pathways. Int J Mol Med 2018;41:2117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang C, Zhang Z, Chang Z, Mao G, Hu S, Zeng A, et al. miR-193b-5p regulates chondrocytes metabolism by directly targeting histone deacetylase 7 in interleukin-1beta-induced osteoarthritis. J Cell Biochem 2019;120:12775–84. [DOI] [PubMed] [Google Scholar]
- 49.Higashiyama R, Miyaki S, Yamashita S, Yoshitaka T, Lindman G, Ito Y, et al. Correlation between MMP-13 and HDAC7 expression in human knee osteoarthritis. Mod Rheumatol 2010;20:11–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khan NM, Haqqi TM. Epigenetics in osteoarthritis: Potential of HDAC inhibitors as therapeutics. Pharmacol Res 2018;128:73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Im GI, Choi YJ. Epigenetics in osteoarthritis and its implication for future therapeutics. Expert Opin Biol Ther 2013;13:713–21. [DOI] [PubMed] [Google Scholar]
- 52.Carpio LR, Westendorf JJ. Histone Deacetylases in Cartilage Homeostasis and Osteoarthritis. Curr Rheumatol Rep 2016;18:52. [DOI] [PubMed] [Google Scholar]
- 53.Wang X, Song Y, Jacobi JL, Tuan RS. Inhibition of histone deacetylases antagonized FGF2 and IL-1beta effects on MMP expression in human articular chondrocytes. Growth Factors 2009;27:40–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Culley KL, Hui W, Barter MJ, Davidson RK, Swingler TE, Destrument AP, et al. Class I histone deacetylase inhibition modulates metalloproteinase expression and blocks cytokine-induced cartilage degradation. Arthritis Rheum 2013;65:1822–30. [DOI] [PubMed] [Google Scholar]
- 55.Saito T, Nishida K, Furumatsu T, Yoshida A, Ozawa M, Ozaki T. Histone deacetylase inhibitors suppress mechanical stress-induced expression of RUNX-2 and ADAMTS-5 through the inhibition of the MAPK signaling pathway in cultured human chondrocytes. Osteoarthritis Cartilage 2013;21:165–74. [DOI] [PubMed] [Google Scholar]
- 56.Chen WP, Bao JP, Tang JL, Hu PF, Wu LD. Trichostatin A inhibits expression of cathepsins in experimental osteoarthritis. Rheumatol Int 2011;31:1325–31. [DOI] [PubMed] [Google Scholar]
- 57.Ho A, Cho CS, Namkoong S, Cho US, Lee JH. Biochemical Basis of Sestrin Physiological Activities. Trends Biochem Sci 2016;41:621–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laubach JP, Schjesvold F, Mariz M, Dimopoulos MA, Lech-Maranda E, Spicka I, et al. Efficacy and safety of oral panobinostat plus subcutaneous bortezomib and oral dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma (PANORAMA 3): an open-label, randomised, phase 2 study. Lancet Oncol 2021;22:142–54 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are included in the manuscript and supplemental material.