

Abstract
Regulatory T cells exert inhibitory function under various physiological conditions and adopt diverse characteristics following environmental cues. Multiple subsets of regulatory T cells expressing master transcription factors of helper T cells such as RORγt, T-bet, Gata3 and PPARγ have been characterized, but the molecular mechanism governing the differentiation of these subsets remains largely unknown. Here we report that the atypical IκB protein family member Bcl-3 suppresses RORγt+ Treg accumulation. The suppressive effect of Bcl-3 was particularly evident in the mouse immune tolerance model of anti-CD3 therapy. Using conditional knockout mice, we illustrate that loss of Bcl-3 specifically in Tregs was sufficient to boost RORγt+ Treg formation and resistance of mice to DSS-induced colitis. We further demonstrate the suppressive effect of Bcl-3 on RORγt+ Treg differentiation in vitro. Our results reveal a novel role of NF-κB signaling pathways in regulatory T cell subset differentiation that may have clinical implications in immunotherapy.
Keywords: NF-κB, Treg, colitis, tolerance
INTRODUCTION
Bcl-3 is an atypical member of the IκB family of proteins and regulates NF-κB activity in a context-dependent manner.1–6 NF-κB transcription factors are homo- or heterodimers composed of members of the Rel-related protein family. These factors are activated by a vast array of stimuli, in particular stress- and pathogen-related signals as well as cytokines. Classic IκB family members such as the prototypical IκBα inhibit transactivation by NF-κB complexes primarily by retaining them in the cytoplasm and preventing them from binding to DNA. In contrast, Bcl-3 can constitutively enter the nucleus where it regulates NF-κB target gene expression by associating mainly with NF-κB1 p50 or NF-κB2 p52 homodimers to either promote or inhibit NF-κB activity depending on the physiological context. Although the molecular mechanisms underlying this process remain poorly understood, it is abundantly clear from studies of Bcl3 gene-targeted mice that Bcl-3 plays critical roles in vivo in regulating diverse aspects of both innate and adaptive immune responses.7–15
With regard to adaptive immunity, NF-κB plays an essential role in regulatory T cell development and function. It has been demonstrated that enhancing NF-κB activity by introducing a constitutively active IKKβ transgene in T cells leads to increased numbers of Foxp3+ cells by a mechanism involving direct enhancement of Foxp3 transcription by NF-κB.16 Early studies using total KO mice showed that c-Rel deficiency led to a reduced number of Tregs whereas mice lacking NF-κB1 (p50) were largely normal.17 With the recent generation of conditional knockout mice, it has been demonstrated that loss of RelA specifically in Foxp3+ cells induced severe inflammation whereas c-Rel seems dispensable in mature Tregs for controlling autoimmunity.18 However, c-Rel deficient Tregs exhibited reduced inhibitory effects in anti-tumor responses,19 which may provide an important target for cancer therapy. Interestingly, p100/NF-κB2 conditional KO mice showed expansion of Tregs with impaired suppressive function and displayed autoimmunity around 12 months of age, whereas mice lacking RelB in Tregs did not exhibit significant changes.20
“Master transcription factors” of Th effector cells, such as T-bet, GATA3 and RORγt, were also found to be expressed in different Treg subsets. These Treg subsets may utilize these transcription factors to mimic the behavior of the T helper cell subset they inhibit in a given tissue location.21 RORγt+ Tregs co-expressing Foxp3 are mostly found in the intestine where they play a very important inhibitory role for immune responses under various conditions.22, 23 Removal of RORγt+ Tregs by specific depletion of RORγt in Foxp3-expressing cells exacerbated disease progression in different mouse models. Generation of RORγt+ Tregs is microbiota-dependent.22, 23 In intestine, RORγt+ Tregs can be induced by oral antigen in Rag mice receiving OT-II cells or in Treg-depleted mice, indicating a peripheral origin.22, 24 By contrast, RORγt+ Tregs in draining LNs of experimental autoimmune encephalomyelitis (EAE) mice are derived primarily from thymic Tregs.25
The molecular mechanisms governing the differentiation of subsets of regulatory T cells, including the possible role of NF-κB, remain unknown. Here we report that Bcl-3 suppresses RORγt+ Treg differentiation in vivo both in the mouse immune tolerance model of anti-CD3 therapy and in the mouse model of DSS-induced colitis.
RESULTS
Increased RORγt+ regulatory T cell formation in Bcl-3 deficient mice
To study the potential role of Bcl-3 in regulatory T cells, we first analyzed cells from spleens and mesenteric lymph nodes (MLN) from WT and Bcl-3-deficient mice under homeostatic conditions. It has been reported that the overall development of T and B cells is largely normal in Bcl-3-deficient mice, with some abnormality in germinal center formation upon challenge.7 Consistent with this, we did not observe a significant difference between WT and Bcl-3-deficient animals in the percentage of Foxp3+ regulatory T cells among CD4+ cells in either spleen or MLN. There was an increase in the absolute number of Tregs in KO spleen and a decrease in the number of Tregs in KO MLN due the slightly larger spleen and smaller MLN in Bcl-3 deficient animals. Furthermore, the percentage of Foxp3+ regulatory T cells was slightly higher in the lamina propria (LP) of the colons of KO mice, but we did not detect a significant difference in Treg number possibly due to the high variance in total CD4+ T cells recovered from the colon (Supplementary figure 1).
We also tested the effect of complete Bcl-3 deficiency on the distribution of the RORγt+ subset of Foxp3+ Tregs under homeostatic conditions. Consistent with previous reports, there were few RORγt+ Tregs in the spleens and MLNs of wild type mice. However, we observed an increase of the RORγt+ regulatory T cell population in both the MLNs and lamina propria of the colon, but not in the spleens, of Bcl-3 deficient mice compared to WT mice (Figure 1), suggesting that Bcl-3 normally suppresses the accumulation of this particular Treg subset selectively in these tissues.
Figure 1. RORγt+ regulatory T cells are increased in mesenteric lymph node and lamina propria of colon of Bcl-3-deficient mice under homeostatic conditions.
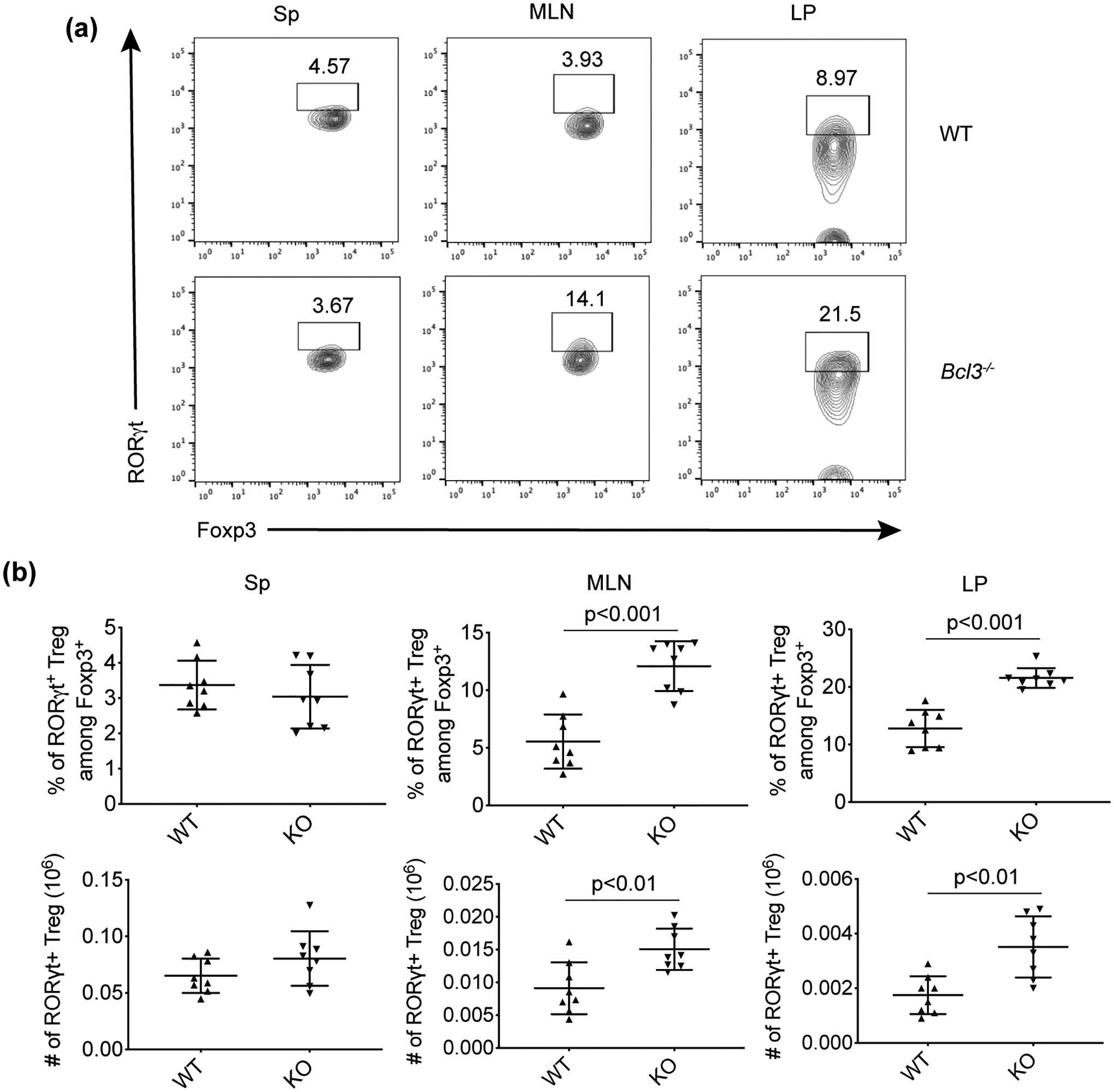
(a) Representative flow cytometric analyses of CD4+Foxp3+ cells from spleen (Sp), mesenteric lymph node (MLN) and lamina propria (LP) of colon of naïve wild-type (WT) and total Bcl-3-deficient (KO) mice littermates. (b) Summary of flow cytometric analyses from (a) (n = 8 from three experiments). Data represent means ± SD.
α-CD3 treatment exacerbated the suppressive effect of Bcl-3 on RORγt+ regulatory T cell formation
We next extended our study of Bcl-3 regulation of Treg development to the mouse model of anti-CD3-induced immune tolerance. Previous studies have shown that a single dose of α-CD3 leads to a selective increase of regulatory T cells and can successfully induce immune tolerance.26 However, administration of this antibody also causes severe side effects; it initially induces polyclonal T cell activation, which at first results in a severe diarrheal syndrome with intestinal damage. When we injected both wild type and Bcl-3 knockout mice with α-CD3, then sacrificed the animals four days later, we observed that the percentage of Foxp3+ regulatory T cells in MLN of Bcl-3 deficient mice was increased compared with wild type control animals (Figure 2). The levels after anti-CD3 treatment of both WT and KO mice were elevated compared to homeostatic levels (Figure 2, Supplementary figure 1). We also observed higher frequencies of RORγt+ Tregs in all α-CD3-treated mice compared to levels observed under homeostatic conditions. The frequency of RORγt+ Tregs in MLN after anti-CD3 treatment was greater in Bcl-3 KO mice than in WT mice (Figure 2). To explore whether the increase of RORγt+ Tregs in Bcl-3 KO MLN is TNF-dependent, we treated TNF KO and Bcl-3/TNF double KO mice with α-CD3. FACS analysis showed that loss of Bcl-3 still led to a dramatic increase in RORγt+ regulatory T cells even when TNF was absent, indicating that the inhibitory role of Bcl-3 on RORγt+ Treg accumulation was TNF-independent (Supplementary figure 2).
Figure 2. Loss of Bcl-3 enhanced the percentage of RORγt+ regulatory T cells upon α-CD3 treatment.
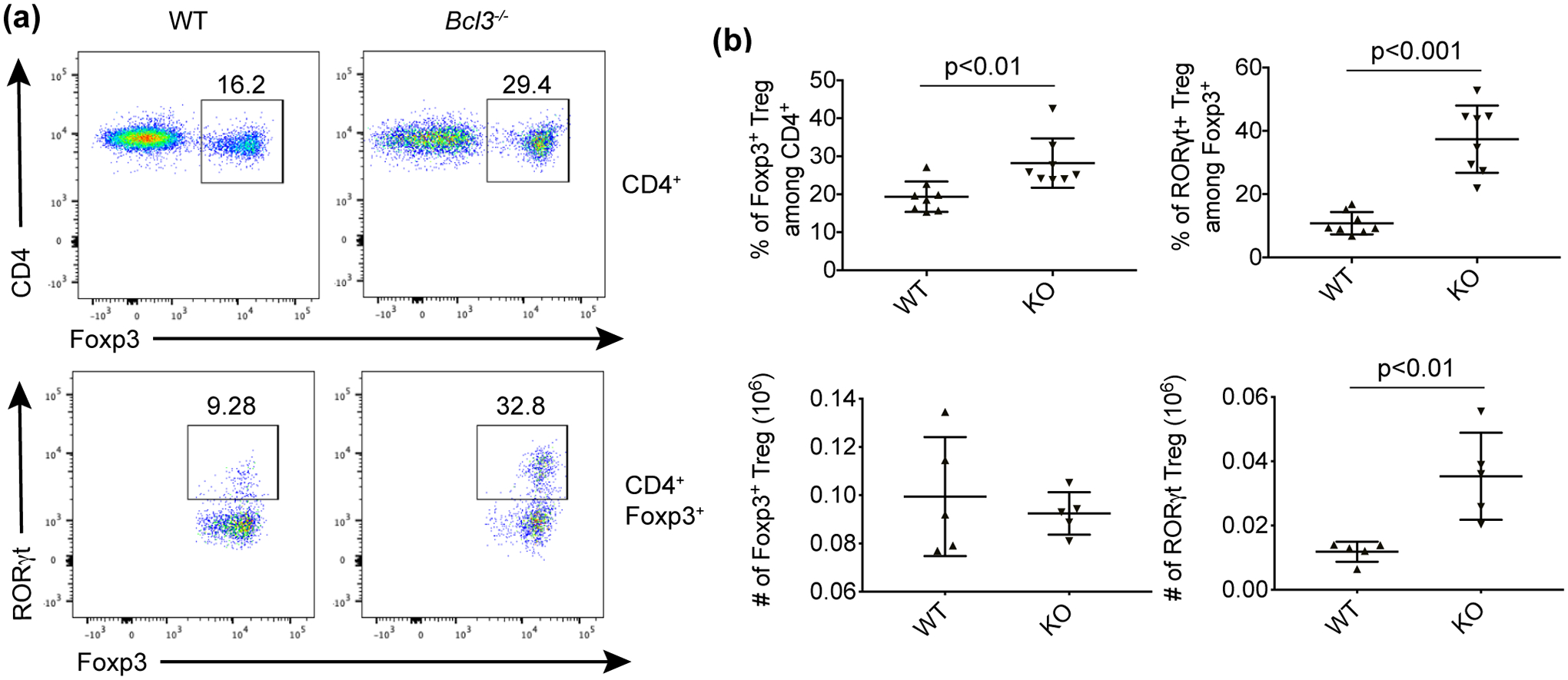
Wild-type (WT) and total Bcl-3-deficient (KO) mice littermates were injected i.p. with CD3-specific antibody and sacrificed on day 4. (a) Representative flow cytometric analyses of cells from mesenteric lymph node. (b) Summary of flow cytometric analyses as shown in (a) (n ≥ 5 from four experiments). Data represent means ± SD.
Loss of Bcl-3 specifically in Foxp3+ cells enhances RORγt+ regulatory T cell formation
We hypothesized that the effect of Bcl-3 on regulatory T cells by α-CD3 treatment is T cell intrinsic. To test this, we treated Bcl3Flox/Flox (Bcl3F/F) CD4-cre mice and littermate controls with α-CD3 for 4 days. We observed a dramatic increase of RORγt+ Tregs when Bcl-3 was specifically deleted in T cells under both homeostatic conditions (Supplementary figure 3) and after α-CD3 treatment (Figure 3). On the other hand, there was no significant difference in the percentage of total Foxp3+ Tregs in the mice. These results indicated that the effect of Bcl-3 on RORγt+ Treg accumulation in response to α-CD3 treatment is T cell intrinsic.
Figure 3. Loss of Bcl-3 specifically in CD4+ T cells led to accumulation of RORγt+ regulatory T cells upon α-CD3 treatment.
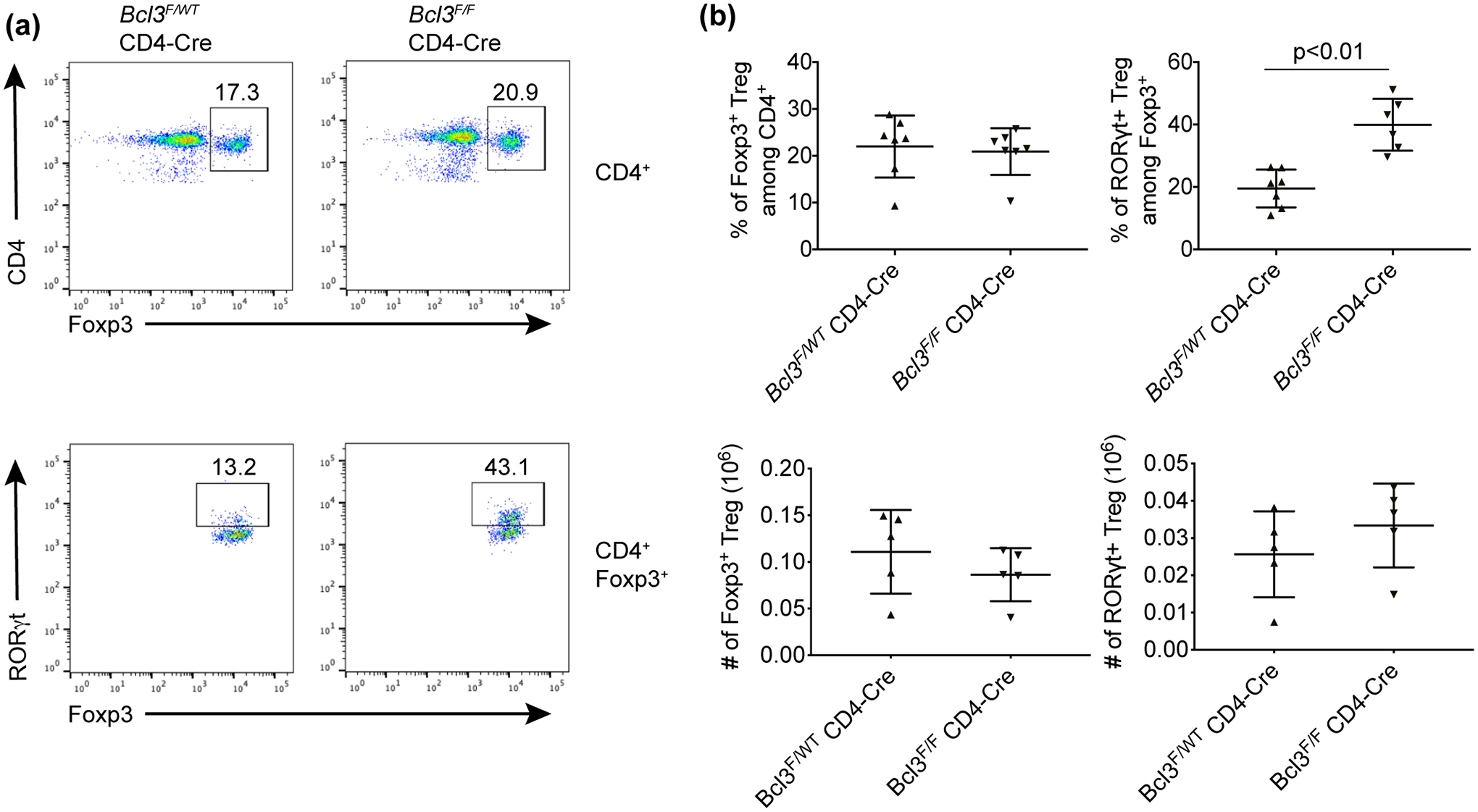
Bcl-3F/F CD4-Cre conditional KO and control Bcl-3WT/F CD4-Cre mice were injected i.p. with CD3-specific antibody and sacrificed on day 4. (a) Representative flow cytometric analyses of cells from mesenteric lymph node. (b) Summary of flow cytometric analyses as shown in (a) (n ≥ 5 from three experiments). Data represent means ± SD.
Next we investigated whether loss of Bcl-3 specifically in Foxp3+ Tregs is sufficient for the observed increase of RORγt+/Foxp3+ cells. Consistent with this, loss of Bcl-3 specifically in Foxp3+ cells also led to a significant increase of RORγt+ Tregs under homeostatic conditions (Supplementary figure 4). We then treated Bcl3F/F Foxp3-cre mice and littermate controls with α-CD3 for 4 days and analyzed cells from the MNL of these mice by FACS. The results were comparable to what was seen in mice lacking Bcl-3 in all T cells after deletion of the gene using CD4-cre. There was no significant difference in the percentage of total Foxp3+ cells among CD4+ T cells, but the percentage of the RORγt+/Foxp3+ subset among Foxp3+ Tregs was greatly increased (Figure 4).
Figure 4. Loss of Bcl-3 specifically in Foxp3+ regulatory T cells led to accumulation of RORγt+ regulatory T cells upon α-CD3 treatment.
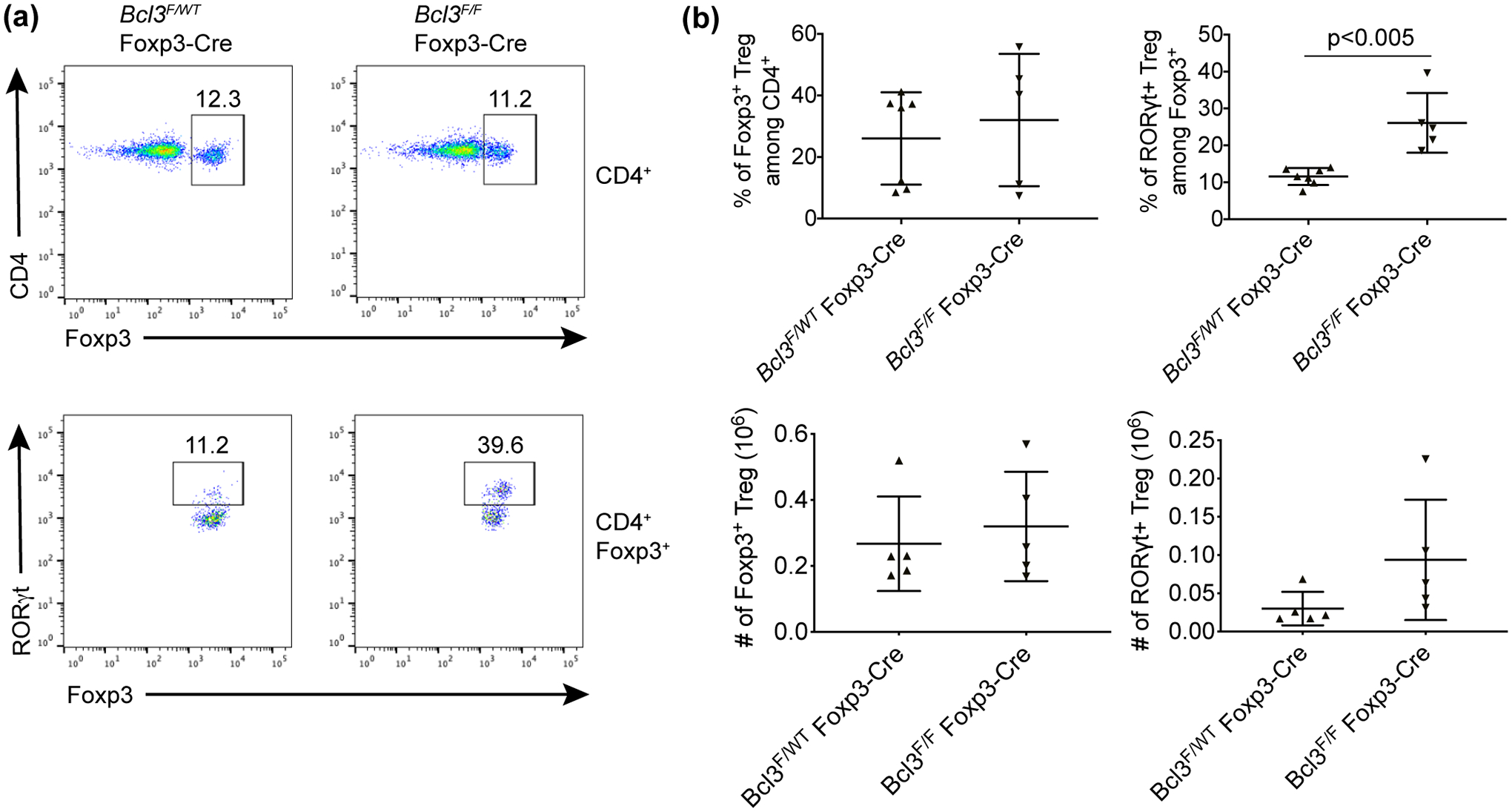
Bcl-3F/F Foxp3-Cre conditional KO mice and control Bcl-3WT/F Foxp3-Cre littermates were injected i.p. with CD3-specific antibody and sacrificed on day 4. (a) Representative flow cytometric analyses of cells from mesenteric lymph node. (b) Summary of flow cytometric analyses as shown in (a) (n ≥ 5 from two experiments). Data represent means ± SD.
We then studied the effect of loss of Bcl-3 in only Foxp3-expressing cells in the DSS-induced colitis model. Both Bcl3F/F Foxp3-cre mice and littermate controls were given 3% DSS in drinking water for 5 days, and body weights of these mice were monitored daily. Our results showed that conditional knockout of Bcl-3 in regulatory T cells resulted in notably ameliorated loss of body weight, increased colon length and lower histology score compared to control littermates, consistent with enhanced Treg activity when Bcl-3 was specifically deleted in these cells (Figure 5a and Supplementary figure 6a, b). We further checked Tregs in the lamina propria (LP) of the DSS-treated mice and found that the percentage of RORγt+/Foxp3+ cells was also increased in the LP of conditional knockout animals (Figure 5b, c). In addition, we measured the expression level of surface markers GITR, CTLA4 and Nrp1 on Tregs and did not observe a significant difference between WT and Bcl-3 conditional KO mice under either homeostatic conditions (Supplementary figure 5) or after DSS treatment (Supplementary figure 6c, d). The Nrp1 staining of MLN Tregs indicated that the majority of expanded KO RORγt+/Foxp3+ cells were pTregs; the results for lamina propria Tregs were equivocal.
Figure 5. Bcl-3 Foxp3-Cre conditional KO mice are more resistant to DSS treatment.
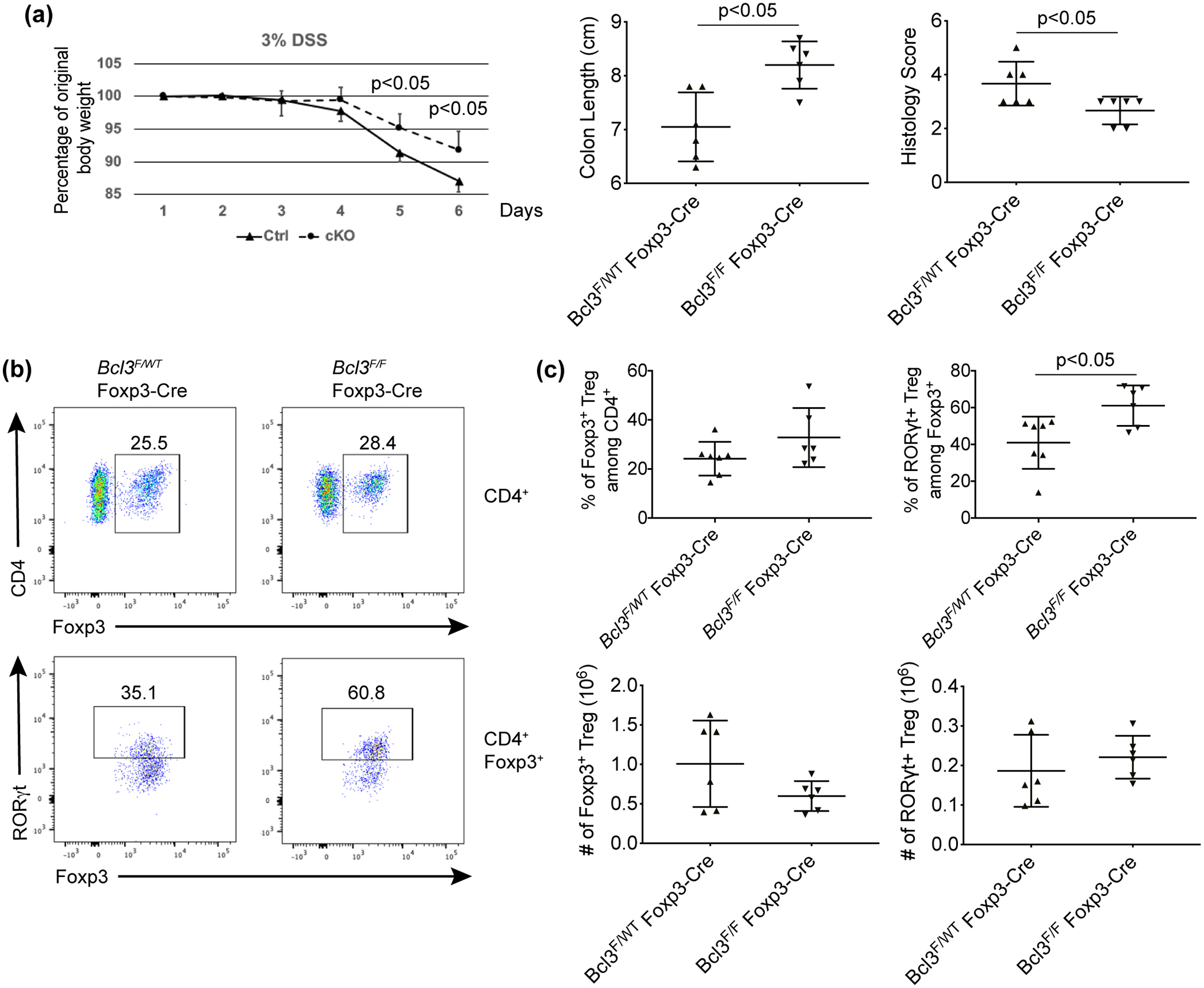
(a) Bcl-3F/F Foxp3-Cre conditional KO mice (cKO) and control (Ctrl) Bcl-3WT/F Foxp3-Cre littermates were treated with 3% DSS for 5 days and weighed daily. After sacrifice, colon lengths and histology scores were assessed. (b) Representative flow cytometric analyses of cells from lamina propria of colons of mice in (a). (c) Summary of flow cytometric analyses from (b) (n ≥ 6 in each of two experiments). Data represent means ± SD.
Bcl-3 suppresses RORγt+ regulatory T cell differentiation in vitro
Since loss of Bcl-3 specifically in regulatory T cells led to an increased percentage of RORγt+/Foxp3+ cells, it is reasonable to hypothesize that Bcl-3 may directly inhibit RORγt+ regulatory T cell differentiation. We first performed in vitro iTreg differentiation but failed to detect a significant difference in RORγt+ expression between WT and KO differentiated Tregs (Supplementary figure 7a). We then FACS sorted regulatory T cells from spleens of WT and Bcl-3 knockout mice. Unlike in MLN, neither WT nor Bcl-3 knockout spleen has significant amounts of RORγt+/Foxp3+ cells, and there is no difference between the two genotypes (see above). As expected Tregs isolated from spleens of WT and KO mice did not show a difference in their inhibitory activity in vitro since they do not express RORγt (Supplementary figure 7b). Tregs isolated from spleen were then subjected to culture in vitro with cytokines able to drive Th17 cell differentiation, including IL-6, TGFβ, IL-1, IL-21 and IL-23, attempting to mimic an intestinal inflammatory environment. About 40% of wild type regulatory T cells acquired RORγt expression after 3 days of culture in vitro. In comparison, the percentage of RORγt+ cells lacking Bcl-3 was about 60% in these cultures (Figure 6a, b). Our results demonstrate that Bcl-3 appears to inhibit the differentiation of Foxp3+ regulatory T cells into RORγt+/Foxp3+ cells.
Figure 6. Bcl-3 suppresses RORγt+ regulatory T cell differentiation in vitro.
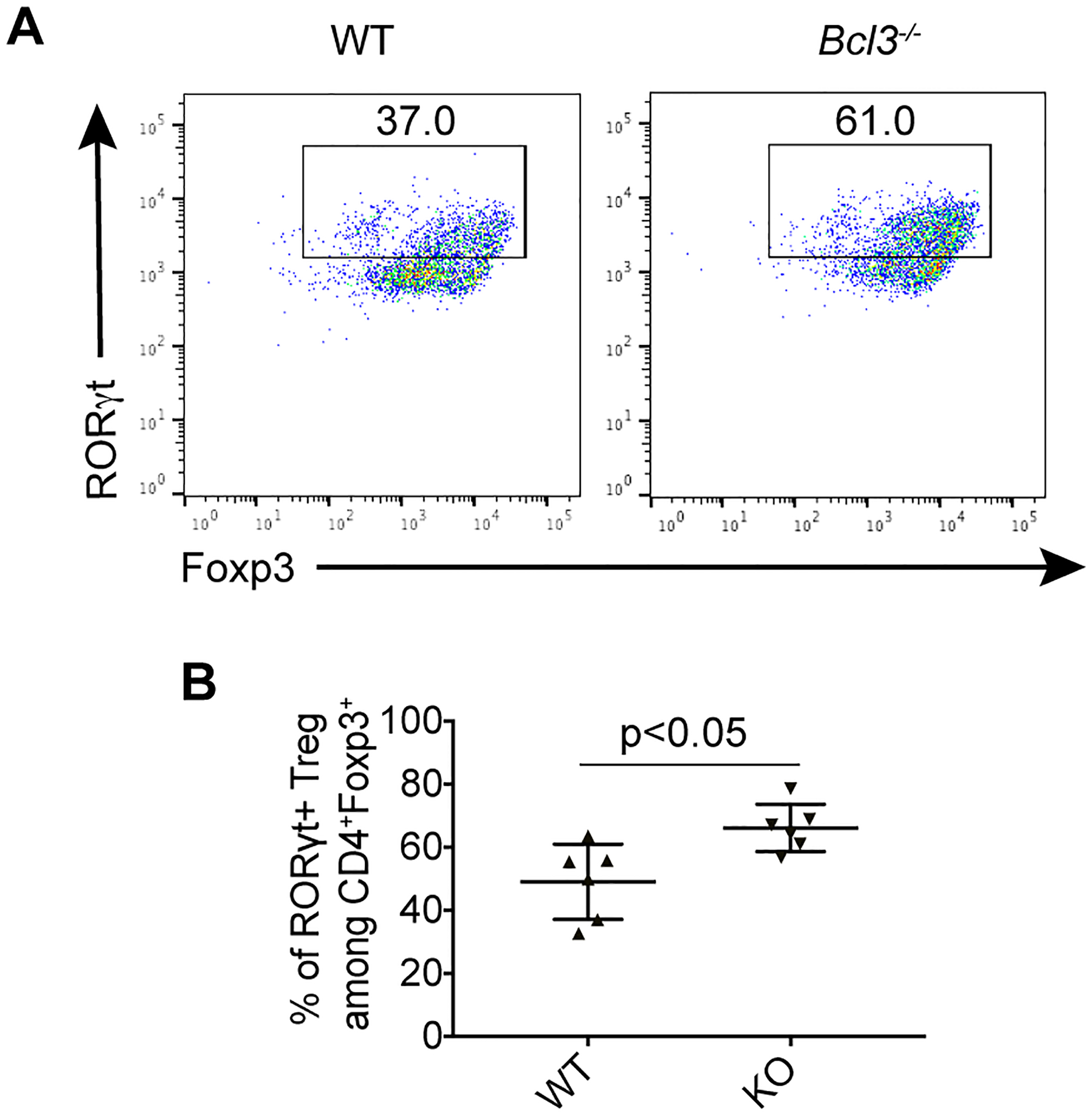
(a) Representative flow cytometric analyses of Tregs sorted from wild-type (WT) and total Bcl-3-deficient (KO) littermates and cultured under Th17 conditions for 3 days. (b) Summary of percentage of RORγt + cells from (a) (n = 6 from three experiments). Data represent means ± SD.
DISCUSSION
Although NF-κB signaling is essential for the development of regulatory T cells under both homeostatic and inflammatory conditions, its role in the development and function of subsets of regulatory T cells has not been previously fully addressed. Previous research demonstrated that conditional deletion of RelA in Tregs led to decreased RORγt+ Treg subset while NF-κB2 knockout Tregs showed increased Il-17a and Rorc expression.18, 20, 27 Here we report that the atypical IκB protein family member Bcl-3 specifically suppresses RORγt+ Treg accumulation in vivo and in vitro. The majority of expanded Bcl-3 KO RORγt+/Foxp3+ cells in MLN were Nrp1− pTregs, however the results for lamina propria Tregs were not as clear. The suppressive effect of Bcl-3 was particularly evident in vivo in the mouse immune tolerance model of anti-CD3 therapy. Loss of Bcl-3 specifically in Tregs was sufficient for enhanced RORγt+ Treg accumulation and increased resistance of mice to DSS-induced colitis. Our in vitro analysis further supports the notion that Bcl-3 directly suppressed RORγt+ Treg accumulation at the level of cell differentiation. We thus demonstrate a novel role of Bcl-3 and NF-κB signaling in regulatory T cell subset differentiation, which may have clinical implications for Treg cell immunotherapy in the gut.
CD3-specific antibodies induce immune tolerance and are used in the clinic to enhance the efficacy of transplantation and to treat autoimmune diseases. Previous reports have shown that both Treg and Tr1 cells play very important roles in α-CD3-induced immune tolerance.28 CD3-specific antibodies deplete pathogenic T cells, and apoptotic T cells engulfed by phagocytes can further promote Treg differentiation.26 Systemic administration of α-CD3 antibodies initially induces polyclonal T cell activation, which results in a severe diarrheal syndrome with intestinal damage, peaking around 2 days after injection. Thereafter IL-10-producing Tregs and Tr1 cells are critical for recovery. We show here that Bcl-3 expression inhibited α-CD3-induced Treg accumulation, especially RORγt+ Tregs, indicating an inhibitory role of Bcl-3 in CD3 antibody-specific-induced immune tolerance. It would be of interest to study the role of Bcl-3 as well as RORγt+ Tregs in patients undergoing such immunotherapy.
We have previously reported that Bcl-3 is critical for CD4 T cell pathogenicity and plasticity.14 Bcl-3 deficient Th1 cells can readily convert to RORγt+ Th17-like cells especially in Rag1−/− recipients in the T cell transfer-induced colitis model. Bcl-3 appeared to govern Th1 cell plasticity through direct regulation of RORγt expression. Consistent with our previous results, we show here that Bcl-3 also inhibits RORγt expression specifically in regulatory T cells. However, it remains unclear how Bcl-3 silences RORγt at the molecular level. There is increasing evidence indicating that NF-κB activity promotes RORγt expression in both Tconv and Treg cells, which implies that Bcl-3 might restrain RORγt expression by modulating NF-κB activity in Tregs.20, 27, 29 Our previous study showed that Bcl-3 associated with regions in the Rorc regulatory domain in Th1 cells, which contain NF-κB binding sites critical for c-Rel- and RelA-induced expression of RORγt during Th17 differentiation. Further investigation is needed to look into whether the same mechanism also applies to Tregs. These results also suggest that Bcl-3 may be critical for suppressing RORγt expression not just in Tregs but in different cell types which normally do not express RORγt but are exposed to Th17 cytokines. Another possible mechanism is that Bcl-3 may be essential for an inhibitory epigenetic modification of the RORγt gene locus. For example, Bcl-3 may either indirectly modulate epigenetic modification by affecting NF-κB binding to DNA, or it may directly regulate histone modification through association with HDACs.30
Our study suggests that Bcl-3-deficient regulatory T cells may be more suppressive than their wild type counterparts. Consistent with our results, it has been reported that Bcl-3 overexpression in T cells limited the inhibitory activity of Tregs.31 Another study showed that RORγt+ Tregs were more suppressive than RORγt− Tregs in the naïve T cell transfer-induced colitis model, decreasing the recruitment of effector T cells into the colon.25 Nevertheless, another report concluded that RORγt+ Tregs were as suppressive as RORγt− Tregs in the nephrotoxic serum nephritis (NTN) mouse model; they were less effective in suppressing IL-2 production but exhibited increased levels of IL-10 in vitro.32 Our data suggest that the increased accumulation of RORγt+ Tregs due to loss of Bcl-3 contributed to the increased inhibitory function of Bcl-3-deficient regulatory T cells. However, additional work will be needed to define the specific molecular changes in RORγt+ regulatory T cells that are mediated by Bcl-3 to modulate Treg function.
METHODS
Mice
All Bcl-3-deficient mice were described previously.7, 12, 14, 33 Mice were housed in National Institute of Allergy and Infectious Diseases facilities, and all experiments were done with approval of the National Institute of Allergy and Infectious Diseases Animal Care and Use Committee and in accordance with all relevant institutional guidelines.
CD3 specific antibody and DSS treatments
For CD3-specific antibody-induced immune tolerance, 8 to 12-week-old mice were injected i.p. once with 20 μg of anti-CD3 (145–2C11, BioXCell, West Lebanon, NH, USA) then sacrificed for Treg analysis four days later. For DSS-induced colitis, 8 to 12-week-old mice were given 3% DSS (MW 36 – 50 kDa, MP Biomedicals, Irvine, CA, USA) in the drinking water for five days. Body weights of these mice were monitored daily and the mice were euthanized when they lost more than 20% of the original body weight. Histology of DSS treated colon was scored by a combination of inflammatory cell infiltration (score 0–3) and tissue damage (score 0–3) as previously described.34 Littermate controls were used in all experiments.
Flow cytometry
Spleen and lymph node samples were forced through 100 μm filters to prepare single-cell suspensions. Dead cells were removed by gradient centrifugation with lymphocyte M (Cedarlane, Burlington, NC, USA) and live cells were stained with surface antibodies for FACS analysis. To isolate lymphocytes from colon, less than 0.1 cm colon pieces were cut and digested at 37°C with 3 mg mL−1 dispase II (Thermo Fisher Scientific, Waltham, MA, USA), 1 mg mL−1 collagenase D (Roche) and 0.1 mg mL−1 DNase I (Roche) for 1 h until no visible pieces were present. The digested tissue was passed through a 100 μm filter, followed by centrifugation on a Percoll gradient.
For intracellular staining, the cells were first stained with antibodies for surface marker expression, then permeabilized and stained with antibodies for intracellular protein for 1 h using Foxp3 Staining Buffer Set (Thermo Fisher Scientific, Waltham, MA, USA). Data were collected in a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA) and analyzed using FlowJo software (BD, Franklin Lakes, NJ, USA). Antibodies to the following markers were used: CD4 (RM4–5, Thermo Fisher Scientific), CD3 (145–2C11, Biolegend, Dedham, MA, USA), TCRβ (H57–597, BD Biosciences), RORγt (AFKJS-9, Thermo Fisher Scientific), CD25 (PC61, BD Biosciences), Foxp3(FJK-16s, Thermo Fisher Scientific).
T cell differentiation
For RORγt+/Foxp3+ cell differentiation, Tregs were isolated from spleens of 8 to 12 weeks old healthy mice by FACS sorting and seeded at a concentration of 2×105 cells per well in 96-well plates. 1 μg mL−1 anti-CD3 (145–2C11) and 2 μg mL−1 anti-CD28 (37.51) were added to the media along with 20 ng mL−1 IL-6, 5 ng mL−1 TGFβ, 10 ng mL−1 IL-1, 50 ng mL−1 IL-21, 20 ng mL−1 IL-23, 10 μg mL−1 anti-IL-12 (C18.2), 10 μg mL−1 anti-IFNγ (XMG1.2) and 10 μg mL−1 anti-IL-4. For iTreg differentiation, naïve Tconv cells were sorted and cultured with 20 ng mL−1 IL-2, 10 ng mL−1 TGFβ, 10 μg mL−1 anti-IL-12 (C18.2) and 10 μg mL−1 anti-IFNγ (XMG1.2). Cells were cultured for 3 days before FACS analysis. All cytokines and antibodies noted above were purchased from PeproTech (Rocky Hill, NJ, USA) and BioXCell, respectively, except anti-IL-12 (C18.2) (Thermo Fisher Scientific).
Statistical Analysis
All data are expressed as the mean ± SD. Differences between groups were evaluated using an unpaired Student’s t-test. P-values were considered to be statistically significant when less than 0.05.
Supplementary Material
ACKNOWLEDGMENTS
We greatly appreciate the constructive inputs provided by all members of the Siebenlist laboratory. This research was supported by the Intramural Research Program of NIAID, NIH.
REFERENCES
- 1.Franzoso G, Bours V, Park S, Tomita-Yamaguchi M, Kelly K, Siebenlist U. The candidate oncoprotein Bcl-3 is an antagonist of p50/NF-kappa B-mediated inhibition. Nature 1992; 359: 339–342. [DOI] [PubMed] [Google Scholar]
- 2.Bours V, Franzoso G, Azarenko V, et al. The oncoprotein Bcl-3 directly transactivates through kappa B motifs via association with DNA-binding p50B homodimers. Cell 1993; 72: 729–739. [DOI] [PubMed] [Google Scholar]
- 3.Fujita T, Nolan GP, Liou HC, Scott ML, Baltimore D. The candidate proto-oncogene bcl-3 encodes a transcriptional coactivator that activates through NF-kappa B p50 homodimers. Genes Dev 1993; 7: 1354–1363. [DOI] [PubMed] [Google Scholar]
- 4.Schuster M, Annemann M, Plaza-Sirvent C, Schmitz I. Atypical IκB proteins - nuclear modulators of NF-κB signaling. Cell Commun Signal 2013; 11: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palmer S, Chen YH. Bcl-3, a multifaceted modulator of NF-kappaB-mediated gene transcription. Immunol Res 2008; 42: 210–218. [DOI] [PubMed] [Google Scholar]
- 6.Hinz M, Arslan SC, Scheidereit C. It takes two to tango: IκBs, the multifunctional partners of NF-κB. Immunol Rev 2012; 246: 59–76. [DOI] [PubMed] [Google Scholar]
- 7.Franzoso G, Carlson L, Scharton-Kersten T, et al. Critical roles for the Bcl-3 oncoprotein in T cell-mediated immunity, splenic microarchitecture, and germinal center reactions. Immunity 1997; 6: 479–490. [DOI] [PubMed] [Google Scholar]
- 8.Zhang X, Wang H, Claudio E, Brown K, Siebenlist U. A role for the IkappaB family member Bcl-3 in the control of central immunologic tolerance. Immunity 2007; 27: 438–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruan Q, Zheng SJ, Palmer S, Carmody RJ, Chen YH. Roles of Bcl-3 in the pathogenesis of murine type 1 diabetes. Diabetes 2010; 59: 2549–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pene F, Paun A, Sonder SU, et al. The IkappaB family member Bcl-3 coordinates the pulmonary defense against Klebsiella pneumoniae infection. J Immunol 2011; 186: 2412–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kreisel D, Sugimoto S, Tietjens J, et al. Bcl3 prevents acute inflammatory lung injury in mice by restraining emergency granulopoiesis. J Clin Invest 2011; 121: 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tassi I, Claudio E, Wang H, et al. The NF-κB regulator Bcl-3 governs dendritic cell antigen presentation functions in adaptive immunity. J Immunol 2014; 193: 4303–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tassi I, Rikhi N, Claudio E, et al. The NF-κB regulator Bcl-3 modulates inflammation during contact hypersensitivity reactions in radioresistant cells. Eur J Immunol 2015; 45: 1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang W, Wang H, Claudio E, et al. The oncoprotein and transcriptional regulator Bcl-3 governs plasticity and pathogenicity of autoimmune T cells. Immunity 2014; 41: 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meguro K, Suzuki K, Hosokawa J, et al. Role of Bcl-3 in the development of follicular helper T cells and in the pathogenesis of rheumatoid arthritis. Arthritis Rheumatol 2015; 67: 2651–2660. [DOI] [PubMed] [Google Scholar]
- 16.Long M, Park SG, Strickland I, Hayden MS, Ghosh S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity 2009; 31: 921–931. [DOI] [PubMed] [Google Scholar]
- 17.Deenick EK, Elford AR, Pellegrini M, Hall H, Mak TW, Ohashi PS. c-Rel but not NF-kappaB1 is important for T regulatory cell development. Eur J Immunol 2010; 40: 677–681. [DOI] [PubMed] [Google Scholar]
- 18.Oh H, Grinberg-Bleyer Y, Liao W, et al. An NF-κB Transcription-Factor-Dependent Lineage-Specific Transcriptional Program Promotes Regulatory T Cell Identity and Function. Immunity 2017; 47: 450–465 e455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grinberg-Bleyer Y, Oh H, Desrichard A, et al. NF-κB c-Rel Is Crucial for the Regulatory T Cell Immune Checkpoint in Cancer. Cell 2017; 170: 1096–1108 e1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grinberg-Bleyer Y, Caron R, Seeley JJ, et al. The Alternative NF-κB Pathway in Regulatory T Cell Homeostasis and Suppressive Function. J Immunol 2018; 200: 2362–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fang D, Zhu J. Dynamic balance between master transcription factors determines the fates and functions of CD4 T cell and innate lymphoid cell subsets. J Exp Med 2017; 214: 1861–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohnmacht C, Park JH, Cording S, et al. The microbiota regulates type 2 immunity through RORγt+ T cell. Science 2015; 349: 989–993. [DOI] [PubMed] [Google Scholar]
- 23.Sefik E, Geva-Zatorsky N, Oh S, et al. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science 2015; 349: 993–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pratama A, Schnell A, Mathis D, Benoist C. Developmental and cellular age direct conversion of CD4+ T cells into RORγ+ or Helios+ colon Treg cells. J Exp Med 2020; 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang BH, Hagemann S, Mamareli P, et al. Foxp3+ T cells expressing RORγt represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal Immunol 2016; 9: 444–457. [DOI] [PubMed] [Google Scholar]
- 26.Perruche S, Zhang P, Liu Y, Saas P, Bluestone JA, Chen W. CD3-specific antibody-induced immune tolerance involves transforming growth factor-β from phagocytes digesting apoptotic T cells. Nat Med 2008; 14: 528–535. [DOI] [PubMed] [Google Scholar]
- 27.Vasanthakumar A, Liao Y, Teh P, et al. The TNF Receptor Superfamily-NF-κB Axis Is Critical to Maintain Effector Regulatory T Cells in Lymphoid and Non-lymphoid Tissues. Cell Rep 2017; 20: 2906–2920. [DOI] [PubMed] [Google Scholar]
- 28.Huber S, Gagliani N, Esplugues E, et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3− and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 2011; 34: 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruan Q, Kameswaran V, Zhang Y, et al. The Th17 immune response is controlled by the Rel-RORγ-RORγ T transcriptional axis. J Exp Med 2011; 208: 2321–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Viatour P, Dejardin E, Warnier M, et al. GSK3-mediated BCL-3 phosphorylation modulates its degradation and its oncogenicity. Mol Cell 2004; 16: 35–45. [DOI] [PubMed] [Google Scholar]
- 31.Reissig S, Tang Y, Nikolaev A, et al. Elevated levels of Bcl-3 inhibits Treg development and function resulting in spontaneous colitis. Nat Commun 2017; 8: 15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kluger MA, Meyer MC, Nosko A, et al. RORγt+Foxp3+ Cells are an Independent Bifunctional Regulatory T Cell Lineage and Mediate Crescentic GN. J Am Soc Nephrol 2016; 27: 454–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franzoso G, Carlson L, Poljak L, et al. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med 1998; 187: 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang W, Wang H, Ha HL, et al. The B-cell tumor promoter Bcl-3 suppresses inflammation-associated colon tumorigenesis in epithelial cells. Oncogene 2016; 35: 6203–6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.