

ABSTRACT
Autoimmune haemolytic anaemia (AIHA) is an acquired heterogenous clinical entity with variable presentations like acute haemolysis or mild, chronic haemolysis compounded with acute exacerbation in winters or fatal uncompensated haemolysis. A step-wise approach to the diagnosis and characterisation of AIHA should be undertaken, firstly the diagnosis of haemolysis followed by the establishment of immune nature with the aid of direct agglutination tests (DAT). Simultaneously the other causes of immune haemolysis need to be excluded too. In light of advancements in diagnostics, a wide array of investigations can be used like absolute reticulocyte count, bone marrow responsiveness index to establish the evidence of haemolysis, sensitive gel technology, enhanced DAT assays, e.g., modified DAT with low ionic strength saline solution (LISS) at 4°C, DAT assays utilizing reagents such as anti-IgA and anti-IgM and DAT by flowcytometry, to detect RBC bound autoantibodies (Abs) and monospecific DAT to establish immune causes of haemolysis and characterisation of the autoantibodies. The compensatory role of bone marrow and synchronous pathologies like clonal lymphoproliferation, dyserythropoiesis, fibrosis are important factors in the evolution of the disease and aid in the customisation of treatment modalities. The laboratory work up should aim to diagnose underlying diseases like chronic lymphoproliferative disorders, autoimmune disorders and infectious diseases. Also, tests like autoimmune lymphoproliferative syndromes (ALPS) screening panel and Next-generation sequencing (NGS) panel for RBC membrane disorders, RBC enzymopathies, and congenital dyserythropoietic aneamia have found their place. It is incumbent upon the clinicians to use the all-available diagnostic modalities for the accurate diagnosis, prognostication and customisation of the therapy.
Keywords: Direct antiglobulin test, enhanced DAT, warm autoimmune haemolytic anaemia cold agglutinin syndrome
Introduction
Autoimmune haemolytic anaemia (AIHA) is a heterogeneous disease with myriad presentations from mild/compensated disease to severe disease which can be life-threatening.[1] Immune haemolysis is mediated through the Abs directed against antigens on the erythrocyte membrane with/without complement fixation.[1,2] Immune haemolysis triggers compensatory responses like increased erythropoietin levels leading to compensatory marrow hyperplasia. The landscape of AIHA has evolved over the years; today apart from conventional therapies like steroids, immunosuppressants, and splenectomy, the calibrated use of bone marrow stimulating agents/immunomodulating agents are being practised in the diverse settings of clinical trials and routine clinical practice.[1] The knowledge of the pathogenesis of AIHA has grown manifold, beyond the role of Abs, complement, and antibody-dependent cell-mediated cytotoxicity. The body of evidence regarding pathogenesis implicates decreased CD4+ T-regs, imbalance of T-helper 1/2 cytokines, and amplified activity of immune-competent cells like cytotoxic CD8+ T lymphocytes, macrophages and natural killer cells.[1] The compensatory roles of bone marrow and synchronous pathologies like clonal lymphoproliferation, dyserythropoiesis, and fibrosis are important variables recognized in the evolution of the disease and the customization of treatment modalities on a case-to-case basis.[1,3,4]
AIHAs are classified as primary or secondary. The secondary AIHAs occur in the settings of lymphoproliferative disorders, solid tumours, immunodeficiencies, transplants, autoimmune conditions, various infectious diseases, and drugs in which various immunologic mechanisms are implicated.[1] AIHAs are also classified as warm (75% of all the cases), cold (15% of all the cases), and mixed (< 5% of the cases) based on the thermal amplitude of the implicated auto Abs.[5] The systematic review by Hill et al.[6] conducted in 2019 deduced that there is ambiguity in the definitions and diagnostic criteria of AIHA used by various researchers. In the majority of the studies the Direct Antiglobulin test (DAT) positivity was deemed essential for the diagnosis of AIHA. The entities exhibiting immune haemolysis with DAT positivity like delayed haemolytic transfusion reaction (DHTR), haemolytic disease of the newborn (HDFN) especially in paediatric population drug-induced immune haemolysis, alloimmune haemolysis secondary to solid organ/hemopoietic stem transplantation have to be excluded too, to establish the diagnosis of AIHA.[6] However, the DAT positive report should also be viewed with caution, as DAT positive results can be associated with other pathologies, like passive deposition of immune complexes or Abs on RBCs in hepatic disease, malignancy, autoimmune disorders like systemic lupus erythematosus, renal diseases, secondary to drug therapies with IV Immunoglobulin (IV Ig), antithymocyte globulin. In absence of these causes, diagnosis of AIHA can be rendered without a doubt.[6,7]
Also, there is an inherent lacuna in this definition of AIHA as it overlooks DAT-negative AIHA.[7] AIHA affects both the paediatric and adult populations, the estimated annual incidence in the paediatric population is approximately 0.8 per 100,000 individuals under 18 years old which is lower than in the adult population.[7]
Case 1: Secondary Warm Autoimmune Haemolytic Anaemia (WAIHA) with Chronic Lymphoproliferative Disorder (CLPD)
We present a case of a 62-year-old male, diagnosed as a case of mantle cell lymphoma, presented with anaemia with haemoglobin (Hb): 7.2 gm/dl [12.0–15.5]), total leucocyte count: 18 × 109/L (3.7–9.7 × 109/L) total platelet count (TPC): 100 × 109/L, (179–373 × 109/L), Lactate dehydrogenase (LDH): 344 U/L [122-222]), peripheral smear showed macrocytic picture with polychromasia, leucocytosis, and 15% lymphoma cells, normoblastaemia. In flow cytometry, lymphoma cells expressed CD19, CD20, FMC-7, CD5, and Cyclin D1, but lacked expression of CD23 and CD10.
The air-dried, giemsa-stained smears of aspirate of the right cervical lymph node showed a monotonous population of small intermediate-sized sized lymphoid cells with an enlarged, nucleus with an irregular nuclear outline coarse chromatin, scant cytoplasm, and absence of large or “transformed” lymphoid cells.
The excision biopsy lymph node specimen measuring 3.5 cm × 3.0 cm and on gross examination showed a homogenous tan cut surface. On microscopic examination, it showed diffuse effacement of the nodal architecture with the presence of small to medium-sized monomorphic cells with an irregular nuclear outline, clumped chromatin, and single micro to inconspicuous nucleoli. [Figure 1a, b]
Figure 1.
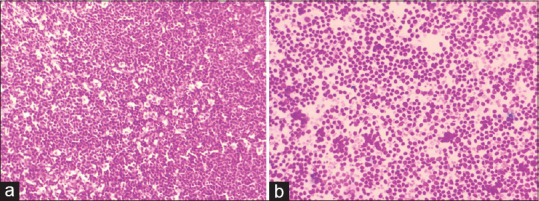
Micrograph of histology, (a) and cytology (b) of MCL. (a) H and E stained slide of mantle cell lymphoma, lymph node biopsy (10 × 10). Showing diffuse effacement of the nodal architecture with the presence of small to medium-sized monomorphic cells. (b) H and E stained slide of Mantle Cell Lymphoma, Lymph Node Biopsy (40 × 10). Showing diffuse effacement of the nodal architecture with the presence of small to medium-sized monomorphic cells with an irregular nuclear outline, clumped chromatin, and single micro to inconspicuous nucleoli
Bone marrow and bone marrow biopsy showed nodular and paratrabecular arrangement of small to medium-sized monomorphic lymphoid cells with irregular nuclear borders, clumped chromatin, and inconspicuous nucleoli. On immunohistochemistry, lymphoma cells expressed CD20, CD5, and Cyclin D-1. The blood sample received for blood typing was typed as B Rh-positive with 1+ positive auto control at room temperature (RT), 3+ positive at 37°C/Coombs phase and negative at 4°C (by conventional tube technique). Polyspecific DAT (IgG + C3d) showed 3+ positivity and monospecific DAT positive 3+ with IgG and C3d both. IAT was 2+ positive (DAT and IAT by column agglutination technique). Antibody screening (AS) (BIO RAD Diamed) with three cell panels and antibody identification (AI) (BIO RAD Diamed) with 11 cell panels showed pan agglutination of uniform strength 2+ at 37°C (in Coomb s phase). The packed red blood cell concentrate (PRBC) transfusion was withheld in line with restrictive transfusion strategies based on AABB guidelines, as the patient was hemodynamically stable. However, erythrocyte transfusions should be considered in anaemic patients with cardiopulmonary symptoms regardless of the Hb levels. In such patients, special compatibility procedures are mandated to rule out the presence of a coexisting allo Ab. The auto Abs are also directed against major erythrocyte antigens, e.g., Rh, Rh-related antigens, Band 3, and glycophorin; hence, Rh negative PRBC units are preferred. The compatibility test should include the removal of auto abs from the patient’s serum by auto adsorption with either autologous RBCs or with selected panels of RBCs with known antigen phenotype leaving behind/unmasking the coexistent allo Ab if present This is followed by AS with RBC panels with known RBC antigenic expression on an adsorbed serum to look for allo Ab. Lastly the crossmatch is done to identify compatible units. In cases with severe anaemia, the availability of autologous erythrocytes maybe not be enough to perform an autologous adsorption test.[8] Another option that can be used is the transfusion of phenotypically matched PRBC to the patient, which requires the determination of the antigen profile of the patient’s erythrocytes using typing sera, providing options for safe transfusions.[8]
Case 2: Primary Mixed AIHA with Broad Thermal Amplitude Abs
A 22-year-old female patient presented with gradual onset of fatigue, with a history of abrupt fall of haemoglobin over the duration of one month preceded by fever for one day with arthralgias without joint effusion, before and after the onset of fever. The patient had significant negative histories of pregnancy and drug intake. Laboratory test results with reference range provided in brackets) were significant for Hb (ranging from 6.7 to 5.6 gm/dL [12.0–15.0]), MCV; 104.0fl [78.5-96.4]), absolute reticulocyte count: 145 X 109/L [40.6-111.8], LDH; 1049 U/L [122-222]), total bilirubin (4.02 mg/dL [0.1-1], indirect bilirubin: 3.39 mg/dl), and blood smear showing polychromasia. The blood sample was received for blood group typing. The case showed naked eye RBC agglutination with a discrepancy in forward and Reverse Blood Grouping at room temperature. So fresh patient EDTA sample was collected under warm conditions. The RBC and Serum were separated immediately by centrifugation at 3000 rpm for 3 min. The cells were washed 3 times with warm normal saline at 37°C. Forward and reverse blood grouping was done at 4°C, room temp and 37° C by tube method which showed discrepant results at 4° C and room temp. However, at 37°C, blood Group was confirmed as A RhD positive. Autocontrol was positive at 4°C, room temp and 37°C/Coombs phase suggesting the presence of auto Ab. [Table 1] Polyspecific DAT (IgG + C3d) showed 4+ positive (Column Agglutination), Monospecific DAT positive (4+) for IgG, IgM, C3d (Column Agglutination). AS (BIO RAD Diamed) with three cell panel and AI (BIO RAD Diamed) with 11 cell panel showed pan agglutination of uniform strength 2+ at 37°C (in Coomb s phase) and at 4°C (in saline phase). IgM antibody titre done by double dilution tube method in saline phase at 4°C was 16. Both warm (IgG) and cold (IgM) autoantibodies with high thermal amplitude were found in this case, so suggestive of mixed AIHA.
Table 1.
Discussion of blood grouping at different temperature conditions
| Temp °C | Forward grouping | Reverse grouping | BG: | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||||
| Anti A | Anti B | Anti D1 | Anti D2 | NS | A cell | B cell | O cell | AC | ||
| 4 °C | 3+ | 3+ | 3+ | 3+ | 3+ | 2+ | 3+ | 2+ | 2+ | invalid |
| RT | 3+ | 3+ | 3+ | 3+ | 3+ | 2+ | 3+ | 2+ | 2+ | invalid |
| 37°C | 3+ | negative | 3+ | 3+ | weak | negative | 3+ | negative | weak | A+ |
The patient was started on prednisolone therapy of 60 mg daily for 4 weeks along with folate supplementation till haemoglobin reached 12 gm/dl; thereafter, the dose of prednisolone was tapered by 20 mg every week until a dose of 20 mg daily was reached, followed by staggered tapering over 8 weeks. Subsequently, the patient was followed up for trends of C-reactive protein, LFT, Hb, and Reticulocyte Count [Graph 1].
Graph 1.
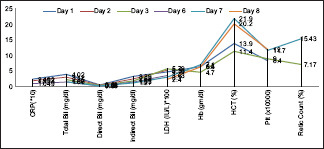
Graphical representation of laboratory parameters pre and post treatment with steroids
The patient attained a complete response within 3 months and was followed for one year with no evidence of relapse.[8]
Case 3: Cold AIHA Secondary to Viral Infection
We report a case of a 42-year-old Indian male patient presenting with dyspnoea, recent onset of fatigue, symptoms of cold intolerance, and acrocyanosis. The blood sample received for hemogram revealed significant anaemia with Hb: ranging from 6.7 to 5.6 gm/dl [13.3–17.2 gm/dl]), MCV; 130.0*** fl [81.2-94.0]), mean corpuscular haemoglobin (MCH):***pg (27.1-32.5), mean corpuscular haemoglobin concentration (MCHC):***(32.5–36.7 g/dL), MCV, MCH & MCHC were flagged by the counter, corrected leucocyte count: 9.96 × 109/L (3.7–9.7 × 109/L), TPC: 130 × 109/L (179–373 × 109/L), absolute reticulocyte count (119 × 109/L [55.1–140.7 X109/L]), LDH (1049 U/L [122–222]), total bilirubin (4.02 mg/dL [0.1–1]), indirect bil (3.39 mg/dl), and blood smear showing polychromasia. The peripheral smear examination showed marked erythrocyte agglutination, polychromasia, and five nucleated red blood cells (nrbcs)/100 wbcs counted. [Figure 2] The absence of abnormal/premature cells of lymphoid/myeloid cells was remarkable. The flagging of MCV, MCHC, and MCH is in line with the findings of erythrocyte agglutination. Consequently, the sample was received for blood group typing, DAT, and cross matching for transfusion. Autoagglutination was present in the sample at RT. Fresh sample was collected in the EDTA vial and incubated at 37°C for half an hour. Cell washing was done 5–6 times with warm saline at 37°C C. In this way, serum and cell grouping was performed and the blood group came out to be AB positive. Autocontrol by conventional tube technique at 4°C was positive but negative at 37°C/coombs phase. We performed cold agglutinin titres which came out to be 1024. The monospecific DAT (column agglutination) was positive for C3d, and negative for IgG. The PCR test for mycoplasma pneumonia, cytomegalovirusm and Epstein Barr virus were negative. Serum complement C3 and C4 levels were reduced. Serum protein immunofixation was positive for IgM; bone marrow aspiration and bone marrow biopsy were unremarkable effectively ruling out the presence of a lymphoproliferative process. The patient attained remission with four doses of rituximab and improved on clinical and laboratory parameters.[8]
Figure 2.
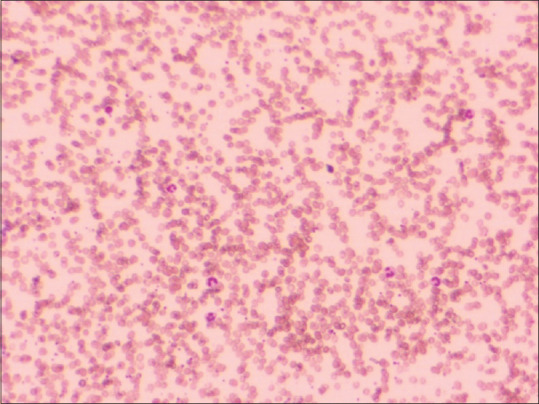
Leishman stained slide (10 × 10). Micrograph showing RBC agglutination in peripheral smear
Case 4. Case of Idiopathic WAIHA
A 42-year-old Indian male presented with recent onset of fatigue to the medical OPD. Physical examination showed marked pallor, dyspnoea on rest, and hepatomegaly. Laboratory results (with reference ranges provided in brackets) were significant for haemoglobin (Hb):5.9 g/dL [13.3–17.2 gm/dl], mean cell volume (MCV; 107.0 fl [81.2–94.0 fl]), absolute reticulocyte count (175 X109/L [55.1–140.7 × 109/L]), total platelet count: 194 × 109/L, (179–373 × 109/L), LDH 260 U/L [122–222]), total bilirubin (1.2 mg/dL [0.1–1]), and blood smear showing macrocytic picture with presence of spherocytes, polychromasia 2 nrbcs/100 was counted. [Figure 3] The patient was typed as A Rh-positive. Since the patient had symptomatic anaemia with an urgent requirement for transfusion, and crossmatching was performed with several random A positive and O positive packed red cells, but no compatible unit was detected. So detailed workup was done. Autocontrol (conventional tube technique) at 37°C/coombs phase found to be positive and negative at room temperature and at 4°C. Polyspecific DAT (IgG + C3d) by Diamed Bio Rad Gel Technique was found to be positive. Monospecific DAT was positive for IgG and C3d. He received two ‘least incompatible’ A Rh-positive non-leuco reduced packed red cell units over the period of two days after informed consent. The transfusions were closely monitored clinically and by laboratory parameters.[9]
Figure 3.
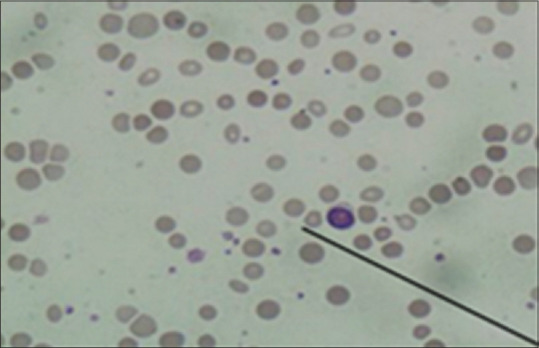
Leishman stained slide (10 × 10). Micrograph showing spherocytes on peripheral smear
Case 5: WAIHA Secondary to Hodgkin’s Lymphoma with Congestive Heart Failure
A 16-year-old male presented to the emergency room with gradual onset of fatigue and shortness of breath at rest. Physical examination showed marked pallor, right cervical lymphadenopathy, and raised jugular venous pressure. There is significant negative history of transfusions, and ingestion of unknown drugs. Laboratory results (with reference ranges provided in brackets) were significant for Hb: 1.9 g/dL ([11.0–14.3]), MCV: 92.0 fl ([80.8–86.6]), absolute reticulocyte count (ARC):119 × 109/L ([39.0–100.0]), LDH: 260 U/L ([122–222]), total bilirubin: 1.2 mg/dL [0.1–1], and blood smear showing polychromasia. The patient was typed as A Rh-negative with auto abs, DAT showed 2+ positive by tube, and direct antiglobulin test with poly-specific Coomb’s reagent (IgG + C3d) was positive. The patient showed pan reactive AS with all three reagent cells Indirect Antiglobulin test (IAT) with warm thermal amplitude of these antibodies. In view of life-threatening anaemia with features of chronic heart failure, with an urgent requirement for transfusion, detailed phenotyping was not done and crossmatching was performed with several random A Rh-negative packed red cell, but no compatible unit was detected. He received three ‘least incompatible’ A Rh-negative non-leuco reduced packed red cell units over three days after informed consent. The transfusions were eventless and an adequate increase in Hb was reported. Besides, he was also started on diuretics, but steroids were withheld as investigations like fine needle aspiration cytology (FNAC) and excision biopsy were pending. On day 3, post-FNAC and excision biopsy were performed. On examination, 4 cm × 3 cm matted lymph nodes with rubbery feel were palpated. The steroid therapy was initiated, and the patient was followed up by the increasing trend of ARC.[10] The giemsa stained FNAC smears show polymorphous population of eosinophils, plasma cells, and histiocytes with the presence of mononuclear and bi nucleate cells with prominent eosinophilic nucleoli with ample amphophilic cytoplasm. The excision biopsy showed H&E-stained sections with enlarged lymph with total effacement of lymphoid architecture, presence of polymorphous population of eosinophils, plasma cells, and histiocytes with the presence of mononuclear and bi nucleate cells with prominent nucleoli with ample cytoplasm consistent with Reed–Sternberg cells and mononuclear variants. [Figure 4] Immunostains positively stained abnormal cells for CD15 and CD 30 and negatively for CD20, CD45, and CD3.
Figure 4.
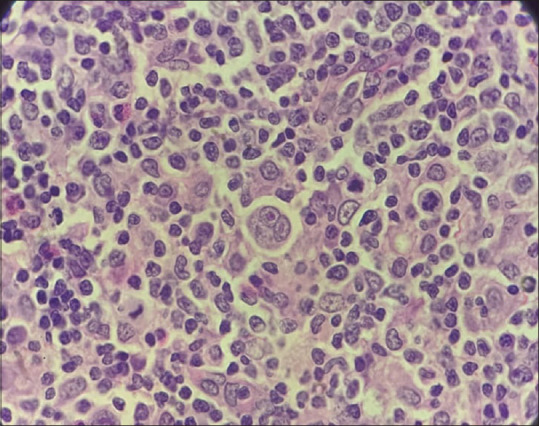
H and E stained slide of mantle cell lymphoma, lymph node biopsy (40 × 10)
Micrograph showing total effacement of lymphoid architecture, presence of polymorphous population of eosinophils, plasma cells, and histiocytes with the presence of mononuclear and bi nucleate cells with prominent nucleoli with ample cytoplasm consistent with Reed–Sternberg cells and mononuclear variants
Discussion
Clinical presentation: The classical presentation in WAIHA is insidiously developing anaemia with clinical symptoms like fatigue, weakness, dyspnoea on exertion, and dizziness with association with less frequent symptoms like bleeding, fever, cough, and symptoms related to aetiology.[9] Whereas patients with cold AIHA (primary/secondary) show chronic disease with symptoms like fatigue and pallor, punctuated with acute exacerbations during cold weather. The symptoms during acute exacerbation include Raynaud’s phenomenon, acrocyanosis, and features of acute haemolysis, e.g., haemoglobinuria and haemoglobinemia secondary to cold exposure.[9]
Patients with mixed-type AIHA also have a chronic presentation with intermittent severe exacerbations without temporal association with cold exposure, and accompanying acrocyanosis and Raynaud’s phenomenon.[9]
The basic and advanced laboratory, serological, molecular and radiological investigations to be carried out in suspected AIHA patients to arrive at a diagnosis, characterisation of the disease, ruling out other causes of immune haemolysis, investigate for secondary/associated disorders or diseases, and for the prognostication and titration of the treatment are discussed in detail [Tables 2 and 3]
Table 2.
Basic and advanced laboratory, molecular and radiological investigations indicated in AIHA
| Laboratory and Radiological Investigations required | Remark[1,6,7] |
|---|---|
| Complete Blood Count with Hemoglobin (Hb), RBC count & RBC indices. | Hb: Decreased |
| Mean Corpuscular Volume (MCV): N/Increased. | |
| Normocytic/Macrocytic picture. | |
| Rarely thrombocytopenia: Evans Syndrome (ES) in the Pediatric population. | |
| Plasma Hb | Increased. |
| Reticulocyte count and other reticulocyte indices | Reticulocyte count>3%. |
| Reticulocytosis is a common finding. | |
| Absolute reticulocyte count (ARC), and bone marrow responsiveness index (BMRI) are used. | |
| Reticulocytopenia in AIHA can be present in cases secondary to bone marrow infiltration by lymphoproliferative disorder or Parvovirus B19 infection. | |
| Peripheral smear examination | Increased polychromatophils for age |
| Normocytic/Macrocytic picture with micro spherocytes in w AIHA±/RBC agglutinates and rouleaux in cold agglutinin syndrome (CAS)/red cell fragments/teardrop cells (suggestive of bone marrow infiltration). | |
| Presence of nucleated RBCs | |
| Serum bilirubin and liver function tests (LFT) | Normal/Increased. |
| *Normal: in cases of milder haemolysis. | |
| LFT: Specifically in infants to rule out AIHA with giant cell hepatitis (GCH). | |
| Serum lactate dehydrogenase enzyme (LDH) | Increased. |
| *LDH can be increased in hepatic disease too. | |
| Serum hemopexin and haptoglobin | Decreased. |
| *Haptoglobin can be increased in hepatic disease too. | |
| Urine routine examination | Evidence of urinary RBCs and haemoglobin. |
| Increased urobilinogen | |
| Urine for hemosiderin (Ferricyanide test) | Evidence of urinary hemosiderin after a week. |
| Peripheral T-cell subsets | If ES is suspected. |
| Serum creatinine and blood urea nitrogen (BUN) | If ES is suspected. |
| Serum immunoglobulins and electrophoresis with immunofixation* | Detection of underlying disorders |
| Serum HIV, HBV, HCV | Detection of underlying disorders |
| Anti-dsDNA, ANA, anti-extractable nuclear antigens, anti-β 2glycoprotein antibodies, and lupus-like anticoagulants. | Detection of underlying disorders. |
| * Specifically in teenage females. | |
| Bone marrow aspiration and biopsy | Indicated if CAD features in history, examination, haemogram or smear suggesting possible marrow infiltration (presence of teardrop cells) |
| Evidence of bone marrow compensation, ±dyserythropoiesis, fibrosis, and clonal lymphoproliferation | |
| *Concurrent thrombocytopenia and/or neutropenia, unusual or prolonged reticulocytopenia, lymphadenopathy, or organomegaly without evidence of concurrent EBV infection. | |
| Infectious screening | A serological test for Epstein–Barr virus and/or Mycoplasma pneumonia if IgM Abs are detected. |
| Dependent on symptoms, travel history, and age | |
| Parvovirus, haematinic | If reticulocytopenia |
| Osmotic gradient ektacytometry | Increased Omin and decreased EImax. |
| Autoimmune lymphoproliferative syndromes (ALPS) screening panel | Follow-up testing as needed with next-generation sequencing on ALPS or PID gene panels |
| Next-generation sequencing (NGS) panel for RBC membrane disorders, RBC enzymopathies, and congenital dyserythropoietic anaemia. | To rule out secondary disorders. |
| Flow cytometry | To rule out paroxysmal nocturnal haemoglobinuria (PNH) & lymphocyte subpopulations. |
| EMA-binding, high-performance liquid chromatography, erythrocyte enzyme activities (G6PD, PKD and other rare ones) | To rule out other causes of haemolysis: congenital membrane and enzyme defects, hemoglobinopathies, thrombotic and mechanical microangiopathies (prosthetic heart valves, rheumatic endocarditis) |
| Tests for allo and autoantibodies in serum, eluate, identification of antibody specificity, immunoabsorbance techniques, and extended RBC genotyping. | To rule out DHTRs, HDFN, and passenger lymphocyte syndrome. |
| Computed tomography of chest, abdomen, and pelvis | CAD (Cold agglutinin disease) at diagnosis and in relapsed/refractory WAIHA |
Table 3.
Additional serological investigations required in selected patients with AIHA
| Investigations required | Possible outcome[1,7] |
|---|---|
| DAggT (Double agglutination test) | If DAT positive for C3d±IgG |
| Cold antibody titre | If DAggT positive |
| Red cell eluate | If monospecific: DAT-negative with a strong suspicion of AIHA. |
| Donath Landsteiner | If DAT is positive for C3d±IgG and i) DAggT-negative or insignificant CAs and ii) Age, 18 years or haemoglobinuria or cold-associated symptoms or atypical serology |
| The thermal range of cold Abs | If the clinical significance of cold auto agglutinin is unclear. |
| Enhanced DAT assays: a.) modified DAT with LISS at 4 °C . b.) DAT assays utilizing reagents such as anti-IgA and anti-IgM (Double DAT) c.) DAT by flowcytometry to detect RBC-bound Abs |
In case of suspicion of DAT negative AIHA. |
| IAT | To look for the presence of coexisting allo Abs. |
A significantly increased LDH and RBC fragments on peripheral smear, along with the presence of urinary hemosiderin are indicators of an intravascular haemolytic process.[7]
DAT negative AIHA
The causes of DAT-negative AIHA are enumerated in the decreasing order of prevalence:
Red blood cell-bound IgG molecules, below the threshold of detection of the DAT, in the event of ongoing in vivo haemolysis
Low-affinity IgG Abs which may be washed off the red cells during the washing phase, hence missed on conventional DAT.
IgA auto Abs or rarely monomeric IgM not detected by conventional DAT.[9,11]
To increase the sensitivity of diagnosis of AIHA, following modifications of DAT are available. e.g., micro-column (gel), solid-phase, polybrene, flow cytometry methods, as well as enzyme-linked antiglobulin, immunoradiometric, and mitogen-stimulated tests.[11]
The modified technique of washing red cells with cold saline or LISS may be effective in detecting low-affinity antibodies because low-affinity Abs is eluted during the washing step in the conventional tube technique.[11] The sequence of investigations required for the diagnosis of DAT-negative AIHA is discussed in flowchart shown in Figure 5.
Figure 5.

Flow chart for the sequence for serological diagnosis of DAT negative AIHA[11]
Reticulocytes in AIHA
In HA cases associated with iron/Vitamin B12 deficiency, infections, or autoimmune conditions, the compensatory reticulocytosis response can be deficient or absent. The association of reticulocytopenia with AIHA was observed in 39% of paediatric cases and about 20% of adult cases, despite erythroid hyperplasia in the marrow. This finding can be attributed to immune attacks on late-stage erythroid precursors or due to delayed bone marrow response.[7,12,13] The reticulocytopenia with AIHA may present as a clinical emergency with increased transfusion demand and poor prognosis as observed by Fattizo B et al. in 13 severe, refractory cases with fatal outcomes. In comparison with ARC, bone marrow responsiveness index (BMRI) is a better parameter, with a cut-off value <121; it discriminates haemolysis with synchronous reticulocytopenia with sensitivity and specificity of 90% and 65%, respectively, in congenital dyserythropoietic Anemias (CDA) patients[14]
BMRI = Patient’s absolute reticulocyte count × (patient’s Hb/normal Hb for age and sex)[12,14]
Conclusion: AIHA is a heterogeneous group of disorders wherein the aetiology remains elusive, but the molecular mechanisms underlying haemolysis and the associated complications are fairly well understood now. The categorization of AIHA as cold or warm or mixed type has a significant bearing on the treatment options available. The treatment should be individualized with an emphasis on the bone marrow picture, e.g., presence/absence of dyserythropoiesis, fibrosis, degree of bone marrow compensation, and clonal lymphoproliferation. It is needless to say that various pathologies associated with secondary AIHA cases like autoimmune diseases, malignancies, and immunodeficiencies have additional immune-mediated haemolytic processes as compared to the primary disease. With the evolution of molecular testing, the bracket of primary AIHA is getting shortened and the distinction between primary and secondary disorders seems to be more and more arbitrary.
Finally, there is the increasing emergence of complex and severe entities, particularly AIHA developing after haemopoietic stem cell transplantation and AIHA associated with novel anticancer drugs such as checkpoint inhibitors, which represent a clinical challenge for complications and fatal outcomes.
Lastly, there is the emergence of a new set of severe cases in the setting of AIHA secondary to HSCT and AIHA secondary to newer anticancer agents like checkpoint inhibitors, which pose a challenge for management. The increasing laboratory and molecular testing have enabled better risk stratification of the patients and diagnosis of underlying associated clinical syndromes.
Abbreviations
WAIHA : Warm autoimmune haemolytic anaemia.
HA : Haemolytic anaemia.
Abs : Antibodies.
RBC : Red blood cells.
DAT : Direct antiglobulin test.
CLPD : Chronic lymphoproliferative disorder.
CTT : Conventional tube test.
LISS : Low ionic strength solution
CAD : Cold agglutinin disease.
CAS : Cold agglutinin syndrome.
Hb : Haemoglobin.
MCV : Mean cell volume.
LDH : Lactate dehydrogenase.
MCH : Mean corpuscular haemoglobin.
MCHC : Mean corpuscular haemoglobin concentration.
PRBC : Packed red blood cell concentrate
AS : Antibody screening.
AI : Antibody identification.
bil : Bilirubin
LFT : Liver function tests.
FNAC : Fine needle aspiration cytology.
MCL : Mantle cell lymphoma.
ALPS : Autoimmune lymphoproliferative syndromes.
DHTR : Delayed haemolytic transfusion reactions.
HDFN : Haemolytic disease of newborn.
BMRI : Bone marrow responsiveness.
ARC : Absolute reticulocyte count.
ES : Evans syndrome.
NGS : Next-generation sequencing.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Barcellini W, Fattizzo B. The changing landscape of autoimmune hemolytic anemia. Front Immunol. 2020;11:946. doi: 10.3389/fimmu.2020.00946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaur P, Basu S, Kaur R, Kaur G. Immune hemolytic anemia: A report of two cases. J Lab Physicians. 2009;1:22–4. doi: 10.4103/0974-2727.54803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fattizzo B, Zaninoni A, Gianelli U, Zanella A, Cortelezzi A, Kulasekararaj AG, et al. Prognostic impact of bone marrow fibrosis and dyserythropoietic in autoimmune hemolytic anemia. Am J Hematol. 2018;93:E88–91. doi: 10.1002/ajh.25020. doi: 10.1002/ajh. 25020. [DOI] [PubMed] [Google Scholar]
- 4.Fattizzo B, Barcellini W. Autoimmune cytopenias in chronic lymphocytic leukemia: Focus on molecular aspects. Front Oncol. 2020;9:1435. doi: 10.3389/fonc.2019.01435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park SH. Diagnosis, and treatment of autoimmune hemolytic anemia: Classic approach and recent advances. Blood Res. 2016;51:69–71. doi: 10.5045/br.2016.51.2.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hill QA, Hill A, Berentsen S. Defining autoimmune hemolytic anemia: A systematic review of the terminology used for diagnosis and treatment. Blood Adv. 2019;3:1897–906. doi: 10.1182/bloodadvances.2019000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hill A, Hill QA. Autoimmune hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2018;2018:382–9. doi: 10.1182/asheducation-2018.1.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adult values established by reference range studies performed by VCU Medical Centre Hematology Laboratory [Google Scholar]
- 9.Chaudhary R, Das SS. Autoimmune hemolytic anemia: From lab to bedside. Asian J Transfus Sci. 2014;8:5–12. doi: 10.4103/0973-6247.126681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pediatric Reference Intervals. 5th ed. AACC Press; 2005. [Google Scholar]
- 11.Takahashi T. Direct antiglobulin test-negative autoimmune hemolytic anemia. Acta Haematol. 2018;140:18–9. doi: 10.1159/000489253. [DOI] [PubMed] [Google Scholar]
- 12.Barcellini W, Fattizzo B. Clinical applications of hemolytic markers in the differential diagnosis and management of hemolytic anemia. Dis Markers 2015. 2015 doi: 10.1155/2015/635670. 635670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aladjidi N, Leverger G, Leblanc T, Picat MQ, Michel G, Bertrand Y, et al. New insights into childhood autoimmune hemolytic anemia: A French national observational study of 265 children. Haematologica. 2011;96:655–63. doi: 10.3324/haematol.2010.036053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Russo R, Gambale A, Langella C, Andolfo I, Unal S, Iolascon A. Retrospective cohort study of 205 cases with congenital dyserythropoietic anemia type II: Definition of the clinical and molecular spectrum and identification of new diagnostic scores. Am J Hematol. 2014;89:E169–75. doi: 10.1002/ajh.23800. [DOI] [PubMed] [Google Scholar]