

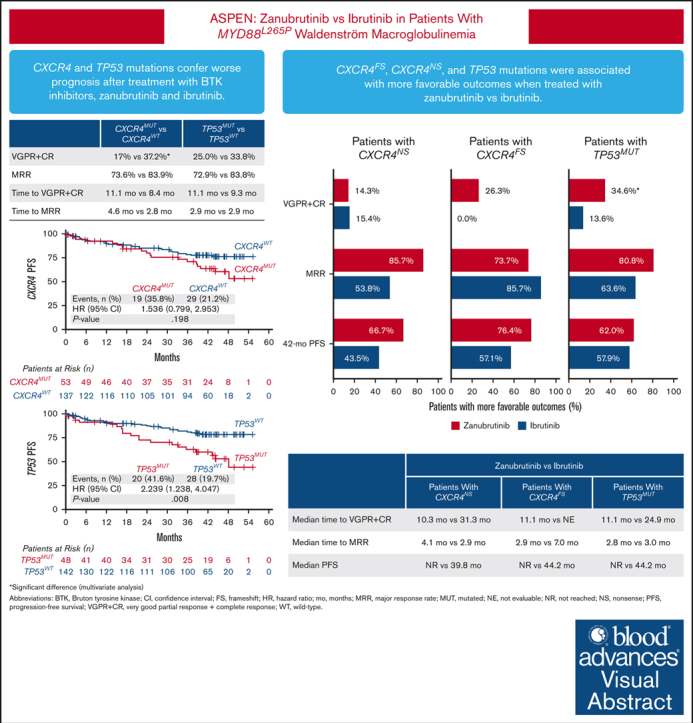
Key Points
-
•
Patients with Waldenström macroglobulinemia and mutations in CXCR4 or TP53 had poorer prognosis after treatment with BTKis.
-
•
Patients with CXCR4 or TP53 mutations had more favorable outcomes when treated with zanubrutinib vs ibrutinib.
Visual Abstract
Abstract
The phase 3 ASPEN trial (NCT03053440) compared Bruton tyrosine kinase inhibitors (BTKis), zanubrutinib and ibrutinib, in patients with Waldenström macroglobulinemia (WM). Post-hoc biomarker analysis was performed using next-generation sequencing on pretreatment bone marrow samples from 98 patients treated with zanubrutinib and 92 patients treated with ibrutinib with mutated (MUT) MYD88 and 20 patients with wild-type (WT) MYD88 treated with zanubrutinib. Of 329 mutations in 52 genes, mutations in CXCR4 (25.7%), TP53 (24.8%), ARID1A (15.7%), and TERT (9.0%) were most common. TP53MUT, ARID1AMUT, and TERTMUT were associated with higher rates of CXCR4MUT (P < .05). Patients with CXCR4MUT (frameshift or nonsense [NS] mutations) had lower very good partial response (VGPR) and complete response rates (CR; 17.0% vs 37.2%, P = .020) and longer time to response (11.1 vs 8.4 months) than patients with CXCR4WT treated with BTKis. CXCR4NS was associated with inferior progression-free survival (PFS; hazard ratio [HR], 3.39; P = .017) in patients treated with ibrutinib but not in those treated with zanubrutinib (HR, 0.67; P = .598), but VGPR + CR rates were similar between treatment groups (14.3% vs 15.4%). Compared with ibrutinib, patients with CXCR4NS treated with zanubrutinib had a favorable major response rate (MRR; 85.7% vs 53.8%; P = .09) and PFS (HR, 0.30; P = .093). In patients with TP53MUT, significantly lower MRRs were observed for patients treated with ibrutinib (63.6% vs 85.7%; P = .04) but not for those treated with zanubrutinib (80.8% vs 81.9%; P = .978). In TP53MUT, compared with ibrutinib, patients treated with zanubrutinib had higher VGPR and CR (34.6% vs 13.6%; P < .05), numerically improved MRR (80.8% vs 63.6%; P = .11), and longer PFS (not reached vs 44.2 months; HR, 0.66; P = .37). Collectively, patients with WM with CXCR4MUT or TP53MUT had worse prognosis compared with patients with WT alleles, and zanubrutinib led to better clinical outcomes.
Introduction
Waldenström macroglobulinemia (WM) is a B-cell malignancy characterized by lymphoplasmacytic bone marrow infiltration of monoclonal immunoglobulin M–secreting cells that constitutively activate the B-cell receptor signaling complex.1 A critical component of the B-cell receptor signaling pathway is Bruton tyrosine kinase (BTK), an important regulator of B-cell proliferation and survival.2 Myeloid differentiation factor 88 (MYD88) and C-X-C chemokine receptor type 4 (CXCR4) are the most frequently mutated (MUT) genes in patients with WM.3 A point mutation in MYD88 that switches leucine to proline at amino acid position 265 (MYD88L265P) is present in >90% of patients, and this activating mutation triggers tumor cell growth via BTK signaling.4 Mutations in CXCR4 (CXCR4WHIM) that are similar to germ line mutations detected in patients with warts, hypogammaglobulinemia, infection, and myelokathexis syndrome–like symptoms have been found in up to 40% of patients with WM and are thought to promote cell survival signaling and confer ibrutinib resistance.5 The mutational status of MYD88 and CXCR4 affects the efficacy of BTK inhibitors (BTKis) in patients with WM. Patients with wild-type (WT) MYD88 had lower major response rate (MRR) and shorter overall survival (OS) than patients with MYD88L265P when treated with ibrutinib.6,7 Patients with CXCR4WHIM mutations treated with ibrutinib had lower response rates and shorter progression-free survival (PFS) than patients with CXCR4WT, and patients with nonsense (NS) mutations in this gene had lower rates of major response and worse PFS than patients with frameshift (FS) mutations.8 Additionally, mutations or deletions, often in the DNA binding domain, of tumor protein P53 (TP53) have been reported in <14% of patients with WM3,9, 10, 11 and are associated with worse survival outcomes.9,10
BTKis have led to substantial improvements in outcomes for patients with WM, reflected by the approval of ibrutinib and zanubrutinib by the US Food and Drug Administration. The ASPEN trial is a phase 3 randomized, open-label, multicenter study comparing zanubrutinib with ibrutinib treatment for patients with WM.12 Although the primary efficacy end point was not met, the study’s long-term follow-up confirmed that patients who received zanubrutinib treatment had a higher very good partial response and complete response (VGPR + CR) rate over time (zanubrutinib, 36% vs ibrutinib, 25%; descriptive P = .07) and lower rates of atrial fibrillation, diarrhea, hypertension, localized infection, hemorrhage, muscle spasms, pneumonia, and adverse events leading to discontinuation or death than those treated with ibrutinib. Patients with MYD88WT treated with zanubrutinib also demonstrated major responses, with MRRs of 63% and 65.4% in 2 separate studies,13 respectively, compared with none reported in patients treated with ibrutinib.14
Although BTKis are effective therapies for many patients, not all patients may have clinical benefit. Many patients with WM and disease progression during covalent BTKi treatment acquire mutations in BTK at the covalent BTKi binding site (BTKCYS481) or its downstream mediator, PLCG2.15 These mutations lead to reactivation of downstream signaling pathways and cytokine release, resulting in resistant BTKWT cells.16 In addition to resistance mutations in BTK and PLCG2, other genetic alterations that may be associated with BTK include deletions on chromosomes 6q and 8p, which disrupt BTK, MYD88/NF-κB, and apoptotic signaling, as well as recurring mutations in ubiquitin ligases, innate immune signaling, and TLR/MYD88 pathway regulators.17,18
The objectives of this post-hoc biomarker analysis were (1) to evaluate baseline genetic alterations in patients with WM enrolled in the ASPEN study and their association with the efficacy of ibrutinib and zanubrutinib treatment and (2) to explore acquired mutations potentially conferring resistance to BTKis.
Materials and methods
Patients
The phase 3 ASPEN study (NCT03053440) enrolled patients with MYD88MUT WM (confirmed by qualitative MYD88 allele–specific polymerase chain reaction with a limit of detection [LOD] of 0.2% to 0.5%19) to compare responses to zanubrutinib (n = 102) vs ibrutinib (n = 99). In total, 28 patients without MYD88 mutations (MYD88WT) were enrolled in a separate cohort 2. Details of the study design have been described previously.12,19 Overall, 190 patients with MYD88MUT (zanubrutinib, n = 98, and ibrutinib, n = 92) and 20 patients with MYD88WT provided residual DNA from pretreatment bone marrow aspirate (BMA) samples without B-cell enrichment for baseline genetic alterations testing. In addition, 5 patients with disease progression after zanubrutinib treatment (3 with MYD88MUT and 2 with MYD88WT) gave informed consent to provide BMA samples at the time of clinical progression to explore resistance mechanisms. The trial was approved by the institutional review board or independent ethics committee at each study site and conducted in accordance with applicable regulatory requirements, the principles of the Declaration of Helsinki, and Good Clinical Practice guidelines of the International Conference on Harmonization. All patients provided written informed consent.
Biomarker assessments
MYD88 mutational status for patient enrollment was assessed by a qualitative allele-specific polymerase chain reaction with an LOD of 0.2% to 0.5%, as previously described.19 To detect other baseline genetic alterations, next-generation sequencing (NGS) was performed using the PredicineCARE panel, a Clinical Laboratory Improvement Amendments–certified NGS assay, which was validated to have a high sensitivity (LOD: ∼0.1%-0.25%), representing 152 genes. To examine mutations that may be associated with resistance to zanubrutinib, sequencing was performed using the PredicineHeme panel containing 106 hematologic malignancy–related genes, including whole exons of BTK and PLCG2, validated to have assay sensitivity of ∼0.1% to ∼0.25%. A variant was considered a true mutation only when (1) at least 3 distinct fragments (at least 1 of which is double stranded) contained the mutation and (2) the variant allele frequency (VAF) was ≥0.25% and hot-spot variants had an allele frequency of ≥0.1%. Mutations annotated as benign, likely benign, or common germ line variants were filtered out based on OncoKB,20 1000 genomes,21 ExAC,22 gnomAD,23 and KAVIAR,24 with population allele frequency of >0.5%. Finally, the hematopoietic expansion–related variants, including those in DNMT3A, ASXL1, and TET2, and specific alterations within ATM (residue 3008), GNAS (residue 201, 202), or JAK (residue 617) were excluded. The clinical efficacy end points used for biomarker analysis included response rate (VGPR + CR, major response), time to response, and PFS assessed by investigator according to response criteria in the National Comprehensive Cancer Network WM guidelines and modified Owen criteria25 as of data cutoff date, 31 October 2021.
To evaluate the dosage effect of TP53 mutations, patients were classified into 4 subgroups based on TP53MUT VAF and deletion status: (1) TP53WT (mutation and deletion not detected), (2) TP53MUT with VAF between 0.25% and <1%, (3) TP53MUT with VAF between 1% and <10%, and (4) TP53MUT with VAF of ≥10% or deletion present. For patients with multiple TP53 mutations, the variant with the maximum VAF was used for classification. The clinical efficacy end points were compared among subgroups.
Statistical analysis
Fisher exact tests were used to evaluate the correlations among different mutations and between mutation status and treatment status (treatment naïve vs relapsed/refractory) or treatment arms. Univariate and multivariate logistic regression models were used to compare the response rates between the mutation statuses. Covariates in the multivariate logistic regression models include mutation status of CXCR4 (WT, NS, and FS or WT and MUT), TP53 (WT and MUT), and TERT (WT and MUT) and the treatment arm to account for treatment differences. Odds ratios (MUT vs WT) and P values were estimated.
The median PFS was analyzed using the Kaplan-Meier method. Hazard ratio (HR) and P values of PFS between mutation statuses (MUT vs WT) were estimated using a Cox regression model with treatment arm and CXCR4, TP53, and TERT mutation status as covariates. P values ≤ .05 were considered statistically significant without multiplicity adjustment.
Results
Baseline genetic alteration profiling from 210 patients with WM treated with BTKi
Across 210 patients with WM, including 190 patients with MYD88MUT (zanubrutinib, 98; ibrutinib, 92) and 20 patients with MYD88WT (all zanubrutinib), baseline genetic profiling revealed 329 genetic alterations in 52 genes from 124 patients with enrichment in genes involved in DNA damage response, cell cycle, chromatin remodeling, and kinase pathways (supplemental Table 1). In 86 patients, genetic alterations were not observed, and the percentage of bone marrow–infiltrated CD20+ cells in these patients was not different from patients with detectable genetic alterations. Although MYD88L265P mutations were the most common, mutations in CXCR4, TP53, ARID1A, and TERT were also observed, with mutation rates of 25.7%, 24.8%, 15.7%, and 9.0%, respectively. Mutation rates of the remaining 48 genes were ≤4.3% (Figure 1; supplemental Table 2). Post-hoc analysis revealed FS and NS mutations were most common in CXCR4, and patients in the zanubrutinib arm had a higher rate of CXCR4MUT than patients in the ibrutinib arm (33.7% vs 21.7%; P = .08). Specifically, CXCR4FS mutations were more prevalent in patients in the zanubrutinib arm compared with those in the ibrutinib arm of the study (19.4% vs 7.6%; P = .02; supplemental Table 4). FS and NS mutations were also common in patients with ARID1AMUT; ARID1AMUT were more often detected in patients with CXCR4MUT compared with in those with CXCR4WT (44.4% vs 5.8%; P < .001; supplemental Table 3). Mutations in TP53 consisted of missense, truncation, and splice site mutations, with the majority localized in the DNA binding domain. TP53 mutations were more common in patients with CXCR4MUT compared with patients with CXCR4WT (35.2% vs 21.2%; P < .05; supplemental Table 3). TERT mutations were localized to promoter regions, including −57A>C, −124C>T, and −146C>T, and were detected more frequently in patients with CXCR4MUT compared with in those with CXCR4WT (24.1% vs 3.9%; P < .001; supplemental Table 3).
Figure 1.

Baseline genetic alteration landscape in 210 patients with WM treated with BTKis. DNA mutation profile of patients with WM and the distribution of mutations among different study cohorts by mutation type and treatment status (TN and RR). Each column represents 1 patient, and each row represents 1 gene (represented by the gene symbol). Mutation rates of each gene are shown on the left. Mutation type, treatment status, and best overall response are color coded as shown in the figure legend. BM, bone marrow; BOR, best overall response; MR, minor response; PD, progressive disease; RR, relapsed/refractory; SD, stable disease; TN, treatment naïve.
Response to BTKi therapy was inferior in patients with mutations in CXCR4 and TP53
To evaluate the association between genetic alterations and clinical efficacy of the BTKis zanubrutinib and ibrutinib in MYD88MUT WM, a pooled analysis of patients with MYD88MUT (n = 190) was conducted. Lower rates of VGPR + CR and longer times to response were generally observed in patients with the common mutations compared with in patients with respective WT alleles. Univariate analysis revealed that patients with CXCR4MUT and TERTMUT had significantly lower VGPR + CR and major response, respectively (Table 1, Table 2, Table 3) than patients with WT alleles; CXCR4MUT and TERTMUT cooccurred in 13 patients (supplemental Table 3). In patients with CXCR4MUT, TP53MUT, and TERTMUT, a dosage-dependent effect on PFS was observed; patients harboring mutations in these genes with VAF of ≥1% trended toward less favorable outcomes than patients with the respective WT alleles (supplemental Figure 1). Because TP53MUT and TERTMUT were associated with higher rates of CXCR4MUT, we performed multivariate analyses including CXCR4, TP53, and TERT mutational status and treatment as covariates (supplemental Table 3). Compared with patients with WT alleles, patients with CXCR4MUT had significantly lower VGPR + CR rates (17.0% vs 37.2%; P = .020; Table 1, Table 2) and patients with TP53MUT had worse PFS (P = .008; Figure 2).
Table 1.
Response assessment by CXCR4, TP53, TERT, and ARID1A mutational status in patients with WM with MYD88MUT
| CXCR4WT (n = 137) | CXCR4MUT (n = 53) | CXCR4FS (n = 26) | CXCR4NS (n = 27) | TP53WT (n = 142) | TP53MUT (n = 48) | TERTWT (n = 171) | TERTMUT (n = 19) | ARID1AWT (n = 158) | ARID1AMUT (n = 32) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Best overall response, n (%) | ||||||||||
| CR | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| VGPR | 51 (37.2) | 9 (17.0) | 5 (19.2) | 4 (14.8) | 48 (33.8) | 12 (25.0) | 58 (33.9) | 2 (10.5) | 52 (32.9) | 8 (25.0) |
| PR | 64 (46.7) | 30 (56.6) | 15 (57.7) | 15 (55.6) | 71 (50.0) | 23 (47.9) | 85 (49.7) | 9 (47.4) | 77 (48.7) | 17 (53.1) |
| MR | 16 (11.7) | 10 (18.9) | 4 (15.4) | 6 (22.2) | 16 (11.3) | 10 (20.8) | 20 (11.7) | 6 (31.6) | 21 (13.3) | 5 (15.6) |
| SD | 3 (2.2) | 3 (5.7) | 2 (7.7) | 1 (3.7) | 4 (2.8) | 2 (4.2) | 5 (2.9) | 1 (5.3) | 6 (3.8) | 0 (0.0) |
| PD | 2 (1.5) | 1 (1.9) | 0 (0.0) | 1 (3.7) | 2 (1.4) | 1 (2.1) | 2 (1.2) | 1 (5.3) | 2 (1.3) | 1 (3.1) |
| NE | 1 (0.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.7) | 0 (0.0) | 1 (0.6) | 0 (0.0) | 0 (0.0) | 1 (3.1) |
| VGPR or better | 51 (37.2) | 9 (17.0) | 5 (19.2) | 4 (14.8) | 48 (33.8) | 12 (25.0) | 58 (33.9) | 2 (10.5) | 52 (32.9) | 8 (25.0) |
| Major response | 115 (83.9) | 39 (73.6) | 20 (76.9) | 19 (70.4) | 119 (83.8) | 35 (72.9) | 143 (83.6) | 11 (57.9) | 129 (81.7) | 25 (78.1) |
| Time to response, median (min, max), mo | ||||||||||
| VGPR or CR | 8.4 (1.9, 50.0) | 11.1 (2.8, 46.0) | 11.1 (2.8, 26.0) | 13.9 (9.4, 46.0) | 9.3 (1.9, 50.0) | 11.1 (3.0, 46.9) | 9.3 (1.9, 50.0) | 34.1 (22.2, 46.0) | 9.8 (1.9, 49.9) | 10.7 (2.8, 46.0) |
| Major response | 2.8 (0.9, 49.8) | 4.6 (1.0, 49.8) | 6.6 (1.8, 49.8) | 3.7 (1.0, 38.7) | 2.9 (0.9, 49.8) | 2.9 (1.0, 13.8) | 2.8 (0.9, 49.8) | 5.6 (1.8, 22.2) | 2.8 (0.9, 49.8) | 3.0 (1.0, 38.7) |
Patients with CXCR4MUT, TP53MUT, and TERTMUT trended toward lower VGPR + CR rate, lower MRR, and/or longer median time to response than patients with the respective WT alleles.
max, maximum; min, minimum; MR, minor response; NE, not evaluable due to discontinuation before first assessment; PD, progressive disease; SD, stable disease.
Table 2.
Statistical analysis for rates of VGPR or better by CXCR4, TP53, TERT, and ARID1A mutational status in patients with WM with MYD88MUT
| Univariate analysis∗ |
Multivariate analysis† |
Multivariate analysis‡ |
||||
|---|---|---|---|---|---|---|
| Odds ratio (95% CI) | P value | Odds ratio (95% CI) | P value | Odds ratio (95% CI) | P value | |
| CXCR4MUT | 0.345 (0.156, 0.765) | .009 | 0.370 (0.160, 0.855) | .020 | - | - |
| CXCR4FS | 0.401 (0.143, 1.130) | .084 | - | - | 0.379 (0.130, 1.101) | .075 |
| CXCR4NS | 0.293 (0.096, 0.896) | .031 | - | - | 0.360 (0.112, 1.156) | .086 |
| TP53MUT | 0.653 (0.311, 1.368) | .259 | 0.780 (0.360, 1.690) | .529 | 0.781 (0.360, 1.692) | .530 |
| TERTMUT | 0.229 (0.051, 1.026) | .054 | 0.353 (0.075, 1.671) | .189 | 0.355 (0.075, 1.687) | .193 |
Patients with CXCR4MUT, TP53MUT, and TERTMUT trended toward lower VGPR + CR rate, lower MRR, and/or longer median time to response than patients with the respective WT alleles.
CI, confidence interval.
To compare the response rates, univariate logistic regression models were performed, and the odds ratio (95% CI) and the corresponding P values are shown.
Odds ratio (95% CI) and P values were estimated using a multivariate logistic regression model with treatment arm and CXCR4 (WT and MUT), TERT (WT and MUT), and TP53 (WT and MUT) mutational status as covariates to account for the correlations among the mutations and the treatment differences between treatment arms. WT is the reference group. The same models were performed to further compare the response rates between CXCR4WT and CXCR4NS or CXCR4FS (CXCR4 [WT, FS, and NS]).
MYD88 status was assessed by polymerase chain reaction–based assay, with a total of 190 patients with MYD88MUT WM.
Table 3.
Statistical analysis for major response rate by CXCR4, TP53, TERT, and ARID1A mutational status in patients with WM with MYD88MUT
| Univariate analysis∗ |
Multivariate analysis† |
Multivariate analysis‡ |
||||
|---|---|---|---|---|---|---|
| Odds ratio (95% CI) | P value | Odds ratio (95% CI) | P value | Odds ratio (95% CI) | P value | |
| CXCR4MUT | 0.533 (0.249, 1.142) | .106 | 0.718 (0.308, 1.677) | .444 | - | - |
| CXCR4FS | 0.638 (0.230, 1.768) | .387 | - | - | 0.737 (0.251, 2.167) | .579 |
| CXCR4NS | 0.454 (0.177, 1.167) | .101 | - | - | 0.701 (0.242, 2.027) | .512 |
| TP53MUT | 0.520 (0.239, 1.132) | .100 | 0.625 (0.276, 1.411) | .258 | 0.626 (0.277, 1.415) | .260 |
| TERTMUT | 0.269 (0.099, 0.729) | .010 | 0.345 (0.118, 1.014) | .053 | 0.348 (0.117, 1.038) | .058 |
Patients with CXCR4MUT, TP53MUT, and TERTMUT trended toward lower VGPR + CR rate, lower MRR, and/or longer median time to response than patients with the respective WT alleles.
Abbreviations are explained in Table 2.
To compare the response rates, univariate logistic regression models were performed, and the odds ratio (95% CI) and the corresponding P values are shown.
Odds ratio (95% CI) and P values were estimated using a multivariate logistic regression model with treatment arm and CXCR4 (WT and MUT), TERT (WT and MUT), and TP53 (WT and MUT) mutational status as covariates to account for the correlations among the mutations and the treatment differences between treatment arms. WT was the reference group. The same models were performed to further compare the response rates between CXCR4WT and CXCR4NS or CXCR4FS (CXCR4 [WT, FS, and NS]).
MYD88 status was assessed by polymerase chain reaction–based assay, with a total of 190 patients with MYD88MUT WM.
Figure 2.

Kaplan-Meier curves of PFS in patients with WM with MYD88MUT in relation to CXCR4, TP53, and TERT mutational status. Pooled analysis of patients with MYD88MUT, including 98 treated with zanubrutinib and 92 treated with ibrutinib. Kaplan-Meier curves of PFS were presented according to the mutational status of (A) CXCR4, (B) TP53, and (C) TERT. PFS in patients with CXCR4MUT, TP53MUT, and TERTMUT trended toward less favorable outcomes than in patients with the respective WT alleles. HR and P values were estimated using a Cox regression model with CXCR4 (WT and MUT), TP53 (WT and MUT), TERT (WT and MUT) mutational status and treatment arms as covariates. WT is the reference group.
Of patients with MYD88MUT WM, 22 of 190 (11.6%) patients had TP53MUT at VAF of <1% whereas 26 of 190 (13.7%) patients had TP53MUT at VAF of ≥1% or had a deletion of TP53. Patients with TP53MUT at VAF of ≥1% or TP53 deletion had higher rates of CXCR4NS (supplemental Table 5). A dosage effect of TP53 mutations in patients treated by both zanubrutinib and ibrutinib was observed. Compared with patients with TP53WT, patients with TP53MUT VAF of <1% treated with ibrutinib had lower rates of VGPR or better and major response, but patients with TP53MUT VAF of ≥1% or deletion had much lower rates of response and 42-month PFS (supplemental Table 6; supplemental Figure 3). Similarly, a lower 42-month PFS rate was observed in patients with TP53MUT VAF of <1% treated with zanubrutinib, and PFS was even lower in patients with TP53MUT VAF of ≥1% or deletions (supplemental Table 6; supplemental Figure 3). Because patients with TP53MUT VAF of ≥1% or deletion had higher rates of CXCR4NS and trended toward less favorable outcomes, a 1% VAF cutoff was used to assess TP53MUT associations with PFS. In both BTKi treatment groups, PFS in this population was inferior compared with patients with TP53WT or TP53MUT VAF of <1% (ibrutinib: HR, 3.792; P = .008; zanubrutinib: HR, 2.239; P = .140; supplemental Figure 3).
TERTMUT were detected in 19 patients (zanubrutinib: n = 10; ibrutinib: n = 9). Patients with TERTMUT, especially those with PFS events, had high rates of CXCR4 or TP53 comutations (supplemental Table 7) and less favorable PFS compared with patients with TERTWT (Table 1, Table 4, Table 5).
Table 4.
Response assessment by CXCR4, TP53, and TERT mutational statuses in patients treated with ibrutinib with MYD88MUT
| Patients with MYD88MUT treated with ibrutinib (n = 92) | |||||||
|---|---|---|---|---|---|---|---|
| CXCR4WT (n = 72) | CXCR4FS (n = 7) | CXCR4NS (n = 13) | TP53WT (n = 70) | TP53MUT (n = 22) | TERTWT (n = 83) | TERTMUT (n = 9) | |
| VGPR or better, n (%)∗ | 22 (30.6) | 0 | 2 (15.4) | 21 (30.0) | 3 (13.6) | 23 (27.7) | 1 (11.1) |
| OR (95% CI) | - | 0.14 (0.00, 3.23) | 0.64 (0.13, 3.08) | - | 0.44 (0.12, 1.55) | - | 0.52 (0.07, 3.79) |
| P value | - | .223 | .579 | - | .202 | - | .525 |
| Major response, n (%)∗ | 61 (84.7) | 6 (85.7) | 7 (53.8) | 60 (85.7) | 14 (63.6) | 70 (84.3) | 4 (44.4) |
| OR (95% CI) | - | 1.06 (0.10, 10.36) | 0.33 (0.07, 1.41) | - | 0.29 (0.09, 0.95) | - | 0.23 (0.04, 1.22) |
| P value | - | .958 | .135 | - | .040 | - | .085 |
| Time to VGPR or better, median (min, max), mo | 11.3 (2.0, 49.9) | - | 31.3 (16.6, 46.0) | 11.4 (2.0, 49.9) | 24.9 (5.6, 46.9) | 11.4 (2.0, 49.9) | 46.0 (46.0, 46.0) |
| Time to major response, median (min, max), mo | 2.8 (0.9, 49.8) | 7.0 (2.8, 41.5) | 2.9 (1.2, 13.6) | 2.9 (0.9, 49.8) | 3.0 (1.0, 13.8) | 2.8 (0.9, 49.8) | 10.3 (2.9, 13.8) |
| PFS† | |||||||
| Event-free rate at 42 months, % | 74.6 | 57.1 | 43.5 | 72.1 | 57.9 | 68.4 | 74.0 |
| Median, mo | NE | 44.2 | 39.8 | NE | 44.2 | NE | 48.2 |
| HR (95% CI) | - | 2.08 (0.70, 6.16) | 3.39 (1.23, 9.31) | - | 2.36 (1.10, 5.09) | - | 0.44 (0.10, 1.81) |
| P value | .185 | .017 | .027 | - | .257 | ||
Response rates, time to response, and PFS were compared according to the mutational status of CXCR4, TP53, and TERT genes in 92 patients with MYD88MUT WM treated with ibrutinib and 98 patients with MYD88MUT WM treated with zanubrutinib, respectively. MYD88 status was assessed by a polymerase chain reaction–based assay, with a total of 190 patients with MYD88MUT WM.
CI, confidence interval; max, maximum; min, minimum; NE, not estimable; OR, odds ratio.
Odds ratio and P values were estimated using a logistic regression model with CXCR4 (WT, NS, and FS), TP53 (WT and MUT), and TERT (WT and MUT) mutational status as covariates.
Median PFS was estimated by Kaplan-Meier method; HR and P values were estimated using a Cox regression model with CXCR4 (WT, FS, and NS), TP53 (WT and MUT), and TERT (WT and MUT) mutational statues as covariates. WT is the reference group.
Table 5.
Response assessment by CXCR4, TP53, and TERT mutational statuses in patients treated with zanubrutinib with MYD88MUT
| Patients with MYD88MUT treated with zanubrutinib (n = 98) | |||||||
|---|---|---|---|---|---|---|---|
| CXCR4WT (n = 65) | CXCR4FS (n = 19) | CXCR4NS (n = 14) | TP53WT (n = 72) | TP53MUT (n = 26) | TERTWT (n = 88) | TERTMUT (n = 10) | |
| VGPR or better, n (%)∗ | 29 (44.6) | 5 (26.3) | 2 (14.3) | 27 (37.5) | 9 (34.6) | 35 (39.8) | 1 (10.0) |
| OR (95% CI) | - | 0.51 (0.16, 1.66) | 0.24 (0.04, 1.26) | - | 1.27 (0.46, 3.52) | - | 0.25 (0.02, 2.26) |
| P value | - | .269 | .093 | - | .636 | - | .219 |
| Major response, n (%)∗ | 54 (83.1) | 14 (73.7) | 12 (85.7) | 59 (81.9) | 21 (80.8) | 73 (83.0) | 7 (70.0) |
| OR (95% CI) | - | 0.66 (0.18, 2.36) | 1.52 (0.25, 9.01) | - | 1.01 (0.29, 3.47) | - | 0.47 (0.09, 2.39) |
| P value | - | .524 | .639 | - | .978 | - | .362 |
| Time to VGPR or better, median (min, max), mo | 6.5 (1.9, 42.0) | 11.1 (2.8, 26.0) | 10.3 (9.4, 11.1) | 6.5 (1.9, 42.0) | 11.1 (3.0, 26.0) | 6.7 (1.9, 49.8) | 22.2 (22.2, 22.2) |
| Time to major response, median (min, max), mo | 2.8 (0.9, 28.5) | 2.9 (1.8, 49.8) | 4.1 (1.0, 38.7) | 2.8 (0.9, 49.8) | 2.8 (1.0, 5.6) | 2.8 (0.9, 49.8) | 3.7 (1.8, 22.2) |
| PFS† | |||||||
| Event-free rate at 42 months, % | 81.3 | 76.4 | 66.7 | 84.6 | 62.0 | 83.4 | 37.5 |
| Median, mo | NE | NE | NE | NE | NE | NE | 25.0 |
| HR (95% CI) | - | 0.62 (0.17, 2.25) | 0.67 (0.15, 2.88) | - | 2.20 (0.81, 5.98) | - | 5.78 (1.75, 19.13) |
| P value | .473 | .598 | .120 | - | .004 | ||
Response rates, time to response, and PFS were compared according to the mutational status of CXCR4, TP53, and TERT genes in 92 patients with MYD88MUT WM treated with ibrutinib and 98 patients with MYD88MUT WM treated with zanubrutinib, respectively. MYD88 status was assessed by a polymerase chain reaction–based assay, with a total of 190 patients with MYD88MUT WM.
CI, confidence interval; max, maximum; min, minimum; NE, not estimable; OR, odds ratio.
Odds ratio and P values were estimated using a logistic regression model with CXCR4 (WT, NS, and FS), TP53 (WT and MUT), and TERT (WT and MUT) mutational status as covariates.
Median PFS was estimated by Kaplan-Meier method; HR and P values were estimated using a Cox regression model with CXCR4 (WT, FS, and NS), TP53 (WT and MUT), and TERT (WT and MUT) mutational statues as covariates. WT is the reference group.
Among 20 patients with MYD88WT, 4 patients with TP53MUT had a lower MRR (50%) and none achieved VGPR or CR, compared with patients with TP53WT who had 62.5% MRR and a 25% VGPR + CR rate (Table 6). Generally, patients with TP53MUT tended to have less favorable PFS than patients with TP53WT; the 12-month PFS for TP53MUT vs TP53WT was 25% vs 75%, respectively (supplemental Figure 4).
Table 6.
Response assessment by TP53 mutation status in patients with MYD88WT
| Total (n = 20) | TP53WT (n = 16) | TP53MUT (n = 4) | |
|---|---|---|---|
| Best overall response, n (%) | |||
| CR | 1 (5.0) | 1 (6.3) | 0 (0.0) |
| VGPR | 3 (15.0) | 3 (18.8) | 0 (0.0) |
| PR | 8 (40.0) | 6 (37.5) | 2 (50.0) |
| MR | 4 (20.0) | 3 (18.8) | 1 (25.0) |
| SD | 3 (15.0) | 2 (12.5) | 1 (25.0) |
| PD | 1 (5.0) | 1 (6.3) | 0 (0.0) |
| VGPR or better, n (%) | 4 (20.0) | 4 (25.0) | 0 (0.0) |
| Major response, n (%) | 12 (60.0) | 10 (62.5) | 2 (50.0) |
| Time to response, median (min, max), mo | |||
| VGPR or CR | 9.6 (2.8, 22.1) | 9.6 (2.8, 22.1) | - |
| Major response | 3.4 (1.8, 44.6) | 3.3 (1.8, 44.6) | 4.3 (3.0, 5.5) |
Among 20 patients with MYD88WT, 4 patients with TP53MUT had a lower major response rate (50%), and none achieved VGPR or CR, compared with TP53WT patients.
max, maximum; min, minimum; MR, minor response; PD, progressive disease, SD, stable disease.
Zanubrutinib demonstrated deeper and faster responses, as well as favorable PFS, compared with ibrutinib in patients with CXCR4NS, CXCR4FS, or TP53MUT
To evaluate the response to different BTKis in patients with MYD88MUT with CXCR4MUT, TP53MUT, and TERTMUT, multivariate analysis was conducted separately for patients treated with zanubrutinib and those treated with ibrutinib. As has been reported previously, patients with CXCR4MUT in the ASPEN study had a lower VGPR + CR rate, lower MRR, and longer time to response than those with CXCR4WT, independent of treatment. However, patients with CXCR4MUT treated with zanubrutinib demonstrated deeper and faster responses, as well as more favorable PFS compared with patients treated with ibrutinib.
To address whether CXCR4 mutation type affected the patients’ response to BTKis, we compared treatment responses in patients with CXCR4NS vs CXCR4FS. Patients with either CXCR4NS or CXCR4FS had lower VGPR + CR response rates than patients with CXCR4WT, regardless of treatment, but patients treated with zanubrutinib trended toward a higher VGPR + CR rate than those treated with ibrutinib with CXCR4FS and CXCR4WT (Table 4, Table 5). Furthermore, relative to patients with CXCR4WT, patients with CXCR4NS had lower MRR with ibrutinib treatment but not with zanubrutinib (Table 4, Table 5). A logistic regression model with treatment group and TERT (WT and MUT) and TP53 (WT and MUT) mutational status as covariates was used to compare VGPR + CR rate and MRR between zanubrutinib and ibrutinib in patients with CXCR4NS and CXCR4FS. Compared with ibrutinib, patients with CXCR4FS treated with zanubrutinib had a more favorable VGPR + CR rate (26.3% vs 0%; P = .06) although MRR was similar (73.7% vs 85.7%). In patients with CXCR4NS, the VGPR + CR rates were similar between zanubrutinib and ibrutinib treatment groups (14.3% vs 15.4%). Patients treated with zanubrutinib had a more favorable MRR (85.7% vs 53.8%; P = .09). Patients with CXCR4NS and CXCR4FS had longer times to response to both ibrutinib and zanubrutinib than patients with CXCR4WT, but zanubrutinib treatment led to faster response compared with ibrutinib in all CXCR4 subgroups (Table 4, Table 5). Compared with CXCR4WT, CXCR4NS was significantly associated with inferior PFS (HR, 3.39; P = .02), and patients with CXCR4FS treated with ibrutinib trended toward a less favorable PFS (HR, 2.08; P = .185). PFS was not significantly affected by CXCR4 mutational status in patients treated with zanubrutinib (CXCR4NS: HR, 0.67; P = .60; CXCR4FS: HR, 0.62; P = .473; Table 5). Patients with either CXCR4NS or CXCR4FS treated with zanubrutinib had similar PFS, but PFS in patients with these mutations treated with ibrutinib was less favorable (supplemental Figure 2). In patients receiving ibrutinib, the median PFS (months) by CXCR4NS, CXCR4FS, and CXCR4WT was 39.8, 44.2, and not reached (NR), respectively, and NR in all subpopulations receiving zanubrutinib. Compared with ibrutinib treatment, zanubrutinib treatment was associated with trends toward more favorable PFS in patients with CXCR4NS (P = .09), CXCR4FS (P = .07), and CXCR4WT (P = .32; supplemental Figure 2).
In addition to detecting high rates of TP53MUT in the ASPEN study population, we further observed that, in patients treated with ibrutinib, these mutations were significantly associated with lower MRR (P = .04) and a trend toward a lower VGPR + CR rate (P = .20) than TP53WT. In patients treated with zanubrutinib, MRR (P = .98) and VGPR + CR rates (P = .64) were similar between patients with TP53MUT and TP53WT (Table 5). A logistic regression model with treatment group and TERT (WT and MUT) and CXCR4 (WT, FS, and NS) mutational status as covariates was used to compare the VGPR + CR rate and MRR between zanubrutinib and ibrutinib in patients with TP53MUT WM. Compared with ibrutinib, zanubrutinib treatment resulted in a better VGPR + CR rate (34.6% vs 13.6%; P < .05) and a more favorable MRR (80.8% vs 63.6%; P. =.11) in TP53MUT. Compared with patients with TP53WT, patients with TP53MUT had a longer time to response among both patients treated with ibrutinib and those treated with zanubrutinib, although patients treated with zanubrutinib responded faster (Table 4, Table 5). Similarly, patients with TP53MUT had worse PFS compared with patients with TP53WT in both zanubrutinib and ibrutinib treatment groups. However, in patients treated with ibrutinib, TP53MUT was associated with significantly inferior PFS vs TP53WT (HR, 2.36; P = .027) compared with patients treated with zanubrutinib (HR, 2.20; P = .120; Table 4, Table 5); the median PFS in patients with TP53MUT was not reached in the zanubrutinib treatment group and was 44.2 months in the ibrutinib treatment group (HR, 0.66; P = .37; Table 4, Table 5). In addition, 3 patients harbored both a TP53 deletion and mutation. One patient treated with ibrutinib had disease progression after 2.8 months of treatment, whereas 2 patients treated with zanubrutinib achieved VGPR and PR but progressed after 35.6 months and 16.9 months of treatment, respectively.
Patients with TP53MUT treated with ibrutinib exhibited a dosage effect on response rate by VAF; patients with TP53MUT with VAF of ≥10% or TP53 deletion had no VGPR + CR and a lower MRR (supplemental Table 6). This dosage effect on response rate was not observed in patients with TP53MUT treated with zanubrutinib. PFS analysis revealed that patients with TP53MUT at VAF of ≥1% or a TP53 deletion had worse PFS when treated with ibrutinib (HR, 3.792; P = .008) relative to patients treated with zanubrutinib (HR, 0.491; P = .22; supplemental Figure 3). In addition, patients with TP53MUT at VAF of ≥1% or TP53 deletion achieved 35.3% VGPR rate with zanubrutinib vs 0% VGPR with ibrutinib treatment (supplemental Table 6). Collectively, these data demonstrate that TP53 mutations confer a worse prognosis in patients treated with BTKi, but zanubrutinib had a more favorable outcome than ibrutinib in this higher risk population.
In summation, the adverse impact of CXCR4MUT and TP53MUT on response and PFS was more marked in patients treated with ibrutinib than patients treated with zanubrutinib.
Resistance mutations analysis
To begin to explore how acquired mutations may confer resistance to BTKis, we sequenced BMA samples from 5 patients with WM who progressed after achieving a response on zanubrutinib (3 with MYD88MUT and 2 with MYD88WT) to analyze the mutational status of BTK and other hematological malignancy–related genes. The median treatment duration was 27.9 months (range, 10.2-34.5 months), and paired baseline progression samples were available for 4 of 5 patients. BTKC481, a mutation associated with resistance, was identified in 1 patient with progressive disease after zanubrutinib treatment but unknown at baseline (supplemental Table 9). Additionally, 4 of 5 (80%) of progressive patients had TP53 mutations, and 2 of 5 (40%) had mutations within the TERT promoter (supplemental Table 9).
Discussion
In this follow-up to the ASPEN study, we examined NGS data and the mutational status of 210 patients with WM to document the association of common mutations with treatment outcomes in patients treated with either ibrutinib or zanubrutinib. Data from the ASPEN study suggest that, overall, patients with MYD88MUT with CXCR4MUT and TP53MUT have a worse response to BTKi treatment compared with patients with WT alleles and that patients with these mutations have generally better treatment outcomes with zanubrutinib.
In our study population, an NGS panel with sensitivity of 0.1% to 0.25% in nonenriched BMA samples identified mutation rates of CXCR4 and ARID1A comparable to previous studies,3,8 indicating that a highly sensitive NGS panel may compensate for nonenrichment of samples. However, compared with previous studies that observed a <14% mutation rate in TP53,3,9, 10, 11 in our study, the TP53 mutation rate (24.8%) was considerably higher. This observation may potentially be because the high-sensitivity assay used in this study detected a high rate (11.6%) of low-frequency (VAF < 1%) mutations compared with less sensitive assays such as Sanger sequencing, whole-exome sequencing, whole-genome sequencing, or targeted sequencing.
A previous study reported that patients with CXCR4NS who were treated with ibrutinib had worse odds of major response and worse PFS than patients with CXCR4FS and CXCR4WT.26 CXCR4 mutations may potentially activate cell survival pathways, therefore bypassing BTK to promote ibrutinib resistance. In cell culture, CXCR4NS and CXCR4FS mutations reportedly cause impaired membrane internalization, leading to prolonged phosphorylation of AKT1 and MAPK1 and, thus, prolonged survival signals for cancer cells.27 In our study, we also found that patients with CXCR4NS mutations had worse clinical outcomes than patients with CXCR4FS or CXCR4WT in treatment with ibrutinib, but response to zanubrutinib was not affected by CXCR4 mutation type. This may suggest that zanubrutinib, a more potent BTKi because of prolonged exposure to, or sustained occupancy on, BTK, can potentially offset the deleterious effects of CXCR4 mutations.28
Our current study, consistent with a previous report,9 found that TP53MUT is commonly associated with MYD88 and CXCR4 mutations. We demonstrated that patients with TP53 VAF <1% have similar PFS to patients with TP53WT, whereas patients with TP53 VAF between 1% and 10% or >10% fared worse. This observation suggests that low-frequency (VAF<1%) TP53 mutations may be small subclones detected with a highly sensitive NGS panel and of less clinical significance. The NGS assay used in this study has a validated limit of detection of 0.1% to 0.25% VAF. Furthermore, variant calls were made using a strict bioinformatic analysis pipeline defined by at least 3 distinct fragments containing mutations and VAF of ≥0.25% or hot-spot VAF of ≥0.1%. Thus, the low-frequency mutations observed in this study are unlikely to be false positives. Three patients harbored both TP53 deletions and mutations and all of them had disease progression after 16.9 months (range, 2.8-35.6 months) after BTKi treatment (2 treated with zanubrutinib). These data indicate that deficiencies in TP53 lead to a poor prognosis in patients with WM treated with BTKis, although validation in additional WM populations is warranted. Our finding that TP53 mutations have a significant negative impact on MRR and PFS in patients treated with ibrutinib, but not in patients treated with zanubrutinib, suggests that more potent BTKis, such as zanubrutinib, could improve the suboptimal response of patients with WM with TP53MUT.
Although mutations in CXCR4, ARID1A, and TP53 have previously been reported in WM, we also identified TERT promoter mutations in 19 patients with MYD88MUT. Although patients with TERTMUT had high rates of CXCR4 or TP53 comutations, no associations between TERTMUT and clinical response were observed (Table 1, Table 2, Table 3; Figure 2). Although more PFS events were observed in patients with TERTMUT treated with zanubrutinib, this may be because of a higher rate of cooccurring CXCR4MUT and TP53MUT in these patients than in those in the ibrutinib treatment arm (60% vs 22.2%). Because of unsorted BMA samples used in this study, we are not able to confirm whether the TERT mutations that we observed were derived from tumor cells or clonal hematopoiesis of indeterminate potential. Subsequent studies to validate TERT mutations in additional WM populations or using single-cell sequencing to elucidate its functional role in WM pathogenesis are needed to determine whether these mutations represent a risk factor or have prognostic value in WM.
The main limitation of this study is the small sample size of patients available for analysis with each mutation type. Further comparisons of zanubrutinib and ibrutinib in patients with these risk mutations from other clinical studies or real-world data are warranted. A second potential limitation is that baseline BMA samples were not enriched for tumor cells before NGS sequencing. A third limitation is that, because paired baseline and disease progression samples were only available for 4 patients treated with zanubrutinib, comprehensive analysis of potential zanubrutinib resistance mechanisms in patients with WM is limited. Finally, it is unclear whether TERT mutations are pathogenic, and additional studies to validate and elucidate their function in WM pathogenesis are needed.
In conclusion, patients with WM harboring mutations in CXCR4 and TP53 were found to have poorer prognosis after treatment with BTKis than those without mutations in these genes, and patients with these mutations who received zanubrutinib had favorable outcomes compared with those who received ibrutinib. Thus, inclusion of the mutational status of MYD88, CXCR4, and TP53 in genomic-based treatment algorithms may be valuable when assessing BTKi treatment options for patients with WM. Our data further suggest that zanubrutinib can be effective independent of MYD88, CXCR4, and TP53 mutational status.
Conflict-of-interest disclosure: C.S.T. receives research funding from Janssen, AbbVie, and BeiGene; and receives honoraria from Janssen, AbbVie, BeiGene, LOXO, and AstraZeneca. S.D. is an employee of, and has equity ownership in, BeiGene; has acted as a consultant/advisor for Janssen, BeiGene, and Sanofi; and has received research funding from BeiGene. W.J. has acted as a consultant/advisor for AbbVie, AstraZeneca, BeiGene, Lilly, Roche, and Takeda; and has received research funding from AbbVie, AstraZeneca, Janssen, BeiGene, Lilly, Roche, Merck, MorphoSys, and Takeda. H.-P.L. has acted as a consultant/advisor for BeiGene. G.C. has received research funding from BeiGene, AstraZeneca, and Glycomimetics. R.G.O. has received honoraria from Janssen-Cilag, BeiGene, and AstraZeneca; and has acted as a consultant/advisor for BeiGene and Janssen-Cilag. P.M. has received consulting fees from BeiGene, Janssen, Astellas, AbbVie, Roche, Novartis, Gilead, IQVIA, and Otsuka; has received honoraria from Janssen, AbbVie, and Roche; and has served on an advisory board for The Australasian Leukaemia and Lymphoma Group. B.E.W. has received research funding from Roche and Incyte. R.G.-S. has received research funding from Gilead and Astellas; has received loyalties from IVS; has received consulting fees from Janssen, Incyte, and BeiGene; and has received honoraria from Millenium/Takeda, Janssen, Incyte, Amgen, BeiGene, AstraZeneca, and Pfizer. H.M. has received honoraria from AstraZeneca, BeiGene, and Janssen. A.T. has received honoraria from Janssen, AbbVie, AstraZeneca, and BeiGene; and has received travel support from Janssen, AbbVie, BeiGene, and AstraZeneca. J.J.C. has acted as a consultant/advisor for AbbVie, BeiGene, Cellectar, Janssen, and PharmaCytics; and has received research funding from AbbVie, AstraZeneca, BeiGene, Loxo Oncology, Pharmacyclics, and TG Therapeutics. J.C. has received travel expenses from Janssen. C.F.D.L.R. has received honoraria from Janssen, BeiGene, Bristol Myers Squibb, Amgen, and GlaxoSmithKline; has acted as a consultant/advisor for Janssen, BeiGene, Bristol Myers Squibb, Amgen, GlaxoSmithKline, and Pfizer; has received research funding from Janssen, Amgen, and Bristol Myers Squibb; and has received travel expenses from Janssen, BeiGene, Bristol Myers Squibb, Amgen, and GlaxoSmithKline. D.B. has acted as a consultant/advisor for Roche, Takeda, Janssen-Cilag, Gilead Sciences, and Novartis; has received research funding from Roche, Janssen-Cilag, Genmab, and MorphoSys; and has received travel expenses from Roche, Takeda, and Gilead Sciences. E.L. has acted as a consultant/advisor for Pharmacyclics; and has received research funding from BeiGene, Bristol Myers Squibb, and Janssen. J.M. has received consulting fees from BeiGene and Pharmacyclics; has received honoraria from BeiGene; and has participated in a data safety monitoring board or advisory board for BeiGene. M. Trněný has received honoraria from Takeda, Novartis, Janssen, AbbVie, Gilead Sciences, Roche, Bristol Myers Squibb, Amgen, and MorphoSys; has acted as a consultant/advisor for Roche, Bristol Myers Squibb, Amgen, Gilead Sciences, Novartis, MorphoSys, Incyte, Takeda, AbbVie, and Janssen; and has received travel funding from Gilead Sciences, Bristol Myers Squibb, Janssen, Takeda, Roche, and AbbVie. M.C.M. has acted as a consultant/advisor for Janssen-Cilag; and has served on the speaker’s bureau for Bristol Myers Squibb, Janssen-Cilag, and Medscape. C.B. has received honoraria from Roche/Genentech, Janssen, BeiGene, Novartis, Pfizer, Incyte, AbbVie, Gilead Sciences, Celltrion, MorphoSys, Regeneron, and Sobi; has received research funding from Roche/Genentech, Janssen, Celltrion, Merck Sharp & Dohme, Pfizer, and Amgen; and has served on the speaker’s bureau for Roche/Genentech, Janssen, BeiGene, Novartis, Pfizer, Incyte, AbbVie, Gilead Sciences, Celltrion, MorphoSys, Regeneron, Sobi, and Lilly. V.L. has received consulting fees from BeiGene, AstraZeneca, and Merck Sharp & Dohme. S.P.T. has received research funding from BeiGene; has received consulting fees from BeiGene, Janssen, and Pharmacyclics/AbbVie; and has received honoraria from BeiGene, Pharmacyclics/AbbVie, and Janssen. J.T. has served on an advisory board for BeiGene. W.Y.C. has equity in Bristol Myers Squibb. M.D. has received honoraria from BeiGene, Amgen, Bristol Myers Squibb, Takeda, and Janssen. B.W., Y.Y., Z.S., W.Y.C., J.S., H.A., and A.C. are employees of, and have equity ownership in, BeiGene. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank the study patients, their supporters, the investigators, and clinical research staff at the study centers. Assistance with medical writing and editorial support, under the direction of the authors, was provided by Lisa McGraw and Elizabeth Hermans, who are employees of, and holders of stocks/shares in, BeiGene Co.
Authorship
Contribution: B.W., Y.Y., Z.S., W.Y.C., H.A., and A.C. designed the research, analyzed the data, generated figures, and assisted in drafting the manuscript; J.S. provided statistical data analysis; C.S.T, S.O., S.D., W.J., H.-P.L., G.C., R.G.O., P.M., B.E.W., R.G.-S., H.M., S.M., A.T., J.J.C., J.C., C.F.D.L.R., D.B., E.L., J.M., M.M., T.S., M. Tani, M. Trněný, M.C.M, C.B., V.L., S.P.T., and J.T. enrolled patients and collected clinical data; and all authors contributed to data interpretation and reviewed the manuscript.
Footnotes
All authors had access to the original data for the analyses described here. On request, and subject to certain criteria, conditions, and exceptions, BeiGene, Ltd will provide access to individual deidentified participant data from BeiGene-sponsored global interventional clinical studies conducted for medicines (1) for indications that have been approved or (2) in programs that have been terminated. BeiGene will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data requests may be submitted to datadisclosure@beigene.com.
The full-text version of this article contains a data supplement.
Supplementary Material
References
- 1.Gertz MA. Waldenström macroglobulinemia: 2019 update on diagnosis, risk stratification, and management. Am J Hematol. 2019;94(2):266–276. doi: 10.1002/ajh.25292. [DOI] [PubMed] [Google Scholar]
- 2.Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol Cancer. 2018;17(1) doi: 10.1186/s12943-018-0779-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hunter ZR, Xu L, Yang G, et al. The genomic landscape of Waldenström macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123(11):1637–1646. doi: 10.1182/blood-2013-09-525808. [DOI] [PubMed] [Google Scholar]
- 4.Yang G, Zhou Y, Liu X, et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenström macroglobulinemia. Blood. 2013;122(7):1222–1232. doi: 10.1182/blood-2012-12-475111. [DOI] [PubMed] [Google Scholar]
- 5.Kaiser LM, Hunter ZR, Treon SP, Buske C. CXCR4 in Waldenström’s macroglobulinema: chances and challenges. Leukemia. 2021;35(2):333–345. doi: 10.1038/s41375-020-01102-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenström macroglobulinemia. Blood. 2014;123(18):2791–2796. doi: 10.1182/blood-2014-01-550905. [DOI] [PubMed] [Google Scholar]
- 7.Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenström’s macroglobulinemia. N Engl J Med. 2015;372(15):1430–1440. doi: 10.1056/NEJMoa1501548. [DOI] [PubMed] [Google Scholar]
- 8.Castillo JJ, Moreno DF, Arbelaez MI, Hunter ZR, Treon SP. CXCR4 mutations affect presentation and outcomes in patients with Waldenström macroglobulinemia: a systematic review. Expert Rev Hematol. 2019;12(10):873–881. doi: 10.1080/17474086.2019.1649132. [DOI] [PubMed] [Google Scholar]
- 9.Gustine JN, Tsakmaklis N, Demos MG, et al. TP53 mutations are associated with mutated MYD88 and CXCR4, and confer an adverse outcome in Waldenström macroglobulinaemia. Br J Haematol. 2019;184(2):242–245. doi: 10.1111/bjh.15560. [DOI] [PubMed] [Google Scholar]
- 10.Poulain S, Roumier C, Bertrand E, et al. TP53 mutation and its prognostic significance in Waldenström's macroglobulinemia. Clin Cancer Res. 2017;23(20):6325–6335. doi: 10.1158/1078-0432.CCR-17-0007. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Gali VL, Xu-Monette ZY, et al. Molecular and genetic biomarkers implemented from next-generation sequencing provide treatment insights in clinical practice for Waldenström macroglobulinemia. Neoplasia. 2021;23(4):361–374. doi: 10.1016/j.neo.2021.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dimopoulos M, Sanz RG, Lee H-P, et al. Zanubrutinib for the treatment of MYD88 wild-type Waldenström macroglobulinemia: a substudy of the phase 3 ASPEN trial. Blood Adv. 2020;4(23):6009–6018. doi: 10.1182/bloodadvances.2020003010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trotman J, Opat S, Gottlieb D, et al. Zanubrutinib for the treatment of patients with Waldenström macroglobulinemia: 3 years of follow-up. Blood. 2020;136(18):2027–2037. doi: 10.1182/blood.2020006449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Treon SP, Meid K, Gustine J, et al. Long-term follow-up of ibrutinib monotherapy in symptomatic, previously treated patients with Waldenström macroglobulinemia. J Clin Oncol. 2021;39(6):565–575. doi: 10.1200/JCO.20.00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu L, Tsakmaklis N, Yang G, et al. Acquired mutations associated with ibrutinib resistance in Waldenström macroglobulinemia. Blood. 2017;129(18):2519–2525. doi: 10.1182/blood-2017-01-761726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen JG, Liu X, Munshi M, et al. BTKCys481Ser drives ibrutinib resistance via ERK1/2 and protects BTKwild-type MYD88-mutated cells by a paracrine mechanism. Blood. 2018;131(18):2047–2059. doi: 10.1182/blood-2017-10-811752. [DOI] [PubMed] [Google Scholar]
- 17.Jiménez C, Chan GG, Xu L, et al. Genomic evolution of ibrutinib-resistant clones in Waldenström macroglobulinaemia. Br J Haematol. 2020;189(6):1165–1170. doi: 10.1111/bjh.16463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guerrera ML, Tsakmaklis N, Xu L, et al. MYD88 mutated and wild-type Waldenström’s macroglobulinemia: characterization of chromosome 6q gene losses and their mutual exclusivity with mutations in CXCR4. Haematologica. 2018;103(9):e408–e411. doi: 10.3324/haematol.2018.190181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tam CS, Opat S, D'Sa S, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood. 2020;136(18):2038–2050. doi: 10.1182/blood.2020006844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakravarty D, Gao J, Phillips S, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017;2017(1):1–16. doi: 10.1200/PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.1000 Genomes Project Consortium. Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karczewski KJ, Weisburd B, Thomas B, et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017;45(D1):D840–d845. doi: 10.1093/nar/gkw971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glusman G, Caballero J, Mauldin DE, Hood L, Roach JC. Kaviar: an accessible system for testing SNV novelty. Bioinformatics. 2011;27(22):3216–3217. doi: 10.1093/bioinformatics/btr540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Owen RG, Kyle RA, Stone MJ, et al. Response assessment in Waldenström macroglobulinaemia: update from the VIth International Workshop. Br J Haematol. 2013;160(2):171–176. doi: 10.1111/bjh.12102. [DOI] [PubMed] [Google Scholar]
- 26.Castillo JJ, Xu L, Gustine JN, et al. CXCR4 mutation subtypes impact response and survival outcomes in patients with Waldenström macroglobulinaemia treated with ibrutinib. Br J Haematol. 2019;187(3):356–363. doi: 10.1111/bjh.16088. [DOI] [PubMed] [Google Scholar]
- 27.Cao Y, Hunter ZR, Liu X, et al. CXCR4 WHIM-like frameshift and nonsense mutations promote ibrutinib resistance but do not supplant MYD88L265P-directed survival signalling in Waldenström macroglobulinaemia cells. Br J Haematol. 2015;168(5):701–707. doi: 10.1111/bjh.13200. [DOI] [PubMed] [Google Scholar]
- 28.Tam CS, Trotman J, Opat S, et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood. 2019;134(11):851–859. doi: 10.1182/blood.2019001160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.