

Abstract
Muscle‐invasive bladder cancer (MIBC) is a disease characterized by molecular and clinical heterogeneity, posing challenges in selecting the most appropriate treatment in clinical settings. Considering the significant role of CD4+ T cells, there is an emerging need to integrate CD4+ T cells with molecular subtypes to refine classification. We conducted a comprehensive study involving 895 MIBC patients from four independent cohorts. The Zhongshan Hospital (ZSHS) and The Cancer Genome Atlas (TCGA) cohorts were included to investigate chemotherapeutic response. The IMvigor210 cohort was included to assess the immunotherapeutic response. NCT03179943 was used to evaluate the clinical response to a combination of immune checkpoint blockade (ICB) and chemotherapy. Additionally, we evaluated genomic characteristics and the immune microenvironment to gain deeper insights into the distinctive features of each subtype. We unveiled four immune‐molecular subtypes, each exhibiting distinct clinical outcomes and molecular characteristics. These subtypes include luminal CD4+ Thigh, which demonstrated benefits from both immunotherapy and chemotherapy; luminal CD4+ Tlow, characterized by the highest level of fibroblast growth factor receptor 3 (FGFR3) mutation, thus indicating potential responsiveness to FGFR inhibitors; basal CD4+ Thigh, which could benefit from a combination of ICB and chemotherapy; and basal CD4+ Tlow, characterized by an immune suppression microenvironment and likely to benefit from transforming growth factor‐β (TGF‐β) inhibition. This immune‐molecular classification offers new possibilities for optimizing therapeutic interventions in MIBC.
Keywords: CD4+ T cells, immune‐molecular subtype, immunotherapy, muscle‐invasive bladder cancer, platinum‐based chemotherapy
This study defined an immune‐molecular subtyping classification with specific genomic characteristics, immune phenotypes, oncogenic pathways, gene expression, and clinical outcomes based on integrating CD4+ T cells and molecular subtypes, which is hoped to serve as a foundation for developing tailored therapeutic approaches for MIBC patients.
Abbreviations
- BC
bladder cancer
- BCR
B cell receptor
- EMT
epithelial–mesenchymal transition
- FGFR3
fibroblast growth factor receptor 3
- FPKM
fragments per kilobase of transcript per million mapped reads
- GSEA
gene set enrichment analysis
- GZMB
granzyme B
- ICB
immune checkpoint blockade
- IFN‐γ
interferon‐γ
- IHC
immunohistochemistry
- MHC
major histocompatibility complex
- MIBC
muscle‐invasive bladder cancer
- MSI
microsatellite instability score
- NMIBC
non‐muscle‐invasive bladder cancer
- OS
overall survival
- PD‐L1
programmed cell death‐ligand 1
- SD
stable disease
- TCGA
The Cancer Genome Atlas
- TCR
T cell receptor
- TEX
exhausted T cells
- TGF‐β
transforming growth factor‐β
- TMA
tissue microarray
- TMB
tumor mutation burden
- TNB
tumor neoantigen burden
- TRM
tissue‐resident memory T cells
- UC
urothelial carcinoma
- ZSHS
Zhongshan Hospital
1. INTRODUCTION
Muscle‐invasive bladder cancer (MIBC) presents a complex and diverse landscape at the molecular and clinical levels, contributing to its high morbidity and mortality. 1 While cisplatin‐based chemotherapy has been the standard treatment for MIBC, over 50% of patients cannot receive this therapy due to renal function impairment, limitations in performance status, or comorbidities. 2 , 3 Immune checkpoint blockade (ICB) has demonstrated remarkable survival benefits in recent years, but only about 20% of patients achieve significant improvements. 4 , 5 Given the relatively low response rate and the high cost associated with immunotherapy, there is a pressing need to develop a more precise and personalized classification to guide treatment selection.
Numerous research teams have presented molecular classifications for bladder cancer (BC). Various gene expression‐based schemes have emerged, encompassing the entire spectrum of BC. These classifications have significantly advanced our comprehension of biology in BC. 6 , 7 In a broad sense, these molecular subtypes can be categorized into two main groups: luminal and basal subtypes. Several studies have underscored the clinical significance of molecular stratification in MIBC, suggesting that specific MIBC subtypes might exhibit enriched responses to chemotherapy and immunotherapy. 8 , 9 , 10 With the increasing prominence of immunotherapy, it is imperative to incorporate tumor immune features into the classification system.
Studies have indicated that various immune cell subsets could serve as valuable indicators for predicting immunotherapy response. 11 , 12 , 13 Recently, the role of CD4+ T cells in the tumor immune microenvironment has drawn increasing attention. It has been observed that CD4+ T cells can bolster CD8+ T cell‐mediated killing in ICB‐induced tumor immunity. 14 Consistently, our previous studies revealed the importance of different CD4+ T cell subsets in orchestrating immune responses. 15 , 16 , 17 Considering its crucial role in tumor immunity, incorporating CD4+ T cells with the existing molecular subtypes may aid in selecting appropriate treatments for MIBC patients.
In this study, we integrated CD4+ T cells with molecular subtypes to establish a novel immune‐molecular classification in MIBC. This innovative classification holds the potential to significantly enhance patient selection for both current and emerging therapies, leading to more personalized treatment strategies for MIBC patients.
2. MATERIALS AND METHODS
2.1. Study cohorts
Four independent cohorts of 895 MIBC patients were analyzed in this study including the Zhongshan Hospital (ZSHS) cohort, The Cancer Genome Atlas (TCGA) cohort, the IMvigor210 cohort, and NCT03179943 (Figure S1A).
The ZSHS cohort involved 215 patients who underwent radical cystectomy from 2002 to 2014 at ZSHS, Fudan University. A total of 81 patients were excluded due to non‐urothelial carcinoma (n = 13), non‐muscle‐invasive bladder cancer (NMIBC, n = 60), and tissue detachment on the tissue microarray (TMA) (n = 1). Finally, we enrolled 141 patients and 69 individuals received chemotherapy. Informed consent was obtained from all patients, and the study was approved by the Clinical Research Ethics Committee of Zhongshan Hospital.
The Cancer Genome Atlas cohort initially included 412 patients from TCGA database. Twenty‐one patients were excluded for various reasons, such as insufficient follow‐up information or RNA sequencing data (n = 7), NMIBC pathologic diagnoses (n = 4), and receiving preoperative therapy (n = 10). Ultimately, TCGA cohort consisted of 391 patients, including 124 patients who underwent chemotherapy.
The IMvigor210 cohort consisted of 348 patients from the IMvigor210 trial, a single‐arm phase II study designed to investigate the clinical effect of atezolizumab in patients with locally advanced or metastatic urothelial carcinoma (UC). 4 Both expression data and relevant clinical data in the IMvigor210 cohort were downloaded from http://research‐pub.gene.com/IMvigor210CoreBiologies/.
NCT03179943 is a single‐arm phase II study designed to test the combination of guadecitabine and atezolizumab in patients with metastatic UC who had experienced primary or secondary progression on ICB and were either not suitable for or had already received platinum‐based chemotherapy. 18 Fifteen of them with pretreatment RNA sequencing data were included. Additional clinicopathological characteristics of these cohorts are listed in Tables S1 and S2.
2.2. Processing of transcriptomic and genomic data
Transcriptomic data were obtained simultaneously with the collection of clinical information. We used fragments per kilobase of transcript per million mapped reads (FPKM) to calculate the expression of genes and normalized mRNA expression by log2(FPKM+1) before analysis. Pathway signature scores were calculated by the mean arithmetic value of log2(FPKM+1) based on the related gene set expression that was reported previously (detailed gene lists are listed in Table S3). 19 , 20 , 21 , 22 , 23 , 24 The total nonsilent somatic mutation per megabase (mut/Mb), which was defined as tumor mutation burden (TMB), was acquired from https://portal.gdc.cancer.gov/. The decomposition of 30 cosmic mutational signatures was performed using the “deconstructSigs” package. We grouped cosmic mutational signatures with similar etiology into nine signature clusters for further analysis: APOBEC mutational signature consisted of signature 2 and signature 13; aging mutational signature consisted of signature 1 and signature 5; tobacco mutational signature consisted of signature 4 and signature 29; homologous recombination defect (HRD) mutational signature consisted of signature 3 and signature 8; defective mismatch repair (dMMR) mutational signature consisted of signatures 6, 14, 15, 20, 21, and 26; polymerase ε (POLE) mutational signature consisted of signature 10; alkylating mutational signature consisted of signature 11; and ultraviolet (UV) mutational signature consisted of signature 7. Signature clusters were generated through the arithmetic summation of the individual signatures mentioned above. Others mutational signature is obtained by subtracting the other 8 mutational signatures from 1. The copy number variation (CNV) data were acquired from cBioPortal for Cancer Genomics. The OncoKB dataset (https://www.oncokb.org/) was matched with TCGA cohort to delineate novel therapeutic targets. The tumor neoantigen burden (TNB) data were evaluated using the methods described in a previous study. 6 The involved genes for gene set enrichment analysis (GSEA) were downloaded from https://gsea‐msigdb.org (detailed sources of GSEA pathways are listed in Table S4). Gene alterations were identified as either nonsense, missense, frameshift, splice‐site, in‐frame deletion variants, or deleterious homozygous deletions and amplifications.
2.3. Immunohistochemistry (IHC) and assay methods
Tissue microarray was constructed and IHC was performed following previously established procedures. 25 Formalin‐fixed and paraffin‐embedded tumor tissues from patients with MIBC were collected from the ZSHS. Each sample was subjected to hematoxylin and eosin staining for assessment, and representative areas were marked on the paraffin blocks. Two tissue cores measuring 1.0 mm from distinct regions were used to build the TMAs. TMA sections were deparaffinized using xylene and then hydrated with distilled water. Following a 30‐min inhibition of endogenous peroxidase with 3% H2O2, the sections underwent a 5‐min heat treatment in unmasking solution (0.01 M sodium citrate buffer, pH = 6) using a pressure cooker. Subsequently, TMA sections were incubated with 10% normal goat serum for 30 min. Anti‐CD4 antibody (Ab67001, diluted 1:50; Abcam) was applied overnight at 4°C in a humid chamber to detect CD4+ T cells in the tissue. After removing the primary antibody, positive staining was revealed using the DAB detection system. Afterward, the sections underwent counterstaining with hematoxylin, dehydration, mounting, and observation. Digital images of TMAs were scanned by NanoZoomer‐XR (Hamamatsu) and Image Pro Plus 6.0 under a high‐power magnification field. The number of CD4+ T cells per high‐power field (HPF, ×200 magnification) was defined as a density score (total number of positive cells per HPF) by two pathologists who were blinded to the clinicopathological data, and the average of the five fields was taken. The cutoff value for CD4+ T cells in each cohort was the median value. Antibodies used for immunostaining are listed in Table S5.
2.4. Statistical analysis
Statistical p‐values were conducted using two‐way tests, and detailed statistical tests are shown in corresponding figure legends. Log‐rank test was conducted for Kaplan–Meier analysis to compare overall survival (OS) between different subgroups. The Pearson chi‐squared test or Fisher exact test was applied for the analysis of categorical variables, and Mann–Whitney U test was performed for continuous variables.
All statistical analyses were conducted using IBM SPSS Statistics 26.0, Graph pad Prism 8.0.1, GSEA 4.1.0, and R software 4.1.2. p ≤ 0.05 was considered statistically significant.
3. RESULTS
3.1. Interplay of CD4 + T cells and molecular subtypes impacts on OS and chemotherapeutic response in MIBC
Though CD4+ T cells and molecular subtypes alone could predict prognosis neither in TCGA (Figure S2A) nor in the ZSHS cohort (Figure S2B), CD4+ T cells had prognostic value in patients with basal subtype (Figure S2C,D). Therefore, we classified patients into four subgroups based on the CD4+ T cells and molecular subtypes: luminal CD4+ Thigh, luminal CD4+ Tlow, basal CD4+ Thigh, and basal CD4+ Tlow. Kaplan–Meier analysis was conducted to demonstrate the clinical outcome of this stratification. We found patients in the basal CD4+ Tlow group possessed the worst prognosis (TCGA cohort, Log rank p = 0.003; ZSHS cohort, Log rank p = 0.087; Figure 1A). For patients receiving chemotherapy, better OS was observed in patients with high CD4+ T cells, while the molecular subtypes showed no difference in either TCGA or the ZSHS cohort (Figure S3A,B). Furthermore, patients with high infiltration of CD4+ T cells could benefit more from chemotherapy in the luminal subtype rather than the basal subtype (Figure S3C,D). After dividing people into four subgroups, we observed that the luminal CD4+ Thigh subgroup could significantly lead to improved OS in patients receiving chemotherapy in TCGA (Log rank p = 0.066, Figure 1B) and the ZSHS cohorts (Log rank p < 0.001, Figure 1B).
FIGURE 1.
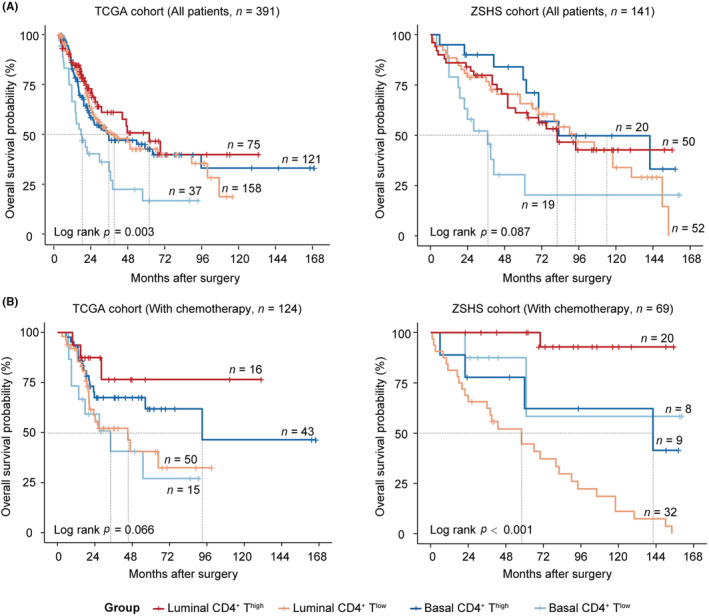
The interplay of CD4+ T cells and molecular subtypes impacts on OS and chemotherapeutic response in MIBC. (A) Kaplan–Meier analysis of OS in TCGA cohort (left) and the ZSHS cohort (right), stratified by CD4+ T cells and molecular subtypes. (B) Kaplan–Meier analysis of OS in patients receiving chemotherapy, performed in TCGA cohort (left) and the ZSHS cohort (right), stratified by CD4+ T cells and molecular subtypes. Log‐rank test was conducted for Kaplan–Meier analysis. MIBC, muscle‐invasive bladder cancer; OS, overall survival; ZSHS, Zhongshan Hospital.
3.2. Patients with luminal subtype and high CD4 + T cells show superior response to PD‐L1 blockade in MIBC
We further investigated the relationship between the immune‐molecular subtypes and PD‐L1 blockade. In the context of atezolizumab treatment, patients with the luminal subtype demonstrated improved OS, while no significant association was observed between patients with different expression levels of CD4+ T cells and treatment outcomes (Figure S4A,B). However, patients with high CD4+ T cells showed better OS in patients with luminal subtype (Log rank p = 0.038, Figure 2A) rather than basal subtype (Log rank p = 0.470, Figure 2A). We then assessed the predictive value to PD‐L1 blockade of this stratification. A notable association between patients in the luminal CD4+ Thigh subgroup and improved OS was observed (Log rank p = 0.021, Figure 2A). Additionally, this subgroup exhibited the highest objective response rate (ORR, 46.4%, Figure 2B). Among patients with metastatic UC who had previously experienced primary or secondary progression on a checkpoint inhibitor, the basal CD4+ Thigh subgroup demonstrated enhanced OS when treated with a combination therapy of guadecitabine and atezolizumab (Figure 2C). Moreover, all patients who responded as stable disease (SD) were present within this particular subgroup (Figure 2C).
FIGURE 2.
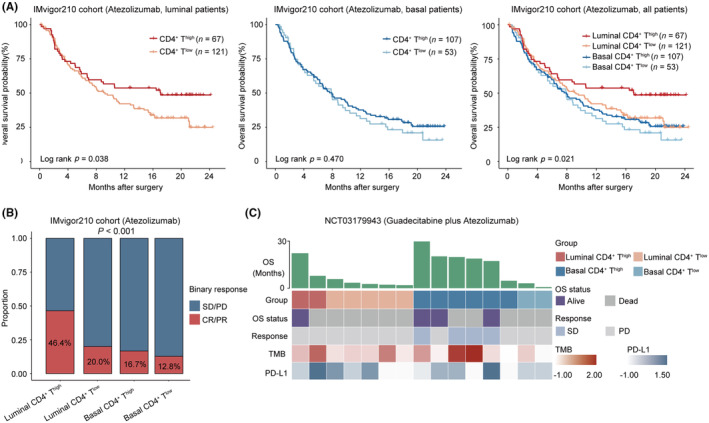
Patients with luminal subtype and high CD4+ T cells show superior response to PD‐L1 blockade in MIBC. (A) Kaplan–Meier analysis of OS in the IMvigor210 cohort according to high/low CD4+ T cells in luminal subtype (left), basal subtype (middle), and four subgroups based on CD4+ T cells and molecular subtypes in all patients (right) after treatment of atezolizumab. (B) Analysis of response to atezolizumab across four subgroups based on CD4+ T cells and molecular subtypes. (C) Heatmap demonstrating clinical information of four subgroups based on CD4+ T cells and molecular subtypes with response to guadecitabine plus atezolizumab in NCT03179943. Log‐rank test was conducted for Kaplan–Meier analysis. Mann–Whitney U test and Pearson's chi‐square test were also applied. CR, complete response; MIBC, muscle‐invasive bladder cancer; OS, overall survival; PD, progressive disease; PR, partial response; SD, stable disease; TMB, tumor mutation burden.
3.3. Genomic characteristics of MIBC stratified by CD4 + T cells and molecular subtypes
We further assessed the genomic characteristics across the stratification based on CD4+ T cells and molecular subtypes. The analysis suggested a higher TMB (p < 0.001, Figure 3A) and microsatellite instability score (MSI, p < 0.001, Figure 3B) in the luminal CD4+ Thigh subgroup. Moreover, the results indicated elevated levels of both APOBEC and POLE mutational signatures in the luminal CD4+ Thigh subgroup (Figure 3C). These mutational features have previously been identified as biomarkers associated with the response to immunotherapy. 26 , 27 The basal CD4+ Tlow subgroup exhibited the highest proportion of homozygous deletion of 9p21.3 (43.2%, Figure 3D), which confers “cold” tumor immune phenotypes according to a previous report. 28 The luminal CD4+ Thigh subgroup exhibited the highest amplification of the 11q13.3 locus (21.3%, Figure 3D). Gene alteration analysis revealed that FGFR3 (30.1%, p < 0.001) and ELF3 (19.0%, p = 0.001) mutations were more frequent in the luminal CD4+ Tlow subgroup, while BRCA1 (16.2%, p = 0.001) mutations were more common in the basal CD4+ Tlow subgroup. On the other hand, mutations in PPARG (32.0%, p < 0.001) were enriched in the luminal CD4+ Thigh subgroup (Figure S5). The expression of FGFR3 and PPARG were in accordance with the mutations of FGFR3 and PPARG, with higher expression of FGFR3 observed in the luminal CD4+ Tlow subgroup and higher expression of PPARG observed in the luminal CD4+ Thigh subgroup (Figure S6A). Notably, patients in the luminal CD4+ Tlow subgroup were found to have a high prevalence of FGFR3 mutation and FGFR3‐TACC3 fusion, categorized as level 1 of actionability defined by the OncoKB precision oncology knowledge database, suggesting potential benefits from FGFR inhibitors (Figure 3E).
FIGURE 3.
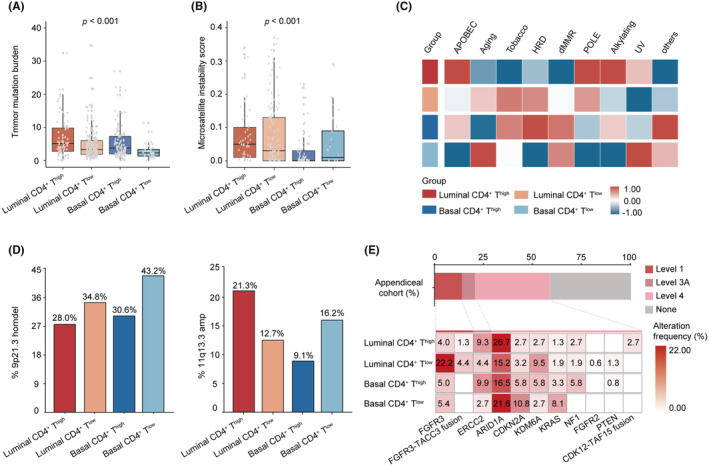
Genomic characteristics of MIBC stratified by CD4+ T cells and molecular subtypes. (A) The distribution of TMB among four subgroups based on CD4+ T cells and molecular subtypes in TCGA cohort. (B) The distribution of MSI among four subgroups based on CD4+ T cells and molecular subtypes in TCGA cohort. (C) Heatmap for mutational signatures in TCGA cohort. (D) Prevalence of 9p21.3 homdel (left) and 11q13.3 amp (right) in four subgroups based on CD4+ T cells and molecular subtypes in TCGA cohort. (E) Highest levels of therapeutic actionability in MIBC stratified by CD4+ T cell infiltration and molecular subtypes in TCGA cohort. Data were analyzed by the Mann–Whitney U test and Pearson's chi‐square test. MIBC, muscle‐invasive bladder cancer; MSI, microsatellite instability score; TMB, tumor mutation burden.
3.4. Distinct tumor immune microenvironments stratified by CD4 + T cells and molecular subtypes
We proceeded to investigate the immune composition of different subgroups. Notably, the luminal CD4+ Thigh subgroup exhibited a significantly higher TNB (p < 0.001, Figure 4A). Moreover, pathways related to immunogenic neoepitope presentation, such as MHCI, MHCII, TCR, and BCR, were hyperactivated in the luminal CD4+ Thigh subgroup (Figure 4B). Interestingly, we observed distinct compositions of CD4+ T helper cell subsets between the luminal CD4+ Thigh and basal CD4+ Thigh subgroups. The luminal CD4+ Thigh subgroup demonstrated a higher presence of Th1 and Th17 cells, whereas the basal CD4+ Thigh subgroup showed more Treg and Th2 cells (Figure 4C). Importantly, each subgroup presented distinct immunological phenotypes. The luminal CD4+ Thigh subgroup exhibited an immune‐inflamed phenotype characterized by T cell activation and IFN‐γ‐related gene signatures. On the other hand, the basal CD4+ Thigh subgroup displayed an immune equilibrium phenotype with an abundance of immune‐inflamed signatures alongside immune suppression signatures. The luminal CD4+ Tlow subgroup was linked to an immune desert phenotype, and the basal CD4+ Tlow subgroup presented an immune suppression phenotype with the highest level of immune suppression and TGF‐β signatures, as well as TGF‐β pathway activity (Figure 4D, Figure S6B). The same immunological phenotypes were observed in the IMvigor210 cohort as well (Figure S7). IHC conducted in the ZSHS cohort confirmed these results. Although there was no significant difference in CD8+ T cells between the luminal CD4+ Thigh and basal CD4+ Thigh subgroups, the luminal CD4+ Thigh subgroup exhibited a higher presence of CD103+CD8+ tissue‐resident memory T cells (TRM), while the basal CD4+ Thigh subgroup had more TIGIT+CD8+ exhausted T cells (TEX) than the luminal CD4+ Thigh subgroup (Figure 4E). Furthermore, patients in the luminal CD4+ Thigh subgroup displayed higher numbers of PD‐L1+, IFN‐γ+, and GZMB+ cells, whereas the basal CD4+ Thigh subgroup had elevated levels of TGF‐β+ cells (Figure 4F,G).
FIGURE 4.
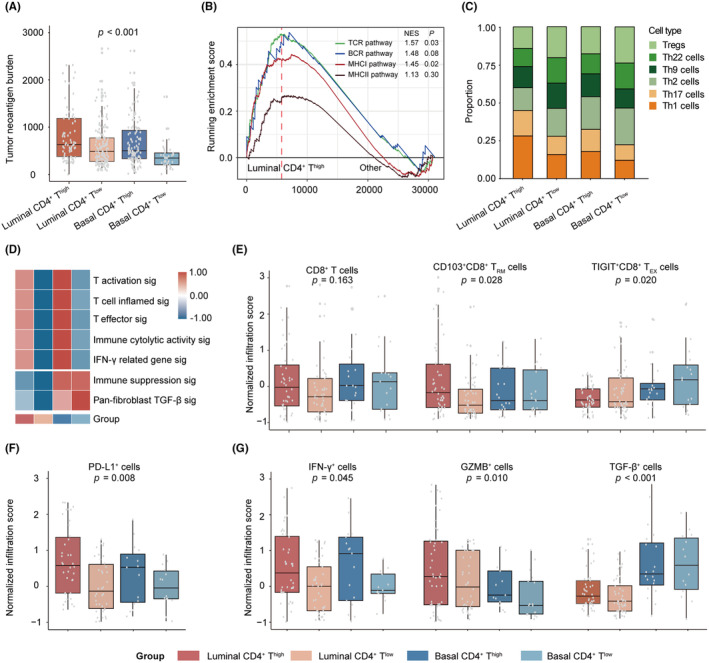
Distinct tumor immune microenvironments stratified by CD4+ T cells and molecular subtypes. (A) The distribution of TNB among four subgroups based on CD4+ T cells and molecular subtypes in TCGA cohort. (B) Gene set enrichment analysis of the antigen‐presenting pathways in four subgroups in TCGA cohort. (C) The distribution of different CD4+ T cell subsets in four subgroups in the ZSHS cohort. (D) Heatmap for immune functional signatures in four subgroups in TCGA cohort. (E) The distribution of different CD8+ T cell subsets in four subgroups in the ZSHS cohort. (F) The distribution of PD‐L1+ cells in four subgroups in the ZSHS cohort. (G) The distribution of effector cells in four subgroups in the ZSHS cohort. Data were analyzed by the Mann–Whitney U test and Pearson's chi‐square test. TNB, tumor neoantigen burden; ZSHS, Zhongshan Hospital.
4. DISCUSSION
Transcriptome profiling facilitates the intrinsic molecular subtypes of BC, which may help to identify a more precise patient stratification with increased sensitivity to various therapeutic options. Several teams have identified distinct molecular subtypes characterized by diverse histopathological features in MIBC, including LUND University, 29 University of North Carolina (UNC), 30 MD Anderson (MDA) Cancer Center, 31 and TCGA. 6 , 32 While these subtypes were established from largely independent datasets using unique transcriptional platforms, they exhibit numerous similarities. 7 Notably, a strong overlap has been observed between certain subtypes. 33 At a higher level, these subtypes represent a division into basal and luminal subtypes, with each system having specific subclassifications. 9 The basal subtype is distinguished by the expression of basal keratin markers such as keratin 14 (KRT14) and keratin 5 (KRT5). Gene pathways associated with cell survival and cell movement were also found to be enriched in this subtype. In contrast, the luminal subtype exhibited elevated levels of uroplakins including uroplakin 2 (UPK2) and uroplakin 3A (UPK3A). Alterations in FGFR3 and tuberous sclerosis 1 (TSC1) were significantly enriched in this subtype. 30 , 31 , 34
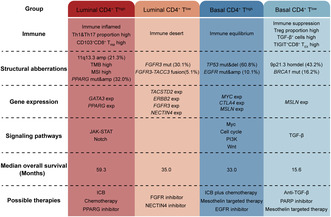
Immune checkpoint blockade has demonstrated significant efficacy in patients with unresectable and metastatic BC as second‐line treatment and in platinum‐ineligible patients as first‐line treatment. 35 , 36 Established evidence indicated that markers associated with the immune microenvironment, such as CD8 expression, PD‐L1 expression on tumor or immune cells, have shown their predictive value of response to ICB. 37 , 38 , 39 , 40 Given the growing importance of immunotherapy, it is essential in clinical practice to incorporate immune features into the molecular classification system to pinpoint the optimal patient population for ICB. Furthermore, while several studies have supposed the potential responses to chemotherapy and immunotherapy in certain subtypes, the diversity of published MIBC classifications has posed challenges in integrating these subtypes into guiding standard management of patients with MIBC. 4 , 9 , 39 In this study, we identified four immune‐molecular subtypes with specific genomic characteristics, immune phenotypes, oncogenic pathways, and clinical outcomes based on integrating CD4+ T cells and molecular subtypes (Figure 5). In addition, this study gives plausible explanations for heterogeneous responses to immunotherapy among the four subtypes according to their specific features.
FIGURE 5.
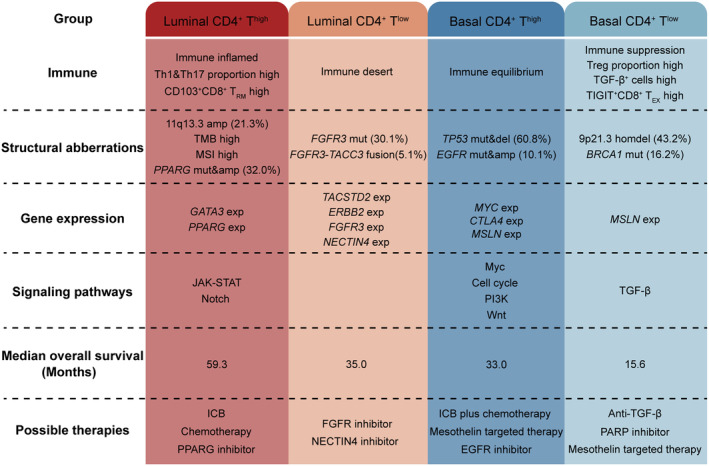
Overview of molecular characteristics and potential therapeutic implications per subtype for muscle‐invasive bladder cancer (MIBC). ICB, immune checkpoint blockade.
Accumulating evidence has linked responses to immunotherapy with T‐cell infiltration. 41 , 42 Our analysis suggested that the four subtypes harbored different immune phenotypes. Among the high‐T‐cell‐infiltration phenotypes, the luminal CD4+ Thigh subtype was characterized by the immune‐inflamed phenotype. The finding was in line with previous reports that patients in this subtype presented the best response to immunotherapy and chemotherapy. 19 , 23 , 43 , 44 , 45 However, the basal CD4+ Thigh subtype, which also possessed high T cell‐inflamed signatures, did not demonstrate superior outcomes from immunotherapy. It might be attributed to the enrichment of the immune suppression signature in this subtype, and the T cell‐inflamed infiltration signature may be counterbalanced by a higher level of immune suppression signature.
The function of CD4+ T cells relies on the composition of various CD4+ T cell subsets, including Th1, Th2, Th9, Th17, Th22, and Treg cells, which are controlled by different transcription factors to produce specific cytokines and thus exert different functions. 46 Th1 cells participate in antitumor immunity through IFN‐γ and tumor necrosis factor (TNF) production, Th2‐mediated immunity supports tumor growth, and Tregs are known to inhibit the function of CD8+ cytotoxic lymphocytes. 47 , 48 , 49 , 50 Although the role of Th9 and Th17 cells remains controversial among various cancers, it was confirmed that Th17 cells were associated with an antitumor immune contexture and Th9 cells were correlated with a tumor‐promoting environment in MIBC. 16 , 17 , 51 In our study, despite possessing high infiltration of CD4+ T cells in both the luminal CD4+ Thigh and basal CD4+ Thigh subtypes, the composition of CD4+ T cell subsets was starkly different. More Th1 and Th17 cells were observed in the luminal CD4+ Thigh subgroup and more Treg and Th2 cells in the basal CD4+ Thigh subtype. This may partially explain the different response rates to immunotherapy in these two subtypes.
The luminal CD4+ Tlow and basal CD4+ Tlow subtypes, which displayed non‐T cell‐inflamed phenotype, both displayed a low response rate to immunotherapy. However, the mechanism that regulates the immune microenvironment might differ in these two subtypes. The luminal CD4+ Tlow subtype showed enrichment in FGFR3 expression and FGFR3 mutations. The FGFR3 signaling has been reported to affect pathways (i.e., EMT and TGF‐β) that interfere with the microenvironmental milieu. 52 Furthermore, numerous clinical trials are currently underway to demonstrate the clinical effectiveness of FGFR inhibitors and to investigate the potential utilization of FGFR alterations as a biomarker. Erdafitinib, a tyrosine kinase inhibitor targeting FGFR1–4, has demonstrated antitumor activity in preclinical models and a phase 1 study with patients carrying FGFR alterations. 53 , 54 In the phase 2 study (NCT02365597), Erdafitinib exhibited an ORR of 40% in metastatic UC with selected FGFR3 mutations or fusions. 55 Based on this clinical trial, erdafitinib has been approved by the FDA as the first targeted drug for BC since 2019. 56 More importantly, Erdafitinib demonstrated a significant improvement in both OS and progression‐free survival (PFS) compared with chemotherapy in patients with metastatic UC with susceptible FGFR3/2 alterations who had progression after previous anti‐PD‐1 or anti‐PD‐L1 therapy in a global phase 3 clinical trial (NCT03390504). 57 This noteworthy enhancement emphasizes the significance of molecular testing for FGFR alterations in patients diagnosed with metastatic UC. Considering the high prevalence of FGFR3 mutation and FGFR3‐TACC3 fusion in the luminal CD4+Tlow subgroup, FGFR inhibitors may be a potential therapeutic option for patients in this subgroup.
The basal CD4+ Tlow subtype exhibited the highest proportion of homozygous deletion of 9p21.3. It was reported that 9p21 loss would confer a “cold” tumor immune phenotype characterized by the reduced abundance of tumor‐infiltrating leukocytes and the activation of immunosuppressive signaling, which may explain our clinical findings. 28 Moreover, considering the activation of TGF‐β signaling, it is potentially beneficial for the application of TGF‐β inhibitors in the basal CD4+ Tlow subtype. 58
Regarding the retrospective and exploratory design of our study, this classification still requires further validation by more extensive, multicentered clinical cohorts. Experimental procedures should be carried out to assess the potential value of this classification to certain therapies. Moreover, the detailed mechanism that caused the non‐T cell‐inflamed microenvironment needs further exploration. Despite these limitations, this study defined an immune‐molecular classification based on CD4+ T cells and molecular subtypes. This classification is hoped to serve as a foundation for developing tailored therapeutic approaches for MIBC patients.
AUTHOR CONTRIBUTIONS
Ge Liu: Data curation; formal analysis; investigation; project administration; validation; visualization; writing – original draft. Kaifeng Jin: Data curation; formal analysis; investigation; project administration; validation; visualization; writing – original draft. Zhaopei Liu: Data curation; formal analysis; investigation; project administration; validation; visualization; writing – original draft. Xiaohe Su: Methodology; resources. Ziyue Xu: Methodology; resources. Bingyu Li: Methodology; resources. Jingtong Xu: Methodology; resources. Hailong Liu: Methodology; resources. Yuan Chang: Methodology; resources. Yu Zhu: Methodology; resources. Le Xu: Conceptualization; funding acquisition; investigation; project administration; supervision; writing – review and editing. Zewei Wang: Conceptualization; funding acquisition; investigation; project administration; supervision; writing – review and editing. Yiwei Wang: Methodology; resources. Weijuan Zhang: Conceptualization; funding acquisition; investigation; project administration; supervision; writing – review and editing.
FUNDING INFORMATION
This study was funded by grants from the National Natural Science Foundation of China (82002670, 82103408, 82272930, 82372793, 82373276), China Postdoctoral Science Foundation (BX20230091), Shanghai Municipal Natural Science Foundation (22ZR1413400, 23ZR1411700, 23ZR1440300), Shanghai Sailing Program (21YF1407000), Shanghai Municipal Health Bureau Project (202340127, 202340195, 20224Y0232), Fudan University Shanghai Cancer Center for Outstanding Youth Scholars Foundation (YJYQ201802), and Shanghai Anticancer Association EYAS PROJECT (SACA‐CY22B02, ZYJH202309). All these study sponsors have no roles in the study design and in the collection, analysis, and interpretation of data.
CONFLICT OF INTEREST STATEMENT
The authors have declared no conflicts of interest.
ETHICS STATEMENT AND CONSENT TO PARTICIPATE
This study was approved by the Clinical Research Ethics Committee of Zhongshan Hospital and Fudan University (No. B2015‐030). Written informed consent was obtained from each patient.
Supporting information
FIGURE S1. Flow chart of cohort selection and representative images of CD4+ T cells.
FIGURE S2. Correlation of CD4+ T cells and molecular subtype with clinical outcomes in muscle‐invasive bladder cancer (MIBC) patients.
FIGURE S3. Correlation of CD4+ T cells and molecular subtype with clinical outcomes in patients treated with chemotherapy.
FIGURE S4. Correlation of CD4+ T cells and molecular subtype with clinical outcomes in patients treated with atezolizumab.
FIGURE S5. Gene alterations across subtypes based on CD4+ T cells and molecular subtype in muscle‐invasive bladder cancer (MIBC).
FIGURE S6. Gene expression and oncogenic pathway activity across subtypes based on CD4+ T cells and molecular subtype in muscle‐invasive bladder cancer (MIBC).
FIGURE S7. Expression of gene signatures related to the immune function.
TABLE S1. Patient characteristics in TCGA and the Zhongshan Hospital (ZSHS) cohort.
TABLE S2. Patient characteristics in the IMvigor210 and NCT03179943 cohort.
TABLE S3. Sources and details of gene signatures.
TABLE S4. Sources of gene set enrichment analysis (GSEA) pathways.
TABLE S5. Immunohistochemistry (IHC) antibodies.
ACKNOWLEDGMENTS
We thank Dr. Lingli Chen (Department of Pathology, Zhongshan Hospital, Fudan University, Shanghai, China) and Dr. Yunyi Kong (Department of Pathology, Fudan University Shanghai Cancer Center, Shanghai, China) for their excellent pathological technology help.
Liu G, Jin K, Liu Z, et al. Integration of CD4+ T cells and molecular subtype predicts benefit from PD‐L1 blockade in muscle‐invasive bladder cancer. Cancer Sci. 2024;115:1306‐1316. doi: 10.1111/cas.16119
Ge Liu, Kaifeng Jin, and Zhaopei Liu contributed equally to this work.
Le Xu, Zewei Wang and Weijuan Zhang share co‐corresponding authorship.
DATA AVAILABILITY STATEMENT
Data and materials generated that are relevant to the results are included in this article. Other data are available from the corresponding author Prof. Zhang upon reasonable request.
REFERENCES
- 1. Lobo N, Mount C, Omar K, Nair R, Thurairaja R, Khan MS. Landmarks in the treatment of muscle‐invasive bladder cancer. Nat Rev Urol. 2017;14:565‐574. [DOI] [PubMed] [Google Scholar]
- 2. Witjes JA, Bruins HM, Cathomas R, et al. European Association of Urology guidelines on muscle‐invasive and metastatic bladder cancer: summary of the 2020 guidelines. Eur Urol. 2021;79:82‐104. [DOI] [PubMed] [Google Scholar]
- 3. De Santis M, Bellmunt J, Mead G, et al. Randomized phase II/III trial assessing gemcitabine/ carboplatin and methotrexate/carboplatin/vinblastine in patients with advanced urothelial cancer “unfit” for cisplatin‐based chemotherapy: phase II–results of EORTC study 30986. J Clin Oncol. 2009;27:5634‐5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rosenberg JE, Hoffman‐Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum‐based chemotherapy: a single‐arm, multicentre, phase 2 trial. Lancet. 2016;387:1909‐1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sharma P, Retz M, Siefker‐Radtke A, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single‐arm, phase 2 trial. Lancet Oncol. 2017;18:312‐322. [DOI] [PubMed] [Google Scholar]
- 6. Robertson AG, Kim J, Al‐Ahmadie H, et al. Comprehensive molecular characterization of muscle‐invasive bladder cancer. Cell. 2017;171:540‐556.e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kamoun A, de Reyniès A, Allory Y, et al. A consensus molecular classification of muscle‐invasive bladder cancer. Eur Urol. 2020;77:420‐433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McConkey DJ, Choi W, Shen Y, et al. A prognostic gene expression signature in the molecular classification of chemotherapy‐naïve urothelial cancer is predictive of clinical outcomes from neoadjuvant chemotherapy: a phase 2 trial of dose‐dense methotrexate, vinblastine, doxorubicin, and cisplatin with bevacizumab in urothelial cancer. Eur Urol. 2016;69:855‐862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seiler R, Ashab HAD, Erho N, et al. Impact of molecular subtypes in muscle‐invasive bladder cancer on predicting response and survival after neoadjuvant chemotherapy. Eur Urol. 2017;72:544‐554. [DOI] [PubMed] [Google Scholar]
- 10. Kim J, Kwiatkowski D, McConkey DJ, et al. The cancer genome atlas expression subtypes stratify response to checkpoint inhibition in advanced urothelial cancer and identify a subset of patients with high survival probability. Eur Urol. 2019;75:961‐964. [DOI] [PubMed] [Google Scholar]
- 11. Xu Y, Zeng H, Jin K, et al. Immunosuppressive tumor‐associated macrophages expressing interlukin‐10 conferred poor prognosis and therapeutic vulnerability in patients with muscle‐invasive bladder cancer. J Immunother Cancer. 2022;10:e003416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goubet AG, Lordello L, Alves Costa Silva C, et al. Escherichia coli‐specific CXCL13‐producing TFH are associated with clinical efficacy of neoadjuvant PD‐1 blockade against muscle‐invasive bladder cancer. Cancer Discov. 2022;12:2280‐2307. [DOI] [PubMed] [Google Scholar]
- 13. Jin K, Yu Y, Zeng H, et al. CD103(+)CD8(+) tissue‐resident memory T cell infiltration predicts clinical outcome and adjuvant therapeutic benefit in muscle‐invasive bladder cancer. Br J Cancer. 2022;126:1581‐1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Khan SM, Desai R, Coxon A, et al. Impact of CD4 T cells on intratumoral CD8 T‐cell exhaustion and responsiveness to PD‐1 blockade therapy in mouse brain tumors. J Immunother Cancer. 2022;10:e005293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zeng H, Liu Z, Wang Z, et al. Intratumoral IL22‐producing cells define immunoevasive subtype muscle‐invasive bladder cancer with poor prognosis and superior nivolumab responses. Int J Cancer. 2020;146:542‐552. [DOI] [PubMed] [Google Scholar]
- 16. Wang Z, Zhou Q, Zeng H, et al. Tumor‐infiltrating IL‐17A(+) cells determine favorable prognosis and adjuvant chemotherapeutic response in muscle‐invasive bladder cancer. Onco Targets Ther. 2020;9:1747332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou Q, Zhang H, Wang Z, et al. Poor clinical outcomes and immunoevasive contexture in interleukin‐9 abundant muscle‐invasive bladder cancer. Int J Cancer. 2020;147:3539‐3549. [DOI] [PubMed] [Google Scholar]
- 18. Jang HJ, Hostetter G, Macfarlane AW, et al. A phase II trial of Guadecitabine plus Atezolizumab in metastatic urothelial carcinoma progressing after initial immune checkpoint inhibitor therapy. Clin Cancer Res. 2023;29:2052‐2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ayers M, Lunceford J, Nebozhyn M, et al. IFN‐γ‐related mRNA profile predicts clinical response to PD‐1 blockade. J Clin Invest. 2017;127:2930‐2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Spranger S, Bao R, Gajewski TF. Melanoma‐intrinsic β‐catenin signalling prevents anti‐tumour immunity. Nature. 2015;523:231‐235. [DOI] [PubMed] [Google Scholar]
- 22. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Powles T, Sridhar SS, Loriot Y, et al. Avelumab maintenance in advanced urothelial carcinoma: biomarker analysis of the phase 3 JAVELIN bladder 100 trial. Nat Med. 2021;27:2200‐2211. [DOI] [PubMed] [Google Scholar]
- 24. Nakauma‐González JA, Rijnders M, van Riet J, et al. Comprehensive molecular characterization reveals genomic and transcriptomic subtypes of metastatic urothelial carcinoma. Eur Urol. 2022;81:331‐336. [DOI] [PubMed] [Google Scholar]
- 25. Liu Z, Zhu Y, Xu L, et al. Tumor stroma‐infiltrating mast cells predict prognosis and adjuvant chemotherapeutic benefits in patients with muscle invasive bladder cancer. Onco Targets Ther. 2018;7:e1474317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li HD, Cuevas I, Zhang M, et al. Polymerase‐mediated ultramutagenesis in mice produces diverse cancers with high mutational load. J Clin Invest. 2018;128:4179‐4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shi R, Wang X, Wu Y, et al. APOBEC‐mediated mutagenesis is a favorable predictor of prognosis and immunotherapy for bladder cancer patients: evidence from pan‐cancer analysis and multiple databases. Theranostics. 2022;12:4181‐4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Han G, Yang G, Hao D, et al. 9p21 loss confers a cold tumor immune microenvironment and primary resistance to immune checkpoint therapy. Nat Commun. 2021;12:5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sjödahl G, Lauss M, Lövgren K, et al. A molecular taxonomy for urothelial carcinoma. Clin Cancer Res. 2012;18:3377‐3386. [DOI] [PubMed] [Google Scholar]
- 30. Damrauer JS, Hoadley KA, Chism DD, et al. Intrinsic subtypes of high‐grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci USA. 2014;111:3110‐3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Choi W, Porten S, Kim S, et al. Identification of distinct basal and luminal subtypes of muscle‐invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014;25:152‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cancer Genome Atlas Research Network . Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aine M, Eriksson P, Liedberg F, Sjödahl G, Höglund M. Biological determinants of bladder cancer gene expression subtypes. Sci Rep. 2015;5:10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dadhania V, Zhang M, Zhang L, et al. Meta‐analysis of the luminal and basal subtypes of bladder cancer and the identification of signature immunohistochemical markers for clinical use. EBioMedicine. 2016;12:105‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Balar AV, Galsky MD, Rosenberg JE, et al. Atezolizumab as first‐line treatment in cisplatin‐ineligible patients with locally advanced and metastatic urothelial carcinoma: a single‐arm, multicentre, phase 2 trial. Lancet. 2017;389:67‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rijnders M, de Wit R, Boormans JL, Lolkema MPJ, van der Veldt AAM. Systematic review of immune checkpoint inhibition in urological cancers. Eur Urol. 2017;72:411‐423. [DOI] [PubMed] [Google Scholar]
- 37. Chen DS, Mellman I. Elements of cancer immunity and the cancer‐immune set point. Nature. 2017;541:321‐330. [DOI] [PubMed] [Google Scholar]
- 38. López‐Soto A, Gonzalez S, Folgueras AR. IFN signaling and ICB resistance: time is on tumor's side. Trends Cancer. 2017;3:161‐163. [DOI] [PubMed] [Google Scholar]
- 39. Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD‐L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as second‐line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376:1015‐1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Szabados B, Kockx M, Assaf ZJ, et al. Final results of neoadjuvant Atezolizumab in cisplatin‐ineligible patients with muscle‐invasive urothelial cancer of the bladder. Eur Urol. 2022;82:212‐222. [DOI] [PubMed] [Google Scholar]
- 42. Liu Z, Zhou Q, Wang Z, et al. Intratumoral TIGIT(+) CD8(+) T‐cell infiltration determines poor prognosis and immune evasion in patients with muscle‐invasive bladder cancer. J Immunother Cancer. 2020;8:e000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Petitprez F, de Reyniès A, Keung EZ, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577:556‐560. [DOI] [PubMed] [Google Scholar]
- 44. Ott PA, Bang YJ, Piha‐Paul SA, et al. T‐cell‐inflamed gene‐expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE‐028. J Clin Oncol. 2019;37:318‐327. [DOI] [PubMed] [Google Scholar]
- 45. Zhu AX, Abbas AR, de Galarreta MR, et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat Med. 2022;28:1599‐1611. [DOI] [PubMed] [Google Scholar]
- 46. Knochelmann HM, Dwyer CJ, Bailey SR, et al. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol Immunol. 2018;15:458‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T‐bet, directs Th1 lineage commitment. Cell. 2000;100:655‐669. [DOI] [PubMed] [Google Scholar]
- 48. Facciabene A, Motz GT, Coukos G. T‐regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res. 2012;72:2162‐2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ellyard JI, Simson L, Parish CR. Th2‐mediated anti‐tumour immunity: friend or foe? Tissue Antigens. 2007;70:1‐11. [DOI] [PubMed] [Google Scholar]
- 50. Speiser DE, Chijioke O, Schaeuble K, Münz C. CD4(+) T cells in cancer. Nat Can. 2023;4:317‐329. [DOI] [PubMed] [Google Scholar]
- 51. Angkasekwinai P, Dong C. IL‐9‐producing T cells: potential players in allergy and cancer. Nat Rev Immunol. 2021;21:37‐48. [DOI] [PubMed] [Google Scholar]
- 52. Wang L, Gong Y, Saci A, et al. Fibroblast growth factor receptor 3 alterations and response to PD‐1/PD‐L1 blockade in patients with metastatic urothelial cancer. Eur Urol. 2019;76:599‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tabernero J, Bahleda R, Dienstmann R, et al. Phase I dose‐escalation study of JNJ‐42756493, an Oral pan‐fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2015;33:3401‐3408. [DOI] [PubMed] [Google Scholar]
- 54. Perera TPS, Jovcheva E, Mevellec L, et al. Discovery and pharmacological characterization of JNJ‐42756493 (Erdafitinib), a functionally selective small‐molecule FGFR family inhibitor. Mol Cancer Ther. 2017;16:1010‐1020. [DOI] [PubMed] [Google Scholar]
- 55. Loriot Y, Necchi A, Park SH, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med. 2019;381:338‐348. [DOI] [PubMed] [Google Scholar]
- 56. Marandino L, Raggi D, Giannatempo P, Farè E, Necchi A. Erdafitinib for the treatment of urothelial cancer. Expert Rev Anticancer Ther. 2019;19:835‐846. [DOI] [PubMed] [Google Scholar]
- 57. Loriot Y, Matsubara N, Park SH, et al. Erdafitinib or chemotherapy in advanced or metastatic urothelial carcinoma. N Engl J Med. 2023;389:1961‐1971. [DOI] [PubMed] [Google Scholar]
- 58. Peng H, Shen J, Long X, et al. Local release of TGF‐β inhibitor modulates tumor‐associated neutrophils and enhances pancreatic cancer response to combined irreversible electroporation and immunotherapy. Adv Sci. 2022;9:e2105240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1. Flow chart of cohort selection and representative images of CD4+ T cells.
FIGURE S2. Correlation of CD4+ T cells and molecular subtype with clinical outcomes in muscle‐invasive bladder cancer (MIBC) patients.
FIGURE S3. Correlation of CD4+ T cells and molecular subtype with clinical outcomes in patients treated with chemotherapy.
FIGURE S4. Correlation of CD4+ T cells and molecular subtype with clinical outcomes in patients treated with atezolizumab.
FIGURE S5. Gene alterations across subtypes based on CD4+ T cells and molecular subtype in muscle‐invasive bladder cancer (MIBC).
FIGURE S6. Gene expression and oncogenic pathway activity across subtypes based on CD4+ T cells and molecular subtype in muscle‐invasive bladder cancer (MIBC).
FIGURE S7. Expression of gene signatures related to the immune function.
TABLE S1. Patient characteristics in TCGA and the Zhongshan Hospital (ZSHS) cohort.
TABLE S2. Patient characteristics in the IMvigor210 and NCT03179943 cohort.
TABLE S3. Sources and details of gene signatures.
TABLE S4. Sources of gene set enrichment analysis (GSEA) pathways.
TABLE S5. Immunohistochemistry (IHC) antibodies.
Data Availability Statement
Data and materials generated that are relevant to the results are included in this article. Other data are available from the corresponding author Prof. Zhang upon reasonable request.