

Abstract
Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations in circulating tumor deoxyribonucleic acid (ctDNA) have been reported as representative noninvasive prognostic markers for pancreatic ductal adenocarcinoma (PDAC). Here, we aimed to evaluate single KRAS mutations as prognostic and predictive biomarkers, with an emphasis on potential therapeutic approaches to PDAC. A total of 128 patients were analyzed for multiple or single KRAS mutations (G12A, G12C, G12D, G12R, G12S, G12V, and G13D) in their tumors and plasma using droplet digital polymerase chain reaction (ddPCR). Overall, KRAS mutations were detected by multiplex ddPCR in 119 (93%) of tumor DNA and 68 (53.1%) of ctDNA, with a concordance rate of 80% between plasma ctDNA and tumor DNA in the metastatic stage, which was higher than the 44% in the resectable stage. Moreover, the prognostic prediction of both overall survival (OS) and progression‐free survival (PFS) was more relevant using plasma ctDNA than tumor DNA. Further, we evaluated the selective tumor‐suppressive efficacy of the KRAS G12C inhibitor sotorasib in a patient‐derived organoid (PDO) from a KRAS G12C‐mutated patient using a patient‐derived xenograft (PDX) model. Sotorasib showed selective inhibition in vitro and in vivo with altered tumor microenvironment, including fibroblasts and macrophages. Collectively, screening for KRAS single mutations in plasma ctDNA and the use of preclinical models of PDO and PDX with genetic mutations would impact precision medicine in the context of PDAC.
Keywords: circulating tumor DNA, KRAS, KRAS G12C inhibitor, pancreatic ductal adenocarcinoma, patient‐derived organoid
We explored the potential of screening KRAS mutations in plasma circulating tumor deoxyribonucleic acid (ctDNA) to drive therapeutic approaches for pancreatic ductal adenocarcinoma (PDAC). KRAS mutation in ctDNA had a higher predicitive rate than DNA in tumor, and this was accomplished by investigating the effect of KRAS G12C inhibitor in preclinical models such as patient‐derived xenografts (PDX) and patient‐derived organoids (PDO). The monitoring of KRAS mutation in plasma ctDNA may guide a direction for targeted therapy of PDAC.
Abbreviations
- CI
confidence interval
- CRC
colorectal cancer
- ctDNA
circulating tumor DNA
- ddPCR
droplet digital PCR
- DMEM
Dulbecco's modified Eagle medium
- DMSO
dimethyl sulfoxide
- EpCAM
epithelial cell adhesion molecule
- ERK
extracellular signal‐regulated kinase
- FFPE
formalin‐fixed, paraffin‐embedded
- GFR
growth factor‐reduced
- H&E
hematoxylin and eosin
- HR
hazard ratio
- KRAS
Kirsten rat sarcoma viral oncogene homolog
- NSCLC
non‐small cell lung cancer
- OS
overall survival
- PDAC
pancreatic ductal adenocarcinoma
- PDX
patient‐derived xenograft
- PDXC
PDX‐derived cell
- PDXO
PDX‐derived organoid
- PFS
progression‐free survival
- TME
tumor microenvironment
- αSMA
alpha smooth muscle actin
1. INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is a major cause of cancer‐related deaths, with approximately 432,242 new deaths reported to be attributed to PDAC in 2018 (Global Cancer Observatory 2018 estimates). 1 Despite advancements in the detection and management strategies, the prognosis of patients with PDAC has remained poor over the past 20 years. 2 , 3 Kirsten rat sarcoma viral oncogene homolog (KRAS) is one of the deadliest cancer‐related proteins that plays a pivotal role in the development of most aggressive and lethal human cancers, with mutations occurring in up to 96% of PDACs. 4
KRAS mutations are the most prevalent oncogenic driver mutations in cancers. Members of the RAS family are mutated in approximately 30% of cancers, making them primary therapeutic targets. 5 KRAS has long been considered undruggable because of its small size and relatively smooth surface, with a few deep pockets where molecules can bind, and its rapid and tight binding to guanosine triphosphate in its active state. 6 However, there has recently been a breakthrough in research on KRAS inhibition with the discovery of an allosteric switch II pocket. 7 In particular, selective inhibition of oncogenic KRAS G12C by the strategy of targeting the inactive state using small molecules has shown clinically remarkable tumor regression in patients with non‐small cell lung cancer (NSCLC). 8 , 9 In addition, because the development and therapeutic effects of drugs targeting G12D are spurred sequentially, the introduction of therapeutic agents targeting KRAS is expected to escalate. 10 , 11 , 12 , 13
Circulating tumor deoxyribonucleic acid (ctDNA) is emerging as a potential biomarker in precision medicine. 14 At approximately 166 bp, ctDNA fragments have a somatic genomic alteration similar to that of a tumor DNA. 15 ctDNA has also been detected in various solid malignancies, with ctDNA fractions ranging from less than 0.1% to more than 50%. 16 Moreover, ctDNA analysis is the most noninvasive method for detecting tumor characteristics. 17 In particular, it can serve as a tool to identify genetic treatment targets for cancers for which new biopsies are difficult to obtain, such as PDAC with a low surgical resection rate, or to monitor the treatment response, residual disease, and recurrence. 18 In PDAC, methylation, fragment size, copy number analysis, and mutations in plasma ctDNA have several prognostic implications. 19 These findings suggest that ctDNA abundance reflects the biological characteristics of tumor burden and predicts patient prognosis. 20
Targeting RAS is the most obvious and attractive approach for developing PDAC treatments. 21 , 22 We previously reported that multiplex detection of KRAS mutations in plasma cell‐free DNA is clinically relevant and provides a potential candidate biomarker for the prognosis of PDAC. 23 However, to apply anti‐RAS target therapy detection of specific KRAS mutations is necessary. 18 Here, in order to predict the prognosis of pancreatic cancer, we sought to identify seven mutations found in KRAS codons 12 and 13 of tumor and plasma ctDNA. By examining the selective tumor growth inhibition effect of sotorasib on patient‐derived xenograft (PDX), PDX‐derived cell (PDXC), and PDX‐derived organoid (PDXO), we also proposed that detection of KRAS mutation in plasma ctDNA has the potential to guide direction for targeted therapy.
2. MATERIALS AND METHODS
2.1. Study design and sample collection
The study prospectively enrolled 128 newly diagnosed patients with PDAC who visited the Pancreatobiliary Cancer Clinic at the National Cancer Center, Korea between March 2015 and April 2019. All patients provided written informed consent. The study protocols were approved by the Institutional Review Board (IRB) of the National Cancer Center of Korea (IRB No. NCC2015‐054, NCC2016‐011). Patient blood samples, tumor tissues, formalin‐fixed paraffin‐embedded (FFPE) tissue samples, and clinical data were collected at the National Cancer Center (NCC), Republic of Korea. Tissue samples were provided by the NCC Bio Bank, Republic of Korea. The patients were divided into three clinical stage groups: resectable, locally advanced, and metastatic.
2.2. Sample processing and DNA extraction from tissue and plasma
Up to 10 mL of peripheral blood was collected by venipuncture in collection tubes containing K2 ethylenediaminetetraacetic acid (BD #366643; Becton Dickinson and Company). Plasma from the collected blood was separated within 2 h of drawing the blood to ensure the integrity of the ctDNA. Whole blood was centrifuged at 1600 g for 10 min, and the supernatant was centrifuged again at 16,000 g for 10 min to remove any remaining contaminating cells. The supernatants were immediately stored at −80°C until use. Plasma ctDNA was extracted from 1 to 2 mL plasma using a QIAamp Circulating Nucleic Acid Kit (Qiagen). The final DNA eluent (50 μL) was quantified using a Qubit 2.0 Fluorometer with a Qubit dsDNA HS (High‐Sensitivity) Assay Kit (Life Technologies). Genomic DNA from FFPE (N = 108) and frozen tissue (N = 20) samples was extracted using the GeneRead FFPE DNA Kit (Qiagen) and DNeasy Blood & Tissue Kits (Qiagen), respectively.
2.3. Organoid culture
As previously reported, PDXCs and PDXOs were established from a PDX model generated by orthotopically implanting patient‐derived tumor samples obtained from ultrasound‐guided biopsies of metastatic lesions into the pancreas of athymic nude mice. 24 Single‐cell suspensions were obtained by a combination of mechanical dissociation and enzymatic degradation of the extracellular matrix (gentle MACS Dissociators; Miltenyi Biotec). PDXO was dissociated and mixed with 40 μL growth factor‐reduced (GFR) Matrigel (BD Bioscience) containing 3 × 104 cells/well in a 24‐well plate. After the Matrigel hardened, the organoid growth media was added.
2.4. Detection of KRAS mutations in ctDNA and tissue DNA by droplet digital polymerase chain reaction (ddPCR)
KRAS mutations in plasma ctDNA and genomic DNA from tumor tissues were detected using ddPCR on a QX200 Droplet Digital PCR System (Bio‐Rad Laboratories) with a ddPCR™ KRAS G12/G13 Screening Kit (Bio‐Rad Laboratories), which covers seven common KRAS mutations (G12A, G12C, G12D, G12R, G12S, G12V, and G13D). Analyses were performed using QuantaSoft software (Bio‐Rad Laboratories) as described in a previous study. 23 Single KRAS mutations in tumor tissues and ctDNA were analyzed in all patients using ddPCR to determine the frequency of each of the seven mutations and their correlations with clinical characteristics. A total of 128 (100%) tumor samples that were employed in the KRAS screening multiplex assay and matched tumor and ctDNA for each of the four common single KRAS mutations (G12D, G12V, G12R, and G12C) were compared in 85 patient samples. The threshold for the mutant‐positive droplets was half the amplitude of the positive control. The cutoff for KRAS mutations for the multiplex kit and each of the seven single probes was evaluated by serial dilution of genomic DNA using the mutated cancer cell line or mutated reference standard (Horizon Diagnostics). Human pancreatic cancer cell lines AsPC‐1 (mutated in G12D), CFPAC‐1 (mutated in G12V), KRAS wild‐type reference standard, and mutated reference standard (Horizon Diagnostics) in each of the seven hotspots produced a standard curve with a low‐end fractional abundance of KRAS mutations of 0.01% (Figure S1). The cutoffs of the multiplex kit and single probe were defined as 0.1% and 0.5%, respectively. Thus, samples with a fractional abundance greater than 0.1% and 0.5% were considered positive for the detection of KRAS multiplex mutations and single mutations, respectively.
2.5. In vitro drug response
Sotorasib (Synonyms: AMG‐510) was purchased from MedChemExpress and dissolved in dimethyl sulfoxide (DMSO) (Sigma). Organoids were plated with 20 μL of 10% GFR Matrigel (BD Bioscience) containing 5 × 102 cells/well and seeded in 384‐well plates. After 3 days incubation for organoid formation, the cells were treated with 0.01–100 μM of sotorasib in organoid growth media. Cell viability was measured after 5 days using a Cell‐titer Glo® 3D viability assay kit (Promega Corporation). The luminescence intensity was determined using an Infinite 200 Pro spectrophotometer (Tecan).
2.6. In vivo drug response
All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the NCC Research Institute (NCCRI) (NCC‐16‐247, NCC‐21‐688). The NCCRI is a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International and abides by the guidelines of the Institute of Laboratory Animal Resources (accredited unit‐NCCRI: unit number: 1392). Female Bagg Albino (BALB)/c nude mice aged 5 weeks (Institute of Medical Science, University of Tokyo) were housed in a specific pathogen‐free environment under controlled conditions of light and humidity and were allowed food and water ad libitum. For the in vivo drug response studies, 5 × 106 PDXCs were subcutaneously injected into the flank of BALB/c nude mice. When the average tumor size reached 100 mm3, the mice were randomly divided (n = 8–10/group). The experimental groups included the control group (0.5% hydroxypropyl methylcellulose and 0.2% Tween 80 as vehicle) and the sotorasib group (10, 30 mg/kg of body weight). Tumor volume was monitored every 3–4 days using an ABSOLUTE Digimatic Caliper (Mitutoyo) and calculated using the following formula: (Width2 × Length)/2. After 24 days, the mice were euthanized and the tumors were excised, weighed, and embedded in paraffin.
2.7. Statistical analysis
Baseline clinical characteristics of patients with PDAC were summarized as median and range (min − max) for age, which is a continuous variable, sex, stage, Eastern Cooperative Oncology Group (ECOG) score, tumor location, CA19‐9, and carcinoembryonic antigen (CEA), which are categorical variables, were summarized as frequency and percentage. In the survival analysis, overall survival (OS) was defined as the date of diagnosis to the last follow‐up or death date, and progression‐free survival (PFS) was defined as the date of diagnosis to the date of progression or death. Survival curves were estimated using the Kaplan–Meier method. All survival curve comparisons were analyzed using log‐rank tests. The Cox proportional hazard model was used to investigate the association between the survival outcomes of patients and one or more variables. To identify clinical factors that have an impact on prognosis, a Cox proportional hazard model was employed. Variables with a p‐value less than 0.1 in the univariable model were included in the multivariable model, and those satisfying a criterion p < 0.05 were analyzed using the backward elimination method. Results were considered statistically significant when the p‐value was <0.05. All statistical analyses were performed using the R project for statistical computing (version 4.1.2).
Additional materials and methods are provided in Data S1.
3. RESULTS
3.1. Characteristics of patients with pancreatic cancer
The characteristics of the patients in this study are summarized in Table 1. Of the 128 patients enrolled, 73 were males, with a median age of 65 years and a median follow‐up period of 67.6 months (range, 4.44–79.10 months). Patients with resectable, locally advanced, and metastatic cancers accounted for 50.0% (n = 64), 6.3% (n = 8), and 43.7% (n = 56) of all patients, respectively. The locally advanced and metastatic group had a significantly higher hazard ratio (HR) than that of resectable groups in PFS (HR, 5.53; 95% confidence interval [CI], 3.62–8.47; p < 0.001) and OS (HR, 4.30; 95% CI, 2.82–6.56; p < 0.001).
TABLE 1.
Characteristics of pancreatic ductal adenocarcinoma patients (N = 128).
| Characteristics | N | % |
|---|---|---|
| Status | ||
| Resectable | 64 | 50 |
| Locally advanced | 8 | 6.25 |
| Metastatic | 56 | 43.75 |
| Age | ||
| Mean ± SD | 64.63 ± 9.82 | |
| Median (min–max) | 65 (40–88) | |
| Gender | ||
| Female | 55 | 42.97 |
| Male | 73 | 57.03 |
| Tumor location | ||
| Head or neck or uncinate process | 64 | 50 |
| Body or tail | 64 | 50 |
| Age | ||
| Mean ± SD | 64.63 ± 9.82 | |
| Median (min–max) | 65 (40–88) | |
| ECOG | ||
| 0 | 80 | 62.5 |
| 1, 2, 3 | 48 | 37.5 |
| CEA Baseline (missing = 5) | ||
| Median (min–max) | 83.8 (<5.0–89,100) | |
| Low: CEA Baseline ≤5 | 63 | 51.22 |
| High: CEA Baseline >5 | 60 | 48.78 |
| CA19‐9 Baseline (missing = 1) | ||
| Median (min–max) | 4.8 (0.8–1090.8) | |
| Low: CA19‐9 Baseline ≤37 | 45 | 35.43 |
| High: CA19‐9 Baseline >37 | 82 | 64.57 |
3.2. Frequency of KRAS mutations in tumor DNA and plasma ctDNA using ddPCR
In a total of 128 patients, KRAS mutations were detected in tumor DNA of 119 patients (93%) and ctDNA of 68 patients (53%) using the multiplex kit. Among seven hotspots, only four mutation types were detected, and mutation frequencies of tumors were as follows: G12D (46.1%), G12V (21.1%), and G12R (10.2%) in tumor DNA and G12C (2.3%), G12D and G12V (1.6%), and G12D and G12R (0.8%) in ctDNA, respectively (Figure 1A,B). In ctDNA, when compared with tumor tissue DNA, detected KRAS single mutation frequency was 50.6%, which was similar to the 58.6% for KRAS multiplex ddPCR. Specifically, the concordance rate was high at 87.9% in the metastatic stage, while it reached 62.5% in the resectable stage. The single mutation concordances of G12D, G12V, G12R, and G12C were 94.2%, 92.3%, 100%, and 100%, respectively, in metastatic patients. Resectable cases showed low concordance rates ranging from 53.5% to 100% compared with those of metastatic cases (Figure 1C).
FIGURE 1.
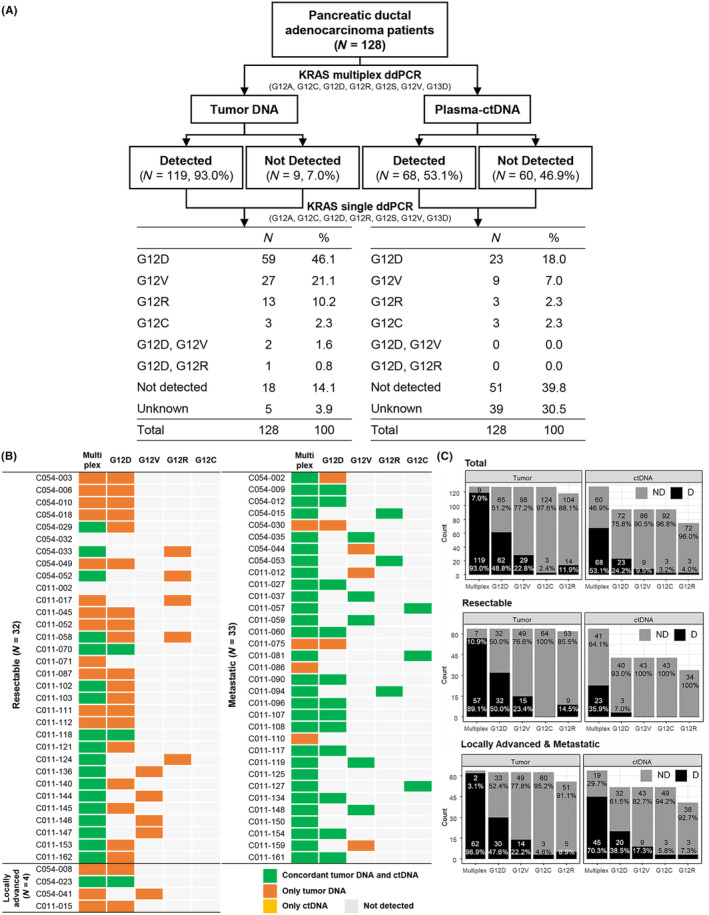
Frequency and concordance of KRAS mutation in tumor DNA and circulating tumor DNA (ctDNA). (A) Scheme of KRAS mutation frequency in multiplex and single droplet digital PCR (ddPCR) in 128 patients with pancreatic ductal adenocarcinoma (PDAC). (B) Concordance of KRAS mutation between tumor DNA and ctDNA in 69 patients. Heatmap indicating mutations detected in both tumor DNA and ctDNA (green), only tumor DNA (orange), and only ctDNA (yellow). (C) Comparison of frequencies of KRAS mutation detected in plasma and tumor DNA. A single KRAS mutation for each G12D, G12V, G12C, and G12R or seven common mutations (multiplex) for G12A, G12C, G12D, G12R, G12S, G12V, and G13D were detected using ddPCR. D, detected; ND, not detected.
3.3. The clinical features of each mutation and the association with survival
In the total set, there were no significant differences in the OS or PFS between the detected and nondetected groups for each mutation. In contrast, for ctDNA, the detected group at each hotspot showed significantly poorer OS (G12D, p < 0.0001; G12V, p < 0.0001; G12R, p = 0.0002; G12C, p = 0.0144) and PFS (G12D, p < 0.0001; G12V, p < 0.0001) than the nondetected group (Figure S2). In addition, the survival difference between the G12C‐detected group, other hotspot (G12D/G12V/G12R)‐detected group, and nondetected group was analyzed. There was no difference between the groups in tumor DNA (Figure 2A), whereas in ctDNA, the G12C‐detected group showed significantly lower OS than the nondetected group. When comparing the OS and PFS survival curves of the other hotspot and nondetected, it was confirmed that there was a significant difference (PFS, p < 0.0001; OS, p < 0.0001; Figure 2B). Status was confirmed to be the only statistically significant factor (HR: 4.79, 95% CI: 2.950–7.776, p < 0.0001). After adjusting the status, it was confirmed that ctDNA detection was a statistically significant factor in OS (HR: 2.640, 95% CI: 1.545–4.510, p = 0.0004) and PFS (HR: 1.768, 95% CI: 1.048–2.981, p = 0.0326) (Table 2).
FIGURE 2.
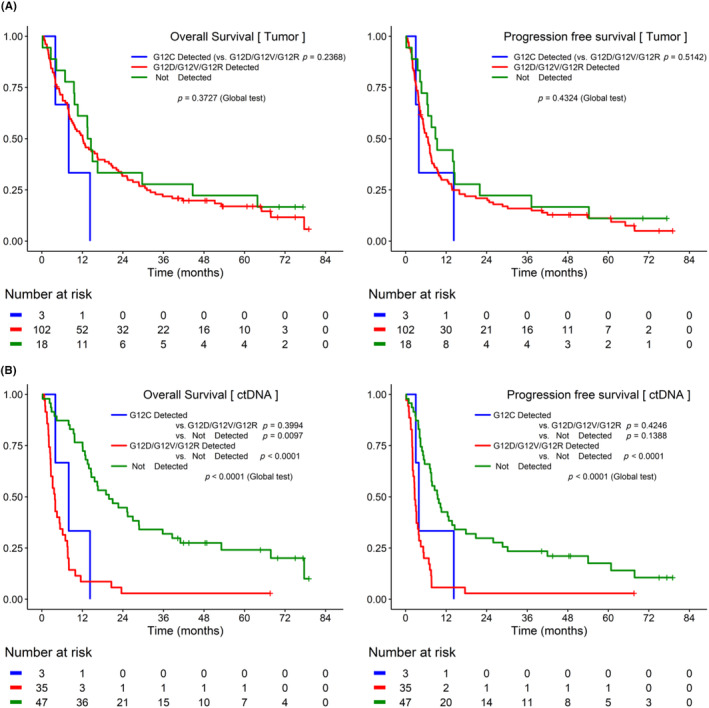
Kaplan–Meier curves of progression‐free survival (PFS) and overall survival (OS) based on KRAS G12C detected in tumor DNA and circulating tumor DNA (ctDNA). (A) In tumor DNA, the difference in survival between the KRAS G12C‐detected group, the other hotspot (KRAS G12D/G12V/G12R)‐detected group, and the nondetected group was analyzed. There was no difference between the groups in tumor DNA. (B) In ctDNA, the KRAS G12C‐detected group showed significantly lower survival rates than the nondetected group.
TABLE 2.
Cox proportional hazard model of overall survival and progression‐free survival with circulating tumor DNA (ctDNA) mutations (N = 95).
| N | Overall survival | Progression‐free survival | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Event | Univariable | Adjusted stage | Event | Univariable | Adjusted stage | ||||||
| HR (95% CI) | p‐Value | HR (95% CI) | p‐Value | HR (95% CI) | p‐Value | HR (95% CI) | p‐Value | ||||
| Status | |||||||||||
| Resectable | 43 | 32 | 1 (ref) | 1 (ref) | 34 | 1 (ref) | 1 (ref) | ||||
| Locally advanced/metastatic | 52 | 51 | 4.790 (2.950–7.776) | <0.0001 | 3.312 (1.901–5.771) | <0.0001 | 52 | 4.392 (2.663–7.244) | <0.0001 | 3.330 (1.869–5.933) | <0.0001 |
| Sex | |||||||||||
| Female | 43 | 38 | 1 (ref) | 39 | 1 (ref) | ||||||
| Male | 52 | 45 | 0.945 (0.613–1.456) | 0.7964 | 47 | 0.999 (0.653–1.528) | 0.9953 | ||||
| Tumor location | |||||||||||
| Head or neck or uncinate process | 49 | 45 | 1 (ref) | 46 | 1 (ref) | ||||||
| Body or tail | 46 | 38 | 0.548 (0.354–0.849) | 0.0071 | 40 | 0.657 (0.428–1.007) | 0.0536 | ||||
| Age | 95 | 83 | 1.005 (0.979–1.032) | 0.7033 | 86 | 0.995 (0.969–1.022) | 0.7259 | ||||
| ECOG | |||||||||||
| 0 | 61 | 53 | 1 (ref) | 55 | 1 (ref) | ||||||
| 1, 2, 3 | 34 | 30 | 1.341 (0.854–2.104) | 0.2023 | 31 | 1.382 (0.887–2.153) | 0.1524 | ||||
| CEA Baseline | |||||||||||
| Low: CEA ≤ 5 | 43 | 33 | 1 (ref) | 35 | 1 (ref) | ||||||
| High: CEA > 5 | 49 | 47 | 2.363 (1.503–3.716) | 0.0002 | 48 | 2.084 (1.336–3.252) | 0.0012 | ||||
| CA19‐9 Baseline | |||||||||||
| Low: CA19‐9 ≤ 37 | 22 | 18 | 1 (ref) | 19 | 1 (ref) | ||||||
| High: CA19‐9 > 37 | 61 | 55 | 1.495 (0.875–2.554) | 0.1414 | 57 | 1.285 (0.764–2.163) | 0.3446 | ||||
| ctDNA Detection | |||||||||||
| Nondetected | 57 | 46 | 1 (ref) | 1 (ref) | 49 | 1 (ref) | 1 (ref) | ||||
| G12D/G12V/G12R/G12C Detected | 38 | 37 | 4.661 (2.900–7.493) | <0.0001 | 2.640 (1.545–4.510) | 0.0004 | 37 | 3.294 (2.095–5.179) | <0.0001 | 1.768 (1.048–2.981) | 0.0326 |
3.4. Application of treatment to patients with KRAS G12C mutation detected in plasma ctDNA
To determine the therapeutic utility of KRAS mutation screening by liquid biopsy, we extracted PDXCs and PDXOs from the patients who had KRAS G12C (C011‐127) or KRAS G12D (C011‐090) single mutations (Figure 1B). The positive cancer cell marker, epithelial cell adhesion molecule (EpCAM) was stained in PDXOs, indicating that they originated from epithelial cancer cells. Both PDXOs had a histological morphology similar to that of PDX tumor tissue (Figure 3A). The fractional abundance of PDX and PDXC as demonstrated by ddPCR for KRAS multiplex or G12C and G12D single mutations was identical to that of the patient, although fractional abundances for mutations appeared to be enriched upon PDX generation. In the ddPCR results for a single mutation, only KRAS G12C was found in patient tumor tissue, plasma, PDX tissue, and PDXC generated from C011‐127 (Figure 3B). In contrast, only KRAS G12D was found in all of those generated from C011‐090 (Figure 3C). These findings suggest that PDXC and PDXO have been validated as effective models that accurately recapitulate the patients' histopathological and genetic features.
FIGURE 3.
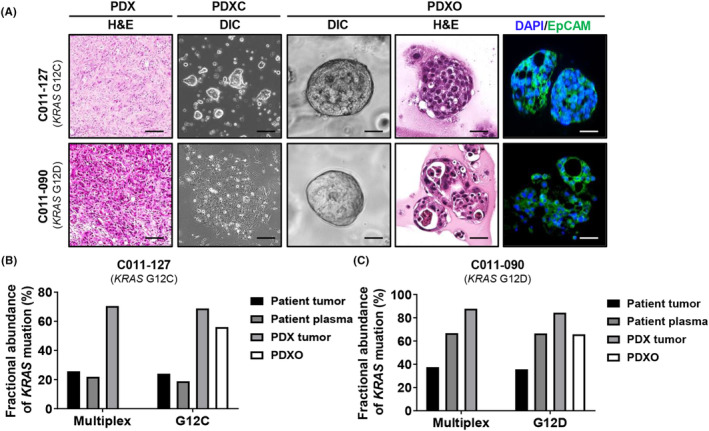
Characterization of patient‐derived xenograft (PDX), PDX‐derived cell (PDXC), and PDX‐derived organoid (PDXO) from percutaneous liver biopsy tissue from a patient with metastatic pancreatic cancer. (A) Representative images of PDX tumor tissue and organoid using hematoxylin and eosin (H&E) staining and immunofluorescence. EpCAM‐positive cells are stained green. The nucleus was stained blue by DAPI staining. Scale bar = 50 μm. (B, C) Comparison of KRAS mutation fractional abundances obtained using droplet digital PCR (ddPCR) with specific primers for G12D or G12C mutation, among organoid and patient, PDX tumor tissues or plasma. A multiplex KRAS mutation detection kit for seven common KRAS mutations (G12A, G12C, G12D, G12R, G12S, G12V, and G13D) was used. Genomic DNA was purified from formalin‐fixed paraffin‐embedded (FFPE) tissue of F0 and F1. Purified DNA was used for ddPCR with KRAS screening.
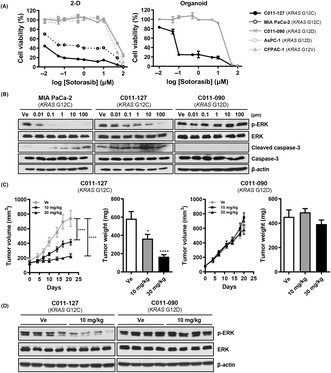
To evaluate the selective antitumor activity of the KRAS G12C inhibitor sotorasib, we tested cell viability using PDXC and PDXO. In this assay, we used the pancreatic cancer cell line MIA PaCa‐2 (KRAS G12C) as a positive control, and AsPC‐1 (KRAS G12D) and CFPAC‐1 (KRAS G12V) as negative controls. Sotorasib showed highly selective cytotoxicity at a concentration less than 1 μM in KRAS G12C‐mutated organoids but induced cell death regardless of KRAS mutation at concentrations higher than 1 μM. C011‐127‐PDXO (KRAS G12C) showed strong cytotoxicity to sotorasib with 0.098 μM compared with C011‐090‐PDXO (KRAS G12D), with a 50% inhibitory concentration of 11.71 μM (Figure 4A). The selective KRAS G12C inhibition effect of sotorasib was also observed in KRAS downstream signaling pathway. Extracellular signal‐regulated kinase (ERK) phosphorylation was decreased only in KRAS G12C‐mutant cells, MIA PaCa‐2, and C011‐127‐PDXC. In addition, C011‐127‐PDXC increased the amount of cleaved caspase‐3 in a sotorasib concentration‐dependent manner, whereas C011‐090‐PDXC showed no change (Figure 4B).
FIGURE 4.
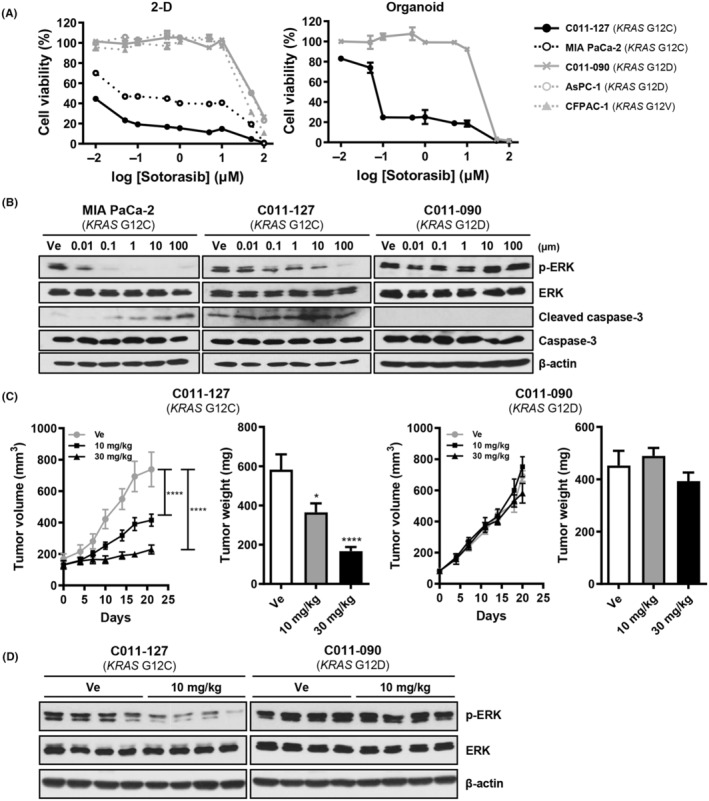
The KRAS G12C inhibitor sotorasib selectively inhibits tumor growth in a patient‐derived xenograft (PDX), PDX‐derived cell (PDXC), and PDX‐derived organoid (PDXO) with KRAS G12C mutation. (A) Drug response curve of sotorasib in 2D culture condition and organoid culture condition with 0.01, 0.1, 1, 10, and 100 μM of sotorasib and vehicle (1% DMSO). The cell viability in 2D was observed after treatment for 72 h and measured using adenosine triphosphate (ATP)‐based assay with CellTiter‐Glo reagent. For the organoids, the ATP was measured after 5 days of treatment. (B) Western blot analysis for KRAS pathway (p‐ERK, ERK) and apoptosis (cleaved caspase‐3) on sotorasib‐treated MIA PaCa‐2, C011‐127‐PDXC (C011‐127‐PDX‐derived cell with KRAS G12C mutation), and C011‐090‐PDXC (C011‐090‐PDX‐derived cell with KRAS G12D mutation) for 24 h. (C) Tumor growth after administration of sotorasib. For the pancreatic ductal adenocarcinoma (PDAC) xenograft mouse model, PDXC was injected into the flanks of BALB/c nude mice aged 5 weeks. Sotorasib was administered daily by oral gavage for 24 days starting at the time of tumor size of approximately 100–150 mm3. The tumor volume was measured twice a week. Tumor weights were measured immediately after euthanasia. (D) Western blot analysis for ERK phosphorylation in C011‐127‐PDX or C011‐090‐PDX tumor tissue after 24 days of administration.
Next, we evaluated the selective tumor growth inhibition effect of sotorasib in vivo. After 10 days of treatment, C011‐127‐PDX (KRAS G12C) showed a significant decrease in tumor growth. Although no changes were observed in C011‐090‐PDX (KRAS G12D), C011‐127‐PDX showed a sustained tumor growth inhibitory effect during the administration period (Figure 4C). After the final administration, the tumor volume decreased 1.9‐fold in the 10 mg/kg treatment group and 3.2‐fold in the 30 mg/kg treatment group compared with that of the vehicle‐treated group. Tumor weight also decreased 1.6‐ and 3.5‐fold at 10 and 30 mg/kg of sotorasib, respectively (Figure 4C). Additionally, C011‐127‐PDX tumor tissue also showed selective inhibition of ERK phosphorylation at the end of administration. By demonstrating the selective inhibition of tumor growth by the KRAS G12C inhibitor, sotorasib, we suggest that the detection of genetic mutations using ctDNA has therapeutic applicability in patients.
3.5. Remodeling of the tumor microenvironment (TME) by KRAS G12C inhibitor
Next, we evaluated histopathological changes of tumor tissue in C011‐127‐PDX, in which the volume was reduced by sotorasib. As a result of the decrease in tumor volume, hematoxylin and eosin (H&E) staining also showed remarkable apoptosis of KRAS G12C tumor cells by sotorasib (Figure 5A). The expression of the proliferation marker Ki‐67 in C011‐127‐PDX decreased in a sotorasib concentration‐dependent manner (Figure 5B). In addition, we also found that changes in immune and stromal cells of the TME as well as tumor cell death occurred (Figure 5A). To elucidate the infiltrating cells, we stained macrophages and fibroblasts using a multiplex immunofluorescent staining. In a sotorasib concentration‐dependent manner, the tumor area in the mass decreased, the stroma area increased, and αSMA+ fibroblasts and macrophages were significantly infiltrated. Macrophages infiltrating the stromal region were more polarized to M1 (F4/80+CD86+) than to M2 (F4/80+CD206+) (Figure 5B). In contrast, C011‐027‐PDX was not affected by sotorasib (Figure 5C). These results indicate that sotorasib induces apoptosis in KRAS G12C tumor cells and changes the TME, recruiting stromal cells such as fibroblasts and macrophages.
FIGURE 5.
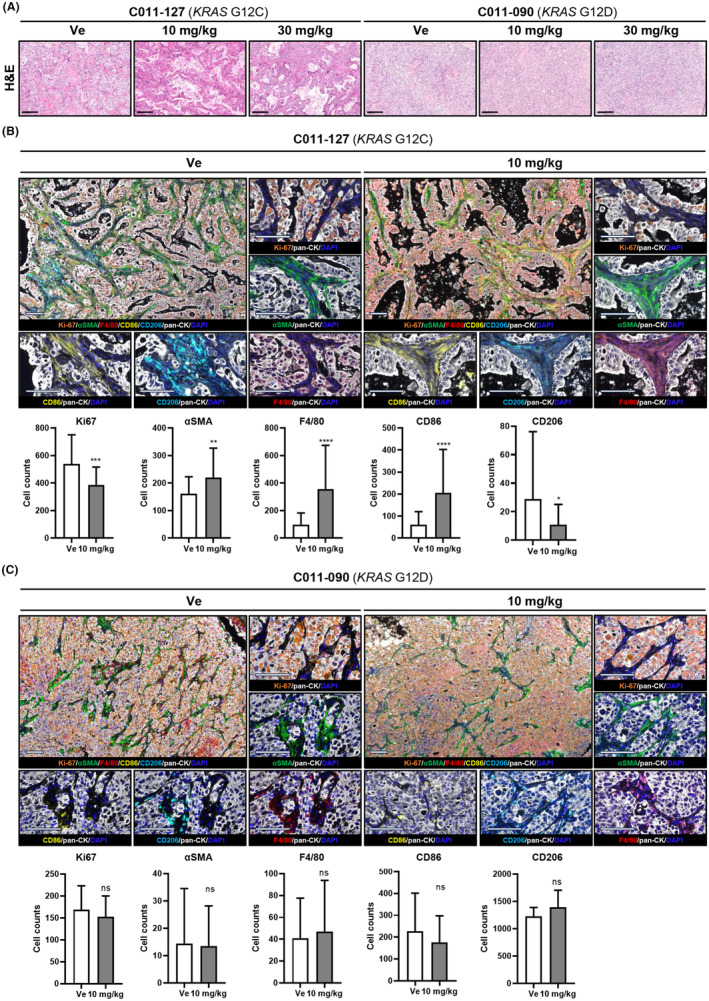
The KRAS G12C inhibitor sotorasib remodels the microenvironment in a patient‐derived xenograft mouse model with KRAS G12C mutation. (A) Representative images of hematoxylin and eosin (H&E) staining. (B, C) Representative images of opal staining for proliferation (anti‐Ki‐67), fibroblast (anti‐αSMA), and macrophages (anti‐F4/80, anti‐CD86, anti‐CD206). All quantification, at least 10 fields of view were averaged per tumor. Objective, 20×. All data are presented as the means ± standard deviation (SD). Statistical significance levels: *p < 0.05, **p < 0.01, ***p < 0.001.
4. DISCUSSION
The presence of KRAS mutations is significantly associated with poor OS, tumor grade, and metastatic stage in cancer. 25 , 26 , 27 Since KRAS mutation is a very common signature that appears in most patients with PDAC, its role as a prognostic biomarker is limited. 22 Nevertheless, ctDNA detection is correlated with the clinical tumor burden and thus could be utilized as a biomarker for monitoring the tumor dynamics and more accurately predicting the clinical prognosis. Specifically, patients with KRAS mutations detected in plasma ctDNA showed worsened OS or PFS, whereas the differences between patients with and without mutations detected in the tumor were not significant, which is consistent with a previous study from large multi‐institutional cohorts. 28 , 29 , 30 Here, we demonstrated that the detection of ctDNA by liquid biopsy had a higher predictive rate of PDAC prognosis than the tumor DNA in 128 patients with PDAC. We presented the clinical relevance of KRAS mutations in blood ctDNA and evaluated the applicability of these therapeutic strategies.
In the present study, we detected ctDNA in the blood of patients with PDAC and analyzed KRAS mutations in seven hotspots using ddPCR. KRAS mutations were found in 93% of tumor tissues, and 53% were detected in plasma, similar to our previous study. 23 The sensitivity of ctDNA for KRAS mutation in tumor tissue was higher at 80% in the metastatic stage than in the resectable stage. Cases of tissue‐ctDNA mismatch may be explained by low tumor burden in surgically resectable cases. 31 , 32 In plasma, ctDNA is generally detected in 12.5%–69% of resectable cases and 40%–86% of unresectable cases. 20 Our study showed similar rates of 53.13% and 70.31%, respectively. In PDAC, somatic KRAS mutations occur mainly at codon 12, and G12D and G12V are prominently observed (approximately 45% and 32%, respectively). 33 In particular, the G12R mutation frequency in this cohort was 3%, lower than that in other groups including COMPIC and International Cancer Genome Consortium (Figure S3). 17
Recently, the importance of KRAS mutation detection in pancreatic cancer has been increasing owing to the successful development of single‐target inhibitors of mutant KRAS. Clinical trials aimed at analyzing the efficacy of Amgen's novel small‐molecule inhibitor sotorasib (Lumakras™) have resulted in its approval for patients with NSCLC harboring KRAS G12C mutation in their tumors. 34 , 35 Mirati Therapeutics' KRAS G12C inhibitor Adagrasib (MRTX849) has shown a successful response in a phase I trial. 36 In addition, development of targeted therapy against KRAS G12D that is effective, easier, and more sensitive for the detection of specific KRAS single mutations in pancreatic cancer patients has become necessary. 10 In this study, we sought to confirm whether KRAS‐targeted therapy can be employed based on the mutation signature detected through ctDNA in plasma, which was prepared by separating cells from PDX created using two patient tissues with KRAS G12C or G12D mutations. Sotorasib substantially and selectively reduced tumor growth in PDXC with the G12C mutation in vitro and in vivo. However, in PDXC with the G12D mutation, no meaningful effect was seen. In the PDX mouse model, fibroblast levels increased in areas where cancer cell levels decreased. This is a common feature of tumor tissues following the administration of chemotherapeutic medicines. 37 Since αSMA+ fibroblasts have a role in suppressing tumor growth in PDAC, 37 the increase in this cancer‐associated fibroblast (CAF) subpopulation may contribute to the antitumor effect and serve as an indicator of drug response. 38 Moreover, CAFs have recently been considered key components regulating immune infiltration in cancer. 39 We observed that sotorasib not only enhances macrophage infiltration but also significantly increases the number of M1 subtype macrophages, which have the unique functions of direct capture, phagocytosis, and tumor cell lysis. There has been no report yet on a direct correlation with increased immune cell infiltration by αSMA+ fibroblasts caused by sotorasib. The previously reported tumor‐suppressive effect of sotorasib by increasing immune cell infiltration into tumor tissue and the synergistic effect of PD‐L1 drug combination with increased CD8+ T cells in an immunocompetent mouse model may have been induced for this reason. 9
Collectively, considering the fact that more than 60% of the pancreatic cancers are diagnosed at advanced stages that are inoperable, it is important to be able to determine the direction of effective target therapy using a noninvasive diagnostic method. We suggest that KRAS mutation screening using ctDNA in blood shows a higher prognostic concordance rate than using DNA in tumors. Furthermore, the applicability of the KRAS G12C inhibitor sotorasib was evaluated using preclinical models including cells, organoids, and mice established from patient‐derived tissues. Additionally, the adoption of patient‐derived preclinical models that reflect the patients' histopathological and genetic features can serve as a steppingstone in overcoming resistance to KRAS target inhibitors, which is currently an unsolved issue. Taken together, this study suggests that non‐invasive monitoring of KRAS mutations using plasma ctDNA may be a more powerful prognostic monitoring tool than monitoring KRAS mutations using tumor DNA and may have the potential to help determine whether KRAS single inhibitors can be used to treat PDAC.
AUTHOR CONTRIBUTIONS
Mi Rim Lee: Investigation; validation; writing – original draft. Sang Myung Woo: Funding acquisition; resources; writing – review and editing. Min Kyeong Kim: Formal analysis; investigation. Sung‐Sik Han: Resources. Sang‐Jae Park: Resources. Woo Jin Lee: Resources. Dong‐eun Lee: Formal analysis. Sun Il Choi: Methodology. Wonyoung Choi: Writing – review and editing. Kyong‐Ah Yoon: Conceptualization. Jung Won Chun: Resources. Yun‐Hee Kim: Supervision; writing – review and editing. Sun‐Young Kong: Funding acquisition; supervision; writing – review and editing.
FUNDING INFORMATION
This study was supported by grants from the NCC (grant number: NCC‐1910190 & NCC‐2212470 & NCC‐2310630) and the National Research Foundation of Korea funded by the Korean government (MIST) (grant number: 2020M3A9A5036362).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
Approval of the research protocol by an IRB: This study conforms to the provisions of the Declaration of Helsinki and was approved by the IRB (IRB No. NCC2015‐054, NCC2016‐011).
Informed Consent: Informed consent was obtained from all patients.
Registry and the Registration No. of the study/trial: N/A.
Animal Studies: All animal studies were reviewed and approved by the IACUC of the NCCRI (NCC‐16‐247, NCC‐21‐688).
Supporting information
Data S1:
ACKNOWLEDGMENTS
The authors express deepest gratitude to research nurse Young Hwa Kang for excellent support in project administration and data collection. This work was supported by the Research Core Center in NCC Korea.
Lee MR, Woo SM, Kim MK, et al. Application of plasma circulating KRAS mutations as a predictive biomarker for targeted treatment of pancreatic cancer. Cancer Sci. 2024;115:1283‐1295. doi: 10.1111/cas.16104
Mi Rim Lee and Sang Myung Woo contributed equally to this work.
Contributor Information
Yun‐Hee Kim, Email: sensia37@ncc.re.kr.
Sun‐Young Kong, Email: ksy@ncc.re.kr.
DATA AVAILABILITY STATEMENT
The raw data used in this article are not available because of the National Cancer Institute's data policy. For access to the dataset the National Cancer Institute should be contacted by mail.
REFERENCES
- 1. Rawla P, Sunkara T, Gaduputi V. Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J Oncol. 2019;10:10‐27. doi: 10.14740/wjon1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Exarchakou A, Papacleovoulou G, Rous B, et al. Pancreatic cancer incidence and survival and the role of specialist centres in resection rates in England, 2000 to 2014: a population‐based study. Pancreatology. 2020;20:454‐461. doi: 10.1016/j.pan.2020.01.012 [DOI] [PubMed] [Google Scholar]
- 3. Bengtsson A, Andersson R, Ansari D. The actual 5‐year survivors of pancreatic ductal adenocarcinoma based on real‐world data. Sci Rep. 2020;10:16425. doi: 10.1038/s41598-020-73525-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388:73‐85. doi: 10.1016/S0140-6736(16)00141-0 [DOI] [PubMed] [Google Scholar]
- 5. Leslie M. Researchers reveal another KRAS inhibitor. Cancer Discov. 2019;9:1152. doi: 10.1158/2159-8290.CD-NB2019-092 [DOI] [PubMed] [Google Scholar]
- 6. Chen H, Smaill JB, Liu T, Ding K, Lu X. Small‐molecule inhibitors directly targeting KRAS as anticancer therapeutics. J Med Chem. 2020;63:14404‐14424. doi: 10.1021/acs.jmedchem.0c01312 [DOI] [PubMed] [Google Scholar]
- 7. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K‐Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548‐551. doi: 10.1038/nature12796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Patricelli MP, Janes MR, Li L‐S, et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016;6:316‐329. doi: 10.1158/2159-8290.CD-15-1105 [DOI] [PubMed] [Google Scholar]
- 9. Canon J, Rex K, Saiki AY, et al. The clinical KRAS (G12C) inhibitor AMG 510 drives anti‐tumour immunity. Nature. 2019;575:217‐223. doi: 10.1038/s41586-019-1694-1 [DOI] [PubMed] [Google Scholar]
- 10. Wang X, Allen S, Blake JF, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J Med Chem. 2021;65:3123‐3133. doi: 10.1021/acs.jmedchem.1c01688 [DOI] [PubMed] [Google Scholar]
- 11. Mao Z, Xiao H, Shen P, et al. KRAS (G12D) can be targeted by potent inhibitors via formation of salt bridge. Cell Discov. 2022;8:1‐14. doi: 10.1038/s41421-021-00368-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang L, Guo Z, Wang F, Fu L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther. 2021;6:1‐20. doi: 10.1038/s41392-021-00780-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kemp SB, Cheng N, Markosyan N, et al. Efficacy of a small‐molecule inhibitor of KrasG12D in immunocompetent models of pancreatic cancer. Cancer Discov. 2023;13:298‐311. doi: 10.1158/2159-8290.Cd-22-1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Corcoran RB, Chabner BA. Application of cell‐free DNA analysis to cancer treatment. N Engl J Med. 2019;74:204‐206. doi: 10.1056/NEJMra1706174 [DOI] [PubMed] [Google Scholar]
- 15. Stroun M, Anker P, Maurice P, Lyautey J, Lederrey C, Beljanski M. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology. 1989;46:318‐322. doi: 10.1159/000226740 [DOI] [PubMed] [Google Scholar]
- 16. Alese OB, Cook N, Ortega‐Franco A, Ulanja MB, Tan L, Tie J. Circulating tumor DNA: an emerging tool in gastrointestinal cancers. Am Soc Clin Oncol Educ Book. 2022;42:1‐20. doi: 10.1200/edbk_349143 [DOI] [PubMed] [Google Scholar]
- 17. Cheng F, Su L, Qian C. Circulating tumor DNA: a promising biomarker in the liquid biopsy of cancer. Oncotarget. 2016;7:48832‐48841. doi: 10.18632/oncotarget.9453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heredia‐Soto V, Rodríguez‐Salas N, Feliu J. Liquid biopsy in pancreatic cancer: are we ready to apply it in the clinical practice? Cancers (Basel). 2021;13:1986. doi: 10.3390/cancers13081986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Keller L, Belloum Y, Wikman H, Pantel K. Clinical relevance of blood‐based ctDNA analysis: mutation detection and beyond. Br J Cancer. 2021;124:345‐358. doi: 10.1038/s41416-020-01047-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sivapalan L, Kocher H, Ross‐Adams H, Chelala C. Molecular profiling of ctDNA in pancreatic cancer: opportunities and challenges for clinical application. Pancreatology. 2021;21:363‐378. doi: 10.1016/j.pan.2020.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Knudsen ES, O'Reilly EM, Brody JR, Witkiewicz AK. Genetic diversity of pancreatic ductal adenocarcinoma and opportunities for precision medicine. Gastroenterology. 2016;150:48‐63. doi: 10.1053/j.gastro.2015.08.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qian ZR, Rubinson DA, Nowak JA, et al. Association of alterations in main driver genes with outcomes of patients with resected pancreatic ductal adenocarcinoma. JAMA Oncol. 2018;4:e173420. doi: 10.1001/jamaoncol.2017.3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim MK, Woo SM, Park B, et al. Prognostic implications of multiplex detection of KRAS mutations in cell‐free DNA from patients with pancreatic ductal adenocarcinoma. Clin Chem. 2018;64:726‐734. doi: 10.1373/clinchem.2017.283721 [DOI] [PubMed] [Google Scholar]
- 24. Choi SI, Jeon A‐R, Kim MK, et al. Development of patient‐derived preclinical platform for metastatic pancreatic cancer: PDOX and a subsequent organoid model system using percutaneous biopsy samples. Front Oncol. 2019;9:875. doi: 10.3389/fonc.2019.00875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Said R, Guibert N, Oxnard GR, Tsimberidou AM. Circulating tumor DNA analysis in the era of precision oncology. Oncotarget. 2020;11:188‐211. doi: 10.18632/oncotarget.27418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hadano N, Murakami Y, Uemura K, et al. Prognostic value of circulating tumour DNA in patients undergoing curative resection for pancreatic cancer. Br J Cancer. 2016;115:59‐65. doi: 10.1038/bjc.2016.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pietrasz D, Pécuchet N, Garlan F, et al. Plasma circulating tumor DNA in pancreatic cancer patients is a prognostic marker. Clin Cancer Res. 2017;23:116‐123. doi: 10.1158/1078-0432.CCR-16-0806 [DOI] [PubMed] [Google Scholar]
- 28. Zhuang R, Li S, Li Q, et al. The prognostic value of KRAS mutation by cell‐free DNA in cancer patients: a systematic review and meta‐analysis. PLoS ONE. 2017;12:e0182562. doi: 10.1371/journal.pone.0182562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wahl SGF, Dai HY, Emdal EF, et al. Prognostic value of absolute quantification of mutated KRAS in circulating tumour DNA in lung adenocarcinoma patients prior to therapy. J Pathol Clin Res. 2021;7:209‐219. doi: 10.1002/cjp2.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Suzuki HI, Onimaru K. Biomolecular condensates in cancer biology. Cancer Sci. 2022;113:382‐391. doi: 10.1111/cas.15232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Patel H, Okamura R, Fanta P, et al. Clinical correlates of blood‐derived circulating tumor DNA in pancreatic cancer. J Hematol Oncol. 2019;12:130. doi: 10.1186/s13045-019-0824-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kirchweger P, Kupferthaler A, Burghofer J, et al. Circulating tumor DNA correlates with tumor burden and predicts outcome in pancreatic cancer irrespective of tumor stage. Eur J Surg Oncol. 2022;48:1046‐1053. doi: 10.1016/j.ejso.2021.11.138 [DOI] [PubMed] [Google Scholar]
- 33. Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91‐100. doi: 10.1016/j.tibs.2013.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lanman BA, Allen JR, Allen JG, et al. Discovery of a covalent inhibitor of KRAS(G12C) (AMG 510) for the treatment of solid tumors. J Med Chem. 2020;63:52‐65. doi: 10.1021/acs.jmedchem.9b01180 [DOI] [PubMed] [Google Scholar]
- 35. Skoulidis F, Li BT, Dy GK, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med. 2021;384:2371‐2381. doi: 10.1056/NEJMoa2103695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jänne PA, Riely GJ, Gadgeel SM, et al. Adagrasib in non‐small‐cell lung cancer harboring a KRAS(G12C) mutation. N Engl J Med. 2022;387:120‐131. doi: 10.1056/NEJMoa2204619 [DOI] [PubMed] [Google Scholar]
- 37. Biffi G, Oni TE, Spielman B, et al. IL1‐induced JAK/STAT signaling is antagonized by TGFbeta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019;9:282‐301. doi: 10.1158/2159-8290.CD-18-0710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nagaria TS, Wang H, Chatterjee D, Wang H. Pathology of treated pancreatic ductal adenocarcinoma and its clinical implications. Arch Pathol Lab Med. 2020;144:838‐845. doi: 10.5858/arpa.2019-0477-RA [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mhaidly R, Mechta‐Grigoriou F. Role of cancer‐associated fibroblast subpopulations in immune infiltration, as a new means of treatment in cancer. Immunol Rev. 2021;302:259‐272. doi: 10.1111/imr.12978 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1:
Data Availability Statement
The raw data used in this article are not available because of the National Cancer Institute's data policy. For access to the dataset the National Cancer Institute should be contacted by mail.