

Abstract
The current state‐of‐the‐art analysis of central nervous system (CNS) tumors through DNA methylation profiling relies on the tumor classifier developed by Capper and colleagues, which centrally harnesses DNA methylation data provided by users. Here, we present a distributed‐computing‐based approach for CNS tumor classification that achieves a comparable performance to centralized systems while safeguarding privacy. We utilize the t‐distributed neighborhood embedding (t‐SNE) model for dimensionality reduction and visualization of tumor classification results in two‐dimensional graphs in a distributed approach across multiple sites (DistSNE). DistSNE provides an intuitive web interface (https://gin-tsne.med.uni-giessen.de) for user‐friendly local data management and federated methylome‐based tumor classification calculations for multiple collaborators in a DataSHIELD environment. The freely accessible web interface supports convenient data upload, result review, and summary report generation. Importantly, increasing sample size as achieved through distributed access to additional datasets allows DistSNE to improve cluster analysis and enhance predictive power. Collectively, DistSNE enables a simple and fast classification of CNS tumors using large‐scale methylation data from distributed sources, while maintaining the privacy and allowing easy and flexible network expansion to other institutes. This approach holds great potential for advancing human brain tumor classification and fostering collaborative precision medicine in neuro‐oncology.
Keywords: brain tumor classification, distributed computing, DataSHIELD, methylome, tSNE, webinterface
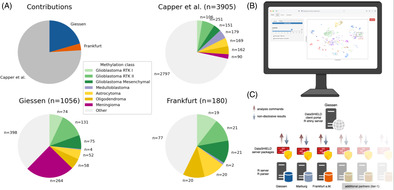
Overview of CNS tumor methylome data and the computational setup (a). Data contribution of the collected data. The most frequent subgroups are color‐coded. (b) R Shiny web application for distributed t‐SNE plotting. Upon user login, raw data can be uploaded to the analysis server via the R Shiny interface. A zoom‐in of the area of interest provides a detailed view of the sample (marked with an asterisk) and its surrounding area. (c) Structure of the distributed t‐SNE across the three sites. A central R client portal manages the computation of the distributed datasets in Frankfurt, Marburg, and Giessen.
1. IMPORTANCE OF THE STUDY
Diagnosing brain tumors requires a delicate balance between histological and molecular methods, with the latter requiring vast datasets for accurate classification. This need for extensive datasets is challenged by rising concerns around digital privacy. We have developed DistSNE as a timely response, offering researchers a secure platform to share and pool DNA methylation data of brain tumors in compliance with strict privacy standards. Simultaneously, this tool facilitates the comparison and classification of novel samples with an extensive, pooled database from various participating institutions. Beyond just refining current diagnostic techniques, DistSNE aims to catalyze collaborative research, paving the way for the discovery of new molecular subgroups, improving CNS tumor classification, and supporting superior patient care in neuro‐oncology.
2. INTRODUCTION
Molecular genetics and high‐throughput genomics have gained increasing importance in tumor diagnostics and therapy in recent years [1]. Specifically, the classification of central nervous system (CNS) tumors relies on genome‐wide DNA methylation analysis, a state‐of‐art high‐throughput epigenome profiling technique, that complements macroscopic, histological examinations, as well as somatic mutation analyses for improved diagnostic, therapeutic, and prognostic performance [2].
Methylation microarray platforms by Illumina have now become widely established standard methods for subtyping CNS tumors [3, 4]. Moreover, these platforms can also identify copy number variations (CNV) [5]. Computational analysis of the methylome profiles using a large reference dataset allows highly accurate determination of CNS tumor methylation classes [6], with the Heidelberg Classifier as one of the most widely used tumor classification tools in neuro‐oncology [7]. More than 130,000 cases have been analyzed so far and more than 98,000 cases were used for the development of the classification according to www.molecularneuropathology.org/mnp/ (accessed on October 27, 2023). To enhance interpretability and complement the random‐forest classification, visualizations of the methylation data by dimensionality reduction can be utilized. The t‐distributed neighborhood embedding (t‐SNE) model is such a method, that reduces the high‐dimensional methylation data to 2D projections, facilitating the grouping and visualization of tumor clusters based on their shared methylation profile.
While such methods enable efficient and standardized tumor classification, the computational setup is currently centralized. Moreover, strict privacy regulations surrounding patient datasets restrict institutes from sharing their patient data with the scientific community for research purposes [8]. While raw data remains local, processed information can be shared and aggregated from different institutes to refine models based on much larger patient cohorts. The Medical Informatics Initiative (MII) was launched to develop infrastructure for the integration of clinical data from patient care and medical research and facilitate data sharing among university hospitals while conforming to privacy regulations [9]. One widely used operating platform for distributed computing is DataSHIELD (Data Aggregation Through Anonymous Summary‐statistics from Harmonized Individual‐level Databases), employed also by MIRACUM, one out of four MII consortia [10, 11, 12]. It has since been extended to facilitate deep learning‐based analyses and Big Data analyses from distributed individual patient data [13, 14]. Recent demonstrations of DataSHIELD‘s usability in large medical informatics projects emphasize its value for the analysis of patient data in a data protection‐compliant way [15, 16]. As a federated meta‐analysis programming library, it has been developed at Newcastle University in cooperation with the Research Institute of the McGill University Health Centre. Used by the EUCAN‐CONNECT project (https://eucanconnect.com) it handles 173 European population‐based cohort studies allowing the investigation of ~2.5 M participants across 30 sites and consortia. Thus, it provides an ideal platform to develop a distributed computing‐based solution for a collaborative, privacy‐compliant CNS tumor classification approach using t‐SNE (DistSNE).
Here, we introduce the DistSNE framework which leverages local, scalable data warehouses from participating sites to gather, normalize, and analyze methylation array data from CNS tumor samples. This synergistic approach enables t‐SNE plot computation for visualization and classification of CNS tumor samples based on DNA methylation data while maintaining high privacy standards for patient data. To facilitate usability, we offer a user‐friendly two‐tier web interface (https://gin-tsne.med.uni-giessen.de/). Participating institutes can perform t‐SNE visualization for their individual tumor sample based on methylome data from all participating institutes (tier‐1). Non‐participating institutes can also classify individual tumor samples by t‐SNE but are restricted to the publicly available reference CNS tumor methylome data (tier‐2). The ability to analyze and interpret large‐scale data from distributed sources while protecting patient privacy holds great promise for the future of precision medicine. We believe that the DistSNE framework will contribute to a better understanding of CNS tumor biology and enable the development of new therapeutic strategies, leading to improved patient outcomes.
3. METHODS
3.1. DNA methylome datasets of CNS tumors
DNA methylome datasets of CNS tumors were collected from three sources. The first sample cohort (n = 3905) was retrieved from the publicly available GSE109381 SuperSeries with 450 k methylation array data (Illumina HumanMethylation 450 BeadChip) from patients. The Giessen Institute of Neuropathology provided 850 k methylation array data (Illumina Infinium MethylationEPIC 850 k) (n = 1056), while the Edinger Institute in Frankfurt provided 850 k methylation array data (n = 180) (Figure 1A). For sample preparation, DNA was extracted and analyzed using the respective DNA methylation array following the manufacturer's instructions. The methylome data was initially classified using the Heidelberg Classifier v11b4 (Frankfurt) or v11b6 (Capper et al., Giessen), respectively, to obtain reference information on methylation class and probability score for each sample.
FIGURE 1.
Overview of CNS tumor methylome data and the computational setup (A). Capper et al. data (GSE109381) were deposited in Marburg (n = 3905) and in‐house datasets were provided by Giessen (n = 1056) and Frankfurt (n = 180). The most frequent subgroups are color‐coded. (B) R Shiny web application for distributed t‐SNE plotting. Upon user login, raw data can be uploaded to the analysis server via the R Shiny interface. The client portal automatically requests the aggregated data from each data warehouse in the background. After successful data retrieval, the t‐SNE plot is displayed on the right site of the interface, available for download. A zoom‐in of the area of interest provides a detailed view of the sample (marked with an asterisk) and its surrounding area. (C) Structure of the distributed t‐SNE across the three sites. A central R client portal manages the computation of the distributed datasets in Frankfurt, Marburg, and Giessen.
To ensure that the distributed dataset encompasses a broad variety of tumor subgroups, we analyzed the datasets at each location for their distribution. The Heidelberg Classifier v11b4/v11b6 has 82 tumor subgroups and nine additional subgroups such as Inflammatory microenvironment, totaling 91 subgroups. The public dataset covered all 91 subgroups, while the Giessen dataset included 69 different tumor subgroups and the Frankfurt dataset covered 28 different tumor subgroups.
3.2. Data preprocessing
We executed the following steps to preprocess methylome array data for t‐SNE computation at each site. (1) Only the intersecting CpGs from the 450 and 850 k methylation arrays were considered. (2) Beta values were computed from the raw signals using normalization references [17]. (3) X and Y chromosomes were excluded from further processing. (4) The 10,000 CpGs with the highest methylation variability across all patient samples and all three datasets were chosen for further computation. These steps were necessary to reduce the dimensionality of the data and to focus on the CpGs that were most informative for the analysis. Data analysis with a login as a tier‐2 client enables tumor classification using the publicly available reference CNS tumor methylome data.
3.3. Distributed t‐SNE computation
DataSHIELD [14] was installed on server instances at each partner site (Giessen, Frankfurt, Marburg), which were integrated into local Data Integration Center (DIC) environments to enable privacy‐protected and distributed data storage and analysis. The Marburg data warehouse hosted the public dataset, while the Giessen and Frankfurt warehouses provided in‐house data. A client portal with a web interface (Figure 1B) aggregated the information, requested from the different instances (Figure 1C), and computed the t‐SNE visualization.
To accelerate t‐SNE computation and reduce input data dimensionality [18], we used principal component analysis (PCA) [19], which involves singular value decomposition (SVD). The implementation of the approach of Iwen and Ong [20] facilitated distributed SVD through DataSHIELD, ensuring that only non‐reversibly transformed data were shared across sites. To ensure a high‐quality assessment, we restricted the samples of each cluster to those with a classification score above 0.9. A fixed seed (42) was used in the experiments to enable reproducible visualization and analysis. The classification of a new sample was determined by matching it with its nearest neighbor as measured by Euclidean distance.
3.4. Computational analyses and statistics
To identify similar samples belonging to the same subgroup it is important to achieve a low intra‐cluster variance as a tight clustering signifies closely similar or related samples. Ideally, a high‐quality cluster should have a low intra‐cluster‐variance, which signifies that similar or related points are closely gathered. The intra‐cluster‐variance describes the area a cluster occupies. Therefore, when increasing the sample size within a cluster while maintaining the intra‐cluster variance, we achieve a higher cluster density and quality. The intra‐cluster‐variance of each cluster was calculated by averaging the Euclidean distance of every point in the cluster to the center. The mean intra‐cluster‐variance over all clusters was computed as follows:
Here, C is the number of clusters and m c is the number of samples of the cluster c. being the center of cluster c.
To evaluate the performance of our approach we compared the classification obtained from the random forest algorithm of the Heidelberg classifier with the nearest neighbor classification for each sample from Giessen and Frankfurt within the distSNE. In the first experiment, we calculated the mean accuracy from 100 runs with varying random initialization. Accuracy was computed as the fraction of correctly assigned classifications (in relation to the random forest classification of Heidelberg) relative to the total number of samples. In the second experiment, we calculated the mean accuracy for all newly added samples in each run. The results were averaged over 100 runs, each varying by the sequence of sample inclusion. We tested for significance between the different approaches using a paired t‐test with Bonferroni correction.
3.5. Hard‐ and software requirements
The central computing instance and participating instances hosting utilized a server instance with four Cores and 64 GB RAM running on Ubuntu 18.04. The preprocessIllumina function of the minfi [17] package as the gold standard was used for raw data normalization from Infinium MethylationEPIC arrays, Ggplot2 [21] for t‐SNE visualization, an R shiny server for the web application, ds.SVD from dsMLpackage [19, 20] for left singular value computation and Rtsne package [18] for t‐SNE computation.
3.6. Data availability
Data will be made available upon reasonable request. To demonstrate the usability of the web interface, we provide two sample datasets for upload under https://doi.org/10.5281/zenodo.10048012.
3.7. Ethics approval
The experimental studies were authorized by the ethics committee of Justus‐Liebig‐University Giessen (AZ 138/18 and 07/09) and the ethics committee of Goethe‐Universität Frankfurt (UCT‐Project‐No: SNO‐19‐2020).
4. RESULTS
4.1. Distributed t‐SNE computation on the DistSNE web interface
We developed the user‐friendly, two‐tier R shiny web‐based analysis suite, DistSNE, enabling users to upload individual samples for t‐SNE analysis (Figures 1B, S1, and S2). Tier‐1 institutes, which actively participate in the distributed network, share their data via a local DataSHIELD instance. Non‐participating Tier‐2 institutes have access only to the publicly available reference CNS tumor methylome data. The DistSNE analysis results are displayed within 2 min (Figures 1B, S1, and S2), and the web interface offers a close‐up of the region for a more detailed view of the surrounding classes with the uploaded sample marked with an asterisk for easy identification. The resulting images are available for download. Upon uploading the two coupled *.idat files from a sample to the DistSNE web‐based application. Each instance computes SVD over local datasets and returns aggregated information to the central instance in Giessen, which computes the t‐SNE, including the uploaded sample. We established a web application that offers an intuitive environment for collaborative, visually assisted CNS tumor analysis.
4.2. The DistSNE classification accuracy is comparable to the classification accuracy of a centralized approach
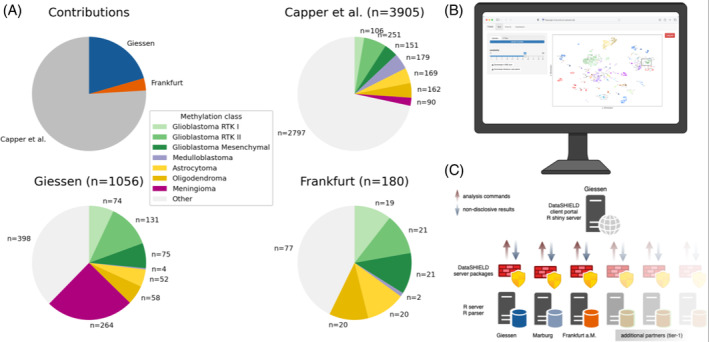
To validate the efficacy of a t‐SNE computation through a federated approach we compared the accuracy of the DistSNE subgroup classification with a centralized classifier in a three‐stage setup using all study samples and the public reference data. Performance was evaluated by comparing the classification results from the t‐SNE (nearest neighbor) with the random forest algorithm of the Heidelberg classifier. The mean accuracy was calculated by dividing the number of correctly assigned samples by the total number of samples across 100 runs. First, we establish a performance baseline by testing the t‐SNE algorithm locally on a single data server in a centralized approach. Second, we applied the distributed approach in a real‐world setting across three locations. Third, we scaled up the analysis to six virtual servers to determine the impact of a distributed analysis (DistSNE) on classification performance (Figure 1C). In all setups, we observed a consistent mean accuracy of ≈82.9% substantiating that the ability of DistSNE to maintain classification performance when accessing data in a distributed manner.
We further conducted a more detailed analysis of the variability introduced by the seed, that is, the randomly selected sample that initiates the DistSNE computation. Through 100 runs with random seed using all collected data we measured a ±0.29% standard deviation, demonstrating the robustness and reliability of the DistSNE approach. The performance most concordant with the results from the Heidelberg classifier was an accuracy of 84.9%. This accuracy is comparable with the reported performance of other predictive algorithm such as the Heidelberg classifier with 88% accuracy for samples with a score above 0.9 [7], further attesting to the efficacy of DistSNE.
4.3. DistSNE improves cluster density and accuracy
The computation of the DistSNE across all sites in the 2nd stage yielded a comprehensive and densely distributed map of data points, where the public reference data laid the foundation and was effectively complemented by the additional datasets from Giessen and Frankfurt (Figure 2A). This denser mapping combined with the enriched visualization facilitated more distinct demarcation of existing groups, enhancing the resolution and distinction of subgroups. These findings underscore the potential of DistSNE to boost the quality of visual mapping but also to potentially uncover previously unidentified clusters. To show that the increased cluster density achieved through DistSNE indeed resulted in a higher classification performance we compared the classification accuracy between the reference data set and the expanded collective data set of a federated approach. We performed a t‐SNE incorporating the data provided by Gießen and Frankfurt either individually (fixed approach) or incrementally (incremental approach). The classification accuracy of this projection was determined by the nearest neighbor classification. We ran the analysis using a Monte Carlo method with 20 permutations and a random order of addition to account for any potential influence of the order of sample addition. The results show that the incremental approach improves classification performance compared to the fixed approach, which in contrast displays saturation (Figure 2B), highlighting that a higher sample number achieved through the federated approach improves classification performance.
FIGURE 2.
DistSNE analysis of the collective data and accuracy measurements. (A) Distributed t‐SNE of the three locations Giessen (blue), Frankfurt (orange), and Marburg (Capper data) (grey). The clusters of IDH mutated gliomas (bottom) and of meningiomas (left lower) are marked with rectangles. (B) Accuracy of the fixed model (gray) and the incremental model (blue) for all gathered samples. The number of samples is visualized on the right y‐axis by the dotted lines for the fixed and incremental approach. Classification accuracy for all groups significantly improves with a higher sample number (incremental vs. fixed; p < 0.001, paired t‐test with Bonferroni correction).
Similarly, the lower intra‐cluster‐variance (see methods) of ≈60.20 for the collective methylome data across all three sites as compared to ≈61.28 for the Capper et al. data indicates that with increasing sample size, as achieved through distributed access to additional datasets, the cluster density concurrently increases. This is essential as a tight clustering is pivotal for obtaining high‐quality clusters and enhanced predictive strength which facilitates the precise identification of similar or related samples by nearest‐neighbor.
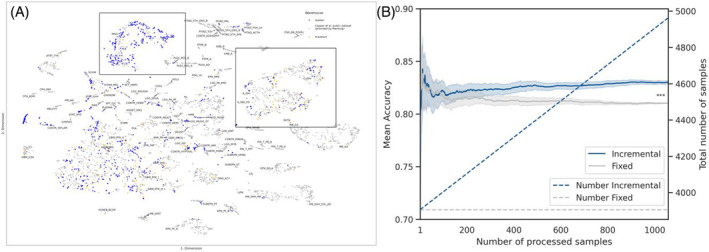
The significance of the DistSNE approach in enhancing cluster quality can be further exemplified by the improved distinction of the IDH‐mutated astrocytoma and oligodendroglioma groups. As an example, we plotted the density map of those subgroups from the dataset (A_IDH, A_IDH_HG, O_IDH, Figure 3A–C).
FIGURE 3.
The DistSNE approach enhances cluster analysis quality. Two‐dimensional kernel density estimations and corresponding scatter plots of selected glioma classes reveal the qualitative advantage of using the federated dataset. Samples from Giessen and Frankfurt are labeled blue and from Capper et al. in gray. (A) The projection with the collective data (B) displays a higher group density than the Capper et al. dataset (C) analysis allowing for better subclass distinction.
4.4. Improvement of cluster quality and meningioma subclassification with DistSNE
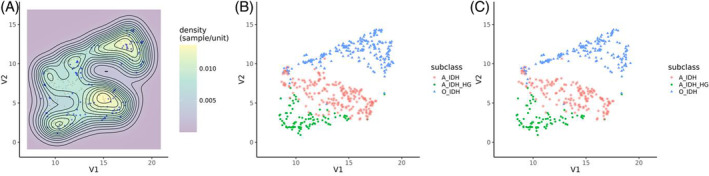
We next wanted to test whether DistSNE can identify new molecular subgroups apart from recognizing existing molecular subtypes. We used meningioma as our test case due to the limitations of the V11 classifier in distinguishing meningioma subtypes (meningioma subclassification was only introduced in the V12 classifier). On integrating federated meningioma data into the reference dataset, we noticed the formation of visually distinct groups suggestive of potential new meningioma subtypes by unsupervised clustering (Figure 4A–C). Notably, these emerging clusters aligned well with the known subtypes from the V12 classifier (see color legend in Figure 4B, C). When we incorporated data from all contributing locations, the clusters exhibited increased density and delineation and continued to overlap with the V12 classification. We want to especially highlight the benign‐2 subclass, where a larger dataset closes the gap in the cluster of benign‐2 (Figure 4B), when compared to the Capper reference dataset (Figure 4C). Concomitantly, the intermediate‐A subclass becomes clearly delineated as a cluster. These findings underscore the efficacy of DistSNE in not only validating established molecular subtypes but also its potential to discover novel ones, in particular with access to additional federated datasets.
FIGURE 4.
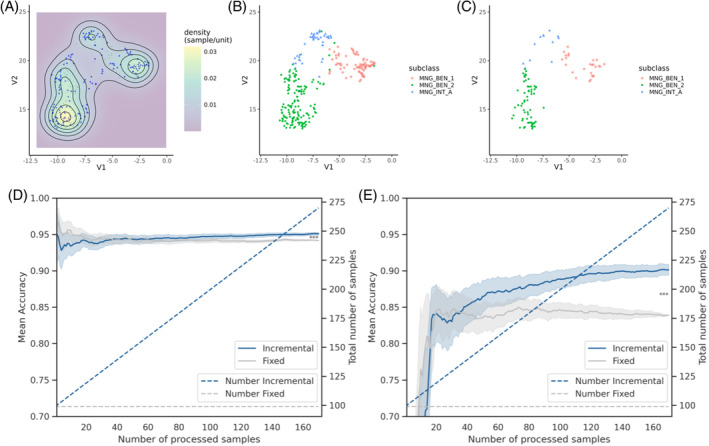
Meningioma subclassification and accuracy improvement with DistSNE. (A) Two‐dimensional kernel density estimation. Meningioma samples from Giessen and Frankfurt are labeled blue and from Capper et al. in gray. (B) Corresponding scatter plot with all data from the federated approach or (C) Capper et al. On integrating federated meningioma data, three distinct clusters can be observed that align with the V12 classification of meningioma subtypes (see color legend). (D) Classification accuracy for all meningioma samples and (E) of the meningioma subgroup intermediate A. The growing number of samples is visualized on the right y‐axis by the dotted lines for the fixed and incremental approach. Classification accuracy improves with a higher sample number (incremental vs. fixed; p < 0.001, paired t test with Bonferroni correction). See text for details.
Next, we evaluated the impact of database expansion via DistSNE on classification accuracy using the fixed or incremental approach for the large cohort of meningioma samples. The incremental approach, which gradually adds more samples to the analysis, improved the projection modestly across all meningioma samples (0.9%). However, a marked improvement (6.3%) was observed when determining the subgroup Intermediate A (Figure 4D, E), which partially overlaps with the benigns 1 and 2 clusters (see Figure 4A–C).
Collectively, these results show that increasing overall cohort size through the federated DistSNE computation significantly enhances classification accuracy, for groups with fewer sample numbers. Coupled with the improved classification accuracy and visualization quality, DistSNE has the potential to identify new subgroups through federated database expansion.
5. DISCUSSION
In this study, we present a distributed t‐SNE analysis (DistSNE) framework for DNA methylation‐based classification of CNS tumors, aiming to improve cluster quality and potentially identify novel tumor subgroups. DistSNE effectively visualizes tumor samples across sites and offers a user‐friendly web interface for researchers to perform distributed t‐SNE analysis on DNA methylation data while preserving data privacy. This approach addresses the growing need for privacy‐preserving methods in cancer research, particularly in multi‐center studies and collaborative projects. Importantly, the DistSNE method has the potential to facilitate the discovery of new molecular subgroups and improve CNS tumor classification by allowing researchers to pool their data, benefiting the neuropathology community.
The analysis of DNA methylation patterns has emerged as a powerful tool for the reliable classification of CNS tumors, as evidenced by the inclusion of genome‐wide DNA methylation profiling as essential or desirable diagnostic criteria for classifying various CNS tumors in the WHO Classification of Tumors of the Central Nervous System (CNS) 5th edition [2]. The aim of our study was to develop a distributed t‐SNE analysis framework for DNA methylation‐based classification of CNS tumors that allows better visual mapping and potentially higher subgroup resolution by accessing additional methylation data from various sites. Our study demonstrates that the federated DistSNE computation improves classification with higher sample numbers and improves cluster quality and density when compared to datasets achieved from public sources [7]. Importantly, DistSNE maintained classification accuracy equivalent to that of a centralized method while preserving data privacy in line with previous studies on other medical datasets [22, 23]. The increased resolution could lead to the discovery of novel clusters and a deeper understanding of tumor biology [24, 25, 26, 27]. This suggests that our federated DistSNE computation approach may be beneficial in large‐scale studies and consortium‐based research projects where data privacy and protection, became a major concern in the era of big data especially in medical research [28, 29]. Researchers often face challenges in sharing sensitive patient data due to ethical and legal constraints. The DistSNE framework offers a viable solution for researchers to perform distributed t‐SNE analysis on DNA methylation data in a privacy‐preserving manner across multiple locations using DataSHIELD [30].
The DistSNE web interface provides an accessible platform for researchers to submit their samples for analysis and receive t‐SNE visualizations and classifications, or interactively explore t‐SNE plots and identify tumor subgroups. This user‐friendly interface enables rapid visualization and analysis of individual tumor samples, aiding in the diagnosis and treatment of CNS tumors. The DistSNE method can be further improved and extended to support more participating institutes and datasets, increasing the overall dataset's diversity, and potentially revealing novel molecular subgroups. Importantly, by maintaining their own DataSHIELD server, institutes can ensure full data autonomy and adherence to strict data protection standards, as sample data stays within their IT framework. This approach has the potential to facilitate collaborative research efforts and lead to new discoveries in the field of CNS tumor classification and molecular subgroup identification as previously achieved through centralized approaches both in the field of CNS and other tumors (e.g., TCGA [31], ICGC [32]). Thus, the DistSNE method may be a valuable tool for the neuro‐oncology community, as it provides a privacy‐preserving, accurate, and efficient way of analyzing and classifying CNS tumors. By enabling researchers and clinicians to visualize and classify tumors without sharing sensitive patient data, DistSNE can facilitate multicentric clinical studies and promote collaboration across institutions [33].
In addition to these benefits of the DistSNE framework, there are potential limitations to consider. While the DistSNE approach preserves data privacy, the computation of large‐scale molecular data as obtained in cancer studies may require significant computational resources and coordination between participating sites [34, 35]. Moreover, potential issues related to data harmonization and standardization may need to be addressed to ensure accurate and consistent results across different datasets. However, as opposed to patient data that has been gathered in an unstructured manner in clinical routine, large‐scale molecular data are usually present in a highly structured manner which will ease a high‐throughput analysis as presented by DistSNE. Future work may focus on extending this framework to other large‐scale, but highly structured omics data types, such as transcriptomics [36], proteomics [37], and genomics [38], to further enhance our understanding of cancer biology and improve patient stratification for personalized treatments [39, 40].
6. CONCLUSION
Here, we have developed a new privacy‐compliant method for analyzing diagnostic DNA methylation array data. Our approach enables sharing the information content of large‐scale data without disclosing the data itself, facilitating the calculation of a distributed t‐SNE across multiple sites. By pooling data from multiple sources, the resulting dataset is much larger and provides a more precise assessment of new cases. This approach can identify new cases that cannot be confidentially assigned to known subtypes, potentially uncovering new subgroups. Involving additional institutes and sites can further enhance the power of this analysis in a simple and collaborative manner, making it an effective tool for brain tumor classification through federated computing and advancing precision medicine in neuro‐oncology.
AUTHOR CONTRIBUTIONS
Kai Schmid: Conceptualization, methodology, investigation, writing, original draft. Jannik Sehring: Methodology, investigation, writing, review & editing. Attila Németh: investigation, writing, review & editing. Patrick N. Harter: Investigation. Katharina J. Weber: Investigation. Abishaa Vengadeswaran: Server implementation. Holger Storf: Investigation. Christian Seidemann: Investigation. Kapil Karki: Server implementation. Patrick Fischer: Server implementation. Hildegard Dohmen: Investigation. Carmen Selignow: Investigation. Andreas von Deimling: Investigation, writing, review & editing. Stefan Grau: Investigation. Uwe Schröder: Investigation. Karl H. Plate: Investigation. Marco Stein: Investigation. Eberhard Uhl: Investigation. Till Acker: investigation, writing, review & editing, supervision. Daniel Amsel: investigation, writing, review & editing, supervision.
FUNDING INFORMATION
This work was supported by the MIRACUM consortium of German Federal Ministry of Education and Research (Grant Number: BMBF FKZ 01ZZ1801 and BMBF FKZ 01ZZ2017 for the junior research group AI‐RON). Additional support was provided by the Innovations‐ und Strukturentwicklungsbudget Hesse.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
FIGURE S1. t‐SNE plot from the DistSNE web app with the color coding for each tumor entity. The classification result of a Rosette‐forming glioneuronal tumor is shown in the marked area.
FIGURE S2. t‐SNE plot from the DistSNE web app with the color coding for each tumor entity. The classification result of a methylation class Glioblastoma, IDH‐wildtype, subtype RTK1 tumor is shown in the marked area.
ACKNOWLEDGMENT
The authors thank the BMBF for their generous funding. Open Access funding enabled and organized by Projekt DEAL.
Schmid K, Sehring J, Németh A, Harter PN, Weber KJ, Vengadeswaran A, et al. DistSNE: Distributed computing and online visualization of DNA methylation‐based central nervous system tumor classification. Brain Pathology. 2024;34(3):e13228. 10.1111/bpa.13228
Contributor Information
Till Acker, Email: till.acker@patho.med.uni-giessen.de.
Daniel Amsel, Email: daniel.amsel@patho.med.uni-giessen.de.
DATA AVAILABILITY STATEMENT
The preprocessing pipeline is available as a specifically tailored Docker container:
https://hub.docker.com/repository/docker/kaischmid/disttsne.
REFERENCES
- 1. Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: a precision oncology knowledge base. JCO Precision Oncol. 2017;1:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella‐Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro‐oncology. 2021;23(8):1231–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Priesterbach‐Ackley LP, Boldt HB, Petersen JK, Bervoets N, Scheie D, Ulhøi BP, et al. Brain tumour diagnostics using a DNA methylation‐based classifier as a diagnostic support tool. Neuropathol Appl Neurobiol. 2020;46(5):478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pickles JC, Fairchild AR, Stone TJ, Brownlee L, Merve A, Yasin SA, et al. DNA methylation‐based profiling for paediatric CNS tumour diagnosis and treatment: a population‐based study. Lancet Child Adolesc Health. 2020;4(2):121–130. [DOI] [PubMed] [Google Scholar]
- 5. Moran S, Arribas C, Esteller M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics. 2016;8(3):389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kumar R, Liu APY, Orr BA, Northcott PA, Robinson GW. Advances in the classification of pediatric brain tumors through DNA methylation profiling: from research tool to frontline diagnostic. Cancer. 2018;124(21):4168–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation‐based classification of central nervous system tumours. Nature. 2018;555(7697):469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chico V. The impact of the general data protection regulation on health research. Br Med Bullet. 2018;128(1):109–118. [DOI] [PubMed] [Google Scholar]
- 9. Semler SC, Wissing F, Heyder R. German medical informatics initiative. Methods Inf Med. 2018;57(S01):e50–e56. 10.3414/ME18-03-0003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wallace SE, Gaye A, Shoush O, Burton PR. Protecting personal data in epidemiological research: DataSHIELD and UK law. Public Health Genomics. 2014;17(3):149–157. [DOI] [PubMed] [Google Scholar]
- 11. Gaye A, Marcon Y, Isaeva J, LaFlamme P, Turner A, Jones EM, et al. DataSHIELD: taking the analysis to the data, not the data to the analysis. International Journal of Epidemiology. 2014;43(6):1929–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Budin‐Ljøsne I, Burton P, Isaeva J, Gaye A, Turner A, Murtagh MJ, et al. DataSHIELD: an ethically robust solution to multiple‐site individual‐level data analysis. Public Health Genomics. 2015;18(2):87–96. [DOI] [PubMed] [Google Scholar]
- 13. Lenz S, Hess M, Binder H. Deep generative models in DataSHIELD. BMC Med Res Methodol. 2021;21(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marcon Y, Bishop T, Avraam D, Escriba‐Montagut X, Ryser‐Welch P, Wheater S, et al. Orchestrating privacy‐protected big data analyses of data from different resources with R and DataSHIELD. PLoS Computational Biology. 2021;17(3):e1008880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jaddoe VWV, Felix JF, Andersen A‐MN, Charles M‐A, Chatzi L, Corpeleijn E, et al. The LifeCycle project‐EU child cohort network: a federated analysis infrastructure and harmonized data of more than 250,000 children and parents. European Journal of Epidemiology. 2020;35(7):709–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peñalvo JL, Mertens E, Ademović E, Akgun S, Baltazar AL, Buonfrate D, et al. Unravelling data for rapid evidence‐based response to COVID‐19: a summary of the unCoVer protocol. BMJ Open. 2021;11:e055630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fortin J‐P, Triche Jr TJ, Hansen KD. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics. 2017;33(4):558–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krijthe JH. Rtsne: T‐distributed stochastic neighbor embedding using Barnes‐hut implementation. R package version 0.13. 2015. https://github.com/jkrijthe/Rtsne
- 19. Pearson K. Principal components analysis. Lond Edinb Dubl Phil Mag J Sci. 1901;6(2):559–572. [Google Scholar]
- 20. Iwen MA, Ong BW. A distributed and incremental SVD algorithm for agglomerative data analysis on large networks. SIAM J. Matrix Anal. Appl. 2016;37(4):1699–1718. [Google Scholar]
- 21. Wickham H, Chang W, Wickham MH. Package ‘ggplot2’. create elegant data visualisations using the grammar of graphics. Version 2.1. pp. 1–189. 2016.
- 22. Froelicher D, Troncoso‐Pastoriza JR, Raisaro JL, Cuendet MA, Sousa JS, Cho H, et al. Truly privacy‐preserving federated analytics for precision medicine with multiparty homomorphic encryption. Nat Commun. 2021;12(1):5910. 10.1038/s41467-021-25972-y Erratum in: Nat Commun. 2021 Nov 11;12(1):6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brisimi TS, Chen R, Mela T, Olshevsky A, Paschalidis IC, Shi W. Federated learning of predictive models from federated electronic health records. Int J Med Inform. 2018;112:59–67. 10.1016/j.ijmedinf.2018.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D, et al. New brain tumor entities emerge from molecular classification of CNS‐PNETs. Cell. 2016;164(5):1060–1072. 10.1016/j.cell.2016.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reinhardt A, Pfister K, Schrimpf D, Stichel D, Sahm F, Reuss DE, et al. Anaplastic ganglioglioma: a diagnosis comprising several distinct tumour types. Neuropathol Appl Neurobiol. 2022;48(7):e12847. 10.1111/nan.12847 [DOI] [PubMed] [Google Scholar]
- 26. Capper D, Engel NW, Stichel D, Lechner M, Glöss S, Schmid S, et al. DNA methylation‐based reclassification of olfactory neuroblastoma [published correction appears in Acta Neuropathol. 2018 Sep;136(3):505]. Acta Neuropathol. 2018;136(2):255–271. 10.1007/s00401-018-1854-7 [DOI] [PubMed] [Google Scholar]
- 27. Drexler R, Schüller U, Eckhardt A, Filipski K, Hartung TI, Harter PN, et al. DNA methylation subclasses predict the benefit from gross total tumor resection in IDH‐wildtype glioblastoma patients. Neuro Oncol. 2023;25(2):315–325. 10.1093/neuonc/noac177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scheibner J, Ienca M, Kechagia S, Troncoso‐Pastoriza JR, Raisaro JL, Hubaux JP, et al. Data protection and ethics requirements for multisite research with health data: a comparative examination of legislative governance frameworks and the role of data protection technologies. J Law Biosci. 2020;7(1):lsaa010. 10.1093/jlb/lsaa010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sweeney SM, Hamadeh HK, Abrams N, Adam SJ, Brenner S, Connors DE, et al. Challenges to using big data in cancer [published online ahead of print, 2023 Jan 10]. Cancer Res. 2023;83:1175–1182. 10.1158/0008-5472.CAN-22-1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wolfson M, Wallace SE, Masca N, Rowe G, Sheehan NA, Ferretti V, et al. DataSHIELD: resolving a conflict in contemporary bioscience: performing a pooled analysis of individual‐level data without sharing the data. Int J Epidemiol. 2010;39(5):1372–1382. 10.1093/ije/dyq111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cancer Genome Atlas Research Network , Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, et al. The cancer genome atlas pan‐cancer analysis project. Nat Genet. 2013;45(10):1113–1120. 10.1038/ng.2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. International Cancer Genome Consortium , Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, et al. International network of cancer genome projects [published correction appears in Nature. 2010 Jun 17;465(7300):966. Himmelbaue, Heinz [corrected to Himmelbauer, Heinz]; Gardiner, Brooke A [corrected to Gardiner, Brooke B]; Cross, Anthony [corrected to Cros, Anthony]]. Nature. 2010;464(7291):993–998. 10.1038/nature08987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Karimi S, Zuccato JA, Mamatjan Y, Mansouri S, Suppiah S, Nassiri F, et al. The central nervous system tumor methylation classifier changes neuro‐oncology practice for challenging brain tumor diagnoses and directly impacts patient care. Clin Epigenetics. 2019;11(1):185. 10.1186/s13148-019-0766-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xiao W, Ren L, Chen Z, Fang LT, Zhao Y, Lack J, et al. Toward best practice in cancer mutation detection with whole‐genome and whole‐exome sequencing. Nat Biotechnol. 2021;39(9):1141–1150. 10.1038/s41587-021-00994-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fang LT, Zhu B, Zhao Y, Chen W, Yang Z, Kerrigan L, et al. Establishing community reference samples, data and call sets for benchmarking cancer mutation detection using whole‐genome sequencing. Nat Biotechnol. 2021;39(9):1151–1160. 10.1038/s41587-021-00993-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsimberidou AM, Fountzilas E, Bleris L, Kurzrock R. Transcriptomics and solid tumors: the next frontier in precision cancer medicine. Semin Cancer Biol. 2022;84:50–59. 10.1016/j.semcancer.2020.09.007 [DOI] [PubMed] [Google Scholar]
- 37. Rodriguez H, Zenklusen JC, Staudt LM, Doroshow JH, Lowy DR. The next horizon in precision oncology: Proteogenomics to inform cancer diagnosis and treatment. Cell. 2021;184(7):1661–1670. 10.1016/j.cell.2021.02.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. ICGC/TCGA Pan‐Cancer Analysis of Whole Genomes Consortium . Pan‐cancer analysis of whole genomes [published correction appears in nature. 2023 Feb;614(7948):E39]. Nature. 2020;578(7793):82–93. 10.1038/s41586-020-1969-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wahida A, Buschhorn L, Fröhling S, Jost PJ, Schneeweiss A, Lichter P, et al. The coming decade in precision oncology: six riddles. Nat Rev Cancer. 2023;23(1):43–54. 10.1038/s41568-022-00529-3 [DOI] [PubMed] [Google Scholar]
- 40. Mani DR, Krug K, Zhang B, Satpathy S, Clauser KR, Ding L, et al. Cancer proteogenomics: current impact and future prospects. Nat Rev Cancer. 2022;22(5):298–313. 10.1038/s41568-022-00446-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1. t‐SNE plot from the DistSNE web app with the color coding for each tumor entity. The classification result of a Rosette‐forming glioneuronal tumor is shown in the marked area.
FIGURE S2. t‐SNE plot from the DistSNE web app with the color coding for each tumor entity. The classification result of a methylation class Glioblastoma, IDH‐wildtype, subtype RTK1 tumor is shown in the marked area.
Data Availability Statement
The preprocessing pipeline is available as a specifically tailored Docker container:
https://hub.docker.com/repository/docker/kaischmid/disttsne.