

Abstract
Breast cancer is a highly heterogeneous disease with varied subtypes, prognoses and therapeutic responsiveness. Human leukocyte antigen class I (HLA-I) shapes the immunity and thereby influences the outcome of breast cancer. However, the implications of HLA-I variations in breast cancer remain poorly understood. In this study, we established a multiomics cohort of 1156 Chinese breast cancer patients for HLA-I investigation. We calculated four important HLA-I indicators in each individual, including HLA-I expression level, somatic HLA-I loss of heterozygosity (LOH), HLA-I evolutionary divergence (HED) and peptide-binding promiscuity (Pr). Then, we evaluated their distribution and prognostic significance in breast cancer subtypes. We found that the four breast cancer subtypes had distinct features of HLA-I indicators. Increased expression of HLA-I and LOH were enriched in triple-negative breast cancer (TNBC), while Pr was relatively higher in hot tumors within TNBCs. In particular, a higher Pr indicated a better prognosis in TNBCs by regulating the infiltration of immune cells and the expression of immune molecules. Using the matched genomic and transcriptomic data, we found that mismatch repair deficiency-related mutational signature and pathways were enriched in low-Pr TNBCs, suggesting that targeting mismatch repair deficiency for synthetic lethality might be promising therapy for these patients. In conclusion, we presented an overview of HLA-I indicators in breast cancer and provided hints for precision treatment for low-Pr TNBCs.
Keywords: breast cancer, HLA-I, multiomics, prognostic biomarker, immunity
INTRODUCTION
Breast cancer remains one of the most common cancer and a major cause of cancer-related mortality in women worldwide [1]. As a molecularly heterogeneous disease, breast cancer can be categorized into distinct subtypes defined by the expression of hormone receptor (HR) and human epidermal growth factor receptor 2 (HER2) amplification: HR+HER2−, HR−HER2+, HR+HER2+ and triple-negative breast cancer (TNBC) [2]. Prognosis and therapeutic approaches differ across various subtypes of breast cancer due to distinct biological features. In addition, a plethora of studies have highlighted the value of dynamic tumor-immune interactions in the control of tumor development and the effectiveness of therapeutic intervention [3–5]. Despite significant progress, the understanding of the complex immune system in the entire breast cancer ecosystem is still insufficient. Hence, further exploration of critical regulators within the tumor microenvironment (TME) holds the potential to drive advancements in precise treatment strategies for breast cancer.
Human leukocyte antigen class I (HLA-I) molecules play a crucial role in tumor antigen presentation and antitumor immune response [6]. Hence, HLA-I is widely recognized as a potent factor influencing patients’ chances of survival. As suggested by previous studies, the greater sequence divergence between HLA-I alleles would increase the diversity of tumor neopeptide repertoire available for presentation, thereby contributing to a more effective immune response and better prognosis [7–9]. Conversely, the loss of heterozygosity (LOH) in somatic HLA-I has been identified as a key factor of immune escape and worse prognosis [10, 11]. However, these reports were contrast with the findings of 17 clinical trials conducted across eight tumor types, involving thousands of patients treated with pembrolizumab. In this research, germline HLA genotype and diversity, such as HLA-I heterozygosity, HLA-A and HLA-B supertypes, were not found to be associated with the clinical outcome of all patients or certain tumor types [12]. Consequently, the intricate interplay between HLA diversity and TME, and its subsequent impact on the prognosis of patients remains controversial. In addition, data evaluating the relationship between indicators associated with HLA-I and clinical outcomes are limited, particularly among breast cancer patients.
Herein, we established a large Chinese breast cancer multiomics cohort (n = 1156) with data of both somatic HLA-I alterations and germline HLA-I diversity. We evaluated HLA-I status in each individual, including HLA-I expression, somatic HLA-I LOH, HLA-I evolutionary divergence (HED) and peptide-binding promiscuity (Pr); and delineated their landscape across all breast cancer samples and specific clinical subtypes. To further evaluate their predictive value, we identified the prognostic significance and immunological characteristics of patients with distinct HLA-I phenotypes. Furthermore, we explored the underlying factors leading to worse clinical outcomes in low-Pr patients with matched multiomics data.
METHODS
Cohort and multiomics database
Patients diagnosed with malignant breast cancer were retrospectively recruited. Chinese patients who were treated at Fudan University Shanghai Cancer Center (FUSCC) and enrolled in three datasets (CBCGA cohort, Luminal cohort and FUSCCTNBC cohort) were selected. Detailed information about patient selection was described in our previous studies [13–15]. We integrated these three cohorts to enlarge the sample size for each subtype of breast cancer. Samples with available RNA sequencing data or HLA-I status were included in the present study. In summary, a total of 1156 patients with RNA sequencing (n = 853), whole-exome sequencing (WES, n = 942), copy number variation (CNV, n = 1020) and clinical data (n = 1156) were obtained for further analyses. Clinicopathological characteristics included tumor size, histological type, lymph node status, Ki67 status and ER, PR, HER2 status. Relapse-free survival (RFS) was defined as the duration from diagnosis to the first recurrence or death from any cause. Distant metastasis-free survival (DMFS) was defined as the duration from the date of surgery to the first detection of distant metastasis or death from any cause. All baseline characteristics of patients in this study are shown in Supplementary Table 1. All patients included in this study provided written informed consent before enrollment. Patient samples were collected with ethics approval by the FUSCC Ethics Committee.
HLA genotyping and diversity
HLA-I genotyping was performed using the POLYSOLVER tool to identify the four-digit HLA type from germline normal DNA exome sequencing data of each breast cancer patient [16]. We measured HLA diversity by the mean mRNA expression level of classical HLA-I molecules (HLA-A, HLA-B and HLA-C), HLA-I LOH, HED (a quantifiable measure of HLA-I evolution) and Pr (a quantifiable measure of peptide repertoire breadth of HLA-I).
HLA-I LOH was determined by LOHHLA tools using copy number values at the segment level [17]. The estimated tumor purity and ploidy were adjusted when assessing copy number values by the ACSAT algorithm [17]. LOH of each HLA-I gene was defined as one of the two alleles showing a copy number < 1 and a P value < 0.05. Only patients with LOH at any HLA-I gene (HLA-A, HLA-B or HLA-C) were considered as ‘LOH’, while those without HLA-I LOH loci were ‘non-LOH’. Patients with LOH were subdivided into ‘LOH at all’ (all of HLA-A, HLA-B and HLA-C showed LOH) and ‘LOH at least one’ (one or two of HLA-A, HLA-B and HLA-C showed LOH).
HED was calculated as described previously [7, 18]. Briefly, protein sequences of each individual allele of each patient’s HLA-I genotype were extracted from the IMGT/HLA database. Exons 2 and 3 of each allele, which encode the variable peptide-binding domains, were annotated according to the Ensembl database and then selected. Next, we calculated HED using the Grantham distance metric implemented in Pierini and Lenz [18]. The mean HED of each patient was calculated as the mean of divergences at HLA-A, HLA-B and HLA-C loci.
In regard to the calculation of Pr, only HLA-I alleles that can bind at least 400 different peptides according to the data of peptide-HLA class I interactions in The Immune Epitope Database were further analyzed [19]. First, the Kullback–Leibler divergence (DKL) of peptides with different amino acid lengths was calculated to measure the distance between the amino acid frequency distribution at each position and that in the complete human proteome [20]. Then, peptide-length-specific divergences were averaged by weighting with the relative proportion in the repertoire. Finally, HLA allelic Pr was calculated as the reciprocal value of the average DKL.
Immune microenvironment phenotypes
The abundance of TME cells was estimated as described previously [3]. We generated a microenvironment compendium that contains 364 genes representing 24 specific microenvironment cell subsets. Single-sample gene set enrichment analysis (ssGSEA, ‘GSVA’ function in R) was then performed to evaluate the level of cell infiltration in each sample with expression data. To further determine the optimal number of microenvironment subtypes, we performed Nbclust testing (‘NbClust’ function in R) and k-means (‘kmeans’ function in R) clustering according to the cell composition in the microenvironment.
Survival analysis
The Cox proportional hazard regression model was used to investigate the effect of variables on patient prognosis. HLA-I indicators, including HLA-A/B/C mean mRNA expression level, HLA-I LOH, HED and Pr, were included for analysis. In addition, survival curves were generated using the Kaplan-Meier method and compared using the log-rank test.
Statistical analysis
Student’s t-test, Wilcoxon test and Kruskal-Wallis test were used to compare continuous variables, such as HED score and Pr. Prior to the comparisons, the normality of the distributions was tested with the Shapiro-Wilk test. Pearson’s chi-squared test and Fisher’s exact test were employed for the comparison of unordered categorical variables. Correlation matrices were created with Pearson or Spearman correlation. All tests were two-sided and P < 0.05 was regarded as statistically significant unless otherwise specified. All statistical analyses were performed using R software (version 4.2.1, http://www.R-project.org).
RESULTS
Overview of the study design
In the present study, we aimed to portray the HLA-I landscape in breast cancer and explore its biological significance (Figure 1). With this purpose, we conducted this research in four steps. First, we integrated three cohorts established in FUSCC, including the CBCGA cohort (n = 773), Luminal cohort (n = 354) and FUSCCTNBC cohort (n = 465) [13–15]. In this study, breast cancer patients who met the criteria for assessing HLA-I status were collected. In total, 1156 samples were selected from our multiomics dataset with RNA sequencing (n = 853), WES (n = 942), CNV (n = 1020) and follow-up data (n = 1156) available in Supplementary Figure 1 and Supplementary Table 1. Second, we calculated four HLA-I indicators in the breast cancer cohorts and delineated their patterns in different subtypes, including HLA-I mRNA expression level, somatic HLA-I LOH, germline HED and peptide-binding Pr. HED quantifies HLA-I allele evolution by calculating the sequence divergence between distinct HLA-I alleles, while Pr represents the diversity of peptides that the HLA-I allele can bind. Third, we assessed the prognostic significance of these HLA-I indicators in breast cancer patients with different clinical subtypes. We also demonstrated their ability to reveal tumor immune microenvironment phenotypes. Finally, we explored the potential immune mechanisms and genomic drivers leading to poor prognosis in patients with low Pr.
Figure 1.

Schematic of the study. The framework of the study. First, a large Chinese breast cancer multiomics cohort was established through amalgamating three cohorts from FUSCC. Second, HLA-I status in each sample was calculated, including HLA-I mRNA expression, HLA-I LOH, HED and Pr. Third, the distribution of each HLA-I indicator, their prognostic significance and immunological characteristics within different subtypes were evaluated. Finally, the underlying mechanisms leading to worse clinical outcomes in low-Pr TNBCs were explored through multiomics analysis. Abbreviations: FUSCC, Fudan University Shanghai Cancer Center; WES, whole-exome sequencing; CNV, copy number variation; LOH, loss of heterozygosity; HED, evolutionary divergence of HLA class I genotype; Pr, promiscuity. See also Supplementary Figure 1.
HLA-I landscape in breast cancer
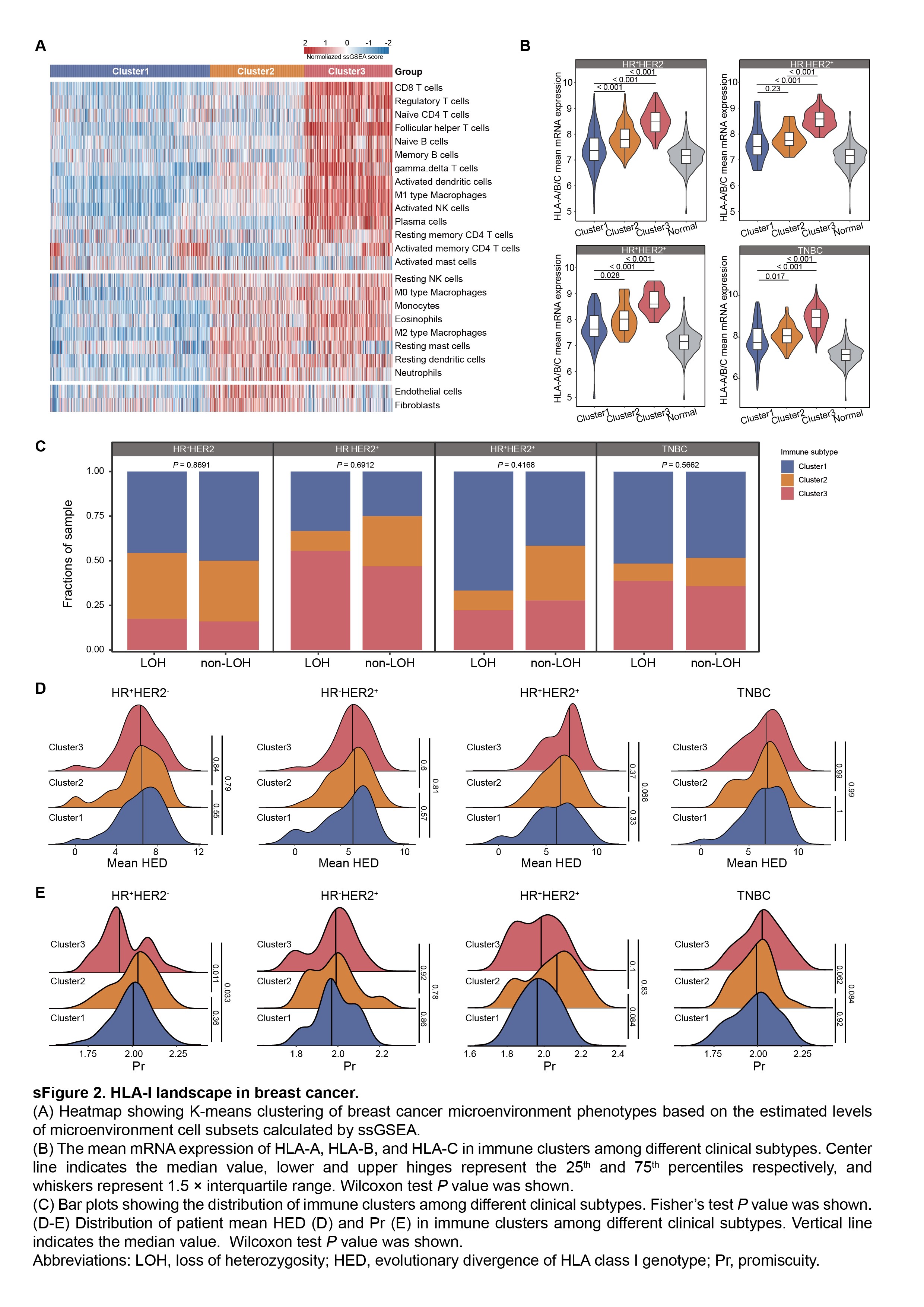
Based on the multiomics dataset, we initially estimated the distribution of four HLA-I-related indicators within the whole breast cancer cohort, as well as among distinct clinical (HR+HER2−, HR−HER2+, HR+HER2+ and TNBC) and immune (Cluster 1, Cluster 2 and Cluster 3, see Methods) subtypes. Samples in cluster 1 exhibited low immune infiltration, and cluster 2 showed moderate immune infiltration distinguished by the infiltration of inactivated immune cells, fibroblasts and endothelial cells, whereas cluster 3 represented the immune-inflamed tumor which was characterized by high immune infiltration (Supplementary Figure 2A).
First, we focused on the HLA-I features of tumor cells, and conducted an evaluation of the HLA-I mRNA expression and scrutinized the HLA-I LOH status (Supplementary Table 2). HLA-I molecules and the mean expression of HLA-A, HLA-B and HLA-C (HLA-A/B/C mean) were significantly higher in TNBC samples and cluster 3 than other subtypes and normal tissues (Figure 2A–C, Supplementary Figure 2B). In addition, 19.9% of samples within 639 patients carried HLA-I genes with LOH (Figure 2D). HLA-I LOH samples were conspicuously enriched within the TNBCs (P = 0.006) and evenly distributed in immune subtypes across all samples and each clinical subtype (Figure 2E–F, Supplementary Figure 2C).
Figure 2.

HLA-I landscape in breast cancer. (A) Heatmap showing the normalized mRNA expression level of HLA-I molecules in each breast cancer sample and normal tissue. The clinical subtypes and immune subtypes were annotated. (B–C) The mean mRNA expression of HLA-A, HLA-B and HLA-C in different clinical subtypes (B) and immune clusters (C). Center line indicates the median value, lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers represent 1.5 × interquartile range. Wilcoxon test P value was shown. (D) Distribution of LOH at all HLA-I alleles, LOH at least one HLA-I allele and non-LOH in the whole breast cancer cohort. (E–F) Bar plots showing the distribution of clinical subtypes (E) and immune clusters (F) among the HLA-I LOH-based subtypes. Fisher’s test P value was shown. (G) Distributions of HED score for each HLA-A, HLA-B and HLA-C genotype. Center line indicates the median value, lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers represent 1.5 × interquartile range. Wilcoxon test P value was shown. (H–I) Distribution of patient mean HED across different clinical subtypes (H) and immune clusters (I). Vertical line indicates the median value. Wilcoxon test P value was shown. (J) Distributions of Pr for each HLA-A, HLA-B and HLA-C genotype. Center line indicates the median value, lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers represent 1.5 × interquartile range. Wilcoxon test P value was shown. (K–L) Distribution of patient Pr score across different clinical subtypes (K) and immune clusters (L). Vertical line indicates the median value. Wilcoxon test P value was shown. Abbreviations: LOH, loss of heterozygosity; HED, evolutionary divergence of HLA class I genotype; Pr, promiscuity. See also Supplementary Figure 2.
In addition, we also assessed a germline HLA feature, HED, which quantified the physiochemical divergence in sequence between HLA-I alleles of each individual. It assumes that an increased HED is correlated with the greater diversity of peptides that a given HLA-I molecule can bind. The divergence in HLA-B alleles was higher relative to HLA-A and HLA-C, in line with the reports that HLA-B was the most polymorphic (P < 0.001, Figure 2G) [21]. Then, the mean HED was calculated as the mean divergence of HLA-A, HLA-B and HLA-C for each patient (Supplementary Table 2). No difference in the mean HED score was observed, neither in clinical subtypes nor in immune clusters (Figure 2H–I, Supplementary Figure 2D). Notably, distinct HLA alleles can exhibit overlapping peptide-binding repertoires. Thus, we further calculated the peptide-binding Pr for a representative set of HLA-I alleles using previously identified HLA-I-peptide interactions. Among these HLA-I alleles, HLA-A carried higher Pr compared with HLA-B and HLA-C (P < 0.001, Figure 2J). The Pr score of each patient was determined by calculating the mean DKL values across HLA-A, HLA-B and HLA-C alleles and subsequently taking the reciprocal value (Supplementary Table 2). However, there was no difference in the Pr score among different clinical subtypes and immune subtypes of breast cancer (Figure 2K–L, Supplementary Figure 2E). To be specific, Pr was relatively higher in cluster 3 among TNBCs (Supplementary Figure 2E).
Prognostic significance of HLA-I indicators in breast cancer
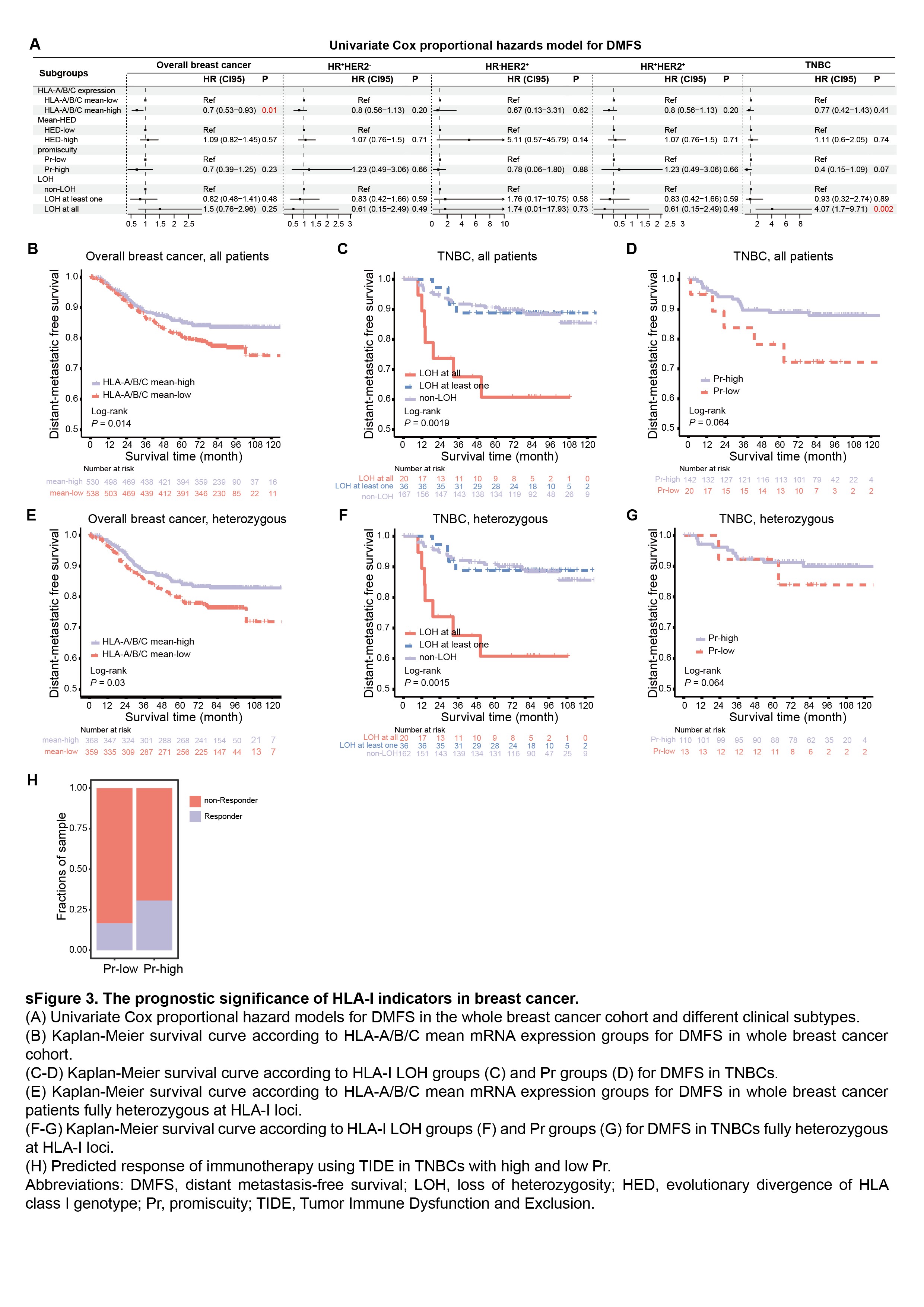
To explore the clinical implications of HLA-I in breast cancer, we performed an analysis to assess the role of the HLA-I status, including both somatic and germline variations, in the clinical course. First, we conducted a Cox proportional hazards model for RFS and DMFS in the breast cancer cohort (Figure 3A, Supplementary Figure 3A). We observed a statistically significant correlation between the expression level of classical HLA-I molecules and clinical outcomes in whole breast cancer samples. In addition, we found that non-LOH and higher Pr were better prognostic indicators in TNBCs. However, HED was not associated with better prognosis, neither across all breast cancer patients nor within each clinical subtype.
Figure 3.

The prognostic significance of HLA-I indicators in breast cancer. (A) Univariate Cox proportional hazard models for RFS in the whole breast cancer cohort and different clinical subtypes. (B) Kaplan–Meier survival curve according to HLA-A/B/C mean mRNA expression groups for RFS in whole breast cancer cohort. (C–D) Kaplan–Meier survival curve according to HLA-I LOH groups (C) and Pr groups (D) for RFS in TNBCs. (E) Kaplan–Meier survival curve according to HLA-A/B/C mean mRNA expression groups for RFS in whole breast cancer patients fully heterozygous at HLA-I loci. (F–G) Kaplan–Meier survival curve according to HLA-I LOH groups (F) and Pr groups (G) for RFS in TNBCs fully heterozygous at HLA-I loci. Abbreviations: RFS, relapse-free survival; LOH, loss of heterozygosity; HED, evolutionary divergence of HLA class I genotype; Pr, promiscuity. See also Supplementary Figure 3.
Furthermore, we confirmed the above findings through the Kaplan-Meier method. Increased expression of the mean HLA-A/B/C was related to favorable clinical outcomes in all breast cancer patients (log-rank, RFS, P = 0.04, Figure 3B; DMFS, P = 0.014, Supplementary Figure 3B), suggesting that interindividual variability in the HLA-I expression may explain the differences in prognosis across patients. In addition, patients with HLA-I LOH showed significantly worse RFS and DMFS than the HLA-I non-LOH group in TNBCs (log-rank, RFS, P = 0.00086, Figure 3C; DMFS, P = 0.0019, Supplementary Figure 3C). In the subgroup of patients with TNBC, the Pr score was associated with favorable RFS (log-rank, P = 0.049, Figure 3D) and DMFS (log-rank, P = 0.044, Supplementary Figure 3D). These effects were also observed when analyzing only fully heterozygous patients (Figure 3E–G, Supplementary Figure 3E–G). Beyond survival, predicted responders to immunotherapy were more likely to be enriched in TNBCs with high Pr (Supplementary Figure 3H), suggesting that high-Pr patients may benefit from immunotherapy.
In conclusion, we uncovered the prognostic significance of classical HLA-I expression in the entire breast cancer cohort, as well as the importance of HLA-I LOH and Pr within TNBCs.
Immunological portrait in HLA-I-based breast cancer groups
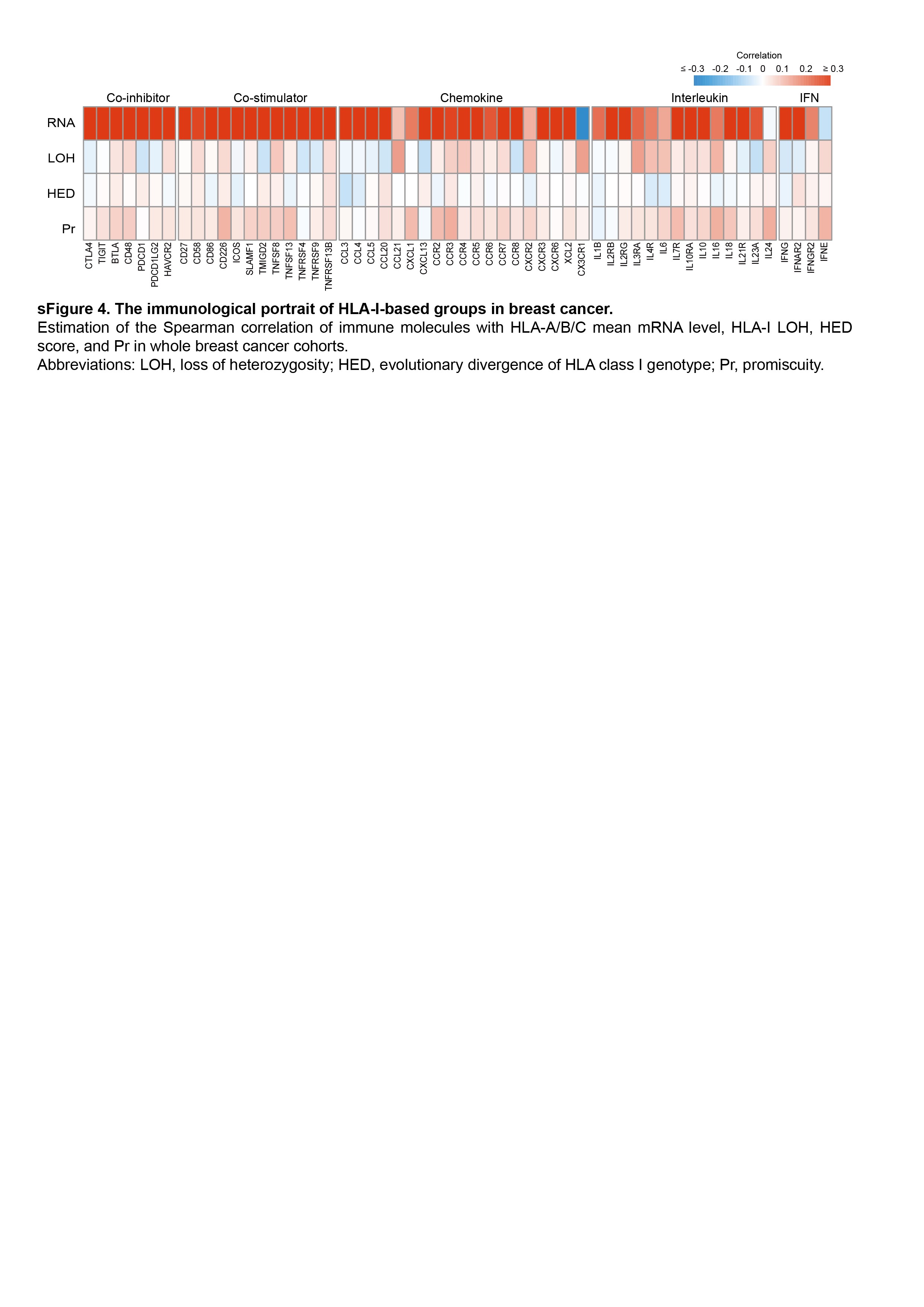
Given the importance of HLA-I in antitumor immune activity, we further examined the immunological features in the subgroup of breast cancer samples with distinct variations in HLA-I. Thus, we separately tested the association of each HLA-I-related indicator with the abundance of immune cells (Figure 4A and B, Supplementary Table 3). In overall breast cancer samples, HLA-I mRNA expression showed a strong association with the infiltration level of immune cells in the TME, including a wide range of innate and adaptive immune cells, which was also observed within the TNBC subtype. However, no notable correlation was displayed between HLA-I LOH, HED, Pr and the composition of immune cells in the TME in our whole breast cancer cohort. Surprisingly, TNBCs with high Pr exhibited a substantial enrichment of antitumor immune cells, such as T cells, dendritic cells and NK cells. These results suggested the potential impact of Pr in shaping the TME of TNBCs. Nevertheless, this association did not remain significant when examined in other clinical subtypes.
Figure 4.

The immunological portrait in HLA-I-based breast cancer groups. (A–B) Estimation of the Spearman correlation of immune cell subsets with HLA-A/B/C mean mRNA level, HLA-I LOH, HED score and Pr in whole breast cancer cohort (A) and TNBCs (B). (C) Estimation of the Spearman correlation of immune molecules with HLA-A/B/C mean mRNA level, HLA-I LOH, HED score and Pr in TNBCs. Abbreviations: LOH, loss of heterozygosity; HED, evolutionary divergence of HLA class I genotype; Pr, promiscuity; TILs, tumor infiltrating lymphocytes; sTIL, stromal tumor infiltrating lymphocyte; iTIL, intratumoral tumor infiltrating lymphocyte. See also Supplementary Figure 4.
The expression of immune molecules was aligned with the above results. Alteration in HLA-I expression was positively related to the level of costimulatory and chemotactic molecules in all breast cancer samples and TNBCs (Figure 4C, Supplementary Figure 4). In addition, the correlation between the expression level of immune molecules and Pr score solely occurred in populations with TNBC (Figure 4C). In contrast, there was no significant difference observed in the levels of immune molecules among patients with different LOH status or varying HED scores, both within the entire breast cancer cohort and across each clinical cluster.
In summary, these results indicated the immunomodulatory role of classical HLA-I mRNA expression level in breast cancer. Intriguingly, we discovered that TNBCs with high Pr displayed the characteristics of a ‘hot’ TME.
Promiscuity is positively correlated with the recruitment of immune cells in TNBCs
To comprehensively understand the potential immunological mechanisms underlying the close correlation between Pr and TME phenotypes in TNBCs, we analyzed the activity of antitumor immunity across a seven-step cancer-immune cycle [22]. As depicted in Figure 5A, the cancer-immune cycle is initiated by the release of tumor antigens from tumor cells (Step 1). Next, antigen-presenting cells such as dendritic cells capture antigens on major histocompatibility complex (MHC) molecules to T cells (Step 2), triggering the priming and activation of T cells (Step 3). Ultimately, the activated T cells traffic to (Step 4) and infiltrate into TME through blood circulation (Step 5), specifically recognizing cancer cells through the interaction between T cell receptor (TCR) and the cognate antigen bound to MHC-I (Step 6), subsequently leading to the antitumor cytotoxic killing (Step 7). The immune activity score of each step for each breast cancer sample is shown in Supplementary Table 4. The overall immune activity in high-Pr TNBCs was significantly higher than that in low-Pr TNBCs (Supplementary Figure 5A). In the early stages of the immune response, it appeared that the processes of tumor antigen release, presentation and activation (Steps 1, 2 and 3) shared similarities between the low-Pr and high-Pr samples (Figure 5B). In line with this result, there were no notable differences in the neoantigen load, diversity or clonality of TCRs between patients with low Pr and high Pr (Supplementary Figure 5B–C). Specifically, the trafficking and infiltration activities of immune cells into tumors (Steps 4 and 5) were notably elevated in high-Pr TNBCs compared with those with low Pr (Figure 5B). We further found that samples with high Pr displayed high expression levels of chemokines and cytokines, such as CCR5 and TNFSF10, contributing to the overall immune cell recruitment potential (Figure 5C, Supplementary Figure 5D). Correspondingly, the levels of antitumor immune cells, including M1 macrophages, B cells and tumor infiltrating lymphocytes (TILs), were significantly higher in high-Pr TNBCs, while immunosuppressive immune cells, such as regulatory T cells, showed the opposite trend (Figure 5D, Supplementary Figure 5E). To validate our findings, we assessed TILs within tumor regions using hematoxylin-eosin staining, and found that TILs infiltration was positively correlated with Pr (Figure 5E, Supplementary Table 5). Furthermore, we observed a higher density of CD8+ cells within tumor tissues with high Pr compared with those with low Pr (Supplementary Figure 5F, Supplementary Table 5). We also performed gene set enrichment analysis (GSEA) to explore the enriched pathways in high-Pr TNBCs (Supplementary Table 6). A series of immune response pathways were upregulated among samples with high Pr (Figure 5F), including lymphocyte migration (normalized enrichment score, NES = 2.48, NOM P value = 0.001), mononuclear cell migration (NES = 2.50, NOM P value = 0.001) and response to chemokines (NES = 2.48, NOM P value = 0.001). In addition, high-Pr patients showed lower exclusion score (Supplementary Figure 5G). To summarize, TNBC patients with high Pr HLA-I alleles exhibit increased expression of chemokines and cytokines in response to the large peptide repertoire breath, thereby possessing a remarkable ability to attract immune cells and stimulate antitumor immunity.
Figure 5.

Pr is positively correlated with the recruitment of immune cells in TNBCs. (A) Schematic view of a seven-step cancer-immune cycle, including release of cancer cell antigens, cancer antigen presentation by antigen presenting cells to immune cells, priming and activation of immune cells, trafficking of immune cells to tumors, infiltration of immune cells into tumors, recognition of cancer cells by T cells through TCR-antigen-MHC I, and killing of cancer cells. (B) The different levels of anticancer immunity across the seven-step cancer-immunity cycle in TNBCs with low and high Pr. Center line indicates the median value, lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers represent 1.5 × interquartile range. Wilcoxon test P value was shown. (C) Volcano plots of enriched chemokines (upper) and cytokines (bottom) in TNBCs with high Pr compared with those with low Pr. (D) Single-sample GSEA scores of immune cell abundance were calculated and compared between the high- and low-Pr TNBC groups. Center line indicates the median value, lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers represent 1.5 × interquartile range. Wilcoxon test P value was shown. (E) The correlation between TIL levels and Pr. (F) Cleveland plot showing the top 20 enriched pathways ordered by NES in the high Pr group. Abbreviations: Pr, promiscuity; TILs, tumor infiltrating lymphocytes; sTIL, stromal tumor infiltrating lymphocyte; iTIL, intratumoral tumor infiltrating lymphocyte; NES, normalized enrichment score; MHC, major histocompatibility complex; TCR, T cell receptor. See also Supplementary Figure 5.
Mismatch repair deficiency enriched in TNBCs with low Pr
As an exploratory analysis, we investigated whether genomic alterations might drive the poor prognosis in low-Pr samples. There were no distinct differences in somatic mutations detected between tumors with high Pr and low Pr (Supplementary Figure 6A). CNV analysis demonstrated that the loss of DNA fragments 3q21 and 12q24, where several genes associated with mismatch repair (MMR) were located, including ALDH2, ALKBH2, ARPC3, MBD4, MCM2 and RAD9B was more frequent in low-Pr TNBCs (Figure 6A–B, Supplementary Table 7). Of the breast cancer-related mutational signatures, only signature 26, which is related to the defective DNA mismatch repair (dMMR), was dominant in low-Pr tumors (Figure 6C). These results all revealed that a worse prognosis in low-Pr TNBCs might be explained by an impaired ability of MMR.
Figure 6.

MMR deficiency is enriched in TNBCs with low Pr. (A) Comparison of the CNVs between the high- and low-Pr groups. The upper plot illustrates the frequency of the amplification, gain, loss and deletion of each gene in each group, and the lower plot illustrates the -log10P value of each gene when comparing the frequency of loss or deletion or gain or amplification between the high- and low-Pr TNBC groups. (B) MMR-related genes at GISTIC peaks were compared between TNBCs with high Pr and those with low Pr. Fisher’s test P value was shown. (C) Heatmap showing the contribution of breast cancer-related mutational signatures to TNBCs with high and low Pr. (D) Cleveland plot showing the top 20 enriched pathways ordered by NES in the low Pr group. (E) The enrichment score of the top-enriched pathway MMR in the low Pr group from GSEA. (F) The correlation between Pr and HRD score. Abbreviations: Pr, promiscuity; LOH, loss of heterozygosity; HRD, homologous recombination deficiency. See also Supplementary Figure 6.
To confirm the findings above, we scrutinized the top enriched pathways in low-Pr TNBCs (Figure 6D, Supplementary Table 6). Pathways associated with chromosomal abnormalities during DNA replication were upregulated in patients with low Pr. We also observed that the MMR pathway was enriched in low-Pr samples (NES = −1.8, NOM P value = 0.005, Figure 6E). In addition, homologous recombination deficiency (HRD) scores, representing the extent of genomic instability, showed a negative association with Pr (Figure 6F). In particular, we further explored the relationship between MMR and immunological characteristics. Based on the enrichment score, we categorized TNBC patients into high- and low-MMR groups, and we observed a higher abundance of antitumor immune cells and upregulated expression of immune molecules in low-MMR patients than in high-MMR patients (Supplementary Figure 6B–C). Therefore, the impaired capacity of MMR may mainly lead to the immunosuppression and worse prognosis in TNBCs carrying low Pr. Synthetic lethal strategies targeting MMR deficiency may be suited for these patients with dMMR. For example, Werner syndrome ATP-dependent helicase (WRN) has been identified as a synthetic lethal target in dMMR cancers [23, 24].
DISCUSSION
HLA class I is a pivotal factor engaged in antitumor activity. In this study, we systematically evaluated the portrait of HLA-I diversity (including classical HLA-I mRNA expression level, HLA-I LOH, HED and Pr) in a large number of breast cancer samples, and further revealed their impact on clinical outcomes and immunological properties. In particular, we uncovered the prognostic power of Pr for TNBC patients and identified MMR as a genomic driver contributing to the poor prognosis in low-Pr TNBCs.
As far as we know, our research is an inaugural exploration to comprehensively assess the clinical value of HLA-I diversity comprising somatic changes and germline variations. Among the four HLA-I-related indicators we included, the classical HLA-I expression level was associated with improved survival in whole breast cancer samples, while HLA-I non-LOH and higher Pr indicated better prognosis in TNBCs. No conclusive association between HED and the prognosis of breast cancer samples was identified in our research. A recent study also reported a similar lack of association between HED and outcomes of patients who did not receive immunotherapy treatment [7]. Specifically, there was no reported association between HLA-I Pr and survival among patients with non-small cell lung cancer [19]. Notably, differences in the genetic ancestry of our research participants compared with those in previous reports arose due to our focus on a population of Chinese patients. Given the acknowledged effect of genetic ancestry on various biological processes, it is reasonable to consider that these differences can contribute to the variations in the obtained results. It is also conceivable that the association between HLA-I-related indicators and clinical outcomes does not manifest in breast cancer but can be detected in other specific tumor types owing to the distinct biological properties and clinicopathologic features among different tumor types [25, 26]. Future evaluations of the impact of HLA-I diversity should be refined by taking these factors into consideration. Notably, in the analyses of outcomes in patients with melanoma and non-small cell lung cancer treated with immune checkpoint inhibitors, higher HED was significantly correlated with better survival, whereas higher Pr yielded worse prognosis, suggesting the influence of HLA-I variations on the response to immunotherapy [7, 9, 19]. The role of HLA-I diversity in sensitivity to immunotherapy in breast cancer needs further exploration in prospective clinical trials.
Intriguingly, we found that MMR pathways were enriched in TNBC samples with low Pr. DNA MMR is a highly conserved biological process that plays a fundamental role in maintaining genome integrity and stability during DNA replication and recombination [27, 28]. Defects in MMR genes and the resulting high microsatellite instability elevate the risk of cancer development. Tumors exhibiting DNA MMR deficiency demonstrate abundant neoantigen accumulation, triggering an activated immune response [29]. However, approximately half of the colorectal cancer patients with MMR deficiency displayed a low level of T-cell infiltration [30]. Downregulation of the cGAS-STING pathway, which was reported as a contributor to reduced T cell infiltration into tumors, was correlated with poor survival in MMR-deficient cancers [31, 32]. In addition, an association has been identified between reduced CD8+ T-cell infiltration and increased glycolysis in tumors with high microsatellite instability, indicating a possible mechanism behind impaired T-cell infiltration [33]. Here, our findings have led to the hypothesis that MMR deficiency could act as a genetic barrier to high HLA-I Pr, potentially correlated to reduced immune cell presence and poorer prognosis in TNBCs. Reactivation of certain immune signals inside the TME might improve the clinical outcome of TNBC patients. Besides, synthetic lethal interactions have been discerned in MMR-deficient cancers. Therefore, synthetic lethal approaches hold the promise of novel therapies for dMMR tumors [23, 24].
There are some limitations in our research. First, we note that the sample size of TNBC cases included in our study was relatively small, particularly among patients with Pr score. Thus, we rigorously tested our results from multiple dimensions to fully validate our findings. Second, HLA diversity has been suggested as a biomarker of response to immunotherapy in previous studies. But still, we continue to have an absence of HLA-I-related data from a comprehensive immunotherapy trial carried out in breast cancer. Here, we predicted the immunotherapy response of patients in our breast cancer cohort by the Tumor Immune Dysfunction and Exclusion website and subsequently tested the influence of Pr on sensitivity to immunotherapy [34]. Finally, the breast cancer cohort with comprehensive HLA genotyping data is scarce, which limits the external validation of our findings. Future assessment of the performance of both germline and somatic HLA-I diversity in forecasting the efficacy of cancer immunotherapy for breast cancer samples warrants careful consideration.
Taken together, with our large-scale cohort, we identified the prognostic value of HLA-I variations in breast cancer. To be specific, low-Pr TNBC patients exhibited MMR defects and worse survival, providing hints for further precision treatment strategies in TNBC.
Key Points
The exploration of characteristics among breast cancer patients with distinct HLA-I status could provide hints for precision treatment.
This research provided an overview of HLA-I indicators in breast cancer utilizing a large multiomics cohort, which will be a valuable resource for the public.
This research highlighted the prognostic value of Pr in TNBC and revealed its correlated genomic features.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We wish to thank all patients and their families, as well as all staff at the study centers.
Author Biographies
Yi Xiao is an attending doctor in Fudan University Shanghai Cancer Center, researching the metabolic rewiring and surgical treatment of breast cancer.
Fenfang Chen and Cheng-Lin Liu are research associates in Fudan University Shanghai Cancer Center in Shanghai and expert in bioinformatics.
Xiao-Hong Ding and Tong Fu study in Fudan University Shanghai Cancer Center and focus on exploring potential therapeutic strategies for breast cancer.
Zhi-Ming Shao and Yi-Zhou Jiang are professors in Fudan University Shanghai Cancer Center, researching the precision treatment for breast cancer.
Contributor Information
Xiao-Hong Ding, Key Laboratory of Breast Cancer in Shanghai, Department of Breast Surgery, Fudan University Shanghai Cancer Center; Fudan University, Shanghai, 200032, P.R. China; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, 200032, P.R. China.
Yi Xiao, Key Laboratory of Breast Cancer in Shanghai, Department of Breast Surgery, Fudan University Shanghai Cancer Center; Fudan University, Shanghai, 200032, P.R. China; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, 200032, P.R. China.
Fenfang Chen, Key Laboratory of Breast Cancer in Shanghai, Department of Breast Surgery, Fudan University Shanghai Cancer Center; Fudan University, Shanghai, 200032, P.R. China.
Cheng-Lin Liu, Key Laboratory of Breast Cancer in Shanghai, Department of Breast Surgery, Fudan University Shanghai Cancer Center; Fudan University, Shanghai, 200032, P.R. China.
Tong Fu, Key Laboratory of Breast Cancer in Shanghai, Department of Breast Surgery, Fudan University Shanghai Cancer Center; Fudan University, Shanghai, 200032, P.R. China; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, 200032, P.R. China.
Zhi-Ming Shao, Key Laboratory of Breast Cancer in Shanghai, Department of Breast Surgery, Fudan University Shanghai Cancer Center; Fudan University, Shanghai, 200032, P.R. China; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, 200032, P.R. China.
Yi-Zhou Jiang, Key Laboratory of Breast Cancer in Shanghai, Department of Breast Surgery, Fudan University Shanghai Cancer Center; Fudan University, Shanghai, 200032, P.R. China; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, 200032, P.R. China.
AUTHOR CONTRIBUTIONS
Y.Z.J., Z.M.S. and Y.X. conceived and designed this project. X.H.D. and F.C. were responsible for the data analysis and interpretation and manuscript writing. C.L.L. and T.F. contributed to methodology. All authors contributed to the review and revision of the manuscript. All authors read and approved the final version of the manuscript.
FUNDING
This work was supported by grants from the National Key Research and Development Project of China (2020YFA0112304), the National Natural Science Foundation of China (82103451, 82373167, 82341003 and 92159301), the Natural Science Foundation of Shanghai (22ZR1479200), the Chinese Society of Clinical Oncology (Y-MSDZD2022-0489), the Shanghai Key Laboratory of Breast Cancer (12DZ2260100), the SHDC Municipal Project for Developing Emerging and Frontier Technology in Shanghai Hospitals (SHDC12021103) and Shanghai Medical Innovation Research Project (22Y11912700). The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
ETHICS STATEMENT
All patients included in the present study were collected after the approval of the research by the FUSCC Ethics Committee. Each patient provided written informed consent before enrollment.
DATA AVAILABILITY
All data reported in this paper can be viewed in The National Omics Data Encyclopedia (NODE, accession numbers: OEP000155 and OEP003049) and the Genome Sequence Archive (GSA) database (accession code: PRJCA017539).
References
- 1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin 2022;72(1):7–33. [DOI] [PubMed] [Google Scholar]
- 2. Waks AG, Winer EP. Breast cancer treatment: a review. JAMA 2019;321(3):288–300. [DOI] [PubMed] [Google Scholar]
- 3. Xiao Y, Ma D, Zhao S, et al. Multi-omics profiling reveals distinct microenvironment characterization and suggests immune escape mechanisms of triple-negative breast cancer. Clin Cancer Res 2019;25(16):5002–14. [DOI] [PubMed] [Google Scholar]
- 4. Chung W, Eum HH, Lee HO, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun 2017;8(1):15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Onkar SS, Carleton NM, Lucas PC, et al. The great immune escape: understanding the divergent immune response in breast cancer subtypes. Cancer Discov 2023;13(1):23–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang K, Halima A, Chan TA. Antigen presentation in cancer - mechanisms and clinical implications for immunotherapy. Nat Rev Clin Oncol 2023;20(9):604–23. [DOI] [PubMed] [Google Scholar]
- 7. Chowell D, Krishna C, Pierini F, et al. Evolutionary divergence of HLA class I genotype impacts efficacy of cancer immunotherapy. Nat Med 2019;25(11):1715–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chowell D, Morris LGT, Grigg CM, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018;359(6375):582–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lu Z, Chen H, Jiao X, et al. Germline HLA-B evolutionary divergence influences the efficacy of immune checkpoint blockade therapy in gastrointestinal cancer. Genome Med 2021;13(1):175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou YF, Xiao Y, Jin X, et al. Integrated analysis reveals prognostic value of HLA-I LOH in triple-negative breast cancer. J Immunother Cancer 2021;9(10):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Montesion M, Murugesan K, Jin DX, et al. Somatic HLA class I loss is a widespread mechanism of immune evasion which refines the use of tumor mutational burden as a biomarker of checkpoint inhibitor response. Cancer Discov 2021;11(2):282–92. [DOI] [PubMed] [Google Scholar]
- 12. Chhibber A, Huang L, Zhang H, et al. Germline HLA landscape does not predict efficacy of pembrolizumab monotherapy across solid tumor types. Immunity 2022;55(1):56–64.e4. [DOI] [PubMed] [Google Scholar]
- 13.Jiang YZ, Ma D, Jin X, et al. Integrated multiomic profiling of breast cancer in the Chinese population reveals patient stratification and therapeutic vulnerabilities, Nat Cancer 2024. [DOI] [PubMed] [Google Scholar]
- 14. Jin X, Zhou YF, Ma D, et al. Molecular classification of hormone receptor-positive HER2-negative breast cancer. Nat Genet 2023;55(10):1696–708. [DOI] [PubMed] [Google Scholar]
- 15. Jiang YZ, Ma D, Suo C, et al. Genomic and transcriptomic landscape of triple-negative breast cancers: subtypes and treatment strategies. Cancer Cell 2019;35(3):428–440.e5. [DOI] [PubMed] [Google Scholar]
- 16. Shukla SA, Rooney MS, Rajasagi M, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol 2015;33(11):1152–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van Loo P, Nordgard SH, Lingjærde OC, et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci U S A 2010;107(39):16910–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pierini F, Lenz TL. Divergent allele advantage at human MHC genes: signatures of past and ongoing selection. Mol Biol Evol 2018;35(9):2145–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Manczinger M, Koncz B, Balogh GM, et al. Negative trade-off between neoantigen repertoire breadth and the specificity of HLA-I molecules shapes antitumor immunity. Nat Cancer 2021;2(9):950–61. [DOI] [PubMed] [Google Scholar]
- 20. Ji S, Zhang Z, Ying S, et al. Kullback-Leibler divergence metric learning. IEEE Trans Cybern 2022;52(4):2047–58. [DOI] [PubMed] [Google Scholar]
- 21. Robinson J, Halliwell JA, Hayhurst JD, et al. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res 2015;43(D1):D423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu L, Deng C, Pang B, et al. TIP: a web server for resolving tumor immunophenotype profiling. Cancer Res 2018;78(23):6575–80. [DOI] [PubMed] [Google Scholar]
- 23. Chan EM, Shibue T, McFarland JM, et al. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 2019;568(7753):551–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wietmarschen N, Sridharan S, Nathan WJ, et al. Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 2020;586(7828):292–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Woroniecka K, Chongsathidkiet P, Rhodin K, et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res 2018;24(17):4175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kluger HM, Zito CR, Turcu G, et al. PD-L1 studies across tumor types, its differential expression and predictive value in patients treated with immune checkpoint inhibitors. Clin Cancer Res 2017;23(15):4270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem 2005;74(1):681–710. [DOI] [PubMed] [Google Scholar]
- 28. Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem 1996;65(1):101–33. [DOI] [PubMed] [Google Scholar]
- 29. Baretti M, Le DT. DNA mismatch repair in cancer. Pharmacol Ther 2018;189:45–62. [DOI] [PubMed] [Google Scholar]
- 30. Cristescu R, Mogg R, Ayers M, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018;362(6411):eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lu C, Guan J, Lu S, et al. DNA sensing in mismatch repair-deficient tumor cells is essential for anti-tumor immunity. Cancer Cell 2021;39(1):96–108.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guan J, Lu C, Jin Q, et al. MLH1 deficiency-triggered DNA hyperexcision by exonuclease 1 activates the cGAS-STING pathway. Cancer Cell 2021;39(1):109–121.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vasaikar S, Huang C, Wang X, et al. Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell 2019;177(4):1035–1049.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jiang P, Gu S, Pan D, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 2018;24(10):1550–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper can be viewed in The National Omics Data Encyclopedia (NODE, accession numbers: OEP000155 and OEP003049) and the Genome Sequence Archive (GSA) database (accession code: PRJCA017539).