

Abstract
A comprehensive analysis of the network of coupled motions correlated to hydride transfer in dihydrofolate reductase is presented. Hybrid quantum/classical molecular dynamics simulations are combined with a rank correlation analysis method to extract thermally averaged properties that vary along the collective reaction coordinate according to a prescribed target model. Coupled motions correlated to hydride transfer are identified throughout the enzyme. Calculations for wild-type dihydrofolate reductase and a triple mutant, along with the associated single and double mutants, indicate that each enzyme system samples a unique distribution of coupled motions correlated to hydride transfer. These coupled motions provide an explanation for the experimentally measured nonadditivity effects in the hydride transfer rates for these mutants. This analysis illustrates that mutations distal to the active site can introduce nonlocal structural perturbations and significantly impact the catalytic rate by altering the conformational motions of the entire enzyme and the probability of sampling conformations conducive to the catalyzed reaction.
Keywords: enzyme catalysis, molecular dynamics
Dihydrofolate reductase (DHFR) catalyzes the reduction of 7,8-dihydrofolate (DHF) to 5,6,7,8-tetrahydrofolate using nicotinamide adenine dinucleotide phosphate (NADPH) as a coenzyme (1). This reduction is an essential step for the biosynthesis of purines, pyrimidines, and amino acids. DHFR has been fostered as a pharmacological target for anticancer drugs and antibacterial agents because of its physiological importance for normal folate metabolism (2). As a result, DHFR has been studied extensively with a wide range of experimental and theoretical approaches. Fig. 1 depicts the 3D structure of DHFR and identifies important loop regions, such as the Met-20 loop (residues 9–24), the βF–βG loop (residues 116–132), and the βG–βH loop (residues 142–150), as well as the adenosine binding domain (residues 38–88).
Fig. 1.

The 3D structure of WT DHFR. The important loop regions are labeled. The substrate and cofactor are depicted in red, and the residues involved in the mutants studied are identified with yellow spheres.
The detailed mechanism for DHFR has been determined from kinetic studies of the Escherichia coli species (1, 3). X-ray crystallographic structures of DHFR in binary and ternary complexes (4) indicate that the enzyme assumes different conformations along the reaction pathway. Furthermore, NMR relaxation experiments (5–7) imply that the binding of the substrate and the coenzyme induces conformational changes of structural elements both in and distal to the active site, including the Met-20 and βF–βG loops. Classical molecular dynamics simulations of the reactant ternary complex with DHF identified correlated and anticorrelated motions involving many of the same spatial regions as implicated by the dynamic NMR measurements (8). These correlations are absent in the product complex with 5,6,7,8-tetrahydrofolate. Mutant DHFR enzymes with reduced activity exhibit a reduction in these correlated motions compared with the wild-type (WT) system (9, 10).
Hybrid quantum/classical molecular dynamics simulations provided evidence of a network of coupled motions extending throughout the protein and ligands (11–13). These coupled motions represent equilibrium, thermally averaged conformational changes along the reaction coordinate. They occur on the millisecond timescale of the hydride transfer reaction. These conformational changes lead to configurations that facilitate hydride transfer through short transfer distances, suitable orientation of the substrate and cofactor, and an appropriate electrostatic environment for charge transfer. Recent simulations of WT DHFR based on alternative approaches identified similar variations in some of these geometrical properties (10, 14, 15). Moreover, subsequent to the identification of Ile-14 as a key player in this network of coupled motions (11, 12), NMR experiments confirmed the catalytic importance of this residue (16). The observed reduction in activity for mutant DHFR enzymes may be attributed to modifications of this network of coupled motions (13). Mutations distal to the active site can cause long-range structural perturbations and influence the conformational motions of the enzyme along the reaction coordinate in a manner that significantly alters the free energy barrier, thereby altering the rates.
This evidence suggests that adequate sampling of favorable equilibrium conformations along the reaction coordinate is critical for hydride transfer in DHFR. The majority of the elucidated coupled motions discovered in the previous simulations (11, 12) were motivated by a genomic analysis used to identify residues conserved across different species and experimental rate measurements of mutant enzymes (11). Despite the conceptual insights gained by these previous studies, this network of coupled motions is not complete or unique. The objective of this work is to map the entire network of coupled motions that facilitate hydride transfer. Specifically, we present a procedure for comprehensively extracting the coupled motions that vary along the reaction coordinate according to a set of prescribed target functions. This procedure combines hybrid quantum/classical molecular dynamics simulations with a rank order correlation analysis method. We apply this approach to the WT DHFR system, as well as to the experimentally studied triple mutant M42F–G121S–S148A and the associated single and double mutants. The resulting identification of coupled motions correlated to hydride transfer provides an explanation for the experimentally measured hydride transfer rates of the mutants and provides guidance for future experimental and theoretical studies of DHFR.
Methods
Hybrid Quantum/Classical Molecular Dynamics. Although the complete catalytic cycle of DHFR involves numerous kinetically observable intermediates (3), in this work we focus on only the hydride transfer reaction from the bound NADPH cofactor to the bound protonated DHF substrate. The potential energy surface is represented by a two-state empirical valence bond (EVB) potential (17). The diagonal elements of the 2 × 2 EVB Hamiltonian are described by the gromos force field (Version 43A1) (18) with the modifications described in ref. 12. The coupling V12 between the valence bond (VB) states (i.e., the off-diagonal element of the Hamiltonian matrix) and the energy adjustment Δ12 to the second VB state are parameterized to reproduce the experimentally determined free energy barriers for the forward and reverse rates of hydride transfer in WT DHFR. We found that the EVB potential is not sufficiently flexible to afford transferability of the parameters for WT to the mutant systems. As a result, V12 and Δ12 are reparameterized for the mutants to reproduce the experimentally determined rates for the mutants relative to the WT DHFR, as given in Table 1. Note that the experimental rates given in Table 1 are for a pH of 7, whereas the experimental rate used previously to parameterize the WT free energy barrier is the pH-independent rate of 950 s–1 (3). Here, we assume that the differences in free energy barriers between the mutant and WT DHFR are independent of pH and that the free energy of reaction is the same for the mutant and WT DHFR. The analysis presented in this work is not expected to be influenced significantly by relatively small differences in the parameters V12 and Δ12. The nuclear quantum effects, such as zero point motion and tunneling, of the transferring hydrogen are included by representing the transferring hydrogen nucleus as a 3D wavefunction (19).
Table 1. Experimentally determined hydride transfer rates khyd at pH 7, corresponding change in the free energy barriers of the mutants relative to WT DHFR ΔΔG‡, and optimized EVB parameters V12 and Δ12.
| DHFR | khyd, s-1 | ΔΔG‡, kcal/mol | V12, kcal/mol | Δ12, kcal/mol |
|---|---|---|---|---|
| WT | 220 | 0.00 | 34.66 | 65.25 |
| M42F | 159 | 0.19 | 34.66 | 65.25 |
| G121S | 3.9 | 2.38 | 27.49 | 67.40 |
| S148A | 157 | 0.20 | 33.46 | 66.92 |
| G121S-S148A | 18 | 1.48 | 29.88 | 65.25 |
| M42F-S148A | 92 | 0.51 | 34.66 | 65.25 |
| M42F-G121S | 2.9 | 2.55 | 28.68 | 69.79 |
| M42F-G121S-S148A | 12 | 1.72 | 28.68 | 63.82 |
The free energy profile is generated as a function of a collective reaction coordinate Λ(R) defined to be the difference between the energies of the two VB states averaged over the ground state vibrational wave function of the transferring hydrogen,
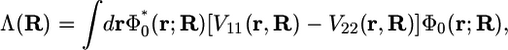 |
[1] |
where V11 and V22 denote the energies of VB states 1 and 2 (i.e., the diagonal elements of the EVB Hamiltonian) and Φ0(r; R) is the wave function representing the hydrogen vibrational ground state. This choice of reaction coordinate has been shown to be physically meaningful for two-state charge transfer processes (20–22). Moreover, for enzymatic reactions described within the framework of transition state (TS) theory, this reaction coordinate leads to estimates of the transmission coefficient that are close to unity (12, 19). The mapping potentials used to sample the entire range of reaction coordinates are defined as linear combinations of the two VB states (17) as follows:
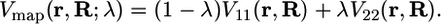 |
[2] |
As the mapping parameter λ is varied from zero to unity, the biasing potential drives the molecular dynamics to sample conformations from the reactant state (RS) to the product state (PS). In this umbrella sampling approach, standard binning techniques are used to generate the segment of the free energy profile for each value of λ. The individual segments are connected by using thermodynamic integration to form the free energy profile corresponding to the unbiased Hamiltonian (19). A similar procedure is used to obtain thermally averaged geometrical properties along the reaction coordinate. A perturbation formula is used to incorporate the nuclear quantum effects into the free energy profile (19).
The system modeled in our WT DHFR simulation includes the protein, a NADPH cofactor, a protonated DHF substrate, and 4,122 explicit simple point charge/extended (SPC/E) water molecules in a truncated octahedral periodic box with a distance of 66.61 Å between opposing square faces. The initial coordinates are taken from a crystal structure of E. coli DHFR complexed with NADPH+ and folate (Protein Data Bank ID code 1RX2) (4). In this complexed state, the Met-20 loop is in the closed conformation, which is thought to be the active form for the hydride transfer step. The reaction studied is the transfer of the pro-R hydrogen on the donor carbon of NADPH to the acceptor carbon of the protonated DHF. The initial coordinates for the mutants (M42F, G121S, S148A, G121S–S148A, M42F–S148A, M42F–G121S, and M42F–G121S–S148A) are obtained by using the mutation utility in swiss-pdbviewer (23) on the above WT crystal structure, where the rotamer that forms the most hydrogen bonds and minimizes the steric clashes was chosen. The periodic boundary box dimensions and the number of solvating water molecules for these mutant systems are comparable to the WT system.
For the WT DHFR simulation, we used a set of 20 mapping parameters and performed 4.5 ns of molecular dynamics for each window. An additional 2 ns of molecular dynamics was performed for the four windows near the TS to obtain 98 ns of total sampling. For the mutants, we used a set of 12 mapping parameters and performed 2.5 ns of molecular dynamics for each window. An additional 2 ns of molecular dynamics was performed for the four windows in the vicinity of the TS and for the two windows near the reactant and product minima for a total of 42 ns of sampling. Although the WT is the more extensively sampled system, our previous statistical analysis of the WT DHFR simulations suggests that longer sampling for the mutants will not impact the qualitative conclusions. By using the method described in ref. 24, we obtained thermally averaged structures for the RS, TS, and PS for WT DHFR and all of the mutants studied. The rms deviations of these structures with respect to the initial crystal structure coordinates are <1.4 Å, confirming that the key structural elements are maintained during the simulations.
The main limitations of these simulations are the potential energy surface and the sampling of configurational space. The EVB potential could be improved by using a more sophisticated off-diagonal element including more parameters, implementing a more accurate molecular mechanical force field for the diagonal elements, and including more than two VB states. However, previous studies indicate that this two-state EVB potential provides a qualitatively reasonable description of DHFR (11–13). The sampling of configurational space could be improved by increasing the length of the molecular dynamics simulations. We investigated the accuracy of our sampling for this application by analyzing the first and second halves of the molecular dynamics data. We found that the results from these partial data sets are similar to those from the entire data set, thereby indicating adequate sampling.
As mentioned in our previous work (11, 12) and pointed out again in ref. 15, the possibility of becoming trapped in local minima is a potential difficulty associated with molecular dynamics simulations. We have sampled a total of 98 ns for the WT DHFR and a total of 42 ns for each mutant DHFR in our simulations (24). Brooks and coworkers (10, 15) performed 10-ns classical molecular dynamics simulations for the reactant ternary complexes of WT DHFR and the G121V and G121S mutants. Swanwick et al. (25) performed a 1-ns classical molecular dynamics simulation of WT DHFR at 300 K and 20 1-ns classical molecular dynamics simulations of WT DHFR and the G121V mutant at 500 K. Given the similarities in the time scales of these simulations (10, 15, 24, 25), the probability of becoming trapped in local minima is similar, although the high-temperature simulations are expected to enhance sampling. The advantage of the approach based on umbrella sampling (11, 12, 24) is that equilibrium ensembles for the entire range of the reaction coordinate, including the TS region, are sampled. Although each mapping potential is sampled for only a few nanoseconds, the data from the different potentials are combined to generate the entire free-energy profile for the chemical reaction. Assuming adequate equilibration for each mapping potential, this procedure provides information about the equilibrium motions occurring on the experimentally determined millisecond timescale of hydride transfer.
Rank Correlation Analysis. Given a matrix of all pairwise atomic distances as a function of the reaction coordinate, our goal is to identify the systematic conformational changes associated with the reaction. For exclusion purposes, we also wish to identify those atomic pairs with distances that do not change during the reaction. Fig. 2 shows two target models of geometrical changes along a 1D reaction coordinate. The SYMM model (Fig. 2 A) describes properties that exhibit a maximum or minimum at the TS. The MONO model (Fig. 2B) describes properties that change monotonically from RS to PS.
Fig. 2.

Target models for the variation of the thermally averaged property as a function of the reaction coordinate. (A) SYMM. (B) MONO.
Here, we present an approach based on rank correlation for extracting conformational changes that are consistent with a given target model. We use Kendall's tau method (26, 27) for this statistical analysis. This nonparametric method enables the detection of correlations between two sets of independent data {xk},{yk} with N data points in each set. In this approach, the correlation is calculated between the ranks of the two data sets, where the rank is determined by numbering the data points in each data set from lowest to highest (i.e., 1, 2,..., N). The algorithm involves a systematic comparison of the relative ordering of the ranks for each pair of data points within each of the two data sets. The value of Kendall's correlation coefficient τ lies between –1 and +1, where complete correlation (τ = 1) indicates that the ranks of the two data sets are identical, and complete anticorrelation (τ =–1) indicates that the ranks of the two data sets are reversed. The significance level is calculated based on the null hypothesis.
We use this procedure to characterize the conformational changes along the reaction coordinate. The data set {xk} is the thermally averaged interatomic distance along the collective reaction coordinate for a pair of atoms, and the data set {yk} is one of the target models given in Fig. 2. Kendall's tau provides an indication of the probability that the distance between a pair of atoms in the enzyme changes according to the chosen target model. In the algorithm, first a 3D matrix D(i,j,k) of thermally averaged interatomic distances is generated, where i and j denote atomic indices and k denotes the discretized reaction coordinate index. For each pair of atoms (i,j), the value of τij is obtained by the application of Kendall's procedure to the data set representing the thermally averaged distance between atoms i and j along the discretized reaction coordinate with the data set representing the target model. The matrix τ with elements τij provides a global picture of the motions most relevant to the progress of the chemical reaction. This approach is not limited to systems modeled with a 1D reaction coordinate but rather is naturally extendable to reaction coordinates of arbitrary dimension.
The advantages of this rank correlation analysis over linear correlation analysis stem from the use of ranks rather than numerical values of the property for the calculation of the correlations (26). The use of ranks effectively coarse grains both data sets before addressing the issue of correlation. As a result, small defects in the data set are averaged out before the analysis. One limitation of Kendall's tau approach is that it does not provide information about the change in magnitude of the property along the reaction coordinate. For example, this approach cannot distinguish a data set that is identical to the target function from a data set that is the target function scaled by a factor of a thousand. To obtain this information, the magnitude of the changes from RS to TS to PS can be determined by evaluating the differences among the values at these points.
A wide range of properties, including interatomic distances, angles, and dihedrals, can be studied with this general rank correlation analysis approach. In this work, we study the variations in the interatomic distances along the reaction coordinate for hydride transfer. In some cases, however, interatomic distances may not be the optimal variables for analyzing the motions of the protein. For processes involving large-scale domain motion, investigation of the variations of a small number of relevant torsional angles may provide a more physically meaningful picture. Alternatively, the atomic indices could be mapped onto the larger structural domains, which could serve as the variables in the rank correlation analysis. In addition to the investigation of variations in geometrical properties, this rank correlation analysis approach could be used to study variations in the electrostatic potential along the reaction coordinate. In general, the choice of the property to be studied will significantly influence the results of the analysis.
Results and Discussion
We have used the rank correlation analysis approach, in conjunction with hybrid quantum/classical molecular dynamics simulations, to extract the variations in the interatomic distances along the reaction coordinate for hydride transfer in WT DHFR. Fig. 3 depicts the correlation matrices τ between heavy atom interatomic distances with the SYMM and MONO target models for WT DHFR. All nonzero matrix elements τij are within the 99% confidence limit according to the calculated statistical significance level. The correlation maps indicate that the chemical reaction is accompanied by a diverse set of distance changes throughout the protein.
Fig. 3.

Correlation maps for WT DHFR using the target models SYMM (A) and MONO (B). Each map depicts the correlation of thermally averaged interatomic distances in the enzyme with the target model along the collective reaction coordinate. The correlations are calculated with Kendall's tau method. Red denotes correlated regions (τ = 1), and blue denotes anticorrelated regions (τ =–1). The two axes are identical and represent the atoms of the enzyme in sequential order. The three sites of mutation are identified on the vertical axis, and three key loop regions are identified on the horizontal axis.
We emphasize that these correlation maps are distinct from previously generated correlation matrices of fluctuations about an average structure for DHFR (8, 9, 24). The degree of correlation in these previous studies refers to the correlation between fluctuations of a pair of atoms about an average structure. These fluctuations are local to the average structure and typically occur on the femtosecond timescale. In contrast, the current rank correlation approach provides a comprehensive statistical analysis of changes in thermally averaged properties along the reaction coordinate. The degree of correlation in the current analysis refers to the correlation between the behavior of thermally averaged interatomic distances and a given target model along the reaction coordinate. These motions occur on the millisecond timescale of the hydride transfer reaction.
The correlation map depicted in Fig. 3A for WT DHFR using the SYMM target model represents variations that exhibit an extremum at the TS. In this case, we observe that similar numbers of pairs of interatomic distances exhibit a maximum at the TS (correlations mapped in red) as exhibit a minimum at the TS (anticorrelations mapped in blue). We observe a modest increase in the interatomic distances between the Met-20 and the βG-βH loops as the reaction progresses from the RS to the TS and then a decrease as the reaction continues toward the PS. The motion of the βG–βH loop relative to the adenosine binding domain shows similar opening and closing motions, although these loops are >20 Å apart. Table 2 provides the values of representative interatomic distances at the RS, TS, and PS to illustrate these trends. As shown by the anticorrelations in Fig. 3A, the relative distances between the βF–βG loop and residues 25–40 that are sequentially adjacent to the Met-20 loop become shorter at the TS and longer again at the PS.
Table 2. Thermally averaged distances between the Cα atoms of specified residue pairs at the RS, TS, and PS for the WT and mutant DHFR enzymes.
| DHFR system | RS | TS | PS |
|---|---|---|---|
| Met-20 loop—βG—βH | |||
| loop* | |||
| WT | 12.72 | 14.05 | 12.51 |
| M42F | 13.75 | 14.94 | 13.39 |
| G121S | 14.36 | 14.06 | 12.45 |
| S148A | 12.68 | 12.34 | 15.64 |
| G121S—S148A | 13.05 | 13.71 | 12.69 |
| M42F—S148A | 14.22 | 14.98 | 13.15 |
| M42F—G121S | 12.67 | 13.78 | 15.52 |
| M42F—G121S—S148A | 13.00 | 13.17 | 12.47 |
| βG—βH loop—ABD† | |||
| WT | 39.80 | 40.19 | 38.85 |
| M42F | 40.01 | 39.51 | 38.85 |
| G121S | 38.78 | 39.33 | 36.53 |
| S148A | 40.72 | 39.85 | 39.93 |
| G121S—S148A | 39.76 | 36.94 | 41.84 |
| M42F—S148A | 41.16 | 39.60 | 39.22 |
| M42F—G121S | 39.48 | 39.87 | 39.89 |
| M42F—G121S—S148A | 41.71 | 40.13 | 36.89 |
All distances are in units of Å.
Residues 20 and 148.
Residues 150 and 68. ABD, adenosine binding domain.
The correlation map in Fig. 3B for WT DHFR using the MONO target model shows similar diversity in the correlation patterns. Many regions of the enzyme increase monotonically in distance relative to the hinge region near residues 88–100. The correlations in the MONO map (red regions in Fig. 3B) between the hinge region and one of the α-helices (residues 23–55), as well as the βF–βG and βG–βH loops, suggest that the distances between these residues and the hinge region increase monotonically along the entire reaction coordinate. In contrast, the distances between the βF–βG and the βG–βH loops and between the residues sequentially adjacent to M42 and the three labeled loop regions decrease monotonically, as shown by the anticorrelations (blue regions) in Fig. 3B. Table 3 provides the values of representative interatomic distances at the RS, TS, and PS to illustrate these trends. We detect few correlations with a constant target model (i.e., no variations in the property along the reaction coordinate).
Table 3. Thermally averaged distances between the Cα atoms of specified residue pairs at the RS, TS, and PS for the WT and mutant DHFR enzymes.
| DHFR system | RS | TS | PS |
|---|---|---|---|
| Hinge region—βF—βG loop* | |||
| WT | 13.48 | 13.87 | 14.17 |
| M42F | 13.23 | 13.72 | 14.29 |
| G121S | 13.38 | 13.95 | 14.20 |
| S148A | 13.31 | 14.15 | 13.71 |
| G121S—S148A | 13.44 | 14.15 | 13.30 |
| M42F—S148A | 13.23 | 13.99 | 13.89 |
| M42F—G121S | 13.87 | 13.56 | 13.56 |
| M42F—G121S—S148A | 13.52 | 13.84 | 14.43 |
| βF—βG loop—βG—H loop† | |||
| WT | 17.48 | 16.33 | 15.91 |
| M42F | 17.57 | 17.52 | 17.65 |
| G121S | 18.14 | 17.68 | 17.55 |
| S148A | 17.65 | 16.84 | 18.48 |
| G121S—S148A | 17.79 | 17.44 | 17.74 |
| M42F—S148A | 17.84 | 17.41 | 17.21 |
| M42F—G121S | 16.96 | 17.84 | 18.43 |
| M42F—G121S—S148A | 18.89 | 17.48 | 17.85 |
All distances are in units of Å.
Residues 93 and 126.
Residues 121 and 148.
As shown in Table 1, the mutation of residues distal to the active site can significantly impact the rate of hydride transfer. Moreover, the experimentally measured hydride transfer rates for the triple mutant M42F–G121S–S148A and the associated single and double mutants exhibit some nonadditivity effects (i.e., the effect of a double or triple mutation is greater than the sum of the effects of the single mutations). The correlation maps in Fig. 3 illustrate that residues 42, 121, and 148 play important roles in the network of coupled motions correlated to hydride transfer. The rank correlation maps of these mutants exhibit differences from those of WT DHFR for distances between pairs of atoms located throughout the enzyme. The correlation maps for the triple mutant, the three double mutants, and the three single mutants are shown in Figs. 4–10, which are published as supporting information on the PNAS web site. Tables 2 and 3 present the thermally averaged distances between representative pairs of atoms at the RS, TS, and PS for the WT and mutant enzymes. These calculations indicate that distal mutations significantly impact the coupled motions correlated to hydride transfer. Moreover, changes in the motions induced by a single mutation are not always observed in the associated double and triple mutants. The participation of these residues in the network of coupled motions provides an explanation for the experimentally observed nonadditivity effects, as well as the significant decrease in the hydride transfer rate.
These observations are consistent with other analyses of the impact of single distal mutations on hydride transfer in DHFR (15, 25). Thorpe and Brooks (15) studied hydride transfer barriers based on snapshots from 10-ns classical molecular dynamics simulations of reactive ternary complexes for WT DHFR and the G121V and G121S mutants. They identified structural changes in the protein that correlate with lower hydride transfer barriers and concluded that the G121V and G121S mutations in DHFR alter the structural ensemble within the protein. Several of the structural changes observed by Thorpe and Brooks (15) were consistent with our previous identification and characterization of the network of coupled motions (11–13). The most probable reasons for discrepancies in some of the details of these motions are differences in the force fields and in the methodology to calculate the hydride transfer barriers. Nevertheless, the extent of agreement between these different analyses is encouraging, and the overall conclusions concerning the impact of distal mutations on the conformational sampling are consistent. Swanwick et al. (25) have performed high-temperature molecular dynamics simulations of WT DHFR and the G121V mutant. They observed differences in the stability and unfolding for the WT and mutant enzymes. They concluded that the mutation causes nonlocal structural effects that may lead to perturbation of the network of coupled motions. These conclusions are consistent with our previous simulations of the G121V mutant (13), in which the distal mutation resulted in nonlocal structural changes that altered the conformational sampling of the entire enzyme. The present calculations on the triple mutant and the associated single and double mutants provide further validation of these fundamental concepts.
Conclusions
We have presented a rank correlation analysis approach for extracting thermally averaged properties that change along a reaction coordinate according to a prescribed target model. The only inputs required for this procedure are the thermally averaged properties along the reaction coordinate and the target model. The rank correlation approach is particularly robust and resistant to defects in the data because ranks rather than numerical values are used for the calculation of the correlations. In our application of this approach to hydride transfer catalyzed by DHFR, we obtained the thermally averaged interatomic distances along the collective reaction coordinate with a hybrid quantum/classical molecular dynamics approach. The rank correlation analysis is straightforward to implement and provides a comprehensive picture of coupled motions that facilitate enzymatic reactions.
Our application of this approach to WT DHFR and to a set of single, double, and triple mutants indicates that each system samples a unique distribution of coupled motions throughout the enzyme. The rank correlation analysis identifies and characterizes the coupled motions correlated to hydride transfer. Note that this analysis is unable to differentiate between motions playing an active role in catalysis and motions responding to alterations caused by catalysis. Moreover, this analysis does not address the impact of the mutations on the stability and folding of the enzyme. Our calculations indicate that residues in the adenosine binding domain and residues in the Met-20, βF–βG, and βG–βH loops play important roles in the network of coupled motions. Experimental kinetic measurements of the triple mutant M42F–G121S–S148A and the associated single and double mutants show that mutations distal to the active site can significantly impact the hydride transfer rate in a nonadditive manner. The participation of these residues in the network of coupled motions correlated to hydride transfer provides an explanation for these experimental results.
This analysis illustrates that site-specific mutations distal to the active site can introduce subtle perturbations that impact the catalytic rate by altering the conformational sampling of the entire enzyme. Because distal regions of the enzyme are coupled to each other through long-range electrostatics and extended hydrogen-bonding networks, the introduction of a site-specific mutation leads to nonlocal structural changes and alters the thermal motions of the entire enzyme. Altering the thermal motions of the enzyme affects the probability of sampling conformations conducive to the catalyzed chemical reaction, thereby impacting the free energy barrier and the rate. These fundamental principles have important implications for protein engineering and drug design. A medically relevant example is the experimentally observed impact of distal mutations on the resistance to HIV type 1 protease inhibitors (28).
Supplementary Material
Acknowledgments
We thank James B. Watney and Alexander Soudackov for helpful discussions. This work was supported by National Institutes of Health Grant GM56207.
Author contributions: K.F.W., S.J.B., and S.H.-S. designed research; K.F.W. and T.S. performed research; K.F.W., S.J.B., and S.H.-S. analyzed data; and K.F.W. and S.H.-S. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: DHF, 7,8-dihydrofolate; DHFR, dihydrofolate reductase; VB, valence bond; EVB, empirical VB; RS, reactant state; PS, product state; TS, transition state.
References
- 1.Miller, G. P. & Benkovic, S. J. (1998) Chem. Biol. 5, R105–R113. [DOI] [PubMed] [Google Scholar]
- 2.Berg, J. M., Stryer, L. & Tymoczko, J. (2002) Biochemistry (Freeman, New York), 5th Ed.
- 3.Fierke, C. A., Johnson, K. A. & Benkovic, S. J. (1987) Biochemistry 26, 4085–4092. [DOI] [PubMed] [Google Scholar]
- 4.Sawaya, M. R. & Kraut, J. (1997) Biochemistry 36, 586–603. [DOI] [PubMed] [Google Scholar]
- 5.Falzone, C. J., Wright, P. E. & Benkovic, S. J. (1994) Biochemistry 33, 439–442. [DOI] [PubMed] [Google Scholar]
- 6.Epstein, D. M., Benkovic, S. J. & Wright, P. E. (1995) Biochemistry 34, 11037–11048. [DOI] [PubMed] [Google Scholar]
- 7.Osborne, M. J., Schnell, J., Benkovic, S. J., Dyson, H. J. & Wright, P. E. (2001) Biochemistry 40, 9846–9859. [DOI] [PubMed] [Google Scholar]
- 8.Radkiewicz, J. L. & Brooks, C. L., III (2000) J. Am. Chem. Soc. 122, 225–231. [Google Scholar]
- 9.Rod, T. H., Radkiewicz, J. L. & Brooks, C. L., III (2003) Proc. Natl. Acad. Sci. USA 100, 6980–6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thorpe, I. F. & Brooks, C. L., III (2003) J. Phys. Chem. B 107, 14042–14051. [Google Scholar]
- 11.Agarwal, P. K., Billeter, S. R., Rajagopalan, P. T. R., Benkovic, S. J. & Hammes-Schiffer, S. (2002) Proc. Natl. Acad. Sci. USA 99, 2794–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agarwal, P. K., Billeter, S. R. & Hammes-Schiffer, S. (2002) J. Phys. Chem. B 106, 3283–3293. [Google Scholar]
- 13.Watney, J. B., Agarwal, P. K. & Hammes-Schiffer, S. (2003) J. Am. Chem. Soc. 125, 3745–3750. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Viloca, M., Truhlar, D. G. & Gao, J. (2003) Biochemistry 42, 13558–13575. [DOI] [PubMed] [Google Scholar]
- 15.Thorpe, I. F. & Brooks, C. L., III (2004) Proteins 57, 444–457. [DOI] [PubMed] [Google Scholar]
- 16.Schnell, J. R., Dyson, H. J. & Wright, P. E. (2004) Biochemistry 43, 374–383. [DOI] [PubMed] [Google Scholar]
- 17.Warshel, A. (1991) Computer Modeling of Chemical Reactions in Enzymes and Solutions (Wiley, New York).
- 18.van Gunsteren, W. F., Billeter, S. R., Eising, A. A., Hunenberger, P. H., Kruger, P., Mark, A. E., Scott, W. R. P. & Tironi, I. G. (1996) Biomolecular Simulation: The gromos96 Manual and User Guide (Hochschulverlag, Zurich).
- 19.Billeter, S. R., Webb, S. P., Iordanov, T., Agarwal, P. K. & Hammes-Schiffer, S. (2001) J. Chem. Phys. 114, 6925–6936. [DOI] [PubMed] [Google Scholar]
- 20.Marcus, R. A. (1964) Annu. Rev. Phys. Chem. 15, 155–196. [Google Scholar]
- 21.Zusman, L. D. (1980) Chem. Phys. 49, 295–304. [Google Scholar]
- 22.Warshel, A. (1982) J. Phys. Chem. 86, 2218–2224. [Google Scholar]
- 23.Guex, N. & Peitsch, M. C. (1997) Electrophoresis 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- 24.Wong, K. F., Watney, J. B. & Hammes-Schiffer, S. (2004) J. Phys. Chem. B 108, 12231–12241. [Google Scholar]
- 25.Swanwick, R. S., Shrimpton, P. J. & Allemann, R. K. (2004) Biochemistry 43, 4119–4127. [DOI] [PubMed] [Google Scholar]
- 26.Press, W. H., Teukolsky, S. A., Vetterling, W. T. & Flannery, B. P. (1992) Numerical Recipes in Fortran 77: The Art of Scientific Computing (Cambridge Univ. Press, New York), 2nd Ed.
- 27.Lehmann, E. L. (1975) Nonparametrics: Statistical Methods Based on Ranks (Holden–Day, San Francisco).
- 28.Boden, D. & Markowitz, M. (1998) Antimicrob. Agents Chemother. 42, 2775–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.