

SUMMARY
We explore the changes in chromatin accessibility and transcriptional programs for cochlear hair cell differentiation from postmitotic supporting cells using organoids from postnatal cochlea. The organoids contain cells with transcriptional signatures of differentiating vestibular and cochlear hair cells. Construction of trajectories identifies Lgr5+ cells as progenitors for hair cells, and the genomic data reveal gene regulatory networks leading to hair cells. We validate these networks, demonstrating dynamic changes both in expression and predicted binding sites of transcription factors (TFs) during organoid differentiation. We identify known regulators of hair cell development, Atoh1, Pou4f3, and Gfi1, and the analysis predicts the regulatory factors Tcf4, an E-protein and heterodimerization partner of Atoh1, and Ddit3, a CCAAT/enhancer-binding protein (C/EBP) that represses Hes1 and activates transcription of Wnt-signaling-related genes. Deciphering the signals for hair cell regeneration from mammalian cochlear supporting cells reveals candidates for hair cell (HC) regeneration, which is limited in the adult.
In brief
Generation of new hair cells after damage to the cochlea is a potential treatment for deafness. Kalra et al. probe the gene expression program of Lgr5+ supporting cells that gives rise to hair cells in organoids, providing clues to gene regulation underlying hair cell differentiation in the postnatal cochlea.
Graphical abstract
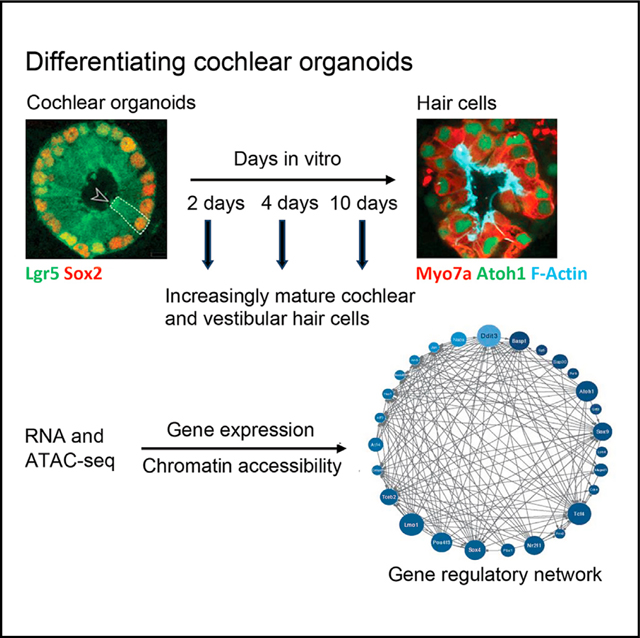
INTRODUCTION
The mouse cochlea contains approximately 3,000 hair cells. Its dimensions and location, and its small number of hair cells, make mechanistic, developmental, and cellular replacement studies difficult. We recently published a protocol to expand and differentiate murine neonatal cochlear progenitor cells into 3D organoids that recapitulate developmental pathways and can generate large numbers of hair cells with intact stereociliary bundles, mechanotransduction channel activity, and molecular markers of native cells, including markers for both inner and outer hair cells.1 We also showed that inner and outer hair cells were segregated into separate organoids based on the hair cell markers prestin and VGlut3,1 suggesting that some of the phenotypic complexity of cochlear hair cell types was represented in this in vitro system.
The relevance of cochlear organoids to in vivo differentiation depends on the fidelity with which they mimic in vivo cell types and regenerative processes.2,3 The organoids held promise for modeling development and differentiation and for higher-throughput screening to identify small molecules and genes that modulate these processes.4 However, the analyses were limited to a small number of known marker genes, which may not fully reveal the cells’ transcriptional states. Moreover, the transcriptional signatures of cells within organoids that do not become hair cells remain little characterized. Here, we perform a comprehensive transcriptional characterization of the cochlear organoids in comparison to in vivo cell types, confirming that the organoids mimic nearly all supporting cell and hair cell subtypes of the in vivo cochlea as well as the utricle. In addition, we analyze chromatin accessibility underlying changes in gene expression and use these epigenetic data to perform gene regulatory network modeling that predicts regulators of hair cell development and differentiation.
RESULTS
Data integration identifies marker genes for cochlear and utricular cell types to allow robust analysis of hair cell differentiation in cochlear organoids
We have recently developed organoids made by expansion of Lgr5+ supporting cells from the cochlear sensory epithelium.1 The conditions for expansion result in organoids highly enriched in Lgr5+ cells (78% of the cells in the organoids). Over the course of 10 days, using the previously described conditions,1 these cells differentiate into hair cells, including inner and outer cochlear hair cells, allowing us to study the phenotypic complexity represented by the organoid system to determine the epigenetic and transcriptional changes underlying the postnatal differentiation of hair cells from supporting cells.
To enable a quantitative comparison of inner ear sensory epithelial cell types, we developed a comprehensive database of genes expressed specifically in each cell type of the mouse cochlea and utricle (Figure 1A). We generated new single-cell RNA sequencing (scRNA-seq) from mouse utricle at postnatal days 2 and 7 (Figures 1B and 1C). Approximately 10,000 dissociated utricular cells were analyzed. We integrated these data with six previously published scRNA-seq datasets to identify marker genes for 71 spatiotemporally distinct cell types and cell states, including subtypes of hair cells, supporting cells, spiral ganglion neurons, and cells of the stria vascularis at multiple time points during pre- and postnatal maturation (embryonic day 14 [E14] to postnatal day 7 [P7]; Figure 1D; Table S1). We also combined all hair cells, supporting cells, spiral ganglion neurons, and strial cells to determine shared molecular signatures among the subtypes of each of those categories (Figure 1D). We calculated correlations among the marker gene signatures from these 71 cell types as validation of the shared and specific molecular signatures between cell types (Figure S1; Table S1). Plotting the expression of the top differentially expressed genes in each cell type confirmed a high degree of specificity, as well as the reproducibility of cell-type-specific expression in closely related cell types (Figure 1D).
Figure 1. Marker genes for cochlear and utricular cell types derived by scRNA-seq integration.

(A) Anatomical organization of transcriptionally defined cell types in the mammalian cochlea and utricle.
(B) Cell types from P2 and P7 mouse utricle were identified by UMAP clustering of scRNA-seq expression data.
(C) Marker genes for the principal cell types obtained by UMAP clustering of the mouse utricle were identified.
(D) Robust marker genes for each cochlear and utricular cell type were defined by integration of multiple scRNA-seq datasets at specific developmental timepoints.5
HC, hair cell; SC, supporting cell; IPhC, inner phalangeal cell; IPC, inner pillar cell; OPC, outer pillar cell; GER, greater epithelial ridge; LER, lesser epithelial ridge; OS, outer sulcus; SGN, spiral ganglion neuron. Ube2c (ubiquitin-conjugating enzyme E2 C) and Oc90 (otoconin-90) are genes previously identified as markers for non-sensory epithelial cells.5
Notably, known markers for cochlear hair cells—many of which were originally identified based on protein abundance—are not always suitable for these analyses, either because their transcript levels are less cell-type specific or because they are also expressed in vestibular hair cells. For instance, a well-known marker of cochlear inner hair cells, VGlut3 (Slc17a8), was also highly expressed in type I and type II utricular hair cells. We concluded that our array of subtype- and cell-state-specific gene profiles would allow us to identify in vitro cell types with a high degree of specificity.
Single-cell analysis of organoids reveals cell clusters resembling multiple subtypes of epithelial cells in the postnatal cochlea
To assess gene expression and chromatin accessibility during the differentiation of Lgr5+ cells to hair cells, we sought to obtain epigenetic and transcriptional signatures of the cells. We analyzed the trajectories of individual cells in the organoids at the single-cell level at various times of differentiation by scRNA-seq, starting from the expanded inner ear progenitors (referred to here as day 0 cells) and differentiating them for 10 days in vitro (referred to here as day 10 cells). We compared the profiles of the differentiating postnatal supporting cells to the complex mosaic of sensory cells in the sensory epithelium. We generated scRNA-seq of a total of 67,162 cells from cochlear organoids at days 0 (4 samples) and 10 (2 samples) of differentiation. Louvain clustering revealed 11 transcriptionally distinct cell types (Figure 2A). As expected, most clusters were differentially abundant at days 0 vs. 10 of differentiation, indicating that differentiation resulted in substantial changes in cell composition. Examination of each cluster’s marker genes (Figure S2; Table S2) and comparison to canonical markers for in vivo cell types indicated that at both time points, organoids were primarily composed of epithelial cells (Epcam), with subsets expressing markers either of hair cells (Atoh1, Pou4f3, and Pvalb) or supporting cells (Figures 2B and 2C).
Figure 2. scRNA-seq characterization of in vitro cell types in cochlear organoids.

(A) Integrated scRNA-seq data from days 0 and 10 of organoid differentiation, labeled by sample and time point, were used to obtain UMAP clusters.
(B) Cells obtained at days 0 and 10 of differentiation are plotted onto the UMAP.
(C) Marker expression is plotted for the epithelial marker Epcam; supporting cell marker Hes1; cochlear interdental cell marker and vestibular supporting cell marker Otoa; and canonical hair cell marker Pou4f3.
(D) Distribution of epithelial markers across UMAP clusters with expression patterns of hair cells, interdental/supporting cells, and GER.
(E) Pairwise correlations of marker genes of in vitro clusters vs. in vivo cochlear and utricular cell types. Color intensity indicates Pearson correlations of cell-type specificity scores for up to 300 genes per cell type. For an interactive version of the trajectory analysis, see https://umgear.org/lgr5org.
Among the organoid clusters that lacked hair cell markers, five were composed primarily of day 0 cells (0, 1, 3, 4, and 9). Of these, clusters 0, 1, 3, and 4 were positively correlated with signatures for several in vivo subtypes of sensory and non-sensory epithelial populations, including greater epithelial ridge (GER), inner phalangeal, inner pillar, prosensory, and inner sulcus, while the signature for cluster 9 was more strongly correlated with mesenchymal cells. Importantly, most of the day 0 organoid cell clusters expressed Lgr5, as well as Notch effector genes such as Hes1 and Id3 (Figures 2C and 2D). The in vivo cell types that shared markers with these clusters also expressed Lgr5 and likely reflect the original cell types from which the organoids were derived. The four remaining non-hair cell clusters (2, 5, 6, and 10) were composed primarily of day 10 cells. The signatures of clusters 2, 5, and 6 were positively correlated with signatures for interdental cells, epithelial cells medial to the organ of Corti in the cochlear duct, and the signature of cluster 10 was correlated with the signatures for supporting cells—Deiters’ and inner pillar—and cells of the stria vascularis. As expected, from days 0 to 10, there was a substantial downregulation of Lgr5 and Notch effector pathways. Instead, day 10 clusters strongly upregulated markers for other epithelial populations in the inner ear, notably Otoa, a marker for interdental cells in the cochlea (Figures 2C and 2D). Overall, these results suggest that many of the cells in organoids retain transcriptional features of supporting cells. However, it is important to note that both the multi-gene signatures and the expression of top markers for in vivo cell types (Figure 2E) indicate that there is not a one-to-one correspondence between these supporting cell-like clusters in organoids, compared to subtypes of in vivo supporting cells and related epithelial populations.
Maturity and diversity of hair cells
Differential expression patterns of genes expressed in maturing hair cells at E14, E16, P1, and P7 suggested a hair cell maturation trajectory from organoid clusters 7–8 (Figure 2E). A differential gene expression analysis revealed upregulation of genes in clusters 7 and 8: mostly immature hair cell genes (embryonic and P1) in cluster 7 and more mature hair cell genes (P7 and older cochlear and vestibular hair cells) in cluster 8 (Figures 3A and 3B). None of the hair cells expressed genes of other Atoh1-dependent lineages such as cerebellar granular precursors, intestinal epithelial cells, or Merkel cells (Figure 3C).
Figure 3. Molecular characterization and pseudotemporal trajectories of hair cells in cochlear organoids.

(A) Volcano plot indicating genes differentially expressed between two clusters of hair cells in organoids: clusters 7 (primarily in day 0 organoids) and 8 (primarily in day 10 organoids).
(B) Aggregate expression of markers specifically expressed in E14, E16, P1, or P7 cochlear hair cells in clusters 7 and 8. Orange indicates high expression, whereas gray indicates low expression.
(C) Aggregate expression of genes expressed in cochlear and non-cochlear Atoh1-dependent cell lineages in clusters 7 and 8.
(D) Monocle pseudotime trajectory delineates transdifferentiation of Lgr5+ supporting cells (cluster 4) to hair cells (clusters 7 and 8). Trajectory labeled by cluster number (left) or pseudotime (right). For an interactive version of the trajectory analysis, see https://umgear.org/lgr5org.
(E) Expression of known marker genes with dynamic expression across pseudotime.
Next, we sought to model the trajectories by which Lgr5+ supporting cells transdifferentiate to hair cells, using pseudotime trajectory analysis of our scRNA-seq data. Using Monocle, we produced a non-branching trajectory from differentially expressed genes (Table S3) in clusters 4, 7, and 8 that followed cells from cluster 4 (the cluster with the highest expression of Lgr5) through cluster 7 (less mature hair cells) to cluster 8 (more mature hair cells) (Figure 3D). We speculated that the small number of hair cells in day 0 organoids arose through activation of hair cell differentiation programs, whereas more numerous and more mature hair cells arose during the 10 day course of organoid differentiation.
Examination of known markers for hair cell development revealed sequential patterns of activation. Lgr5 expression was high at the beginning of the trajectory and then declined to low levels as hair cells matured (Figure 3E). Atoh1, the master regulator of hair cell development, peaked in immature hair cells at the middle of the trajectory. Markers that continue to be expressed in more mature hair cells, including Gfi1, Pou4f3, Myo6, Myo7a, and Pvalb showed increased expression throughout the trajectory. As noted above, Sox2 was co-expressed with hair cell markers in the organoids; its expression was highest at the end of the pseudotime trajectory. Extending this analysis to additional genes with dynamic expression across pseudotime, we identified 6,523 dynamically expressed genes. Clustering these genes with BEAM revealed three gene co-expression clusters (Table S4).
To expand the analysis of clusters 7 and 8, we assessed the expression of hair cell genes that we have previously identified by immunostaining the organoids as part of the validation of the Lgr5+ organoids for hair cell differentiation studies.1 We examined multi-gene signatures and specific marker genes for subtypes of organoid-derived hair cells in all 11 clusters in relation to a list of hair cell-specific genes (Figure S3) derived from both our own and others’ results,6–9 and we reclustered the two hair cell-specific clusters to obtain further resolution (Figure S4). Our list included developmental and postnatal hair cell genes from both vestibular and cochlear hair cells. Although some of the specific markers of differentiated hair cells have low-abundance mRNA and are difficult to detect, the hair cell-specific genes annotated in Figure S3 were preferentially found in clusters 7 and 8. The canonical marker of immature hair cells, Atoh1, was expressed primarily in cluster 7, as were Fgf8 and Lmo1 (Lim domain only 1), both markers for developing and immature hair cells, while markers that continue to be expressed in mature hair cells, including Myo15, Myo7a, Espn, Pvalb, Pou4f3, and Gfi1, were most highly expressed in cluster 8 (Figures 2D and 2E). Surprisingly, some organoid-derived hair cells also expressed signatures of type I and type II utricular hair cells, suggesting that these cells had not assumed a distinct cochlear vs. vestibular identity.10 Vestibular-specific genes, such as Anxa4 and Calb2, were found in both clusters 7 and 8 but with a preference for cluster 7. Indeed, the genes in this list were mostly shared between the two clusters, and the distinctions between hair cell types could not be clearly made because of the overlap between markers and the low abundance of the message.
In summary, immature hair cells show some signs of maturing into distinct types expressing cochlear and vestibular markers. Two clusters—7 and 8—strongly expressed markers of developing, in vivo hair cells. These cells represent 1.6% of the cells in day 0 organoids and 12.4% in day 10 organoids. Based on vestibular and cochlear hair cell marker expression in the organoid cells (Figure S3) and a reclustering of the pooled clusters 7 and 8 (Figure S4), cluster 7 (49.9% of day 0 and 50.1% of day 10 cells) correlated most strongly with signatures of early cochlear hair cells and vestibular hair cells, whereas cluster 8 (99.7% of day 10 cells) correlated with more mature, but still early, cochlear hair cells.
Analysis of chromatin accessibility during hair cell differentiation
To confirm these gene dynamics, we generated mRNA-seq of bulk organoid cells at days 0, 2, 4, and 10 of differentiation (Figure 4A), as well as of P2 hair cells (Atoh1+) and supporting cells (Lgr5+ and Sox2+) (Figure 4B). Principal-component analysis on the day 0, 2, 4, and 10 samples confirmed that the largest component of variation separating the cells was the different time points (Figure 4A). Examination of known markers confirmed activation of hair cell marker genes during differentiation (e.g., Myo7a and Tmc1), accompanied by a decrease in Notch pathway genes (e.g., Notch3 and Jag1) and cell-cycle genes (e.g., Ccnc1, Birc5, and Cdk1; Figure 4C). K-means clustering of the bulk RNA-seq data revealed eight distinct expression patterns (Figure 4D; Table S5). Three of the eight bulk RNA-seq-derived patterns statistically overlapped the three transdifferentiation-related co-expression patterns derived from the scRNA-seq trajectory (groups 1, 3, and 4) (Table S6). Representative genes from these clusters include Ccnd1 and Lgr5 (group 1, decreasing expression during differentiation), Atoh1 and Pax2 (group 4, middle-onset expression), and Myo7a and VGlut3 (Slc17a8) (group 3, late-onset expression) (Figure 4E). Thus, the progression toward more mature hair cells could be seen in the bulk RNA-seq data, where Atoh1 expression decreased, while Myo7a expression continued to increase, over the course of the 10 days of differentiation (Figure 4).
Figure 4. Clustering analysis of bulk RNA-seq data from days 0, 2, 4, and 10.

(A) Principal-component analysis (PCA) of the day 0, 2, 4, and 10 bulk RNA-seq samples and their replicates.
(B) PCA of Atoh1+, Lgr5+, and Sox2+ cells and their replicates from P2 mouse cochlea.
(C) Expression of select genes in day 0, 2, 4, and 10 samples.
(D) K-means clustering of day 0, 2, 4, and 10 bulk RNA-seq samples, showing eight gene expression patterns.
(E) Expression of select genes in groups 1, 3, and 4 from (D).
Functional annotation of gene co-expression modules across datasets revealed biological processes that were robustly enriched at the early, middle, and late phases of transdifferentiation. Significant Gene Ontology (GO) terms (adjusted p value [p.adj] < 0.05) for the early gene co-expression modules included “negative regulation of Notch signaling pathway,” “regulation of inner ear auditory receptor cell differentiation,” and “regulation of mechanoreceptor differentiation.” Significant GO terms for the middle-onset module included “regulation of epithelial cell differentiation” (p = 4.01 × 10−5) and development-related terms. Significant GO terms for the late-onset module included “cilium movement,” “inner ear development,” “hair cell differentiation,” and “inner ear morphogenesis” (Table S7). These gene expression dynamics largely mirror hair cell development processes observed in vivo.
Gene regulatory networks underlying transdifferentiation
The transcriptional regulators involved in transdifferentiation of supporting cells to hair cells remain incompletely characterized. We predicted these regulators by reconstructing a gene regulatory network (GRN) model from our scRNA-seq and mRNA-seq data. Briefly, we applied a random forest GRN reconstruction algorithm, GENIE3,11 to predict target genes for transcription factors (TFs) using scRNA-seq data from clusters 4, 7, and 8. This resulted in a GRN model predicting the regulation of 17,849 genes by 1,024 TFs. To predict key regulators of transdifferentiation, we tested for the enrichment of GENIE3-derived regulons in the three modules (early-, middle-, and late-onset gene co-expression; Figure 4E; Table S6) observed in the bulk RNA-seq-derived patterns that overlapped the transdifferentiation-related co-expression patterns derived from the scRNA-seq trajectory. A total of 67 TFs were reproducibly enriched (p < 0.05) in modules derived from both scRNA-seq and bulk RNA-seq data (Figure 5A). Next, these 67 TFs were used as input to reconstruct a dynamical TF-to-TF GRN along pseudotime using SCODE12 (Figures S5 and 5B). TFs with the most predicted targets in this network include known regulators of hair cell differentiation (e.g., Atoh1, Pou4f3, and Lmo1), as well as TFs that have not previously been implicated in this process (e.g., Ddit3, Basp1, Tcf4, and Sox4).
Figure 5. Gene regulatory network of transcription factors that drive the transdifferentiation of Lgr5+ cochlear progenitor cells to hair cells.

Each TF is colored by its activity (A) in the pseudotime course from Figure 3E, and its size in the diagram (B) reflects the magnitude of its outdegree.
Chromatin accessibility changes associated with transdifferentiation of supporting cells to hair cells in cochlear organoids
We performed assay for transposase-accessible chromatin (ATAC)-seq in cochlear organoids at days 0, 2, and 10 of differentiation to characterize changes in chromatin accessibility during the transdifferentiation of supporting cells to hair cells (n = 2 biological replicates per condition). For comparison, we also generated ATAC-seq of sorted Sox2+ and Lgr5+ supporting cells and Atoh1+ hair cells from in vivo mouse cochlea at postnatal day 1. Peak calling with model-based analysis of chromatin immunoprecipitation sequencing (MACS) identified 44,540 chromatin accessibility peaks that were reproducible across two or more samples (Figure S6; Table S8). 15,123 of these peaks showed at least a nominally significant change in accessibility across groups (one-way ANOVA, p < 0.05). We used these data to explore gene regulatory mechanisms underlying transdifferentiation, focusing on TFs implicated in our gene-expression-based GRN model.
First, we considered the patterns of chromatin accessibility at the promoters of predicted key regulator TFs. The promoter regions for many of these TFs were differentially accessible during transdifferentiation, with varying dynamics. As expected, the promoters of Atoh1 and Pou4f3 were accessible in Atoh1+ sorted hair cells from the in vivo cochlea. The Atoh1 promoter and enhancer were never fully closed in Lgr5+ or Sox2+ supporting cells or day 0 organoids. Accessibility of the Atoh1 enhancer in the organoids increased at day 2 and decreased back to near-basal levels at day 10, concordant with the expression of Atoh1 (Figure 4E), whereas the Pou4f3 promoter became accessible primarily at day 10 (Figures 6A and 6B). By contrast, the promoter regions of several TFs predicted to regulate the early stages of transdifferentiation, including Sox9 and Atf3, showed decreased accessibility from days 0 to 10, as well as in Atoh1+ hair cells vs. Lgr5+ and Sox2+ supporting cells (Figures 6D and 6E). In addition, the promoters of some TFs with dynamic gene expression across transdifferentiation had similar chromatin accessibility throughout this process (e.g., Tcf4; Figure 6C). These results suggest that transdifferentiation involves cis-acting changes in the chromatin states of genes encoding key regulator TFs.
Figure 6. Chromatin accessibility dynamics during transdifferentiation in cochlear organoids.

(A–E) Chromatin accessibility at the promoters of the known and predicted key regulator TFs Atoh1 (A), Pou4f3 (B), Tcf4 (C), Sox9 (D), and Atf3 (E).
(F and G) Average accessibility pattern for peaks within clusters 3 (F) and 1 (G) and enrichment of peaks for sequence motifs recognized by key regulator TFs with dynamic expression in the RNA-based gene regulatory network model.
Next, we characterized trans-acting effects of these TFs on chromatin accessibility. Using k-means clustering, we identified 12 patterns of chromatin co-accessibility across our in vitro and in vivo data (Table S8). Several of these patterns describe increasing or decreasing chromatin accessibility across transdifferentiation in vitro, accompanied by concordant differences of chromatin accessibility in hair cells vs. supporting cells in vivo. For instance, peaks in cluster 3 were upregulated at day 10 of transdifferentiation, as well as in Atoh1+ hair cells (Figure 6F), while peaks in cluster 1 were downregulated at day 10, as well as in Atoh1+ hair cells (Figure 6G). Sequence motif enrichment analysis with HOMER13 predicted TFs that may regulate these patterns (Figure S6; Table S9). Importantly, clusters characterized by dynamic changes in chromatin accessibility across transdifferentiation were enriched for motifs recognized by several of the key regulator TFs from our GRN model, providing independent validation. Specifically, cluster 3 (upregulated in hair cells) was enriched for octamer motifs recognized by Pou4f3, as well as E-box motifs recognized by Atoh1 and Tcf4 (Figure 6F). Cluster 1 (downregulated) was enriched for SRY-box motifs recognized by Sox family TFs, as well as motifs recognized by several activity-dependent factors such as Atf3, Jun, and Junb (Figure 6G). Changes in chromatin accessibility governed by key regulator TFs with dynamic expression may therefore regulate the dynamic activity of thousands of enhancers and promoters in this context.
DISCUSSION
A capacity for transdifferentiation of sensory epithelial supporting cells to hair cells allows the chick to regenerate hair cells in the deafened cochlea. Although cochlear sensory epithelial supporting cells in the adult mammal lack the capacity for regeneration, the cells show the capacity to differentiate into hair cells in the early postnatal period.14–16 Deciphering the signals for reprogramming of mammalian cochlear supporting cells to hair cells would be an important step toward therapies for hair cell regeneration as a treatment for deafness. Here, utilizing an established protocol,1 we generated cochlear organoids from murine Lgr5+ progenitor cells and performed a comprehensive molecular characterization at multiple time points in their differentiation by scRNA-seq, bulk RNA-seq, and ATAC-seq. Transcriptional signatures of maturing hair cells were apparent after 10 days of organoid differentiation, and during the course of differentiation, the cells mimicked nearly all subtypes of supporting cells and hair cells in the newborn cochlea. From these data, we reconstructed a gene regulatory model to gain insight into the transcriptional and epigenetic programs that drive the differentiation of Lgr5+ progenitor cells to a hair cell fate.
Clustering of the cells in the uniform manifold approximation and projection (UMAP) allowed us to identify several groups of cells that were derived largely from the day 0 time point of organoid differentiation and were related to supporting cells and the surrounding epithelium in the cochlear duct. Tracing their lineage to the hair cell clusters showed that cluster 4, which expressed Lgr5 and Sox2, was the primary source of hair cells and allowed us to identify genes in their trajectory from supporting cell to hair cell. The cells at the start of the differentiation protocol correlated best with in vivo inner and outer pillar, inner phalangeal, Deiters’, prosensory, GER, and inner sulcus cells. Cells made by a variant of this protocol were largely comprised of GER cells,17 and organoids grown from human cochlear cells also expressed markers of both supporting cells and hair cells.3,18 Cells at the end of the differentiation protocol correlated best with hair cells.
We validated these networks across transcriptional and epigenomic datasets. Dynamic changes in TF expression were confirmed across bulk RNA-seq and scRNA-seq datasets and in many cases were accompanied by changes in the accessibility of each TF’s promoter. Trans-acting effects of these same TFs on downstream target genes were predicted from TF-gene co-expression, as well as by the enrichment of their sequence motifs in networks of co-accessible chromatin regions. We also integrated the in vitro data with six previous studies of cochlear and utricular cell types and with newly generated in vivo data from intact cochlea and utricle.
We present here a thorough database of robust marker genes for cell types of the cochlea and the utricle, which allowed us to identify cell types involved in the in vitro organoid differentiation protocol. We also present gene expression dynamics during the organoid differentiation protocol, supported by scRNA-seq and bulk RNA-seq data. Our data integration analysis, which yielded robust cell-type-specific marker genes for various cochlear and utricular cell types, allowed a more precise characterization of cells involved in the organoid differentiation protocol. At later time points, hair cells from organoids expressed mixed transcriptional signatures for cochlear and vestibular subtypes. We show that the expanded Lgr5+ cells in the cochlear organoids have the capacity to differentiate into cochlear and vestibular hair cell types spanning an early developmental to postnatal range of maturation. This could be explained by the presence of cochlear and vestibular progenitor cells in the expanded organoids used for differentiation or by the presence in the expanded organoids of a common progenitor that can give rise to both lineages. Further resolution of the progenitors and the resulting hair cells will be required to distinguish between these possibilities.
We previously demonstrated that organoid differentiation yielded cells expressing key hair cell markers, including those of inner and outer hair cells. Our analysis demonstrates that the hair cells reach maturity comparable to in vivo postnatal day 7. Our model identified known regulators of hair cell development, including Atoh1, Pou4f3, and Gfi1. It also predicted roles in postnatal hair cell differentiation for the hair cell-expressed genes Sox4, Tceb2, Nr2f1, and Lmo1. Their expression is consistent with previous findings on these genes. Sox4 restores supporting cell proliferation and hair cell production after hair cell loss,19 Tceb2 is expressed in cochlear hair cells,8 and Nr2f1 (COUP-TFI) knockout results in a significant increase in hair cell number through misregulation of Notch signaling components, including Jag1, and Hes5.20 Lmo1 is a transcriptional regulator that contains two cysteine-rich LIM domains but lacks a DNA-binding domain. Lmo1 is specifically expressed in vestibular and cochlear hair cells.21
Our exploration of the transcriptional network also elucidated the regulation of genes expressed during supporting cell differentiation to hair cells. Ddit3, a cell-cycle-related gene corresponding to endoplasmic reticulum stress, was significantly upregulated. Ddit3 is a member of the CCAAT/enhancer-binding protein (C/EBP) family of TFs22 and acts as a dominant-negative inhibitor by forming heterodimers with other C/EBP members and as an inhibitor of the canonical Wnt signaling pathway by binding to Tcf7l2, impairing its DNA-binding properties and repressing its transcriptional activity.23 Ddit3 loss, moreover, contributes to hearing loss.24 Our network reconstruction revealed that Ddit3 had a high outdegree during differentiation, was most active toward the end of the maturation process, and acted similarly to Pou4f3, Atoh1, and Lmo1 in repressing Hes1 and activating transcription of Wnt-signaling-related genes such as Sox4, Tceb2, Jun, and Junb.22,23 Another TF identified in our transcriptional network, Atf4, is a downstream target of Ddit3. It regulates expression of genes involved in endoplasmic reticulum function, reactive oxygen species production, and cell death.25 Among Atf4-target genes is the C/EBP homologous protein CHOP/GADD153,26 which inhibits canonical Wnt signaling by interfering with binding of β-catenin to its interaction partners.
Our analysis also predicts regulatory factors such as Tcf4, the E-protein and heterodimerization partner of Atoh1. Tcf4 interacts with Atoh1 to induce neural differentiation27 but has not been reported in hair cell maturation literature and was not considered essential, as Atoh1 can interact with other E-proteins depending on cell context.27 Its high level of connection with other network genes indicates an important role in the control of hair cell differentiation. Tcf4 mutations are causal for Pitt-Hopkins syndrome,28,29 which is thought to be due to incomplete maturation or an absence of cortical neurons. Tcf4 recruitment coincides with areas of high transcriptional activity as shown by the occurrence of H3K27Ac marks in regions adjacent to Tcf4 binding.28 This suggests a potentially fundamental role of a known TF and interaction partner with Atoh1.
The progenitors are heterogeneous and consist of cells from GER as well as inner pillar and 3rd Deiters’ and other supporting and non-supporting cells that expand in the GSK3β inhibitor. The results show that Lgr5+ cells act as progenitors to hair cells. They are reprogrammed to hair cells by the combined activity of the γ-secretase inhibitor and the GSK3β inhibitor to inhibit Notch and activate Wnt, which stimulated the expression of several TFs and modeled in vivo postnatal progenitor cell differentiation to hair cells.14,30–33 Their molecular trajectories adhered to the same overall steps as postnatal supporting cells and allowed a determination of epigenetic and transcriptional steps in their reprogramming to hair cells.
Limitations of the study
The depth of the single-cell analysis of gene expression was not sufficient to determine the precise nature of the type I and II hair cells or inner and outer hair cells obtained. Identification of cell types was therefore based on cell-specific markers with sufficient expression for detection in our analysis. We are hoping in future studies to obtain sufficient cells to resolve the progenitors and the key steps in their trajectories to these various hair cell types.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Albert Edge.
Materials availability
The study did not generate new unique reagents.
Data and code availability
The bulk RNA-seq, scRNA-seq, and ATAC-seq data from this study are available on GEO (accession numbers GSE132635, GSE137299, GSE172327, GSE71982, and GSE136196) and in an interactive version on gEAR at umgear.org/Lgr5org (Figure S7).39
This paper does not report original code.
Additional information is available upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cells from the organ of Corti were prepared1 from Atoh1-nGFP,40 Sox2-GFP,41 or Lgr5-GFP42 mice. Inner ear organoids were made by expanding cochlear sensory epithelial cells from newborn mice.
All animal care and procedures complied with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Mouse housing and animal procedures were approved by the Massachusetts Eye and Ear Institutional Animal Care and Use Committee and performed.
We took all necessary steps to minimize animal suffering, including the use of anesthesia and analgesia during surgical procedures and careful monitoring of animal health and welfare. Both male and female animals were included in all analyses.
METHOD DETAILS
Expansion and differentiation of Lgr5+ cochlear progenitor cells
The organ of Corti was dissected in medium supplemented with growth factors and expanded in GSK3β inhibitor, CHIR, and HDAC inhibitor, VPA, which results in the growth of Lgr5+ cochlear hair cell progenitors. Organoids were induced to differentiate in GSK3β inhibitor, CHIR, and γ-secretase inhibitor, LY411575. Generation of hair cells was assayed by flow cytometry for GFP, which relies on Atoh1 enhancer activation in these transgenic mice.40
RNA-sequencing
Organoids were examined by RNA-seq at 4 time points: the start (D0), the early (D2), middle (D4) and late (D10) stages of differentiation. RNA was isolated by published procedures.31,43 The cells at these time points were compared to sorted cells prepared at P2 from Atoh1-nGFP, Sox2-GFP,41 and Lgr5-GFP42 mice, corresponding to hair cells, supporting cells, and the Lgr5+ subset of supporting cells. RNA quality was confirmed, and cDNA synthesis, and library preparation carried out using 2 ng of sheared cDNA. Illumina NextSeq500 Single-End 75 bp (SE75) sequencing was performed to provide an estimated coverage of 20–30 million single-end reads per sample.31
ATAC-sequencing
Concurrently we performed ATAC-seq to query chromatin accessibility under these conditions. We performed these experiments using NextSeq500 Paired-End 40 bp (PE40) sequencing (Dana-Farber Cancer Institute Core). Initial data analysis, including alignment to the mouse genome (Mm10) was performed using Bowtie2 and peak-calling using MACS v2.1.35 Experiments were repeated at least 3 times for each condition. Reads were counted within reproducible peaks and normalized to library size with DiffBind.36 Normalized read counts were log-transformed, and a linear batch effect between replicates was regressed out. Peaks with variable accessibility across groups were calculated using one-way ANOVA. We then applied k-means clustering (k = 30) to the normalized, log-transformed, and batch-corrected read counts of variable peaks. Peaks that were insufficiently correlated with the average expression within each cluster (r < 0.7) were removed, and clusters with strongly correlated average expression (r > 0.85) were combined, resulting in 12 merged clusters. Motif enrichment analysis was performed with HOMER13 using default parameters, comparing the peaks within each cluster to the background of all reproducible peaks in our dataset. Motifs were assigned to the TFs for which they are named, as well as to TFs with similar DNA-binding domains in the TFClass database.44
Single cell RNA-seq of organoids
scRNA-seq of the organoids was performed at D0 and D10 of differentiation to follow gene expression at the single cell level. Organoids were prepared from 6 to 12 newborn ears of both sexes, and more than 5,000 cells were collected for analysis using the 10X Genomics droplet-based single-cell sequencing platform. The cell suspension was diluted to a concentration of 500 cells per mL and immediately captured, lysed, and primed for reverse transcription (RT) using the high throughput, droplet microfluidics Gemcode platform from 10X Genomics with v2 chemistry. Each droplet on the Gemcode co-encapsulates a cell and a gel bead that is hybridized with oligo(dT) primers encoding a unique cell barcode and unique molecular identifiers (UMIs) in lysis buffer. After capture for 6 min on gel beads, the transcriptomes were pooled and reverse transcribed to cDNA. Cell barcodes and UMIs were employed, after sequencing to demultiplex the originating cell and mRNA transcript from the pooled and PCR amplified cDNA. RT-PCR amplification of cDNA, and preparation of a library from 30 ends were conducted according to the manufacturer’s published protocol. We performed 14 cycles of PCR amplification of cDNA. The library was sequenced on an Illumina NovaSeq 6000 with an S2 100 cycle reagent kit at the Broad Institute Sequencing Facility.
Single cell RNA-seq of in vivo cells
scRNA-seq for postnatal day 2 and day 7 mouse utricle was performed as follow: 3 mice (CD-1 background) were euthanized and their temporal bone removed. Utricles were harvested and incubated in thermolysin (Sigma-Aldrich) for 20 min at 37°C. Thermolysin was then replaced with Accutase (Sigma-Aldrich) and the tissue incubated for 3 min at 37°C followed by mechanical dissociation until a single cell suspension was obtained. After inactivation of the Accutase with 5% fetal bovine serum, the cell suspension was filter through a 35μm nylon mesh and processed for scRNA-seq. Dissociated cells were captured into a Chromium Controller (10x Genomics) for droplet-based molecular barcoding. Library preparation was performed using the 10x Single Cell Gene Expression Solution. Libraries from two utricular samples were sequenced across three lanes of an Illumina HiSeq4000 sequencer to produce paired-end 75 bp reads.
Initial scRNA-seq data processing, including demultiplexing, alignment to the mouse genome (mm10), and read counting were performed with cellranger (10X Genomics). The number of genes expressed, the number of UMIs detected, and the percentage of mitochondrial and ribosomal RNA were calculated for quality control. Cells with >5% of UMIs from mitochondrial genes were discarded. We applied the Seurat v3 Standard Workflow37 to integrate cells across replicates, using 7,000 highly variable genes, 3000 anchors, and 50 dimensions. Subsequently, principal component analysis was performed, and cells clustered on a K-nearest neighbor graph based on Euclidean distance using the previously defined PCA dimensionality as the input. Cells were clustered using the Louvain algorithm to optimize the standard modularity function before performing dimensionality reduction via UMAP. Further analysis was performed by re-clustering selected sets of cells followed by differential gene expression analysis to identify unique cell markers.
Comparison of organoid cell clusters to in vivo cell types
Cell clusters in organoids were compared to cell types in the in vivo mouse cochlea based on correlations among shared marker genes. A cell type specificity score was defined for each gene in each cluster.45 Briefly, for each gene detected in >50% of cells from a cluster, we multiplied its enrichment (log2 fold change) by its specificity (percent of relevant cluster expressing the gene/percent of other clusters expressing the gene) to yield a specificity score within each cluster. Specificity scores were calculated for each organoid cell cluster, as well as for each cell type in the mouse cochlea, based on scRNA-seq of E14, E16, P1, and P7 cochlear cell types.5 We then used Pearson’s correlations to quantify the similarity of marker genes for each in vivo vs. in vitro cell cluster.
In addition, we used the projectR R package34 to “score” each cell for the expression of marker genes in each in vivo cell type. For each in vivo cochlear cell type, we defined a set of specifically expressed markers (>50% non-zero counts; p value <0.05). Cells in organoids were then scored based on the combined expression of each set of marker genes, using the projectR function. Similarly, cells in organoids were compared to other cell types that share an Atoh1 lineage, defining scores based on genes specifically expressed in gut cells, Merkel cells, cerebellar granule cell progenitor cells, and hair cells.10
Marker genes for each cell type
By integrating publicly available scRNA-seq data from cochlear and utricular cell types, we derived marker genes for each cell type. Utricular hair cells and cochlear hair cells and neurons were integrated into one expression matrix, and utricular supporting cells, cochlear supporting cells, and stria vascularis cells were integrated into a separate expression matrix. Marker genes were derived by performing differential gene expression analysis on each cell type against all others in that matrix, using the Seurat R package, and calculating a specificity score for each gene.5 Genes with a specificity score for the relevant cell type and either 0 for other cell types or a very low score for other cell types (as necessary for type 1A-C SGNs) were selected as marker genes.
Monocle trajectory construction
We used Monocle, a widely recognized tool for capturing cell lineage trajectories in scRNA-seq data, to reconstruct a pseudotime trajectory to elucidate the gene expression changes during the differentiation of organoids into hair cells.
We focused our analysis on the organoid cluster most similar to greater epithelial ridge and with the highest expression of Lgr5 (cluster 4), as well as the two clusters representing different stages of hair cell development (clusters 7 and 8). Cluster 7 represents less mature hair cells, while cluster 8 consists of more mature hair cells. Trajectory analysis was performed using Monocle version 2.14.0. Cells were ordered along a pseudotime trajectory using Monocle’s orderCells function. This function utilizes differential expression analysis to arrange cells along a pseudotime axis. We used the top 375 differentially expressed genes with a minimum percentage (min.pct) cutoff of 0.25 for each cluster. The number of included differentially expressed genes was restricted to the top 375 to reduce noise from genes that do not exhibit meaningful changes during differentiation. The differentialGeneTest function (fullModelFormulaStr = “sm.ns(Pseudotime)”) was used to calculate the significance of each gene’s expression change over pseudotime. Genes with a q-value <0.01 and detected in at least 200 cells were retained for clustering in pseudotime. With this filter, we aim to focus on genes that are most likely to play an important role in hair cell development.
GO term enrichment
GO term enrichment on the genes supported by the bulk RNA-seq clusters and the pseudotime-derived scRNA-seq clusters was performed using clusterProfiler R package.34 GO terms with a BH adjusted p value <0.05 were reported.
Gene regulatory network reconstruction
A gene regulatory network for the differentiation of organoids to hair cells was derived from scRNA-seq data. As with trajectory analysis, we selected clusters 4, 7, and 8 for this analysis. First, GENIE311 was used to predict target genes for each of 1,186 TFs, the subset of TFs with expression in these cells from a list of 1675 mouse TFs from http://genome.gsc.riken.jp/TFdb/data/tf.name. The GENIE3 output was a list of TFs and their predicted target genes (regulons). We used hypergeometric tests to identify TFs whose predicted target genes were over-represented in each gene co-expression cluster from pseudotime analysis and bulk RNA-seq. We further reconstructed a dynamical model for TF-to-TF interactions during hair cell differentiation using SCODE, which implements an ordinary differential equation model using pseudotime. For this analysis, we selected 58 TFs whose targets were over-represented within gene co-expression clusters whose expression peaked early, middle, or late in hair cell differentiation, consistently in our bulk and single-cell datasets.
QUANTIFICATION AND STATISTICAL ANALYSIS
We applied the Seurat v3 Standard Workflow to integrate cells across replicates. Peaks with variable accessibility across groups were calculated using one-way ANOVA. We applied k-means clustering (k = 30) to the normalized, log-transformed, and batch-corrected read counts of variable peaks. Peaks that were insufficiently correlated with the average expression within each cluster (r < 0.7) were removed, and clusters with strongly correlated average expression (r > 0.85) were combined, resulting in 12 merged clusters. The differentialGeneTest function (fullModelFormulaStr = “sm.ns(Pseudotime)”) was used to calculate the significance of each gene’s expression change over pseudotime.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit polyclonal anti-myosin-7a | Proteus Biosciences | #25–6790; RRID:AB_10015251 |
| Mouse monoclonal anti-Sox2 | Santa Cruz Biotechnology | #sc-365823; RRID:AB_10842165 |
| Rabbit polyclonal anti-neurofilament 200 | Sigma | #N4142; RRID:AB_477272 |
| Donkey anti-rabbit IgG, Alexa Fluor 546 | Thermo Fisher Scientific | # 2534016; RRID:AB_2534016 |
| Donkey anti-mouse IgG, Alexa Fluor 488 | Thermo Fisher Scientific | #141607; RRID:AB_141607 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Alexa Fluor 647 Phalloidin | Invitrogen | # 2620155 |
| Paraformaldehyde | EMS | #15710 |
| DAPI | Thermo Fisher Scientific | #2629482 |
| Mounting Media (Citifluor CFM-3) | EMS | #17979–30 |
| Recombinant human FGF | Peprotech | #100–18 |
| Recombinant human IGF-1 | R&D | #291-G1–200 |
| Laminin from human placenta | Sigma | #L6274 |
| ACCUTASE | Innovative Cell Technology | #AT 104 |
| DMEM-F12 basal medium | Gibco | #11320033 |
| N2 | ThermoFisher Scientific | #17502048 |
| Phospho-vitamin C | Sigma | #49752 |
| Matrigel | Corning | #356234 |
| Trypsin-EDTA | GIBCO | #25200–056 |
| Phosphate-buffered saline | GIBCO | #10010 |
| Polybrene Infection/Transfection Reagent | Sigma | #TR-1003-G |
| Human Wnt3a | R&D | #5036-GMP-010 |
| Antibiotics (Penicillin-Streptomycin) | Gibco | #15–140-163 |
| Antifungal (Amphotericin B) | Gibco | #15290018 |
| BIX01294 | Sigma | #B9311 |
| CHIR99021 | LC Labs | #C-6556 |
| LY411575 | Sigma | #SML0506 |
| VPA | Sigma | #P4543 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Chromium Single Cell 50 Library | 10x Genomics | #PN-1000006 |
| Chromium Next GEM Single Cell | 10x Genomics | #PN-1000263 |
|
| ||
| Deposited data | ||
|
| ||
| Organoids D0, D2, D4, D10 | This paper | GSE132635 |
| Inner hair cells P1, P7 | This paper | GSE137299 |
| Outer hair cells P1, P7 | This paper | GSE137299 |
| Type I HCs P7, P12, P100 | This paper | GSE115934 |
| Type II HCs P7, P12, P100 | This paper | GSE115934, GSE172327 |
| Type 1 SGNs P63, P25–27 | This paper | GSE114759, GSE114997 |
| Type 2 SGNs P63, P25–27 | This paper | GSE114759, GSE114997 |
| Hensen’s E16, P1 | This paper | GSE137299 |
| Inner pillar E16, P1, P2, P7 | This paper | GSE137299, GSE172327 |
| Outer pillar P1, P2, P7 | This paper | GSE137299 |
| Inner phalangeal E16, P1, P2, P7 | This paper | GSE137299 |
| Claudius E16, P1 | This paper | GSE137299 |
| Deiters’ row 1,2 P1, P2, P7 | This paper | GSE137299 |
| Deiters’ row 3 P1, P2, P7 | This paper | GSE137299 |
| Utricular SCs P1, P2 | This paper | GSE71982, GSE172327 |
| Marginal P30 | This paper | GSE136196 |
| Intermediate P30 | This paper | GSE136196 |
| Basal P30 | This paper | GSE136196 |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | Fiji | http://fiji.sc/ |
| Prism Version 8.4.1 | GraphPad | https://www.graphpad.com/features |
| Cell Ranger Version 2.1.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads |
| ProjectR R Package | Yu et al.34 | https://www.bioconductor.org/packages/release/bioc/html/projectR.html |
| MACS version 2.1 | Zhang et al.35 | https://pypi.org/project/MACS2/ |
| DiffBind | Ross-Innes et al.36 | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| HOMER | Heinz et al.13 | http://homer.ucsd.edu/homer/motif/ |
| Seurat v3 Standard Workflow | Butler et al.37 | https://satijalab.org/seurat/ |
| Monocle Version 2.14.0 | Trapnell et al.38 | http://cole-trapnelllab.github.io/monocle-release/monocle3/ |
| GENIE3 | Huynh-Thu et al.11 | https://bioconductor.org/packages/release/bioc/html/GENIE3.html |
Highlights.
Differentiation of Lgr5+ cochlear progenitors to hair cells in organoids
Transcriptional program for postnatal differentiation of hair cells
Differentiation of vestibular and cochlear hair cells
Gene expression compared to developing, newborn, and mature in vivo cells
ACKNOWLEDGMENTS
We thank the Dana-Farber Genomics Core, the Harvard Chan Bioinformatics Core, and the Broad Institute Sequencing Facility for technical assistance and the NIH (grant DC014089), Steven and Deborah Barnes, and the Hearing Restoration Project of the Hearing Health Foundation for funding.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113421.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.McLean WJ, Yin X, Lu L, Lenz DR, McLean D, Langer R, Karp JM, and Edge ASB (2017). Clonal Expansion of Lgr5-Positive Cells from Mammalian Cochlea and High-Purity Generation of Sensory Hair Cells. Cell Rep. 18, 1917–1929. 10.1016/j.celrep.2017.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clevers H. (2016). Modeling Development and Disease with Organoids. Cell 165, 1586–1597. 10.1016/j.cell.2016.05.082. [DOI] [PubMed] [Google Scholar]
- 3.Roccio M, and Edge ASB (2019). Inner ear organoids: new tools to understand neurosensory cell development, degeneration and regeneration. Development 146, dev177188. 10.1242/dev.177188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lenz DR, Gunewardene N, Abdul-Aziz DE, Wang Q, Gibson TM, and Edge ASB (2019). Applications of Lgr5-Positive Cochlear Progenitors (LCPs) to the Study of Hair Cell Differentiation. Front. Cell Dev. Biol 7, 14. 10.3389/fcell.2019.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kolla L, Kelly MC, Mann ZF, Anaya-Rocha A, Ellis K, Lemons A, Palermo AT, So KS, Mays JC, Orvis J, et al. (2020). Characterization of the development of the mouse cochlear epithelium at the single cell level. Nat. Commun 11, 2389. 10.1038/s41467-020-16113-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burns JC, Kelly MC, Hoa M, Morell RJ, and Kelley MW (2015). Single-cell RNA-Seq resolves cellular complexity in sensory organs from the neonatal inner ear. Nat. Commun 6, 8557. 10.1038/ncomms9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.García-Añoveros J, Clancy JC, Foo CZ, García-Gómez I, Zhou Y, Homma K, Cheatham MA, and Duggan A. (2022). Tbx2 is a master regulator of inner versus outer hair cell differentiation. Nature 605, 298–303. 10.1038/s41586-022-04668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu H, Pecka JL, Zhang Q, Soukup GA, Beisel KW, and He DZZ (2014). Characterization of transcriptomes of cochlear inner and outer hair cells. J. Neurosci 34, 11085–11095. 10.1523/JNEUROSCI.1690-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McInturff S, Burns JC, and Kelley MW (2018). Characterization of spatial and temporal development of Type I and Type II hair cells in the mouse utricle using new cell-type-specific markers. Biol. Open 7, bio038083. 10.1242/bio.038083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menendez L, Trecek T, Gopalakrishnan S, Tao L, Markowitz AL, Yu HV, Wang X, Llamas J, Huang C, Lee J, et al. (2020). Generation of inner ear hair cells by direct lineage conversion of primary somatic cells. Elife 9, e55249. 10.7554/eLife.55249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huynh-Thu VA, Irrthum A, Wehenkel L, and Geurts P. (2010). Inferring regulatory networks from expression data using tree-based methods. PLoS One 5, e12776. 10.1371/journal.pone.0012776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsumoto H, Kiryu H, Furusawa C, Ko MSH, Ko SBH, Gouda N, Hayashi T, and Nikaido I. (2017). SCODE: an efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics 33, 2314–2321. 10.1093/bioinformatics/btx194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bramhall NF, Shi F, Arnold K, Hochedlinger K, and Edge ASB (2014). Lgr5-positive supporting cells generate new hair cells in the postnatal cochlea. Stem Cell Rep. 2, 311–322. 10.1016/j.stemcr.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cox BC, Chai R, Lenoir A, Liu Z, Zhang L, Nguyen DH, Chalasani K, Steigelman KA, Fang J, Rubel EW, Cheng AG, and Zuo J. (2014). Spontaneous hair cell regeneration in the neonatal mouse cochlea in vivo. Development 141, 816–829. 10.1242/dev.103036141/4/816 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu L, Lu J, Chiang H, Wu H, Edge ASB, and Shi F. (2016). Diphtheria Toxin-Induced Cell Death Triggers Wnt-Dependent Hair Cell Regeneration in Neonatal Mice. J. Neurosci 36, 9479–9489. 10.1523/JNEUROSCI.2447-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kubota M, Scheibinger M, Jan TA, and Heller S. (2021). Greater epithelial ridge cells are the principal organoid-forming progenitors of the mouse cochlea. Cell Rep. 34, 108646. 10.1016/j.celrep.2020.108646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roccio M, Perny M, Ealy M, Widmer HR, Heller S, and Senn P. (2018). Molecular characterization and prospective isolation of human fetal cochlear hair cell progenitors. Nat. Commun 9, 4027. 10.1038/s41467-018-06334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gnedeva K, and Hudspeth AJ (2015). SoxC transcription factors are essential for the development of the inner ear. Proc. Natl. Acad. Sci. USA 112, 14066–14071. 10.1073/pnas.1517371112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang LS, Alger HM, and Pereira FA (2006). COUP-TFI controls Notch regulation of hair cell and support cell differentiation. Development 133, 3683–3693. 10.1242/dev.02536. [DOI] [PubMed] [Google Scholar]
- 21.Deng M, Pan L, Xie X, and Gan L. (2006). Differential expression of LIM domain-only (LMO) genes in the developing mouse inner ear. Gene Expr. Patterns 6, 857–863. 10.1016/j.modgep.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 22.Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, and Majsterek I. (2016). The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med 16, 533–544. 10.2174/1566524016666160523143937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hennig KM, Fass DM, Zhao WN, Sheridan SD, Fu T, Erdin S, Stortchevoi A, Lucente D, Cody JD, Sweetser D, et al. (2017). WNT/beta-Catenin Pathway and Epigenetic Mechanisms Regulate the Pitt-Hopkins Syndrome and Schizophrenia Risk Gene TCF4. Mol. Neuropsychiatry 3, 53–71. 10.1159/000475666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng C, Wang Y, Guo L, Lu X, Zhu W, Muhammad W, Zhang L, Lu L, Gao J, Tang M, et al. (2019). Age-related transcriptome changes in Sox2+ supporting cells in the mouse cochlea. Stem Cell Res. Ther 10, 365. 10.1186/s13287-019-1437-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie MX, Cao XY, Zeng WA, Lai RC, Guo L, Wang JC, Xiao YB, Zhang X, Chen D, Liu XG, and Zhang XL (2021). ATF4 selectively regulates heat nociception and contributes to kinesin-mediated TRPM3 trafficking. Nat. Commun 12, 1401. 10.1038/s41467-021-21731-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horndasch M, Lienkamp S, Springer E, Schmitt A, Pavenstädt H, Walz G, and Gloy J. (2006). The C/EBP homologous protein CHOP (GADD153) is an inhibitor of Wnt/TCF signals. Oncogene 25, 3397–3407. 10.1038/sj.onc.1209380. [DOI] [PubMed] [Google Scholar]
- 27.Mulvaney J, and Dabdoub A. (2012). Atoh1, an essential transcription factor in neurogenesis and intestinal and inner ear development: function, regulation, and context dependency. J. Assoc. Res. Otolaryngol 13, 281–293. 10.1007/s10162-012-0317-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quevedo M, Meert L, Dekker MR, Dekkers DHW, Brandsma JH, van den Berg DLC, Ozgür Z, van IJcken WFJ, Demmers J, Fornerod M, and Poot RA (2019). Mediator complex interaction partners organize the transcriptional network that defines neural stem cells. Nat. Commun 10, 2669. 10.1038/s41467-019-10502-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teixeira JR, Szeto RA, Carvalho VMA, Muotri AR, and Papes F. (2021). Transcription factor 4 and its association with psychiatric disorders. Transl. Psychiatry 11, 19. 10.1038/s41398-020-01138-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mizutari K, Fujioka M, Hosoya M, Bramhall N, Okano HJ, Okano H, and Edge ASB (2013). Notch inhibition induces cochlear hair cell regeneration and recovery of hearing after acoustic trauma. Neuron 77, 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samarajeewa A, Lenz DR, Xie L, Chiang H, Kirchner R, Mulvaney JF, Edge ASB, and Dabdoub A. (2018). Transcriptional response to Wnt activation regulates the regenerative capacity of the mammalian cochlea. Development 145, dev166579. 10.1242/dev.166579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi F, Hu L, and Edge ASB (2013). Generation of hair cells in neonatal mice by beta-catenin overexpression in Lgr5-positive cochlear progenitors. Proc. Natl. Acad. Sci. USA 110, 13851–13856. 10.1073/pnas.1219952110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi F, Kempfle JS, and Edge ASB (2012). Wnt-responsive lgr5-expressing stem cells are hair cell progenitors in the cochlea. J. Neurosci 32, 9639–9648. 10.1523/JNEUROSCI.1064-12.201232/28/9639 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Orvis J, Gottfried B, Kancherla J, Adkins RS, Song Y, Dror AA, Olley D, Rose K, Chrysostomou E, Kelly MC, et al. (2021). gEAR: Gene Expression Analysis Resource portal for community-driven, multi-omic data exploration. Nat. Methods 18, 843–844. 10.1038/s41592-021-01200-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lumpkin EA, Collisson T, Parab P, Omer-Abdalla A, Haeberle H, Chen P, Doetzlhofer A, White P, Groves A, Segil N, and Johnson JE (2003). Math1-driven GFP expression in the developing nervous system of transgenic mice. Gene Expr. Patterns 3, 389–395. [DOI] [PubMed] [Google Scholar]
- 41.Arnold K, Sarkar A, Yram MA, Polo JM, Bronson R, Sengupta S, Seandel M, Geijsen N, and Hochedlinger K. (2011). Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 9, 317–329. 10.1016/j.stem.2011.09.001S1934-5909(11)00432-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, and Clevers H. (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007. [DOI] [PubMed] [Google Scholar]
- 43.McLean WJ, McLean DT, Eatock RA, and Edge ASB (2016). Distinct capacity for differentiation to inner ear cell types by progenitor cells of the cochlea and vestibular organs. Development 143, 4381–4393. 10.1242/dev.139840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Funk CC, Casella AM, Jung S, Richards MA, Rodriguez A, Shannon P, Donovan-Maiye R, Heavner B, Chard K, Xiao Y, et al. (2020). Atlas of Transcription Factor Binding Sites from ENCODE DNase Hypersensitivity Data across 27 Tissue Types. Cell Rep. 32, 108029. 10.1016/j.celrep.2020.108029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bhaduri A, Andrews MG, Mancia Leon W, Jung D, Shin D, Allen D, Jung D, Schmunk G, Haeussler M, Salma J, et al. (2020). Cell stress in cortical organoids impairs molecular subtype specification. Nature 578, 142–148. 10.1038/s41586-020-1962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The bulk RNA-seq, scRNA-seq, and ATAC-seq data from this study are available on GEO (accession numbers GSE132635, GSE137299, GSE172327, GSE71982, and GSE136196) and in an interactive version on gEAR at umgear.org/Lgr5org (Figure S7).39
This paper does not report original code.
Additional information is available upon request.