

Abstract
A general methodology for calculating the equilibrium binding constant of a flexible ligand to a protein receptor is formulated on the basis of potentials of mean force. The overall process is decomposed into several stages that can be computed separately: the free ligand in the bulk is first restrained into the conformation it adopts in the bound state, position, and orientation by applying biasing potentials, then it is translated into the binding site, where it is released completely. The conformational restraining potential is based on the root-mean-square deviation of the peptide coordinates relative to its average conformation in the bound complex. Free energy contributions from each stage are calculated by means of free energy perturbation potential of mean force techniques by using appropriate order parameters. The present approach avoids the need to decouple the ligand from its surrounding (bulk solvent and receptor protein) as is traditionally performed in the double-decoupling scheme. It is believed that the present formulation will be particularly useful when the solvation free energy of the ligand is very large. As an application, the equilibrium binding constant of the phosphotyrosine peptide pYEEI to the Src homology 2 domain of human Lck has been calculated. The results are in good agreement with experimental values.
Keywords: free energy perturbation, molecular dynamics, Src Homology 2 domain
A problem of central importance in computational biology is the quantitative determination of absolute binding affinities in diverse and complex systems. Predicting the binding free energy of ligands to macromolecules can have great practical values in identifying novel molecules that can bind to target receptors and act as therapeutic drugs (1). Furthermore, molecular recognition phenomena involving various kinds of binding modules linked to cell surface receptors play an essential role in a wide variety of intracellular signal transduction pathways (2–5).
Approaches at different levels of complexity and sophistication have been used to calculate binding free energies in complex biomolecular systems. Screening of large molecular databases of compounds to identify potential lead drug molecules typically relies on very simplified scoring schemes to achieve the needed efficiency (6). The binding free energy may be estimated on the basis of a continuum solvent approximation assuming quadratic fluctuations around a unique configuration (7, 8). The Molecular Mechanics/Poisson–Boltzmann (PB) and Surface Area (MM/PB-SA) method is a popular approach that relies on a mixed scheme combining configurations sampled from molecular dynamics (MD) simulations with explicit solvent, together with free energy estimators based on an implicit continuum solvent model (9). MM/PB-SA shares some similarities with the linear interaction energy method, which also uses averages calculated from explicit solvent simulations within a linear response framework (10). Despite their usefulness, such approximate schemes can be limited, and how to improve the results is unclear because they do not offer a rigorous route to compute the equilibrium binding constant.
In principle, treatments based on MD free energy perturbation (FEP) simulations with explicit solvent molecules offer the most powerful and promising approach to estimate the binding free energies of ligands to macromolecules (11). Nonetheless, although previous studies have provided many of the fundamental elements necessary for the calculation of binding free energy by means of MD (12–17), the computations so far have been limited mostly to fairly small and rigid ligands [e.g., rare gas atom (12, 15), water (14), camphor (13), benzene (15), and a single amino acid (16)], and some aspects require further considerations. The challenges that lie ahead are well illustrated by considering the association of peptide ligands to Src homology 2 (SH2) domains. SH2 domains are highly conserved noncatalytic proteins of ≈100-aa residues, which can bind phosphotyrosine-containing polypeptide sequences with high affinity and specificity. They are found in a wide variety of intracellular signal transduction pathways involving tyrosine kinases (18–22), and inappropriate cellular signaling caused by malfunctions of SH2-mediated process has been linked to many pathologic conditions (e.g., cancer, autoimmune diseases, asthma, allergies, etc.). SH2 domains are highly selective toward the sequence phosphotyrosine-Glu-Glu-Ile (pYEEI) (23), with dissociation constants ranging from micromolar to nanomolar ranges (19, 24; for a review, see ref. 25). These values correspond to an absolute binding free energy in the range of approximately –8 kcal/mol. The determinants of the phosphopeptide selectivity of SH2 domains have been characterized previously by using various empirical approaches (22, 26, 27).
Our ability to understand many aspects concerning the specificity of SH2 domains for phosphotyrosyl peptides of particular sequences would benefit from all-atom MD/FEP computations, although a straightforward application of current approaches appears to be problematic for a number of reasons. First, the sheer magnitude of the electrostatics interactions arising from the doubly charged phosphotyrosine side chain (28) suggests that the standard FEP technique used to compute absolute binding free energies, which consists of reversibly decoupling the ligand from its surrounding (12–17), is essentially impractical. Furthermore, additional difficulties are expected to arise from the significant conformational flexibility of the unbound peptide ligand in solution. Although such difficulties may at first appear to be merely technical, the truth is that available methods to compute a binding constant from simulations with explicit solvent are inadequate or prohibitive. Clearly, some significant extension to the present computational methodologies is needed to tackle the more complex situations that invariably are encountered in biological systems.
Our goal is to address these issues and design an efficient approach for calculating the binding constant of a flexible ligand to a protein. Below, the equilibrium binding constant is derived directly on the basis of configurational ensemble averages. This derivation, which differs from the traditional arguments based on chemical potentials and standard states, is particularly advantageous because of its simplicity and clarity. For instance, various computationally convenient expressions can be obtained readily from the fundamental expression for the equilibrium binding constant. In the present case, the absolute binding free energy is rigorously expressed as the sum of separate contributions corresponding to a step-by-step process describing the association of the ligand with the receptor. The formulation relies on potential of mean force (PMF) techniques designed to avoid the problems associated with large free energies involved in the standard FEP decoupling schemes (12–17). As an illustration of the present treatment, the absolute binding free energy of the phosphotyrosyl peptide Ace-pYEEI to the SH2 domain of Lck kinase is computed. The result of the computations is found to be in good agreement with experimental estimates.
Theoretical Developments
Let us consider a dilute solution in thermodynamic equilibrium comprising receptor proteins and flexible ligands able to associate in a bimolecular fashion. Classically, the equilibrium binding constant Keq of the process L + P ⇌ LP is defined as a function of the concentrations of each species, [LP], [L], and [P], as Keq = [LP]/[L][P]. Let p0 and p1 be the fraction of protein receptor with no ligand or one ligand bound, respectively. Two distinct regions of configurational space can be clearly distinguished without ambiguity: the binding “site” and the “bulk” unbound regions, and [P] = p0[P]tot and [LP] = p1[P]tot, where [P]tot is the total concentration of the receptor in the system. By normalization, there can either be zero or one ligand L bound to the receptor protein P, thus, p0 + p1 = 1. It follows that the binding constant can be expressed as
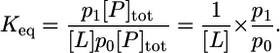 |
[1] |
Assuming that the receptor concentration is sufficiently low, it is possible to consider a single one with its center of mass held fixed at the origin surrounded by a solution of ligands without loss of generality. As will be shown below, the logarithm of the ratio (p1/p0) is related to the reversible work needed to take one ligand molecule from the bulk and carry it to the binding site. Eq. 1 can be written as
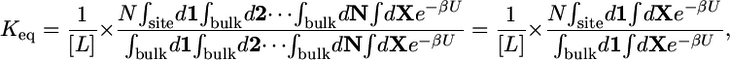 |
[2] |
where U is the total potential energy of the system, 1/β = kBT is the Boltzmann constant times temperature, and {1, 2,... N, X} are the degrees of freedom of the N ligand molecules and the remaining atoms (solvent or protein), respectively. The subscripts site and bulk in the integrals indicate the relevant spatial regions of the configurational space to be included in each integration, representing the bound and unbound states. In Eq. 2, the ligand molecule “1” has been chosen arbitrarily to occupy the binding site, and the factor N accounts for the fact that any ligand could have been chosen. Because the bulk region is isotropic and homogeneous, we have
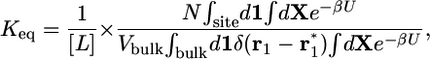 |
[3] |
where r1 is the position of the center of mass of ligand 1 and 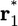 is some arbitrary (fixed) location in the bulk region, far away from the receptor. [The integrals over the (N – 1) remaining ligands have been omitted for the sake of simplicity, assuming low concentration and absence of ligand-ligand interactions.] Because [L] = N/Vbulk, the equilibrium binding constant Keq is
is some arbitrary (fixed) location in the bulk region, far away from the receptor. [The integrals over the (N – 1) remaining ligands have been omitted for the sake of simplicity, assuming low concentration and absence of ligand-ligand interactions.] Because [L] = N/Vbulk, the equilibrium binding constant Keq is
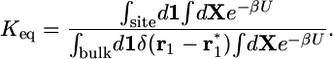 |
[4] |
The denominator and the numerator of Eq. 4 each represent initial and final states of the binding process: the ligand bound to the receptor and the ligand with its center of mass at  in the bulk, respectively (note that all coordinates are expressed relative to the center of mass of the receptor).
in the bulk, respectively (note that all coordinates are expressed relative to the center of mass of the receptor).
The fundamental expression Eq. 4 serves as the cornerstone to develop various computational strategies. In particular, it can be rewritten as
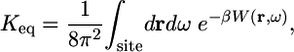 |
[5] |
where W(r, ω) is the protein-ligand solvent-averaged PMF as a function of the relative translation and orientation 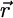 and ω
and ω
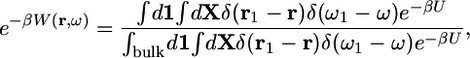 |
[6] |
(by definition W → 0 in the bulk). Although the utility of Eq. 5 is severely limited in all-atom MD simulations with explicit solvent, it is helpful to clarify the significance of approximations based on implicit solvent models such as MM/PB-SA (9). Assuming quadratic fluctuations in the bound state, the dominant contribution to the binding constant may be approximated as
 |
[7] |
where Wmin corresponds to the minimum of the PMF in the bound state, and (Δω/8π 2) and ΔV represent the orientational freedom and translational volume of the bound ligand, respectively (7). In MM/PB-SA (9), a statistical ensemble of configurations of the bound ligand is generated from all-atom MD simulations, and Wmin is estimated from the average value of the interaction free energy calculated by using an implicit continuum solvent model; the quantities Δω and ΔV are estimated from the root-mean-square (rms) fluctuations by using quasiharmonic approximation (29, 30).
The design of a computational method relying on FEP simulations with explicit solvent molecules consists of inserting intermediate states in Eq. 4 such that each individual contribution can be easily calculated. Here, the intermediate states are constructed by introducing various restraining potentials, which are designed to bias the ligand–protein complex toward the configuration it adopts in the bound state. It is useful to first establish a local frame of reference from three centers in each binding partner (Fig. 2), in which the position of the center of mass of the ligand relative to the receptor r1 can be specified by (r1, θ1, φ1) in spherical coordinates, and its orientation can be specified from the three Euler angles (Θ1, Φ1, Ψ1). To restrain the ligand orientation as in the bound complex, we introduce the potential uo(Θ1, Φ1, Ψ1). We also introduce the potential ua(θ1, φ1), designed to restrain the ligand position along a specific axis as in the bound complex. Lastly, we introduce the potential uc, designed to restrain the conformation of the ligand around the average conformation that it adopts when it is bound to the receptor. Although other choices are possible, a simple potential can be constructed on the basis of ξ, the rms deviation (rmsd) of the ligand relative to its average conformation. With these definitions, the equilibrium binding constant Keq in Eq. 4 can be written as
 |
[8] |
 |
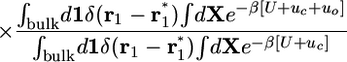 |
 |
Most of the terms in Eq. 8 are dimensionless ratios of configurational integrals corresponding to free energy differences that can be calculated from a standard application of the FEP simulation technique,
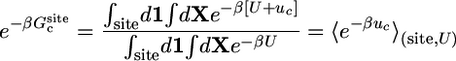 |
[9a] |
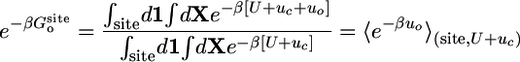 |
[9b] |
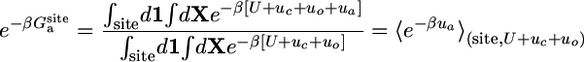 |
[9c] |
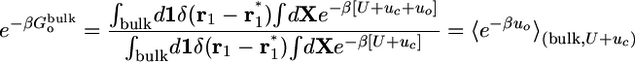 |
[9d] |
 |
[9e] |
One may note that the delta function involving  , when it appears both in the numerator and denominator, does not affect the calculated free energies in the bulk region because it is invariant to translation. The free energy
, when it appears both in the numerator and denominator, does not affect the calculated free energies in the bulk region because it is invariant to translation. The free energy 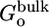 in Eq. 9d can be calculated directly, as an angular integral, because the bulk is isotropic. However, the fourth term in Eq. 8, which involves a ratio of configurational integrals with the bound ligand (numerator) and the ligand held with its center of mass at
in Eq. 9d can be calculated directly, as an angular integral, because the bulk is isotropic. However, the fourth term in Eq. 8, which involves a ratio of configurational integrals with the bound ligand (numerator) and the ligand held with its center of mass at 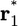 in the bulk by a delta function, requires special attention because it does not correspond to a free energy difference like the other terms. It can be reexpressed as (see Supporting Text, which is published as supporting information on the PNAS web site)
in the bulk by a delta function, requires special attention because it does not correspond to a free energy difference like the other terms. It can be reexpressed as (see Supporting Text, which is published as supporting information on the PNAS web site)
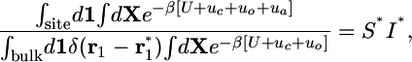 |
[10] |
where S* is an integral over the angles θ1 and φ1,
 |
[11] |
and I* is a one-dimensional (1D) integral over r1
 |
[12] |
defined in terms of the PMF 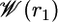 calculated in the presence of the configurational and orientational restraints uc, uo, and ua (see Eq. 3 in Supporting Text). It follows that the binding constant can be expressed as
calculated in the presence of the configurational and orientational restraints uc, uo, and ua (see Eq. 3 in Supporting Text). It follows that the binding constant can be expressed as
 |
[13] |
By definition, Keq has the units of volume (e.g., Å3). Bimolecular equilibrium binding constants are normally expressed in inverse moles per liter, and it is customary to define an absolute binding free energy Gbind ≡ –kBT ln[KeqC°] by assuming a standard state concentration C° of 1 mol/liter (≡ 1/1,661 Å3).
Fig. 2.

Schematic representation of the local reference frame used to define the position and orientation of the ligand relative to the receptor protein and construct the restraining potentials. The spherical coordinate system establishing the position of the ligand relative to the protein are the P3–L1 distance r1, the P2–P1–L1 angle θ1, and the P3–P2–P1–L1 dihedral angle φ1. The Euler angles needed to define the orientation of the ligand relative to the protein are the P3–L1–L2 angle Θ1, the P2–P1–L1–L2 dihedral angle Φ1, and the P1–L1–L2–L3 dihedral angle ψ1.
Computational Methods
The crystallographic structure of the AcpYEEI peptide in complex with the human p56lck SH2 domain from ref. 21. was used as the initial structure for the bound state (Fig. 1). The phosphotyrosine was assumed to be doubly charged as determined experimentally (28). The complex was solvated and equilibrated for 0.7 ns. The resulting structure was used for the PMF calculations and as the reference conformation for the rmsd restraint of the ligand. All MD simulations were generated by using the charmm program (31). The PARAM27 force field (32) was used with the TIP3P water potential (33). The trajectories were generated with periodic boundary conditions in the isobaric-isothermal ensemble at constant pressure of 1 atm (1 atm = 101.3 kPa) and temperature of 300 K. Electrostatic interactions were treated with a particle-mesh Ewald method (34); a grid with roughly one point per Å was used for all systems. Potassium and chloride ions were added both to neutralize the overall systems and simulate an aqueous salt solution at 150 mM.
Fig. 1.

Crystallographic structure of the p56lck SH2 domain in complex with a pYEEI peptide (21).
The coordinate system for specifying the overall relative position and orientation of the ligand with respect to the protein was constructed by choosing three groups of atoms within the protein and in the ligand (see Fig. 2). The three centers, P1, P2, and P3, for the SH2 domain were given by the center of mass of (H208), (D171), and (I183, L202, L205, L165), respectively. The three centers, L1, L2, and L3, for the peptide were given by the center of mass of (pY1, E2, E3, I4), (pY), and (I4), respectively. The spherical coordinate system to establish the position of the ligand relative to the protein comprises, the P3–L1 distance r1, the P2–P1–L1 angle θ1, and the P3–P2–P1–L1 dihedral angle φ1. The Euler angles needed to define the orientation of the ligand relative to the protein are the P3–L1–L2 angle Θ1, the P2–P1–L1–L2 dihedral angle Φ1, and the P1–L1–L2–L3 dihedral angle Ψ1.
The primary tools for the calculation of all of the components in Eq. 13 are FEP simulations (11, 35), umbrella sampling simulations (36), and the weighted histogram analysis method for unbiasing the data from multiple simulations (37–39). Harmonic biasing potentials used are the conformational restraint uc, the orientational restraint uo, and the axis restraint ua. The conformation of the peptide was restrained by using the potential uc(ξ) = kc(ξ[1;1ref])2, where ξ is the rmsd of the ligand relative to its average conformation in the bound state 1ref, which is used as a reference. A force constant of kc = 1.192 kcal/mol·Å2 was used. The orientation of the ligand peptide was restrained by using the harmonic potential 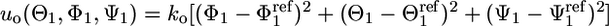 , where (
, where ( ,
, 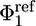 ,
, 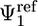 ) corresponds to the average orientation of the bound ligand. The ligand peptide also was restrained to lie along the 1D axis r1 by using the harmonic potential
) corresponds to the average orientation of the bound ligand. The ligand peptide also was restrained to lie along the 1D axis r1 by using the harmonic potential  , where (
, where ( ,
, 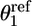 ) corresponds to the average values for the bound ligand. The force constants were ko = ka = 100 kcal/mol per rad2..
) corresponds to the average values for the bound ligand. The force constants were ko = ka = 100 kcal/mol per rad2..
The PMF along the 1D axis r1, 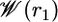 , was calculated by using umbrella sampling simulations. The system was solvated by using a preequilibrated orthorhombic box of water molecules (approximate dimension of 76 × 56 × 56 Å) followed by 200 ps of equilibration before collecting statistics of the distance r1. Umbrella sampling window configurations then were generated in the presence of the biasing radial potential
, was calculated by using umbrella sampling simulations. The system was solvated by using a preequilibrated orthorhombic box of water molecules (approximate dimension of 76 × 56 × 56 Å) followed by 200 ps of equilibration before collecting statistics of the distance r1. Umbrella sampling window configurations then were generated in the presence of the biasing radial potential 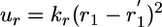 ; 28 windows were simulated with interwindow spacing of 1 Å, and 10 additional windows separated by 0.5 Å for the short distances where the PMF was seen to vary most. Harmonic window potentials
; 28 windows were simulated with interwindow spacing of 1 Å, and 10 additional windows separated by 0.5 Å for the short distances where the PMF was seen to vary most. Harmonic window potentials  were used with force constant values of 0.5 and 5 kcal/mol·Å2. The total umbrella sampling simulation is slightly longer than 3 ns. The integral in Eq. 10 was calculated numerically by using
were used with force constant values of 0.5 and 5 kcal/mol·Å2. The total umbrella sampling simulation is slightly longer than 3 ns. The integral in Eq. 10 was calculated numerically by using  Å as a reference.
Å as a reference.
The free energy 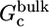 , corresponding to the restriction on the conformation of the ligand free in solution, was obtained by means of a direct integration of the Boltzmann factor after calculating the PMF
, corresponding to the restriction on the conformation of the ligand free in solution, was obtained by means of a direct integration of the Boltzmann factor after calculating the PMF  as a function of ξ, the rmsd relative to the reference conformation 1ref,
as a function of ξ, the rmsd relative to the reference conformation 1ref,
 |
[14] |
A similar expression was used for 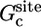 with a PMF wsite(ξ). Harmonic window potentials, kc(ξ – ξ(i))2, with a force constant kc = 1.0 kcal/mol·Å2 were used. The PMF in the bulk was calculated from 20 umbrella sampling simulations separated by 0.5 Å, for a total of 2 ns; a cubic system with 897 water molecules (30 × 30 × 30 Å) was used. The PMF in the bound state was calculated from 20 umbrella-sampling simulations separated by 0.2 Å, for a total of 0.8 ns; a cubic system with 5,490 water molecules (56 × 56 × 56 Å) was used.
with a PMF wsite(ξ). Harmonic window potentials, kc(ξ – ξ(i))2, with a force constant kc = 1.0 kcal/mol·Å2 were used. The PMF in the bulk was calculated from 20 umbrella sampling simulations separated by 0.5 Å, for a total of 2 ns; a cubic system with 897 water molecules (30 × 30 × 30 Å) was used. The PMF in the bound state was calculated from 20 umbrella-sampling simulations separated by 0.2 Å, for a total of 0.8 ns; a cubic system with 5,490 water molecules (56 × 56 × 56 Å) was used.
The free energy terms 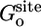 and
and 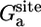 , to the orientational and axial restriction in the corresponding binding site, respectively, were calculated from Eqs. 9b and 9c by using FEP with 10 intermediate values of the thermodynamic coupling parameter λ between 0 and 1, for a total of 0.5 ns. A cubic system with 5,490 water molecules (56 × 56 × 56 Å) was used. The free energy term
, to the orientational and axial restriction in the corresponding binding site, respectively, were calculated from Eqs. 9b and 9c by using FEP with 10 intermediate values of the thermodynamic coupling parameter λ between 0 and 1, for a total of 0.5 ns. A cubic system with 5,490 water molecules (56 × 56 × 56 Å) was used. The free energy term 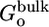 was calculated from Eq. 9d by direct numerical integration over the three Euler angles. The surface element S* was calculated by direct numerical integrations of Eq. 11. The electrostatic contribution to the solvation free energy of the peptide ligand was calculated by using FEP with 10 intermediate values of the thermodynamic coupling parameter λ between 0 and 1, for a total of 1 ns. The peptide was solvated by using the Spherical Solvent Boundary Potential (SSBP) with 421 water molecules (40).
was calculated from Eq. 9d by direct numerical integration over the three Euler angles. The surface element S* was calculated by direct numerical integrations of Eq. 11. The electrostatic contribution to the solvation free energy of the peptide ligand was calculated by using FEP with 10 intermediate values of the thermodynamic coupling parameter λ between 0 and 1, for a total of 1 ns. The peptide was solvated by using the Spherical Solvent Boundary Potential (SSBP) with 421 water molecules (40).
For the MM/PB-SA approximation, the value of Wmin in Eq. 7 is approximated by the average net interaction free energy between the protein and ligand ΔGLP = GLP – GL – GP +ΔELP, where GLP, GP, and GL are the total electrostatic (PB) solvation free energies of the bound complex, isolated protein, and isolated ligand, respectively, and ΔELP is the direct (bare) ligand–protein electrostatic interaction energy. Each energy term was obtained by averaging >40 equilibrium configurations extracted from a MD trajectory of the bound complex. The finite-difference PB calculations were performed with a grid of 0.4 Å by using the pbeq module of the charmm program (31) with the optimized atomic Born radii (41). The dielectric constants of the solvent and protein were 80 and 1, respectively. The salt concentration was taken as 0.15 mM. The binding free energy then was estimated as Gbind ≈ –kBT ln[Δω/8π 2] – kBT ln[C°ΔV] + ΔGLP, where Δω and ΔV are the typical Euler angle and volume of the orientational and translational fluctuations of the ligand in the bound complex.
Results and Discussion
Table 1 summarizes the calculated values of the various contributions to the absolute binding free energy. In Table 2, the calculated binding free energy Gbind is compared with experimental values taken from refs. 24 and 42. It is observed that the result of the calculation is within ≈1 kcal/mol from experimental estimates for closely related sequences (there may be some uncertainty about the experimental value for the AcpYEEI peptide, which was estimated on the basis of a IC50). Given the complexity of the system and the magnitude of the molecular interactions involved, such a good agreement is very satisfying. The dominant contribution to the error in the overall binding free energy arises from the PMF involved in the factor I* given by Eq. 12; all other errors are correspondingly much less important. The error on the radial PMF was estimated by using different fractions of the overall time-series data collected over the course of umbrella sampling simulations (4 × 1/4 of the total). This estimate suggests that the error on the binding free energy is on the order of 2–3 kcal/mol (see also Fig. 5, which is published as supporting information on the PNAS web site). For the sake of comparison, the binding free energy also was calculated according to the MM/PB-SA scheme (9). In this approximation, an ensemble of representative configurations of the bound protein–ligand complex is generated for which the Poisson–Boltzmann continuum electrostatic calculations are performed to obtain the electrostatic free energies of the protein–ligand complex, protein, and ligand only. The overall binding free energy obtained from MM/PBSA is approximately –80 kcal/mol (see Table 3, which is published as supporting information on the PNAS web site) nearly an order of magnitude larger compared with both the experimental values and the result from the free energy simulations with explicit solvent (Table 2). Applications of MM/PB-SA often ignore the translational/orientational factor in Eq. 7 and consider exclusively the ligand–protein interaction free energy, although this treatment is incorrect (see refs. 7 and 29 for discussions). Here, those factors were estimated from the rms fluctuations of the Euler angles and the center of mass of the peptide in the bound state MD trajectories in the harmonic approximation as Δω ≃ 0.045 rad, and ΔV ≃ 5.35 Å3, yielding an orientational free energy contribution of –kBT ln[Δω/8π2] of 4.5 kcal/mol, and a translational free energy contribution of –kBT ln[C°ΔV] of 3.4 kcal/mol, for a total of 7.9 kcal/mol. Clearly, the large inaccuracy arises from the continuum solvent approximation used to compute the interaction free energy and not from those smaller contributions.
Table 1. Computation of the absolute binding free energy.
| Component | Value |
|---|---|
| Gcbulk | 3.70 kcal/mol |
| Gcsite | 1.43 kcal/mol |
| Gobulk | 5.35 kcal/mol |
| Gosite | 0.04 kcal/mol |
| Gasite | 0.40 kcal/mol |
| S* | 22.17 Å2 |
| I* | 3.12 × 1013 Å |
| Keq | 4.13 × 109 Å3 |
| Keq | 2.49 × 106 M-1 |
| Kd | 0.40 μM |
| Gbind | -8.8 kcal/mol |
Table 2. Binding free energy.
One previously undescribed aspect of the present formulation is the introduction of the conformational restraint uc based on the rmsd relative to a reference structure (here the average conformation of the bound ligand). Fig. 3 shows the PMF calculated both in the binding site and in the bulk. Together, these two PMFs help quantify the free energy “cost” arising from the loss of conformational freedom of the ligand when it must adopt a specific conformation in the binding site. Whereas a single specific average conformation of the peptide is strongly favored in the bound state (Fig. 3A), a wide range of conformational states differing from the bound conformation is allowed in the bulk (Fig. 3B). The most frequent conformations of the isolated peptide in solution are ≈4 Å in rmsd relative to the average bound conformation. A closer examination reveals that those correspond to a range of widely different conformations rather than a uniquely identifiable structure, which is indicative of the significant conformational freedom of the peptide in solution. One may note the steep rise in both PMFs at very small rmsd (ξ < 1 Å). This rise is related to the impossibility of quenching all of the thermal fluctuations away from a given reference structure, which is akin to an effective Jacobian associated with the rmsd volume element dξ in the multidimensional configurational space. With the PMF as a function of ξ, the free energies [ and
and  ] can be calculated by direct integration of the Boltzmann factor. The ligand conformation is restrained while it is in the bulk and released once it is in the binding site, yielding the total free energy cost associated with the loss of conformational freedom of the flexible peptide, Gbulkc – Gsitec.
] can be calculated by direct integration of the Boltzmann factor. The ligand conformation is restrained while it is in the bulk and released once it is in the binding site, yielding the total free energy cost associated with the loss of conformational freedom of the flexible peptide, Gbulkc – Gsitec.
Fig. 3.

Calculated PMF w(ξ) of the ligand conformational degrees of freedom as function of the rmsd ξ in the bound (A) and bulk (B) states.
Fig. 4 shows the PMF 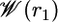 as a function of the distance r1 along the radial axis. The free energy rises steeply near the immediate vicinity of the sharp minimum at 12 Å, the stable bound state, and becomes nearly flat for r1 > 30 Å (continuum electrostatic calculations based on PB confirms that the interactions between the peptide and the SH2 domain are essentially screened out at this distance). The dominant contribution to the overall binding free energy, on the order of –18 kcal/mol, clearly arises from the PMF
as a function of the distance r1 along the radial axis. The free energy rises steeply near the immediate vicinity of the sharp minimum at 12 Å, the stable bound state, and becomes nearly flat for r1 > 30 Å (continuum electrostatic calculations based on PB confirms that the interactions between the peptide and the SH2 domain are essentially screened out at this distance). The dominant contribution to the overall binding free energy, on the order of –18 kcal/mol, clearly arises from the PMF 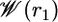 , corresponding to the interactions gained as the peptide moves from the bulk solution into the binding pocket. The integral of the PMF in Eq. 12 is dominated by the contribution from the immediate vicinity of the free energy minimum near 12 Å. For this reason, the results are not affected by the precise definition of the bound state. The strong favorable interaction in
, corresponding to the interactions gained as the peptide moves from the bulk solution into the binding pocket. The integral of the PMF in Eq. 12 is dominated by the contribution from the immediate vicinity of the free energy minimum near 12 Å. For this reason, the results are not affected by the precise definition of the bound state. The strong favorable interaction in 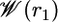 is counter-balanced by unfavorable contributions. Regrouping the various contributions according to their physical significance, one finds that the overall free energy cost associated with the loss of orientational freedom upon binding, [
is counter-balanced by unfavorable contributions. Regrouping the various contributions according to their physical significance, one finds that the overall free energy cost associated with the loss of orientational freedom upon binding, [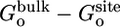 ], is on the order of 5 kcal/mol. Similarly, the free energy cost associated with the loss of internal conformational freedom, [
], is on the order of 5 kcal/mol. Similarly, the free energy cost associated with the loss of internal conformational freedom, [ ], is ≈3 kcal/mol. The magnitude of these unfavorable contributions is significant compared with the value of Gbind. The total cost of the conformational restriction of the peptide is, in fact, slightly overestimated because the rmsd restraining potential is not accounting for the existence of physically equivalent isomers. For example, the Tyr side chain with its phosphate group could adopt other equivalent rotameric states (3 and 2, respectively) that are treated as distinct states by the rmsd restraint. In the bulk, six equivalent rotameric states of the pTyr side chain can be visited, and the PMF wbulk(ξ) overestimates the free energy cost for the reference conformation by kBT ln(6) = 1.07 kcal/mol. In the bound state, these states are not easily sampled because the side chain is tightly surrounded by the protein, and there is no correction.
], is ≈3 kcal/mol. The magnitude of these unfavorable contributions is significant compared with the value of Gbind. The total cost of the conformational restriction of the peptide is, in fact, slightly overestimated because the rmsd restraining potential is not accounting for the existence of physically equivalent isomers. For example, the Tyr side chain with its phosphate group could adopt other equivalent rotameric states (3 and 2, respectively) that are treated as distinct states by the rmsd restraint. In the bulk, six equivalent rotameric states of the pTyr side chain can be visited, and the PMF wbulk(ξ) overestimates the free energy cost for the reference conformation by kBT ln(6) = 1.07 kcal/mol. In the bound state, these states are not easily sampled because the side chain is tightly surrounded by the protein, and there is no correction.
Fig. 4.

Calculated PMF W(r1) as a function of the radial distance r1 between the ligand center of mass relative to the protein.
In the present calculations, the binding constant was expressed as a step-by-step reversible process, and the ligand-receptor PMF reporting the reversible work to translate the ligand from the bulk solution to the bound state is a central quantity. The alternative “double-decoupling” scheme (12–17, 43) augmented by various restraining potentials is particularly advantageous for computing the binding constant of a ligand bound inside a pocket located deeply in the interior of a protein because it does not require any consideration of the pathway leading from the bulk to the binding site. Nonetheless, this method is nearly impractical when the absolute solvation free energy of the ligand is very large because the binding free energy then is computed as the (small) difference between two very large numbers. In such cases, the limited statistical precision of the FEP computations becomes a major hurdle, which is particularly problematic in computing the binding constant of a highly charged ligand such as the pYEEI peptide. The electrostatic contribution to the total solvation free energy of the ligand in bulk water, calculated by using FEP simulation with explicit solvent molecules, is of the order of –773 kcal/mol. This large number contrasts with the binding free energy, which is of the order of –8 kcal/mol (Table 2). Even if the statistical uncertainty of the FEP calculation was only ≈1% of the total solvation free energy, that would still translate into an error that is of the same order of magnitude as the quantity of interest itself. Attempts to compute the binding free energy of the pYEEI peptide with the SH2 domain by using a double-decoupling scheme based on Eq. 20 exhibited errors on the order of 75 kcal/mol (data not shown). To avoid such problems, the binding constant was expressed in terms of the ligand-receptor PMF, without involving the decoupled state.
The idea of estimating binding constants directly from the ligand-receptor PMF is not novel; for example, a radial bimolecular PMF was used in ref. 44. However, an approach based on the true unbiased ligand-receptor PMF would be of little use in practice because sampling over the entire range of possible motions of the ligand, receptor, and solvent would be computationally prohibitive. The present development shows that such extensive sampling is not needed and that the problem of computing the equilibrium binding constant can be rigorously formulated in terms of the PMF of the ligand restrained along the 1D axis r1. The computational efficiency also is improved by introducing the conformational and orientational restraining potentials, uc and uo, which further contribute to reduce the amount of configurational space that needs to be thoroughly sampled. Those two restraining potentials help significantly in the statistical convergence of the restrained PMF calculated along the 1D radial axis r1.
Conclusion
We have presented an efficient PMF-based computational method for calculating the equilibrium association constant between a flexible ligand and a protein receptor from MD simulations with explicit solvent. The result of the calculation is within ≈1 kcal/mol from experimental estimates. Such a good agreement may be partly fortuitous, although it is very encouraging and suggests that accurate computations of absolute binding free energies from all-atom simulations is an achievable goal.
The present PMF-based approach avoids the double-decoupling scheme (12–17), which can lead to significant difficulties in the case of highly charged ligands. Exact and computationally advantageous expressions for the equilibrium binding constant were derived directly from configurational ensemble averages. The derivation is straightforward and has the advantage of avoiding arguments based on chemical potentials invoked in the traditional framework (16, 45). Precision of the results is only limited by the computational requirements for the adequate sampling of configurations, with no uncontrollable approximations or assumptions.
One of the previously undescribed aspects of the present method is the configurational restriction potential based on the rmsd relative to the average structure of the bound state. The introduction of the rmsd into the formulation of the equilibrium binding constant permits a rational and quantitative discussion of the free energy cost associated with the loss of conformational freedom of the ligand to adopt a given bound conformation. From a practical point of view, the rmsd restraining potential essentially transforms a flexible ligand into a relatively rigid one, thereby reducing significantly the difficulties associated with the sampling of a multitude of conformations. In the present case, the configurational restriction was only applied to the flexible pYEEI ligand. Because the SH2 domain is fairly rigid, one expects that its small atomic fluctuations will be readily sampled spontaneously during unbiased MD simulations. Nonetheless, some receptor proteins are known to undergo significant conformational changes upon ligand binding, e.g., the HIV protease (46). The present approach, based on the PMF of the rmsd relative to a reference structure, could be generalized to quantify the importance of conformational flexibility of the receptor.
Supplementary Material
Acknowledgments
We thank Stefan Boresch, Jose Faraldo-Gomez, and Yuqing Deng for their comments on the manuscript. This work was supported by National Institutes of Health Grant CA-93577. The computations were carried out at the Pittsburgh Supercomputing Center with a grant from the National Resource Allocation Committee.
Author contributions: H.-J.W. and B.R. performed research and wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: FEP, free energy perturbation; MD, molecular dynamics; PB, Poisson–Boltzmann; MM/PB-SA, Molecular Mechanics/PB and Surface Area; PMF, potential of mean force; rmsd, rms deviation; SH2, Src homology 2.
References
- 1.Wlodawer, A. (2002) Annu. Rev. Med. 53, 595–614. [DOI] [PubMed] [Google Scholar]
- 2.Vindigni, A. (1999) Combinatorial Chem. High Throughput Scr. 2, 139–153. [PubMed] [Google Scholar]
- 3.Cheng, A. C., Calabro, V. & Frankel, A. D. (2001) Curr. Opin. Struct. Biol. 11, 478–484. [DOI] [PubMed] [Google Scholar]
- 4.Garvie, C. W. & Wolberger, C. (2001) Mol. Cell. 8, 937–946. [DOI] [PubMed] [Google Scholar]
- 5.Pawson, T. & Nash, P. (2003) Science 300, 445–452. [DOI] [PubMed] [Google Scholar]
- 6.Schneider, G. & Bohm, H. J. (2002) Drug Discovery Today 7, 64–70. [DOI] [PubMed] [Google Scholar]
- 7.Luo, H. & Sharp, K. (2002) Proc. Natl. Acad. Sci. USA 99, 10399–10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roux, B. & Simonson, T. (1999) Biophys. Chem. 78, 1–20. [DOI] [PubMed] [Google Scholar]
- 9.Massova, I. & Kollman, P. A. (2000) Perspect. Drug Discovery 18, 113–135. [Google Scholar]
- 10.Brandsdal, B. O., Osterberg, F., Almlof, M., Feierberg, I., Luzhkov, V. B. & Aqvist, J. (2003) Adv. Protein Chem. 66, 123–158. [DOI] [PubMed] [Google Scholar]
- 11.Kollman, P.A. (1993) Chem. Rev. 93, 2395–2417. [Google Scholar]
- 12.Hermans, J. & Shankar, S. (1986) Isr. J. Chem. 27, 225–227. [Google Scholar]
- 13.Helms, V. & Wade, R. C. (1998) J. Am. Chem. Soc. 120, 2710–2713. [Google Scholar]
- 14.Roux, B., Nina, M., Pomès, R. & Smith, J. C. (1996) Biophys. J. 71, 670–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hermans, J. & Wang, L. (1997) J. Am. Chem. Soc. 119, 2707–2714. [Google Scholar]
- 16.Boresch, S., Tettinger, F., Leitgeb, M. & Karplus, M. (2003) J. Phys. Chem. B 107, 9535–9551. [Google Scholar]
- 17.Mann, G. & Hermans, J. (2000) J. Mol. Biol. 302, 979–989. [DOI] [PubMed] [Google Scholar]
- 18.Pawson, T. & Gish, G. D. (1992) Cell 71, 359–362. [DOI] [PubMed] [Google Scholar]
- 19.Bradshaw, J. M., Mitaxov, V. & Waksman, G. (1999) J. Mol. Biol. 293, 971–985. [DOI] [PubMed] [Google Scholar]
- 20.Waksman, G., Shoelson, S. E., Pant, N., Cowburn, D. & Kuriyan, J. (1993) Cell 72, 779–790. [DOI] [PubMed] [Google Scholar]
- 21.Tong, L., Warren, T. C., King, J., Betageri, R., Rose, J. & Jakes, S. (1996) J. Mol. Biol. 256, 601–610. [DOI] [PubMed] [Google Scholar]
- 22.Sheinerman, F. B., Al-Lazikani, B. & Honig, B. (2003) J. Mol. Biol. 334, 823–841. [DOI] [PubMed] [Google Scholar]
- 23.Zhou, S. Y., Shoelson, S. E., Chaudhuri, M., Gish, G., Pawson, T., Haser, W. G., King, F., Roberts, T., Ratnofski, S., Lechleider, R. J., et al. (1993) Cell 72, 767–778. [DOI] [PubMed] [Google Scholar]
- 24.Cousins-Wasti, R. C., Ingraham, R. H., Morelock, M. M. & Grygon, C. A. (1996) Biochemistry 35, 16746–16752. [DOI] [PubMed] [Google Scholar]
- 25.Bradshaw, J. M. & Waksman, G. (2002) Adv. Protein Chem. 61, 161–210. [DOI] [PubMed] [Google Scholar]
- 26.Zvelebil, M. J., Panayotou, G., Linacre, J. & Waterfield, M. D. (1995) Protein Eng. 8, 527–533. [DOI] [PubMed] [Google Scholar]
- 27.Lee, J. K., Moon, T., Chi, M. W., Song, J. S., Choi, Y. S. & Yoon, C. N. (2003) Biochem. Biophys. Res. Commun. 306, 225–230. [DOI] [PubMed] [Google Scholar]
- 28.Bradshaw, J. M. & Waksman, G. (1998) Biochemistry 37, 15400–15407. [DOI] [PubMed] [Google Scholar]
- 29.Swanson, J. M. J., Henchman, R. H. & McCammon, J. A. (2004) Biophys. J. 86, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanner, M. F., Olson, A. J. & Spehner, J. C. (1996) Biopolymers 38, 305–320. [DOI] [PubMed] [Google Scholar]
- 31.Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States, D. J., Swaminathan, S. & Karplus, M. (1983) J. Comp. Chem. 4, 187–217. [Google Scholar]
- 32.MacKerell, A. D., Jr., Bashford, D., Bellot, M., Dunbrack, R. L., Evanseck, J. D., Field, M. J., Fischer, S., Gao, J., Guo, H., Ha, S., et al. (1998) J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- 33.Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. (1983) J. Chem. Phys. 79, 926–935. [Google Scholar]
- 34.Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H. & Pedersen, L. G. (1995) J. Chem. Phys. 103, 8577–8593. [Google Scholar]
- 35.Simonson, T., Archontis, G. & Karplus, M. (1997) J. Phys. Chem. B 101, 8349. [Google Scholar]
- 36.Torrie, G. M. & Valleau, J. P. (1977) J. Comp. Phys. 23, 187–199. [Google Scholar]
- 37.Ferrenberg, A.M. & Swendsen, R.H. (1989) Phys. Rev. Lett. 63, 1195–1198. [DOI] [PubMed] [Google Scholar]
- 38.Kumar, S., Bouzida, D., Swendsen, R. H., Kollman, P. A. & Rosenberg, J. M. (1992) J. Comp. Chem. 13, 1011–1021. [Google Scholar]
- 39.Roux, B. (1995) Comp. Phys. Comm. 91, 275–282. [Google Scholar]
- 40.Beglov, D. & Roux, B. (1994) J. Chem. Phys. 100, 9050–9063. [Google Scholar]
- 41.Nina, M., Beglov, D. & Roux, B. (1997) J. Phys. Chem. B 101, 5239–5248. [Google Scholar]
- 42.Nam, N. H., Ye, G., Sun, G. & Parang, K. (2004) J. Med. Chem. 47, 3131–3141. [DOI] [PubMed] [Google Scholar]
- 43.Jorgensen, W. L., Buckner, J. K., Boudon, S. & Tirado-Rives, J. (1988) J. Chem. Phys. 89, 3742–3746. [Google Scholar]
- 44.Jorgensen, W. L. (1989) J. Am. Chem. Soc. 111, 3770–3772. [Google Scholar]
- 45.Gilson, M. K., Given, J. A., Bush, B. L. & McCammon, J. A. (1997) Biophys. J. 72, 1047–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carlson, H. A. & McCammon, J. A. (2000) Mol. Pharmacol. 57, 213–218. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}