

Abstract
A multiscale simulation of a complex between the lac repressor protein (LacI) and a 107-bp-long DNA segment is reported. The complex between the repressor and two operator DNA segments is described by all-atom molecular dynamics; the size of the simulated system comprises either 226,000 or 314,000 atoms. The DNA loop connecting the operators is modeled as a continuous elastic ribbon, described mathematically by the nonlinear Kirchhoff differential equations with boundary conditions obtained from the coordinates of the terminal base pairs of each operator. The forces stemming from the looped DNA are included in the molecular dynamics simulations; the loop structure and the forces are continuously recomputed because the protein motions during the simulations shift the operators and the presumed termini of the loop. The simulations reveal the structural dynamics of the LacI–DNA complex in unprecedented detail. The multiple domains of LacI exhibit remarkable structural stability during the simulation, moving much like rigid bodies. LacI is shown to absorb the strain from the looped DNA mainly through its mobile DNA-binding head groups. Even with large fluctuating forces applied, the head groups tilt strongly and keep their grip on the operator DNA, while the remainder of the protein retains its V-shaped structure. A simulated opening of the cleft of LacI by 500-pN forces revealed the interactions responsible for locking LacI in the V-conformation.
Keywords: protein–DNA interaction, molecular dynamics, elastic rod model, DNA loop, large-scale protein motion
The lac repressor (LacI) is a celebrated DNA-binding protein that regulates the function of the lac operon (1), a set of genes responsible for the lactose metabolism in Escherichia coli. In the absence of lactose, LacI inhibits the expression of the operon by binding to two of three specifically recognized DNA sites, called operators, and causing the DNA between the operators to fold into a loop (2–4). Depending on which operators are bound, the loop may have a length of either 384 or 75 bp (4, 5). The smaller loop contains the promoter of the lac operon, which includes binding sites for RNA polymerase and the activator protein CAP (1).
Several x-ray structures of LacI, both alone and in complex with DNA, were reported (4, 6, 7). The repressor is a homotetramer folded into a dimer of dimers, two massive “arms” connected at the ends by means of a four-helix bundle (Fig. 1). Each arm consists of a core and a DNA-binding head group domain; the lactose binding sites divide the core domains into two subdomains. This architecture is essential for the function of LacI. The protein binds two operators with its two head groups and holds them close together, enforcing the interoperator loop, while leaving the lactose binding sites inside the core domains open for lactose molecules to enter, disrupt the repressor, and induce the expression of the lac operon. At the same time, the loose connections between the LacI domains imply a great degree of structural flexibility that LacI needs to first span a wide distance in search for the operators and, once bound to them, absorb the fluctuations of the created DNA loop. FRET experiments demonstrated that the V-shaped configuration of LacI observed in the crystal (4), where the protein binds disjoint DNA segments (Fig. 1c), is indeed altered when the protein is bound to a continuous DNA loop (8). It is believed that the arms of LacI rotate around the hinge domain when absorbing pressure from the looped DNA (refs. 6 and 8 and D. Swigon, B. D. Coleman, and W. K. Olson, unpublished data). Conceivably, rotation and tilting of the flexible head group domains also contribute to the conformational change.
Fig. 1.

The structure of the LacI–DNA complex in the multiscale simulation. (a) Overall setup of the simulation. The all-atom structure of the complex between LacI (blue and green protein cartoon) and operator DNA segments (bonds and ribbons) is solvated in a bath of water (blue box) and ions (Na+, yellow spheres; Cl–, pink spheres). The 75-bp-long DNA loop connecting the operators is modeled as an elastic ribbon (gray). The forces of interaction between the loop and the operators (red arrows) are included in the MD simulation. (b) The centerline of the elastic ribbon approximating the DNA loop and the main variables of the continuum model. (c) The all-atom structure of the LacI–DNA complex, built by using available x-ray structures (4, 7, 17). The two symmetrical dimers of which the LacI tetramer is composed are colored green and blue; the monomers in each dimer (1A and 1B, 2A and 2B) are shown with a different degree of opacity. The different domains of the dimers are indicated.
Here we study the structural dynamics of LacI by means of molecular dynamics (MD) simulation of the all-atom model of its complex with DNA. The simulation presents a formidable computational problem, because LacI alone is already a very large protein that, solvated in a water bath, results in a system of >200,000 atoms (10). Including even the short 75-bp-long DNA loop in the model increases the size of the system to at least 700,000 atoms and including the long 384-bp loop increases the size of the system to 2 million atoms. A MD simulation of a system of this size is extremely costly computationally. Moreover, the characteristic motions of the DNA loop in water occur on a much longer time scale than the tens of nanoseconds attainable by MD today. We therefore approach the problem through a multiscale strategy (10–12), describing the different parts of the system at different spatial and temporal resolution. The LacI complex with the operator DNA segments is simulated in solvent by all-atom MD, whereas the 75-bp DNA loop between the operators is described by a continuum model that mimics the observed elastic properties of DNA in solution (13–15) (Fig. 1 a and b). The mechanical properties of the loop are described by means of those of its lowest-energy equilibrium conformation, which is assumed to instantaneously follow the changes in the protein structure. A system of differential equations that yields the equilibrium conformation of the elastic loop and the force that the loop exerts on LacI is solved for a fraction of the computational cost of an all-atom MD simulation of the DNA loop (10, 15); the force then is included in the MD simulation of LacI. The multiscale approach permits us to address the key question regarding the structure–function relationship of LacI: how the structural dynamics of the repressor change under the influence of the strained DNA loop.
Methods
In this section, the simulated model systems and the protocols of the multiscale simulations are described.
All-Atom System. The simulated all-atom structure of the LacI–DNA complex (Fig. 1), deposited to the Protein Data Bank (ID code 1Z04), was built from several Protein Data Bank structures; protein or DNA segments poorly resolved in one structure were taken from the other structures (see ref. 16 for details). The protein head groups in the constructed structure are bound to the 16-bp-long palindromic DNA sequence TTGTGAGCGCTCACAA that is a central part of the symmetrized LacI operator (4, 17). The constructed complex was embedded in a box-shaped (170 × 140 × 100 Å3) bath of water molecules by using the program vmd (18). One hundred eighty water molecules were replaced by 114 Na+ and 66 Cl– ions according to the electrochemical potential around the LacI–DNA complex computed with the DelPhi program (19), resulting in an ionic strength of ≈100 mM and zero net charge. The resulting system of 226,314 atoms (Fig. 1a) was equilibrated in a 2-ns-long MD simulation following a multistep protocol, as described in Supporting Methods, which is published as supporting information on the PNAS web site.
To study large conformational changes during the LacI opening, a system of 314,452 atoms was similarly built by using a larger water box (240 × 140 × 100 Å3; 143 Na+, 95 Cl–) and equilibrated for 1 ns.
MD Protocol. The MD simulations of the constructed system were run with software package namd (20), by using the CHARMM27 force field with a TIP3P water model (21). The system was equilibrated as an NPT ensemble, using the Nosé–Hoover method (22) at a temperature T = 300 K and pressure P = 1 atm (1 atm = 101.3 kPa). Periodic boundary conditions were assumed, and long-range electrostatic forces were computed by using the P3ME method (23). During equilibration, the simulation employed rigid and harmonic constraints on parts of the system as described in Supporting Methods. Planarity of the nucleotide bases at the 5′ and 3′ termini of the operators was enforced by additional harmonic constraints throughout the simulation (10).
The total of 22.4 ns of MD was simulated for the 226,314-atom system on 600 processors at the Pittsburgh Supercomputer Center, with an average production speed of 2.7 ns/day. The 314,452-atom system was simulated for 17 ns on 254 processors at the National Center for Supercomputing Applications (University of Illinois), with an average production speed of 2.5 ns/day.
DNA Loop Model. The 75-bp-long DNA loop connecting the LacI-bound operators was modeled as an elastic ribbon (10, 15, 24) connecting the terminal base pairs of the operators (Fig. 1c). The equilibrium structure of the ribbon was obtained by solving the system of Kirchhoff equations of elasticity, which assume balance of the elastic stress at every cross-section along the loop, thereby minimizing the energy functional (10, 15, 24),
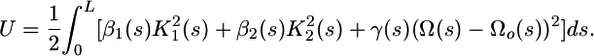 |
[1] |
Here, s is the arclength that parameterizes the elastic loop, L = 258.4 Å is the length of the loop, β1 = 0.8 × 10–19 erg·cm (1 erg = 0.1 μJ), β2 = 3.2 × 10–19 erg·cm, and γ = 3 × 10–19 erg·cm are the elastic moduli of DNA bending and twisting (15, 25, 26), K1, K2, and Ω are the local curvatures and twist, respectively, and Ωo = 34.6° per bp is the intrinsic twist of DNA (10, 15, 24). Electrostatic interaction terms, negligible for this loop at the chosen ionic strength of 100 mM, were excluded from the computations (10). The boundary conditions for the Kirchhoff equations were obtained from the coordinates of the terminal base pairs of each operator in the all-atom model (10, 15).
Multiscale Simulation. In our multiscale approach, the MD simulations included the forces that account for the elastic stress and torque due to the looping of the DNA between the operators. The forces were computed by using Kirchhoff equations and applied to the LacI-bound DNA segments during the simulation as described in ref. 10. The equilibrium structure of the loop and the resulting forces were recomputed every 10 ps of MD with the boundary conditions derived from the last MD snapshot of the protein–DNA structure. The forces had a typical value of several piconewtons (pN).
The multiscale simulations also were performed with additional force terms. These forces were orders of magnitude larger than either the computed elastic forces, the forces that occur in vivo, or those used in single molecule experiments. The purpose of using much larger forces was to bring the dynamics of the LacI–DNA complex within the time scale available to MD simulations (27). One simulation included forces obtained as a continuous time series that obeyed a Gaussian distribution around an average magnitude of 80 pN, which is roughly 10 times the magnitude of the elastic forces in the initial conformation (see Fig. 6, which is published as supporting information on the PNAS web site). The goal was to observe large-scale motions in the LacI–DNA complex and to mimic the thermal fluctuations of the DNA loop. Moreover, the simulation with fluctuating forces also may mimic the in vivo interactions of LacI–DNA complex with its surroundings, suggesting a routine usage of the fluctuating forces in future multiscale simulations. Another simulation applied constant forces of 500 pN to the external base pairs of the operators as shown in Fig. 4a. The goal was to mimic the opening of the LacI cleft as it presumably occurs in vivo or is enforced in single-molecule experiments.
Fig. 4.

Opening of the LacI–DNA complex by an applied external force. (a) Snapshots of the LacI–DNA complex at the following different stages of the simulation: I, 0 ns; II, 1.65 ns; III, 3.4 ns; IV, 6.4 ns; and IV′, 16 ns. The DNA loop is colored according to the local density of the elastic energy (scale is shown in the lower left of the figure). (b) Density plot of the elastic energy along the DNA loop over the course of the simulation. The value of the energy is color-coded according to the same scale as in a. The length is measured in DNA helical steps: h = 3.4 Å. (c) The cleft angle α (violet) and the angle of rotation ϑ of a head group (green), as defined in Fig. 2b, over the course of the simulation. The different stages of the opening, indicated on the plot, are characterized by the difference in the slope  (red lines) as follows: stage I, 2.4° per ns; II, 0.15° per ns; III, 2.7° per ns; and IV to IV′, 4.5° per ns. Each of the snapshots in a shows a characteristic structure from each stage.
(red lines) as follows: stage I, 2.4° per ns; II, 0.15° per ns; III, 2.7° per ns; and IV to IV′, 4.5° per ns. Each of the snapshots in a shows a characteristic structure from each stage.
Observable Monitored. The conformational changes of LacI during the simulation were monitored through a few geometric properties. The rms deviation (RMSD) of atomic coordinates from their initial values was computed for the protein and DNA backbone of each domain of the LacI–DNA complex. The RMSD value computed for a certain domain after the best-fit alignment with its initial structure allows one to assess how well that structure was conserved during the simulation. Typically, MD studies show that structurally stable protein domains fluctuate with an average RMSD of 0.5–3.0 Å from their average or experimental structure (see, e.g., refs. 28–30). Alternatively, the RMSD value computed for a protein domain after the best-fit alignment of a different domain allows one to assess the mobility of those domains with respect to each other. A substantial (>1 Å) difference between the thus-computed RMSD and the individual RMSD of both domains signifies a motion of the domains.
We denote by avRMSD the RMSD value averaged over the entire simulation, implying that the RMSD was observed to stabilize at this average with a standard deviation of <0.5 Å, unless otherwise stated. For the duplicate domains of the same kind (head groups, dimer cores, DNA operators, etc.), the avRMSD value is the average of the two avRMSDs computed for each individual domain.
Three angle parameters are introduced to describe the movements of the LacI domains during the simulation (Fig. 2a). The cleft angle α is defined as the angle between the lines connecting the center of mass of each dimer arm of LacI with the center of mass of the four-helix bundle. Altitudinal and azimuthal angles ϑ1,2 and ϕ1,2 describe the orientation of the longest principal axis of inertia of each of the two head groups in the lab coordinate system. ϑ is the angle between the principal axis and the lab coordinate system vector 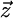 , and ϕ is the angle between the lab coordinate system vector
, and ϕ is the angle between the lab coordinate system vector 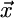 and the projection of the principal axis on the x–y plane of the lab coordinate system (Fig. 2a). The changes in the three angles Δα = α – α0, Δϑ = ϑ – ϑ0, and Δϕ = ϕ – ϕ0 with respect to their values at the end of the equilibration stage are reported below.
and the projection of the principal axis on the x–y plane of the lab coordinate system (Fig. 2a). The changes in the three angles Δα = α – α0, Δϑ = ϑ – ϑ0, and Δϕ = ϕ – ϕ0 with respect to their values at the end of the equilibration stage are reported below.
Fig. 2.

Dynamics of the LacI–DNA complex in the multiscale simulation with fluctuating forces. (a) The principal degrees of freedom of the protein involve the cleft angle α between the two halves of the tetramer and the spherical angles ϑ and ϕ that determine the orientation of each head group domain, as defined in Methods. The head groups are shown in green; the DNA operators are omitted for clarity. (b) Time dependence of the RMSD of various domains of the LacI–DNA complex. (Top) RMSD of each core domain (green and dark green), the two core domains together (blue), and the four-helix bundle (magenta). (Middle) RMSD of a head group (black), the operator bound to that head group (orange), and the same operator if computed after the best-fit alignment of the head group (violet). (Bottom) RMSD of the two core domains (blue) and the two head groups (red, turquoise) computed after the alignment of the core domains. (c) The dynamics of the principal angles α (violet), θ1 (blue), ϕ1 (green), θ2 (red), and ϕ2 (orange).
Results
The work presented here comprises one of the largest protein–DNA simulations to date. In this section, we first describe the equilibration of the constructed model of the LacI–DNA complex, then present the results of the multiscale simulation of that system, and, finally, discuss the forced opening simulation of the LacI–DNA complex.
Equilibration. During the equilibration, the LacI structure was stable; protein domains conserved their secondary and tertiary structure. The structure of the protein core had an avRMSD of 1.27 Å; i.e., the “V” configuration remained mainly undisturbed (〈Δα〉 = 0.05°). The upper core domain of the dimers did not significantly move with respect to the lower core (avRMSD 1.48 Å). The interdomain interactions and the structure of the lactose-binding pockets also remained unchanged. The pockets were filled with, on average, 15 water molecules freely exchanging with the bulk solvent.
The head group structures showed similar stability (avRMSD 1.43 Å). However, the head groups moved with respect to the core of the protein (avRMSD 2.59 Å), with no clear tendency to the direction of that movement. The maximum angles of rotation of the head groups were Δϑ1 = 2.6°, Δϕ1 = 2.0°, Δϑ2 = 3.0°, and Δϕ2 = 2.9° (see Fig. 7, which is published as supporting information on the PNAS web site). The DNA segments preserved their B-DNA structure with a protein-induced kink in the middle (17) (avRMSD 1.75 Å).
Overall, the model complex proved remarkably stable for a hybrid, newly constructed from several Protein Data Bank structures, and allowed further simulation studies.
Multiscale Simulation. Our 7.4-ns multiscale simulation of the equilibrated system included the forces from the elastic model of the DNA loop, as described in Methods. Throughout the simulation, these forces remained in the range of 1–10 pN, which is comparable with observed values of other biomolecular forces, e.g., those generated by protein motors (31). The forces did not change the structure of the protein core, which remained stable throughout the simulation (avRMSD 2.23 Å, Δαmax = 1°; see Fig. 2), but caused the protein head groups to rotate. The rotation followed the direction of the force, with Δϑmax = 10°. One can conclude that the flexible head groups can readily absorb the strain from the DNA loop. However, the configurational changes in the LacI–DNA complex under forces of this scale are slow compared with the time scale of the simulation, and it is impossible to foresee the largest extent of the head group rotation or what other changes may occur in the LacI structure.
A second simulation was performed for 12.8 ns in which fluctuating forces with an average magnitude of 80 pN acted on the operators (see Methods). Similar forces have been shown to be sufficient to disrupt protein structure in vivo and in silico (9, 32). The results of the simulation are presented in Figs. 2 and 3 (see also Movies 1 and 2, which are published as supporting information on the PNAS web site). Despite the strong forces, the domains of LacI mainly conserved their overall structure (avRMSD of only 1.2 Å for individual core domains and 2.2 Å for the whole protein core, 2.5 Å for the head groups, and 2.6 Å for the operators). The core domains did not move significantly with respect to each other; the maximum opening of the cleft was Δαmax = 4°. The whole effect of the applied forces was to drastically increase the motion of the head groups with respect to their dimer cores (avRMSD = 15.1 ± 1.5 Å; Δϑ1,max = 51.4° and Δϕ2,max = 40.7°; see Fig. 2 b and c). It is notable that different spherical angles (Δϑ1,max vs. Δϕ2,max) showed the dominant change for the two head groups, meaning that the head groups rotated in different planes (Figs. 2c and 3a). This result suggests that the head group rotations can occur in a variety of directions, implying even greater conformational flexibility than initially appreciated. The flexibility allows the head groups to absorb the strain of the DNA loop without LacI losing its grip on it.
Fig. 3.

Head group and DNA loop motion during the multiscale simulation with fluctuating forces. (a) The protein structure before (red) and after (blue) the simulation. The colored lines represent the long principal axis of each head group after every 200 ps of the simulation (see the color scheme at the bottom). (b) The structure of the DNA loop drawn after every 200 ps of the simulation and colored by the same scheme as in a. The initial structure of the whole complex (red) and the final structure of the head groups and the operators (blue) also are shown. (c) The distance d between two fluorophores attached to the DNA loop in ref. 8. The red line in the plot shows the change in d during the simulation; the green line shows how d would change if the head groups were immobile with respect to the core domains. The purple band with the dotted line shows the experimental range and the average estimate for d (8).
The change in the structure of the DNA loop during the simulation is shown in Fig. 3b. The loop steadily moved toward lower-energy conformations, its energy (Eq. 1) diminished from 20 kBT to 12 kBT during the simulation. The force of the protein–DNA interaction dropped from 7.2 to 2.0 pN. Thus, a significant relaxation of the loop can be possible solely due to the mobility of the LacI head groups. Interestingly, this finding dissents from the prevailing view that the opening of the LacI cleft is the principal degree of freedom of the protein (ref. 8 and D. Swigon, B. D. Coleman, and W. K. Olson, unpublished data). Edelman et al. (8) measured the distance between fluorophores attached to the DNA loop near the LacI head groups to be ≈50 Å larger than the same distance in the crystal structure (4); this increase in distance was ascribed to the opening of the LacI cleft. However, the corresponding increase in distance measured in our simulation is 52 Å (Fig. 3c), which explains the experimental observation without invoking a significant opening of the cleft.
Opening LacI. To study the opening of the cleft, a third simulation was performed for 16 ns, in which external forces of 500 pN were applied to the outer DNA ends (Fig. 4a). Even under these exceedingly large forces, the structure of all domains of LacI once again remained unchanged (avRMSD of 1.51 Å for the head groups, 1.32 Å for the dimer cores, and 2.17 Å for the operators). Nevertheless, the simulation succeeded in opening the cleft by Δα = 53° (Fig. 4 a and c; see also Fig. 8 and Movie 3, which are published as supporting information on the PNAS web site). The opening was accompanied by an appreciable change in the structure of the four-helix bundle (avRMSD = 3.28 ± 0.54 Å). The head groups yet again rotated (〈Δϑ〉= 48°), absorbing a significant part of the applied force. The DNA loop adapted a more open configuration (Fig. 4a); the overall twist of the loop changed both its value and its sign because of the rotation of the head groups, from ΔΩ = –1.24° per bp to ΔΩ = 0.75° per bp. The elastic energy (Eq. 1) of the loop decreased from 20 kBT to 10 kBT and became more evenly distributed along the loop (Fig. 4b). It is notable that the final energy is only slightly less than that recorded in the previous simulation, indicating that a significant degree of the loop relaxation can be achieved solely by rotation of the head groups.
The simulation revealed four distinct stages of LacI opening, characterized by different rates of change in α (Fig. 4c). During stage I, some initial opening of the cleft took place as α grew steadily at an average rate 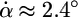 per ns. During stage II, the increase in α slowed down to
per ns. During stage II, the increase in α slowed down to 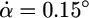 per ns, indicating that certain interactions in the protein resisted further cleft opening. At the same time, stage II is characterized by a jump in the ϑ angles (Fig. 4c), indicating that the head groups of the LacI rotated, absorbing the pulling force. During stage III, the protein resumed its opening motion at the earlier rate (
per ns, indicating that certain interactions in the protein resisted further cleft opening. At the same time, stage II is characterized by a jump in the ϑ angles (Fig. 4c), indicating that the head groups of the LacI rotated, absorbing the pulling force. During stage III, the protein resumed its opening motion at the earlier rate (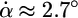 per ns), and during stage IV, it continued the opening at an even faster pace,
per ns), and during stage IV, it continued the opening at an even faster pace, 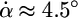 per ns.
per ns.
We identified several interactions responsible for locking LacI in the V configuration when the protein is subject to strain (Fig. 5; see also Movie 4, which is published as supporting information on the PNAS web site). These interactions accounted for 80% of the interaction energy between the dimers, excluding the self-interaction of the four-helix bundle (see Fig. 9, which is published as supporting information on the PNAS web site). The interactions included two salt bridges (E2351B–R3262A and E2352B–R3261A), a charge–dipole interaction between R3511A and the C terminus of an α-helix 2472B–2602B, and the hydrophobic interaction between two α-helices, 2221B–2361B and 2222B–2362B, protruding from each dimer into the cleft. The latter hydrophobic interaction had been suggested to be the most important bond between the cores of the dimers (ref. 4 and D. Swigon, B. D. Coleman, and W. K. Olson, unpublished data). Fig. 5 shows the evolution of these interactions during stages I–IV. During stage I, the attraction on the hydrophobic interface became stronger. At the beginning of stage II, salt bridges E2351B–R3262A and E2352B–R3261A, nonexistent in the initial structure, were formed (Fig. 5c, I and II). During stage III, the electrostatic bonds were ruptured, R3511A switched to bind to the C terminus of an α-helix in its own dimer (Fig. 5c, III and IV), and the α-helices that formed the hydrophobic interface moved apart. By stage IV, all the interactions discussed were lost.
Fig. 5.

Dissociation of the interdimer lock during the LacI cleft opening by a constant force. (a) The lock is located on the outlined interface between the dimer cores and the four-helix bundle; the key elements of the lock are highlighted as described below. (b) Interactions in the lock. (Top) Van der Waals (orange) and electrostatic (lime) energy of interaction between the α-helices 2221B–2361B and 2222B–2362B, connecting the two dimers. (Middle) Electrostatic energy of the salt bridges R3262A–E2591B (blue) and R3261A–E2592B (magenta). (Bottom) Interaction energy between R3511A and the backbone oxygens of either its own dimer (purple) or the other dimer (green). (c) Snapshots of the interface between the dimers at the different stages of the cleft opening, defined in Fig. 4. The interacting helices (see b Top) are highlighted in orange with the tone corresponding to the strength of the interaction. The key salt bridges (b Middle) are shown by dotted lines that are thick for fully formed bridges and thin for broken bridges. R3511A is highlighted in turquoise, and the backbone sections of either dimer, most closely interacting with R3511A in each snapshot, are highlighted with the same colors as the corresponding plots in b Bottom.
These observations suggest the following mechanism of LacI opening. During stage I, some initial opening of the cleft is allowed by the protein structure but results in straining the interdimeric interface and forming the lock that keeps LacI in the V configuration. During stage II, the pulling force is absorbed for a while by the rotating head groups. During stage III, the cost of head group rotation becomes comparable with the energy of the interdimeric interactions, the bonds in the lock rupture, and LacI opens its cleft. During stage IV, the interaction between the dimers is lost, and LacI dimers move independently (Fig. 4c,IV′), held together only by the four-helix bundle acting as a hinge.
Conclusion
In this work, we combined MD simulations with a previously described multiscale approach (10) to study the structural dynamics and large-scale motions of the LacI–DNA complex, a paradigm system of gene regulation. The protein domains in the constructed structure of the complex showed remarkable stability: Throughout the simulations, they conserved their structure and moved with respect to each other like rigid bodies. The protein head group domains showed significant mobility with respect to the protein core and proved to be able to absorb significant strain from the bound DNA loop. We therefore conclude that head group rotation is an important degree of freedom, essential for LacI function. Our simulations of LacI cleft opening identified the key residues that lock the LacI tetramer in the V configuration during repression of the lac operon genes.
The multiscale method applied here furnished much-needed insight into the large-scale structural dynamics of a complex between a multidomain protein and a DNA loop. Because similar complexes form in the genomes of many living organisms (2, 3), our modeling method shows much promise for future applications.
Supplementary Material
Acknowledgments
We thank L. Mahadevan for earlier collaboration on the elastic rod model of DNA and Mu Gao for stimulating discussions. This work was supported by National Institutes of Health Grant PHS5 P41RR05969-04 and National Science Foundation Grant MCB-9982629; computer time was provided by National Resource Allocations Committee Grant MCA93S028.
Author contributions: E.V., A.B., and K.S. designed research; E.V. performed research; E.V. and A.B. contributed new reagents/analytic tools; A.B. contributed new structure; E.V., A.B., and K.S. analyzed data; and E.V., A.B., and K.S. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: MD, molecular dynamics; RMSD, rms deviation; avRMSD, RMSD value averaged over the entire simulation.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 1Z04).
References
- 1.Berg, J. M., Tymoczko, J. L. & Stryer, L. (2002) Biochemistry (Freeman, New York), 5th Ed.
- 2.Matthews, K. S. (1992) Microbiol. Rev. 56, 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schleif, R. (1992) Annu. Rev. Biochem. 61, 199–223. [DOI] [PubMed] [Google Scholar]
- 4.Lewis, M., Chang, G., Horton, N. C., Kercher, M. A., Pace, H. C., Schumacher, M. A., Brennan, R. G. & Lu, P. (1996) Science 271, 1247–1254. [DOI] [PubMed] [Google Scholar]
- 5.Oehler, S., Eismann, E. R., Krämer, H. & Müller-Hill, B. (1990) EMBO J. 9, 973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedman, A. M., Fischmann, T. O. & Steitz, T. A. (1995) Science 268, 1721–1727. [DOI] [PubMed] [Google Scholar]
- 7.Bell, C. E. & Lewis, M. (2000) Nat. Struct. Biol. 7, 209–214. [DOI] [PubMed] [Google Scholar]
- 8.Edelman, L. M., Cheong, R. & Kahn, J. D. (2003) Biophys. J. 84, 1131–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rief, M., Gautel, M., Oesterhelt, F., Fernandez, J. M. & Gaub, H. E. (1997) Science 276, 1109–1112. [DOI] [PubMed] [Google Scholar]
- 10.Villa, E., Balaeff, A., Mahadevan, L. & Schulten, K. (2004) Multiscale Model Simul. 2, 527–553. [Google Scholar]
- 11.Phillips, R., Dittrich, M. & Schulten, K. (2002) Annu. Rev. Mater. Res. 32, 219–233. [Google Scholar]
- 12.Ayton, G., Bardenhagen, S., McMurtry, P., Sulsky, D. & Voth, G. (2001) J. Chem. Phys. 114, 6913–6924. [Google Scholar]
- 13.Schlick, T. (1995) Curr. Opin. Struct. Biol. 5, 245–262. [DOI] [PubMed] [Google Scholar]
- 14.Olson, W. K. (1996) Curr. Opin. Struct. Biol. 6, 242–256. [DOI] [PubMed] [Google Scholar]
- 15.Balaeff, A., Mahadevan, L. & Schulten, K. (1999) Phys. Rev. Lett. 83, 4900–4903. [Google Scholar]
- 16.Balaeff, A., Mahadevan, L. & Schulten, K. (2004) Structure (London) 12, 123–132. [DOI] [PubMed] [Google Scholar]
- 17.Spronk, C. A. E. M., Bovin, A. M. J. J., Radha, P. K., Melacini, G., Boelens, R. & Kaptein, R. (1999) Struct. Folding Des. 7, 1483–1492. [DOI] [PubMed] [Google Scholar]
- 18.Humphrey, W., Dalke, A. & Schulten, K. (1996) J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- 19.Honig, B. & Nicholls, A. (1995) Science 268, 1144–1149. [DOI] [PubMed] [Google Scholar]
- 20.Kalé, L., Skeel, R., Bhandarkar, M., Brunner, R., Gursoy, A., Krawetz, N., Phillips, J., Shinozaki, A., Varadarajan, K. & Schulten, K. (1999) J. Comp. Phys. 151, 283–312. [Google Scholar]
- 21.MacKerell, A. D., Jr., Bashford, D., Bellott, M., Dunbrack, R. L., Jr., Evanseck, J., Field, M. J., Fischer, S., Gao, J., Guo, H., Ha, D., et al. (1998) J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- 22.Martyna, G., Tobias, D. J. & Klein, M. L. (1994) J. Chem. Phys. 101, 4177–4189. [Google Scholar]
- 23.Deserno, M. & Holm, C. (1998) J. Chem. Phys. 109, 7678–7693. [Google Scholar]
- 24.Balaeff, A., Koudella, C. R., Mahadevan, L. & Schulten, K. (2004) Philos. Trans. R. Soc. London A 362, 1355–1371. [DOI] [PubMed] [Google Scholar]
- 25.Hagerman, P. J. (1988) Annu. Rev. Biophys. Biophys. Chem. 17, 265–286. [DOI] [PubMed] [Google Scholar]
- 26.Strick, T. R., Allemand, J.-F., Bensimon, D., Bensimon, A. & Croquette, V. (1996) Science 271, 1835–1837. [DOI] [PubMed] [Google Scholar]
- 27.Isralewitz, B., Gao, M. & Schulten, K. (2001) Curr. Opin. Struct. Biol. 11, 224–230. [DOI] [PubMed] [Google Scholar]
- 28.Karplus, M. & Petsko, G. A. (1990) Nature 347, 631–639. [DOI] [PubMed] [Google Scholar]
- 29.Snow, C. D., Nguyen, H., Pande, V. S. & Gruebele, M. (2002) Nature 420, 102–106. [DOI] [PubMed] [Google Scholar]
- 30.Dominy, B. N. & Brooks, C. L., III (2001) J. Comp. Chem. 23, 147–160. [DOI] [PubMed] [Google Scholar]
- 31.Howard, J. (2001) Mechanics of Motor Proteins and the Cytoskeleton (Sinauer, Sunderland, MA).
- 32.Gao, M., Craig, D., Vogel, V. & Schulten, K. (2002) J. Mol. Biol. 323, 939–950. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.