

Abstract
3-O-Sulfogalactosylceramide (sulfatide) is a sphingolipid constituting about 4% of myelin lipids in the central nervous system. Previously, our group characterized a mouse with sulfatide’s synthesizing enzyme, cerebroside sulfotransferase (CST), constitutively disrupted. Consequently, these constitutive CST knockout (KO) mice were incapable of synthesizing sulfatide. Using these mice, we demonstrated that sulfatide is required for establishment and maintenance of myelin, axoglial junctions and axonal domains and that sulfatide depletion results in structural pathologies commonly observed in Multiple Sclerosis (MS). Interestingly, sulfatide is reduced in regions of normal appearing white matter (NAWM) of MS patients. Sulfatide reduction in NAWM suggests that depletion occurs early in disease development and consistent with functioning as a driving force of disease progression. To more closely model MS, an adult onset disease, our lab generated a “floxed” CST mouse and mated it against the PLP-creERT mouse, resulting in a double transgenic mouse that provides temporal and cell-type specific ablation of the CST gene. Using this mouse, we demonstrate that adult-onset sulfatide depletion has limited effects on myelin structure but results in the loss of axonal integrity including deterioration of domain organization accompanied by axonal degeneration. Moreover, the structurally preserved myelinated axons present with conduction velocities consistent with unmyelinated axons. Together, our findings indicate that sulfatide depletion, which occurs in the early stages of MS progression, is sufficient to drive the loss of axonal function independent of demyelination and that axonal pathology, which is responsible for the irreversible loss of neuronal function that is prevalent in MS, may occur earlier than previously recognized.
Keywords: sulfatide, myelin, axon degeneration, multiple sclerosis
1. Introduction
Multiple Sclerosis (MS), a demyelinating, autoimmune disorder of the central nervous system (CNS) that is mostly commonly diagnosed in the third and fourth decades of life (Gbaguidi et al., 2022; Walton et al., 2020), is characterized by sensory, motor, and cognitive dysfunction. MS affects nearly one million people in the United States(Wallin et al., 2019) and nearly 2.5 million worldwide. Currently, there is no cure for MS and treatments exhibit limited efficacy(Piehl, 2021). While understanding of pathogenesis is largely immune focused (O’Connor et al., 2001; Rodríguez Murúa et al., 2022), the exact disease etiology remains unknown. Although immune activation is a consistent aspect of MS, other factors implicated in disease onset and progression include environment, gender, genetics, and viral exposure (Bjornevik et al., 2022; Olsson et al., 2017).
Although a multifaceted disease, an interesting and consistent observation is the dysregulation of lipid metabolism in the CNS (Podbielska et al., 2022; Wheeler et al., 2008a). Marbois et al. (2000), using ion electrospray mass spectrometry, reported a 25% reduction specifically of the myelin sphingolipid sulfatide in normal appearing white matter (NAWM) compared to non-MS brains confirming a much earlier report of specific sulfatide depletion in MS (Yahara et al., 1982). Additionally, Wheeler (Wheeler et al., 2008a) reported a significant increase in the phospholipid/sulfatide ratio in both normal appearing white and normal appearing grey matter in MS brains as compared to non-MS brains while Moscatelli and Issacson (1969) (Moscatelli & Isaacson, 1969) reported an increase in sphingosine, a metabolite of sulfatide breakdown. Together, these findings indicate that sulfatide depletion in MS occurs prior to demyelination temporally positioning sulfatide loss as a causative event in disease onset and progression.
Myelin is essential for proper transduction of electrical signals in axons by acting as an insulator through its unique lipid rich biochemical composition (Huxley & Stämpeli, 1949). In addition to its structural role, myelin also metabolically supports the axon and maintains overall axonal integrity(Lee et al., 2012). During MS onset, the myelin sheath is attacked by CD4+ myelin reactive T cells resulting in demyelination and/or axonal loss, creating areas of plaque matter; while areas of NAWM supposedly are undisturbed. However, a growing body of literature suggests the NAWM contains axonal pathologies suggesting that the irreversible functional loss that has been attributed to axonal degeneration(Trapp et al., 1998) may precede demyelination in contrast to axonal pathology as a consequence of myelin loss (Gallego-Delgado et al., 2020a; Luchicchi et al., 2021b). Therefore, it is imperative to develop a better understanding of the pathological events that occur in the earliest stages of disease.
To delineate the role that sulfatide plays in both a healthy and diseased CNS, our group generated(Honke et al., 2002) and characterized a mouse with a disruption in the cerebroside sulfotransferase (CST) gene(Ishibashi et al., 2002a; Marcus et al., 2006; A. Pomicter et al., 2013; Shroff et al., 2009), which encodes the enzyme that catalyzes the final step of sulfatide synthesis(Afshin Yaghootfam et al., 2007; Honke et al., 1997). This global, constitutive CST knockout (KO) mouse displays progressive myelin pathologies that are observed in MS brains(Gallego-Delgado et al., 2020a; Luchicchi et al., 2021b; Suzuki et al., 1969; York et al., 2021) including (1) thin, unstable, and uncompacted myelin sheaths (Marcus et al., 2006); (2) ultrastructurally compromised nodal and paranodal domains (Honke et al., 2002; Marcus et al., 2006); (3) abnormal and unstable clustering of nodal and juxtaparanodal ion channels (Ishibashi et al., 2002a) and (4) reduction of major myelin proteins and lipids (Palavicini et al., 2016). Thus, sulfatide is implicated in the establishment and maintenance of myelin and axonal structure and function(Hayashi et al., 2013a). Although a valuable resource for elucidating functions of sulfatide, the constitutive CST KO mouse develops in the absence of the lipid. To overcome potential developmental confounds, we have generated a CST floxed (flanking loxP; CSTfl/fl) mouse capable of age- and cell type- specific sulfatide depletion. We mated the CSTfl/fl mouse with a mouse that expresses tamoxifen-inducible Cre recombinase under the control of the oligodendrocyte (OL)-specific proteolipid protein (PLP) promoter(Belachew et al., 2001; Mallon et al., 2002; Wight & Dobretsova, 2004). Here, we exploit this novel double transgenic mouse to determine the causal relationship between adult onset sulfatide depletion and myelin and axonal structure and function.
2. Materials and Methods
2.1. Animal Model
Male and female CST inducible knockout mice, generated on the c57black6J background (see below), were bred, housed and aged in the Central Virginia Veterans Affairs Health Care Systems AAALAC accredited vivarium on a 12-hour light/dark cycle with food and water provided ad libitum.
2.2. Generation of CST conditional knockout mouse
Using CRISPR technology, we generated cerebroside sulfotransferase (CST) floxed (flanking loxP) mice (Applied StemCell, Inc, Milpitas, CA). Based on transcript sequence information in Ensembl (Transcript ID MGP_C57BL6NJ_T0028206.1), and consistent with published sequence analysis (Hirahara et al., 2000; Honke et al., 2002), the CST gene consists of 3 exons. Exon 1 is non-coding with variable regions termed 1a- 1g. Exons 2 and 3 constitute the coding regions. LoxP sequences were inserted in introns up and down stream of exons 2 and 3 (Fig. 1A). Guide RNAs, two single-stranded oligodeoxynucleotide donors and designed Cas-9 mRNA, were injected into the cytoplasm of C57BL/6J embryos. Resulting pups were screened for loxP sites at the designated locations using PCR followed by sequencing to confirm that the CST gene locus contained the loxP sites in the correct location and orientation (not shown). To specifically target ablation of the CST gene in oligodendrocytes in the CNS, we purchased PlpCreERT mice (Jax Labs; stock # 005975) to induce recombination and mated them to the homozygous CST floxed (CSTfl/fl) line. To generate mice used in all subsequent studies, we mated homozygous CSTfl/fl mice with heterozygous PlpCreERT−/+;CSTfl/fl mice, creating both PLPcreERT−/− (no cre):CSTfl/fl and PlpCreERT−/+;CSTfl/fl. All experimental mice were PlpCreERT−/+;CSTfl/fl (abbreviated as CST-cKO) and PLPcreERT−/−;CSTfl/fl were used as controls (abbreviated as CTL). All mice received tamoxifen treatment (see below).
FIGURE 1. Genetic ablation of the cerebroside sulfotransferase (Cst) gene. (a) Schematic illustrating the Cst gene with loxP sites (solid black triangles) upstream and downstream of coding regions exons 2 and 3.

Three primers (striped triangles) were designed around the loxP sites to detect for genetic ablation: F (forward) primer 1, R (reverse) primer 2, and R (reverse) primer 1. Tamoxifen was administered to both CST-cKO and CTL mice, resulting in Cst gene recombination in CST-cKO mice (left; ablated gene product indicated by red line) with gene sparing (right; intact gene product indicated by red line) in CTL mice. (b) PCR amplification of genomic DNA from the CTL and CST-cKO mice confirmed recombination in CST-cKO mutant mouse brains 6 weeks post tamoxifen injection. (c) Real-Time PCR of CTL and CST-cKO mutant mouse brains 6 weeks post tamoxifen injection revealed ~50% reduction in mRNA expression. (d) Lipidomic analysis of total sulfatide in CST-cKO and CTL brains presented no change in sulfatide levels by 3-months PI but a significant sulfatide loss at 6 and 11 months PI. Unpaired Student’s t-test. All data are presented as mean ± standard error. **p < .005 ****p < .0001.
2.3. Induction of Cre-mediated Recombination with Tamoxifen
Mice were intraperitoneally injected with 60mg/kg of tamoxifen based on previous work from our group (Qui et al., 2021). Tamoxifen was diluted in corn oil, and delivered through intraperitoneal injection into 10-week-old PlpCreERT−/+;CSTfl/fl and PLPcreERT−/−;CSTfl/fl mice for four consecutive days.
2.4. Polymerase Chain Reaction – CST gene ablation assessment
To confirm Cre-mediated recombination, genomic DNA was extracted from corpus callosum from 6-week post tamoxifen injected mice using Qiagen DNeasy Mini kit (Qiagen, Germantown, MD) according to the manufacturer’s instructions. Briefly, Mice were deeply anesthetized using 0.016 mL/gm body weight of a 2.5% solution of avertin (2, 2, 2 tribromoethanol; Sigma-Aldrich; St. Louis, MO; cat#T48402) in 0.9% sodium chloride (Sigma-Aldrich, St. Louis, MO), and transcardially perfused with ice-cold saline for 3 minutes. The brain was harvested; sectioned into 1mm coronal slices which were used for corpus callosum isolation; snap-frozen in liquid nitrogen and stored at −80C. Snap frozen tissue was used for DNA ablation assessment and mRNA quantitation. DNA fragments spanning genomic target sites were amplified by PCR using the following primers:
GAL3ST1 Forward (5’- GATTGTAGCCTTCCGTATGAACCG −3’)
GAL3ST1 Reverse 1 (5’- CGAACTCAACTCAAAGAGAGCAGG −3’) and
GAL3ST1 Reverse 2 (5’- TAATCTCTGCTCTAACCTGGTCGC −3’).
The GAL3ST1 Forward primer targets upstream of the first flanking loxP site and GAL3ST1 Reverse 1 primer targets downstream of the first flanking loxP site; the GAL3ST1 Reverse 2 primer targets downstream of the second flanking loxP site, such that when recombination does not occur, the size of the potential product between Forward and Reverse 2 primers is too large to amplify. However, after cre-mediated recombination, the distance between these primer target sites is shortened and a 432bp band is detected (Fig. 1A). If no recombination occurs, the Forward and Reverse 1 primer will detect a product of 246bp (Fig 1A). Cycling parameters were one cycle at 95°C (3 min), 40 cycles of 95°C (30 s), 60°C (30 s), and 72°C (1 min 30 s), then 72°C (5 min), and a final hold at 4°C. PCR amplified products were analyzed by agarose gel electrophoresis (Fig. 1B).
2.5. Real Time-Polymerase Chain Reaction – CST mRNA quantitation
Total RNA was extracted from isolated corpus callosi using a Qiagen RNeasy Micro kit (Qiagen, Germantown, MD) according to manufacturer’s instructions. Contaminating DNA was eliminated through treatment with Ambion DNase I (Invitrogen Life Technologies, Grand Island, NY). Omniscript Reverse Transcription Supermix (BioRad, Hercules, CA) was used to create cDNA from the isolated RNA (150μL/sample). Quantitative RT-PCR was performed with a CFX96 (BioRad, Hercules, CA) RT-PCR detection system using 1 μL of cDNA, SsoFast Evagreen Supermix (BioRad), and the following primers (20μM):
GAL3ST1 Forward (5’- GCAGCACACTGCTCAACATC −3’)
GAL3ST1 Reverse (5’- ACCAGGCTTCGTGCAAAGTA −3’)
Cyclophilin A Forward (5’- CTAGAGGGCATGGATGTGGT −3’)
Cyclophilin A Reverse (5’- TGACATCCTTCAGTGGCTTG-3’)
Phosphoglycerate kinase 1 Forward (5’- ATGCAAAGACTGGCCAAGCTA −3’)
Phosphoglycerate kinase 1 Reverse (5’-AGCCACAGCCTCAGCATATTT-3’)
Cycling parameters were: 1 cycle at 95°C (5 min), 39 cycles of 95°C (5 s) and 56°C (5 s) followed by a melt curve measurement consisting of 5 s 0.5°C incremental increases from 65°C to 95°C. The fold changes in expression of the CST genes in corpus callosum samples were calculated using the formula RQ 5 22DDCt, using Cyclophilin and Phosphoglycerate kinase 1. For statistical analysis, a student’s t-test was performed using GraphPad Prism software version 9.4.1 for Windows.
2.6. Mass spectrometry
Multidimensional mass spectrometry-based shotgun lipidomics analysis was performed as described in Qiu et al. (2021). Briefly, mice were injected with Avertin and perfused with ice cold saline. The brains were sliced into 1 mm coronal slices and corpus callosi was dissected and flash frozen in liquid nitrogen. Tissue was homogenized and protein concentrations was determined using Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA). In the presence of internal standards, lipids were extracted following a modified procedure of Bligh and Dyer. Lipids were measured using a triple-quadrupole mass spectrometer (TSQ Altis, (Thermo Fisher Scientific, Waltham, MA, USA) equipped with a Nanomate device (Advion Ithaca, NY, USA) and Xcalibur system. Data were processed using ion peak selection, baseline correction, data transfer, peak intensity comparison, 13C deisotoping. Data were analyzed using a custom-programmed Microsoft Excel macro.
2.7. Immunohistochemistry
Tissue was processed as previously described (Clark et al., 2016; Dupree et al., 1999; Shepherd et al., 2012). Briefly, mice were deeply anesthetized using 0.016 mL/gm body weight of a 2.5% solution of avertin in 0.9% sodium chloride, and transcardially perfused with 4% paraformaldehyde (Ted Pella, Redding, CA; cat#18501 ) in 0.1M Millonigs buffer (Dupree et al., 1999; Shepherd et al., 2012). Following perfusion, the brains were cryopreserved in 1X PBS containing 30% sucrose for 48 hours, frozen in Optimal Cutting Temperature compound (Fisher Scientific; Hampton, NH, cat# 23–730-571), and serially sectioned, spanning 1.1 mm anterior to bregma to 2.5 mm posterior to bregma, at 40 μm in a coronal orientation using a Leica CM 1850 cryostat (Leica, Buffalo Grove, IL). Fifteen sets of six sections were collected and placed on ProbeOn Plus slides (Fisher Scientific, Loughborough, UK; cat# 15–188-51) and stored at −80°C. Adapted from Dupree et (1999) and Shepherd et al. (2012) (Dupree et al., 1999; Shepherd et al., 2012), coronally sectioned brains were triple labelled with Caspr at a dilution of 1:500 (rabbit polyclonal, Abcam cat# ab34151), Kv1.1 at a dilution of 1:750 (mouse monoclonal IgG2b, Antibodies Inc cat#75–105) and Nav1.6 at a dilution of 1:250 (mouse monoclonal IgG1, Antibodies Inc cat #75–026 ); each antibody is knock-out validated. After overnight primary antibody incubation at 4°C, the sections were rinsed in PBS, blocked in blocking solution (0.5% triton X-100, 10% cold water fish skin gelatin (Aurion, Netherlands; cat# 900.033) in PBS) as previously described (Benusa et al., 2017; Clark et al., 2016) and incubated with the appropriate, fluorescently tagged Alexa™fluor secondary antibodies (Invitrogen Life Technologies, Grand Island, NY) diluted 1:500. Slides were cover slipped with Vectashield™ (Vector Laboratories, Newark, CA, cat# H-1000) and stored at −80°C until imaged. For immunohistochemistry (IHC) analysis, an n=3–7 mice were used per sex per genotype per timepoint (see Table 1).
TABLE 1.
Sample size breakdown per timepoint
| Timepoint | Genotype | Sex | EM | IHC | EP |
|---|---|---|---|---|---|
| 3 | CTL | Female | 3 | 3 | 3 |
| Male | 3 | 4 | 4 | ||
| CST-cKO | Female | 3 | 3 | 3 | |
| Male | 3 | 4 | 3 | ||
| 6 | CTL | Female | 3 | 3 | 4 |
| Male | 3 | 4 | 3 | ||
| CST-cKO | Female | 3 | 3 | 4 | |
| Male | 3 | 6 | 5 | ||
| 11 | CTL | Female | 3 | 3 | 4 |
| Male | 3 | 4 | 3 | ||
| CST-cKO | Female | 3 | 4 | 3 | |
| Male | 4 | 7 | 5 |
Abbreviation: EM, Electron Microscopy; EP, Electrophysiology; IHC, Immunohistochemistry.
2.8. Image collection and Quantitation
All images were collected using a Zeiss LSM 880 confocal laser scanning microscope (Carl Zeiss Microscopy, LLC; White Plains, NY), housed in the VCU Microscopy and Imaging Facility. Confocal z-stacks, each spanning 3μm, using a pin hole of 1 Airy disc unit and Nyquist sampling, were collected from the corpus callosum at the level of the fornix. Six images were collected per animal and a minimum of 150 nodal regions were quantitatively analyzed per animal. Images were taken with a 63X oil-immersion objective with a numerical aperture of 0.55; optical slice thickness was 0.21 μm using a line scanning average of 4. X, Y, and Z dimensions were 67μm × 67μm × 3μm, respectively. The magnification was digitally increased by a factor of 2 using the zoom feature. The gain and offset were kept consistent for all images. The 405 laser detector gain was approximately 700; the 488 laser set at 750 detector gain, and the 594 laser set at 780 detector gain. For quantitation, criteria were established to quantify overlapping, absent, and/or aberrant placement of nodal protein fluorescent signals. Nodal abnormalities were quantified using ImageJ analysis software by manually marking overlapping fluorescent labels from maximum intensity projection images. All analyses were conducted blinded from experimental group and sex. Fluorescently labelled domains that touched the edge of the image were excluded from analysis. We quantitatively analyzed age-matched CST-cKO and control mice at 3-, 6-, and 11-month post tamoxifen injection. Analysis was performed using a 2-way ANOVA comparing genotypes across time. All graphing and statistical analysis were performed using GraphPad Prism version 9.4.1 for Windows (GraphPad Software, San Diego, CA).
2.9. Electron Microscopy
For ultrastructural quantitation, mice were processed for transmission electron microscopic analyses as previously described by (Dupree, Coetzee, Blight, et al., 1998a; Dupree, Coetzee, Suzuki, et al., 1998; Dupree et al., 1999; Marcus et al., 2006). Briefly, mice were deeply anesthetized, as described above, and transcardially perfused with a solution of 0.1M Millonigs buffer containing 4% paraformaldehyde and 5% glutaraldehyde (EM sciences, Hatfield, PA cat#16310). The perfusions were followed by a 2-week incubation in the same fixative solution at 4°C. Brains were harvested, vibratome- sectioned at 100 μm to generate both coronally and sagittally oriented samples of the corpus callosum at the level of the fornix. Comparable sections were selected and postfixed in 1% osmium tetroxide (EM sciences, Hatfield, PA; cat# 20816–12-0), dehydrated in increasing concentrations of ethanol, followed by incubation in the transition solvent propylene oxide (EM sciences, Hatfield, PA cat# 20412) and embedded in PolyBed epoxy resin (PolySciences, Warrington, PA; cat#08791–500). One micron and ultrathin (70nm) sections were stained with toluidine blue or the heavy metals uranyl acetate and lead citrate, respectively. One-micron sections were imaged using a Nikon ECLIPSE E800M upright light microscope (Nikon Instruments, Melville, NY), which is housed in the VCU Microscopy and Imaging Facility, to confirm region of interest and quality of tissue preservation. The ultrathin sections were used for myelin and axonal analyses and imaged using a JEOL JEM-1400Plus (JEOL USA, Inc, Boston, MA) equipped with a Gatan One View 1095 T and Gatan Microscopy Suite (GMS) 3.3.2 software.
For g-ratio analysis, a minimum of 100 myelinated axons per mouse at the level of the fornix, with a diameter > 0.3 μm (Mason et al., 2001), were used for quantitative analysis. Images were collected at magnification of 7,200X from the sagittally oriented sections. For each myelinated axon, 2 axon diameters (the longest and shortest), and two myelin widths (the widest and the thinnest) were measured using NIH ImageJ (Marcus et al., 2006). Only myelin regions that exhibited no sign of fixation artifact or non-compaction were used for determining myelin thickness. Also, regions containing either the inner or outer tongues were not used for g ratio analyses. To calculate g-ratio, the average diameter for each axon was divided by the average axon diameter plus the sum of the myelin widths. For all EM analysis an n=3–4 was used per sex per genotype per timepoint (see Table 1). Analysis was performed using a 1-way ANOVA for average values per animal, comparing genotypes across time. All graphing and statistical analysis were performed using GraphPad Prism version 9.4.1 for Windows (GraphPad Software, San Diego, CA).
To quantify myelin and axon integrity, the same electron micrographs used for g-ratio analysis were used. These images were used to assess the percent of myelinated versus unmyelinated axons. Only axons with a diameter equal to or greater than 0.3um (Mason et al., 2001; Marcus et al., 2006) were included in the quantitation. Additionally, the prevalence of myelin and axon pathology was quantified for presence of redundant myelin, myelin non-compaction, myelin pulling away from the axon, and axonal degeneration as previously described (Dupree et al., 2015). Analysis was performed using a 2-way ANOVA comparing genotypes across time. All graphing and statistical analyses were performed using GraphPad Prism version 9.4.1 for Windows (GraphPad Software, San Diego, CA).
Coronally oriented ultrathin sections were imaged at 7,2000X for nodal regions. A minimum of 10 nodal regions were imaged per mouse to quantify node of Ranvier length. For statistical testing, the fit of the two models (single variance/covariance matrix or group specific variance/covariance matrices) was compared using a likelihood ratio test (LRT). For all analyses and α = 0.05 was used for statistical significance and SAS v9.4 was used for all analyses. Qualitative assessment of paranodal-axonal junctions (transverse bands) and organization and orientation of paranodal loops were also conducted.
2.10. Electrophysiology
Mice were initially anesthetized with the volatile anesthetic isoflurane followed by an intraperitoneal injection of ketamine (200 mg/kg) and xylazine (20 mg/kg). Mice were then transcardially perfused with ice-cold sucrose artificial cerebrospinal fluid (sucrose-ACSF), which contained (in mM): 206 sucrose, 25 glucose, 25 NaHCO3, 4 MgSO4, 3 KCl, 1.25 NaH2PO4, 1.2 CaCl2 saturated with 95% O2 and 5% CO2. Following perfusion, mice were decapitated and the brain removed to produce ex vivo brain slices. Coronal brain slices of 350μm thickness were sectioned in the same ice-cold sucrose-ACSF using a vibratome (Leica VT1200). Slices between 1.4 mm posterior and 2.5 mm posterior to bregma were collected for recording and maintained in an incubation chamber for at least 30 min before recording. The incubation chamber contained room temperature ACSF (~21° C) consisting of (in mM): 125 NaCl, 25 NaHCO3, 25 glucose, 3 KCl, 1.2 NaH2PO4, 1.2 CaCl2, 1.2 MgSO4 bubbled with 95% O2 and 5% CO2. For recording, slices were transferred to a recording chamber mounted on the stage of an Olympus BX51WI microscope (Olympus Life Science Solutions, Bartlett, TN) equipped with a 10x (0.3 NA) water immersion objective and perfused with oxygenated room temperature ACSF. Where indicated, ACSF was supplemented with 4-aminopyridine (4-AP, 100 μM (Sigma Aldrich; St. Louis, MO cat# 275875)) or tetrodotoxin (TTX 100 uM (Sigma-Aldrich; St. Louis, MO cat# 554412)) as previously described (Bell et al., 2021). For electrophysiology studies, an n = 3–5 mice per sex per genotype per timepoint was used (Table 1).
To stimulate corpus callosal fibers, a bipolar stimulating electrode (cat #30250 matrix electrode; FHC Neural Microtargeting, Bowdoin, ME) was placed approximately 1 mm from a glass micropipette recording electrode (1.65 mm OD, 1.0 mm ID, 8250 capillary glass, King Precision Glass, Claremont, CA). Micropipettes were pulled on a horizontal electrode puller (P1000; Sutter Instruments, Novato, CA) to produce tip diameters of approximately 2 μm that resulted in ~3 MΩ resistances when filled with ACSF. Compound action potentials (CAP) were evoked with a DS3 Isolated Current Stimulator (Digitimer) and measured with a Model 2400 patch clamp amplifier (A-M Systems; Sequim, WA). The resultant extracellular analog signals were converted into a digital signal by a PCI-6040E A/D board (National Instruments, Austin, TX), and stored and analyzed on a PC computer using WCP Strathclyde Software (courtesy of Dr. J Dempster, Strathclyde University, Glasgow, Scotland). CAPs were elicited using a stimulation protocol consisting of five consecutive sweeps, 100μs in duration, with a 10s delay between sweeps ranging from 100μA to 500μA. An average of three recordings from one slice was used to reduce signal to noise ratio. CAPs were analyzed using the WinWCP program (courtesy of Dr. J Dempster, Strathclyde University, Glasgow, Scotland). Graphpad prism (San Diego, CA) was used to graph the ratio of N1 (myelinated axons) to N2 (unmyelinated axons) and for statistical analysis. Analysis was performed using a 2-way ANOVA comparing genotypes across stimulus current. Representative traces were superimposed using Origin2020b software (Origin, Northampton, MA).
3. Results
3.1. Confirmation of gene ablation and lipid reduction following tamoxifen injection of the conditional CST knock-out model
Studies have reported that MS patients present with a significant reduction of the myelin lipid known as sulfatide even in regions of NAWM (Marbois et al., 2000; Yahara et al., 1982). To determine the consequence of adult-onset sulfatide reduction, we generated a conditional CST knockout (CST-cKO) mouse using CRISPR technology. Fig. 1A depicts the modified CST gene with loxP sites inserted in non-coding regions and flanking exons 2 and 3. We designed and positioned primers up- and downstream of the loxP sites to detect ablation. Fig. 1A displays gene products after tamoxifen injection of both the CST-cKO and control with the CST-cKO yielding a PCR product of 432 bp, which is the product size consistent with gene ablation. Fig. 1B demonstrates successful ablation of the CST gene in the CST-cKO mice having both ablated and non-ablated products. The presence of both ablated and non-ablated products is consistent with: 1. starting material of the corpus callous, which also contains non-oligodendrocytic cells and 2. less than 100% ablation efficiency of oligodendrocyte lineage cells. To further assess ablation efficiency, we isolated mRNA. Fig. 1C demonstrates significantly reduced transcript levels for CST message in the CST-cKO corpus callosum compared to message levels of the control animals. Our gene and message analyses demonstrate gene ablation accompanied with >60% message reduction.
Although our findings confirm both appropriate gene ablation and message reduction, these data do not directly assess lipid levels. To quantify sulfatide amounts, we isolated corpus callosi from CST-cKO and control mice at 3-, 6-, and 11-months post tamoxifen injection (PI). These time points were chosen since sulfatide requires ~4 months to turnover(Hayes & Jungawala, 1976; Norton & Cammer, 1984). At 3 months PI, there was no significant difference in relative sulfatide levels between the CST-cKO and control mice (Fig. 1D). By 6 months PI, we observed a significant and consistent reduction of the lipid with sulfatide levels in the CST-cKO mice only reaching 40% of the sulfatide levels in the controls. By 11 months PI, sulfatide levels remained significantly depleted and the mice presented with an additional 10% reduction indicating a sustained and progressive loss of the lipid. Importantly, consistent with our group’s previous analyses(Qiu et al., 2021), these findings confirm sulfatide reduction and reveal no difference in the levels of galactosylceramide, the precursor of sulfatide (Morell & Radin, 1969) at any of the PI time points (data not shown). Taken together, our findings demonstrate that the novel CST floxed mouse provides a unique opportunity to ablate the CST gene and specifically reduce sulfatide levels in the adult CNS.
3.2. Progressive axonal pathology with myelin sparing
Previously, our group reported that in the constitutive CST KO mice, myelin sheaths were significantly thinner than in their wildtype littermates. We also observed a variety of myelin pathologies including uncompacted, degenerating, and redundant myelin profiles (Marcus et al., 2006). To determine if adult-onset sulfatide depletion resulted in similar pathologies, we used EM analysis to calculate g ratios in the corpus callosum. Figure 2A i-vi presents representative electron micrographs for each genotype at each PI time point. Figure 2B compares the average g ratio values obtained for individual mice and individual axons. Using a 1-way ANOVA, there was no significant difference among the genotypes at each timepoint (3m p=0.8938, 6m p=0.9483, 11m p=0.4794). Since sulfatide is reduced in MS and MS is 3 times more prevalent in females than males(Koch-Henriksen & Sørensen, 2010), we further assessed g ratio, with sex as a biological variable. Binning the values per sex, also revealed no sex differences at any of the experimental endpoints (data not shown).
FIGURE 2. No change in extent of myelination in the corpus callosum of CST-cKO mice.
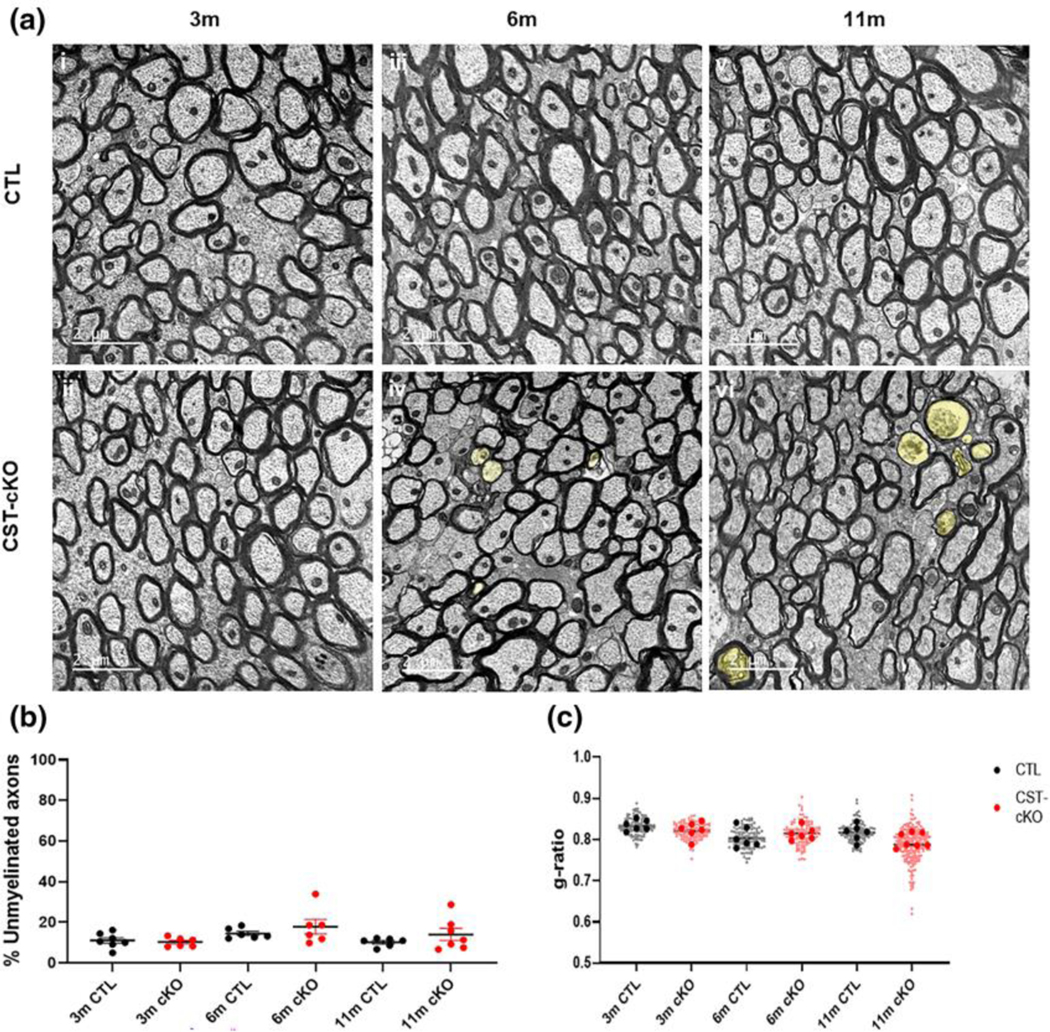
(a) Cross section representative images from the corpus callosum at the level of the fornix from control (CTL; ai, aiii and av) and CST-cKO (aii, aiv and avi) mice following 3- (ai and aii), 6- (aiii and aiv) and 11- (av and avi) months post injection. 3-, 6- and 11- months PI mice presented abundant myelinated axons with no difference in the percent of myelinated versus unmyelinated fibers (b) or in myelin thickness as quantitatively compered by g ratio analysis. (c) Note axonal pathology (yellow shade) in the 6 m- and 11 m-PI tissue. (examples highlighted by yellow shade in Panels aiv and avi). Data are presented as mean ± standard error; data statistically compared by 2-way ANOVA analysis followed by a Tukey’s post-hoc multiple comparison test. Scale bar = 2 μm.
Previous studies have reported that myelin and axon defects are more prone to axons of specific caliber (Thomason et al., 2022). Therefore, we used a similar binning approach (increments of 0.1μm) to assess axonal caliber-specific pathologies; however, no difference in g-ratios, axon caliber, or myelin thickness was detected (data not shown).
To determine if demyelination or myelin instability occurs following adult-onset sulfatide depletion, we compared the percent of myelinated and unmyelinated axons in the corpus callosi of mice with normal and depleted levels of sulfatide. Across all timepoints, there was no significant difference in the percent of unmyelinated fibers between the two genotypes (Fig. 2C) (3m p=0.9891, 6m p=0.6478, 11m p=0.5010). To further assess the consequence of adult onset sulfatide depletion on CNS integrity, we quantified myelin and axonal pathologies including: 1. myelin un-compaction, 2. myelin pulling-away, 3. redundant myelin profiles and 4. degenerating axonal fibers. The compilation of these pathologies revealed a significant increase in the CST-cKO mice reaching statistical significance at 11 months post injection (Fig. 3B) (p=0.0018) indicating a loss of overall myelin/axon integrity following sulfatide depletion. Interestingly, when separating the four pathologies, there was no statistical difference at 11 months PI for any of the myelin pathologies((un-compaction p=0.9541; pulling away (p=0.9996); redundant myelin (p=0.9986) (Fig. 3C–E)). These data imply that physiological sulfatide levels in adulthood are not essential for proper myelin structural maintenance. However, there was a robust and significant difference in the extent of axonal degeneration at 11 months post injection (Fig. 3F) (p=<0.0001) (3m p= 0.9988, 6m p= 0.3068) suggesting that loss of sulfatide facilitates axonal pathology independent of myelin loss.
FIGURE 3. Quantification of myelin and axonal pathologies in the corpus callosum of CST-cKO mice.

(a) Representative image of a healthy axon with an intact, compacted myelin sheath and appropriate array of neurofilament and microtubules (ai). Representative images of various myelin and axonal abnormalities observed in the CST-cKO mouse included (from left to right): axon losing attachment to the myelin sheath (aii vacuolar degeneration; yellow arrows); presence of cytoplasm between compacted wraps of myelin (aiii; un-compaction; yellow star); compact myelin out folding (aiv; redundant myelin; white arrows) and a variety of axonal degenerating profiles (f) including (fi) organelle collapse; red arrow, (fii) presence organelle and cytoskeletal debris from the axon; white star, and (fiii) complete loss of axon exhibited by an empty myelin shell; black star. (b) Quantitative analysis combining all myelin and axonal pathologies revealed a significant increase in myelin/axonal pathologies at 11 m PI but not at 3- and 6- months PI. Separating out the individual pathologies revealed (c-e) no significant difference in myelin ultrastructure between genotypes at any of the timepoints analyzed; however, (g) axonal degeneration was significantly increased in the 11 m CST-cKO mice. All data were statistically compared using a 2-way ANOVA; ** p < .005 ****p < .0001. Scale bar = 1 μm.
3.3. Variation in Nodal Length
Although no significant difference in myelin integrity was observed by our myelin ultrastructural analyses, axonal pathology was abundant. Consistent with sulfatide playing a role in maintaining axonal integrity, our previous analyses of the constitutive CST KO mice revealed abnormal lengths of the nodes of Ranvier (Marcus et al., 2006). Therefore, using a similar approach, we measured the length of the nodes of Ranvier in the conditional CST KO mice at the three PI time points. As shown in Fig. 4C, CST-cKO mice did not have a significant difference in the length of the nodes at 3, 6, or 11 months post injection, (3m control 1.548μm ± 0.175μm vs. 3m CST-cKO 1.236 μm ± 0.106 μm; p=0.8269; 6m control, 1.065μm ± 0.096μm vs. 6m CST-cKO, 1.038μm ± 0.120 μm; p=0.9999; and 11m control, 1.047μm ± 0.111 vs. 1.575μm ±0.479 p=0.3780). Using a variability analysis, we see no significant difference in the variability at 3 or 6 months post injection. (3m p=0.7047 and 6m p=0.8607). However, at 11 months post injection, there was a significant increase in variability (p<0.0001).
FIGURE 4. No change in extent of myelination in the dorsal column of the cervical spinal cord following adultonset sulfatide depletion.

Quantitative analysis of the dorsal column of the spinal cord revealed no difference in the percent of myelinated versus unmyelinated axon (a), or myelin thickness as calculated by g ratios (b). Similar to the corpus callosum, quantitative analysis of the combined axonal and myelin pathologies revealed a significant increase in the CST-cKO mice at PI 11-months but not at 3- and 6-months PI (c). However, when the individual pathologies were compared, myelin pathologies were comparable between genotypes (d-f) while axonal degeneration was significantly more abundant in the CST-cKO at 11-months PI compared to 11-months PI CTL mice (g). All data were statistically compared using a 2-way ANOVA; ** p < .005 ****p < .0001.
To further investigate this increased variability in node length, we focused our attention on the structural integrity of the paranode, which flanks the node of Ranvier defining the nodal gap (Peters, 1966). A prominent structural component of the paranodal junctions are the electron dense transverse bands, which are composed of glial and neuronal adhesion molecules (Schnapp et al., 1976). We and others have previously proposed that a function of the transverse bands is to maintain adherence between the myelin sheath and the axon resulting in the maintenance of myelin integrity and axon-myelin communication, organized axonal domains and overall axonal health (Coman et al., 2006; Dupree, Coetzee, Blight, et al., 1998a; Mierzwa et al., 2010a; Pillai et al., 2009; Rosenbluth et al., 2003; Susuki et al., 2018). Additionally, we reported compromised paranodal junctions in the constitutive CST KO mice (Honke et al., 2002; Marcus et al., 2006). Therefore, we proposed that increased variability in nodal length could be a consequence of compromised junctions as evidenced by a deterioration of transverse bands. In the healthy CNS, EM analysis reveals transverse bands as regularly spaced electron densities that span the periaxonal space of the paranode(Peters et al., 1992) (Fig. 4D) Consistent with our previous analyses of the constitutive CST KO mice and with the loss of axonal integrity, we observed a frequent loss or irregular spacing of these densities that constitute the myelin/axon paranodal junctions in the CST-cKO mice (Fig. 4E).
3.4. Progressive nodal protein domain abnormalities
Our ultrastructural analyses demonstrate a progressive lengthening of the nodal gap and a loss of paranodal integrity. Since the paranodal junctional complexes (i.e. transverse bands) regulate the establishment and maintenance of the distribution of the axonal proteins that are specifically clustered in the node of Ranvier, the paranode and the juxtaparanode, the loss of transverse bands may result in compromised domain organization (Coman et al., 2006; Dupree, Coetzee, Suzuki, et al., 1998; Einheber et al., 2006; Garcia-Fresco et al., 2006; Kojima & Hayashi, 2018; Marcus et al., 2006; Mierzwa et al., 2010a). To determine if the axonal domains are disrupted consequential of adult-onset loss of sulfatide, we immuno-labeled corpus callosum sections from CST-cKO and control mice with Nav1.6, caspr, and Kv1.1 to visualize the node of Ranvier, the paranode, and the juxtaparanode, respectively (Fig. 5A). Common pathologies observed were: 1. binodal sodium channel clusters (Fig 5B), 2. elongation of Nav1.6 and caspr domains (Fig. 5C), 3. mixing of proteins from adjacent domains or (Fig. 5D) or 4. a combination of several pathologies (Fig. 5D). Quantification of domain organization (Fig. 5E) revealed no alterations by 3 months post tamoxifen injection (8.3% for control vs 10.2% for CST-cKO p=0.958 n=7, 150+ nodes per animal), a time point when sulfatide levels remained unaltered; however, by 6 and 11 months post tamoxifen injection, time points when sulfatide levels were reduced (Fig. 1D), the percentage of abnormal domain arrangement was significantly and progressively increased (6 months – 9.6% for control vs 21.8% for CST-cKO; p=0.032 n=6 and 11 month – 20.8% for control vs 81.4% for CST-cKO; p<0.0001 n=7–11, 150+ nodes per animal for each timepoint). These data are consistent with previous findings in the constitutive CST KO mice that demonstrated neuronal nodal domain proteins are mis-localized in the absence of sulfatide (Ishibashi et al., 2002b; Hayashi et al., 2013a)
FIGURE 5. Increase in nodal length variability and loss of transverse bands following adult onset sulfatide depletion.

(a) Normal ultrastructure of a node of Ranvier (bound indicated by black arrows) including nodal length (~1 micron) and flanking paranodal regions with lateral loops closely opposed to the axolemma. (b) CST-cKO mice revealed nodes of Ranvier with varying lengths including abnormally long nodal gaps (nodal bounds indicated by arrows). (c) Overall, node quantitation revealed no significant change in average nodal length between genotypes at any of the time points analyzed; however, a significant difference was observed in length variability in the 11-months post tamoxifen injected CST-cKO mice with nodes of Ranvier presenting both shorter and longer than 1 micron. (d) Calculating the nodal length variance per mouse further confirmed this variability at 11-months PI with a significant increase in length variance with no difference in variance at the earlier PI time points. All data were statistically compared using a 2-way ANOVA. (e) CTL mice revealed equally spaced electron densities, known as transverse bands (arrows), at the interface between the lateral loops of the myelin sheath and the axolemma. (f) In contrast, transverse bands were unevenly spaced and structurally disrupted (arrows) in the CST-cKO. For analysis, the fit of the two models (single variance/covariance matrix or group specific variance/covariance matrices) was compared using a likelihood ratio test. a-b Scale bars = 1 μm. *p < .05 ****p < .0001. E-F Scale bars = 0.5 μm.
3.5. Progressive shift in myelinated axonal function
Action potential conduction is mediated by appropriate clustering of ion channels in specific domains along the axon (H. Wang et al., 1994) (Caldwell et al., 2000). When domain organization is disrupted, axonal function is compromised (Bagchi et al., 2014; Sinha et al., 2006). Based on both our ultrastructural and immunohistochemical data, we proposed that the axons of the corpus callosum were functionally compromised. However, to directly assess axonal function, we employed a well-established electrophysiological approach designed to quantify compound action potentials (CAPs) in both the myelinated and unmyelinated axons(Baker et al., 2002; Reeves et al., 2005; Yamate-Morgan et al., 2019). Typically, a CAP consists of two downward deflections referred to as N1 and N2 (Figure 6A). The CAPs of myelinated axons are represented in the N1 peak, the first downward deflection across time, while the N2 peak represents the CAPs of unmyelinated axons, the second downward deflection. To compare N1 and N2 peaks across time and between genotypes, we have presented the data as a ratio of N1/N2.
FIGURE 6. Mis-localization of axonal domain proteins following adult-onset sulfatide depletion.

(a) An axon from a CTL mouse demonstrates proper ion channel and axonal domain protein organization in the corpus callosum with Nav1.6 (green) localized to the node of Ranvier; caspr (blue) localized to the paranode; and Kv1.1 (red) localized to the juxtaparanode. Common, but not exclusive pathologies, observed included (b) binodal Nav1.6 (green; yellow arrows); (c) elongated Nav1.6 (green; yellow arrows indicating bounds of Nav1.6 distribution); and elongated caspr (blue) with Kv1.1 (red) intermixed (white stars flank region of intermixing). (d) A combination of several pathologies such as binodal Nav1.6 (yellow arrows) and Kv1.1 and caspr intermixing (white stars) was also observed. (e) Quantification of protein organization revealed preservation of protein domain at 3-months PI but a significant loss of axonal domain organization at 6- and 11-month PI. Data are presented as mean ± standard error and analyzed using a 2-way ANOVA; **** p < .0001. Scale bars = 5 μm.
At the 3- month PI timepoint, there is no change in N1/N2 ratio (Fig. 6Ai), which is consistent with our lipidomic data showing no difference in sulfatide levels between genotypes, as well as our IHC data showing no difference in abnormal domain localization. However, consistent with our IHC data, which revealed significant disruption in ion channel domain organization, CAP analyses show that at 6- and 11-month post injection, there is a statistically significant reduction in the N1/N2 ratio in the CST -cKO mice compared to control mice (Fig. 6Bi and 6Ci, respectively). At the 11-month timepoint, the N1 peak is completely absent (Fig. 6Cii) giving a ratio of zero across all stimulation intensities. While previous studies using the CST KO mice quantified conduction velocity in the sciatic nerve (Hayashi et al., 2013b), we provide the first direct evidence of functional loss within the CNS following sulfatide depletion and more interesting, we show a profound impaired function that parallels ion channel redistribution that precedes significant myelin disruption or myelin loss. Figures 2–4 demonstrate that myelin is intact, and Figure 3 shows no increase in myelin pathology, yet, these myelinated axons are functioning as unmyelinated axons at the 11-month timepoint. Collectively, these findings demonstrate that loss of sulfatide in adulthood leads to increased axonal pathology with relative myelin sparing.
4. Discussion
In MS brains, lipid metabolism is disrupted (Moscatelli & Isaacson, 1969) even in regions of NAWM (Wheeler et al., 2008b). More specifically, sulfatide, a prominent glycosphingolipid of the myelin sheath(Norton & Cammer, 1984), is decreased in NAWM while no other myelin proteins or lipids are reduced(Marbois et al., 2000; Yahara et al., 1982) indicating sulfatide loss is independent of demyelination. Previous work from our group (Marcus et al., 2006; A. Pomicter et al., 2013) and our collaborators (Ishibashi et al., 2002b; Shroff et al., 2009) using the constitutive CST KO mouse (Honke et al., 2002) shows a plethora of myelin abnormalities accompanied by axonal abnormalities that are consistent with myelin and axonal pathologies prevalent in MS(A. D. Pomicter et al., 2010; Suzuki et al., 1969; York et al., 2021). Recognizing that CNS pathologies presented in the constitutive CST KO mouse may be consequential of disrupted development, we generated the CST floxed mouse which enables adult-onset sulfatide depletion. Exploiting this novel mouse, we show for the first time that adult onset sulfatide reduction is sufficient to compromise neuronal structure and function independent of myelin loss, yet not sufficient to drive demyelination.
4.1. Adult onset sulfatide depletion is not sufficient to drive demyelination
In our previous work we showed a significant increase in the g ratio of myelinated axons in the CNS of the constitutive CST KO mice at all ages (Marcus et al., 2006). In contrast, our quantitative analyses of the conditional CST KO mice showed no change in g ratios (Fig. 2). We propose that this difference in g ratio data between the 2 CST mutant models reflects 1) a developmental inhibition of myelin synthesis with the absence of sulfatide during the critical period of myelin production and maturation(Foran & Peterson, 1992; Sturrock, 1980) in the constitutive KO mice whereas sulfatide synthesis in the conditional KO animals was not inhibited until 10 weeks of age and 2) the retention of a portion of the sulfatide that was synthesized prior to gene ablation. In addition to thin myelin, our analysis of the constitutive CST KO mice showed that early depletion of sulfatide synthesis resulted in unstable myelin sheaths as evidenced by increased and progressive myelin uncompaction and demyelination (Marcus et al., 2006). Those findings led us to conclude that the absence of sulfatide plays a prominent role in myelin loss observed in MS. However, our current findings from the conditional CST KO mice contrast with our original conclusion. Although myelin that is formed in the absence of the sphingolipid is more vulnerable to demyelination (Marcus et al., 2006), our longitudinal study of the conditional KO mice strongly suggests that myelin synthesized in the presence of sulfatide, but with reduced lipid after maturation, is relatively stable with regard to compaction and myelin loss. Based on our previous work, we assessed a variety of other pathologies including pulling away of the axon from the myelin sheath (Fig. 3B), uncompaction (Fig. 3C), frequency of myelin redundancy (Fig. 3D) and axonal degeneration (Fig. 3E). Although all of these pathologies were observed, the only pathology that was significantly more prevalent in the sulfatide depleted mice was axonal degeneration, suggesting that adult-onset depletion of sulfatide is not sufficient to drive overt dys- or demyelination but is sufficient for the loss of axonal structure and function
4.2. Adult-onset sulfatide depletion induces axonal pathology
Our ultrastructural analysis indicated the absence of demyelination but revealed a significant impact on axon integrity following chronic sulfatide depletion. Axonal degeneration was evidenced by abnormal electron dense bodies within the axon, the loss of organelle structure and the retention of myelin sheaths that lack the axon entirely (Fig 3Av). Oligodendrocytes play important roles in metabolically and structurally supporting axons (Fünfschilling et al., 2012; Lappe-Siefke et al., 2003; Lassetter et al., 2023; Lee et al., 2012; Morrison et al., 2013; Mukherjee et al., 2020). Therefore, it is not surprising that loss of an oligodendrocyte component leads to axonal pathology with myelin sparing, as this has been previously reported (Buscham et al., 2022, p. 5; Griffiths et al., 1998). There are several mechanisms in which progressive loss of sulfatide could lead to axonal degeneration. One possibility is involvement of the immune system. Using the conditional sulfatide KO mouse, our group has demonstrated a significant increase in disease-associated microglial and astrocyte neuroinflammation, particularly within myelin containing regions (Qiu et al., 2021). Ultrastructural analysis revealed astrocytic processes around the myelin sheath and TREM2, which is expressed by microglia and acts as a lipid sensor, is known to interact with sulfatide (Poliani et al., 2015; Y. Wang et al., 2015, p. 2). Since sulfatide is enriched in the outer leaflet of the myelin sheath (Boggs et al., 2008), TREM2 may sense altered lipid stoichiometry and launch and launch an immune response (Cantoni et al., 2015; Poliani et al., 2015).
Previously, our collaborators demonstrated this sulfatide deficient model can induce astrogliosis and microgliosis, in NAWM of MS patients, activated microglia can drive disruption of axo-glial junctions, leading to elongated and mis-localized sodium and potassium channels (Howell et al., 2010). Our findings provide evidence of loss of transverse bands and subsequent sodium and potassium channel redistribution. Transverse bands, which are formed by the binding of neurofascin155 of the myelin sheath to the contactin/caspr1 axonal complex (Charles et al., 2002), tether the myelin sheath to the axon. Previous work from us (Dupree, Coetzee, Blight, et al., 1998b; Dupree et al., 1999; A. D. Pomicter et al., 2010) and others (Charles et al., 2002; Coman et al., 2006; Rasband et al., 1999) has shown that a loss of myelin-axon interaction, particularly at the paranode region, is associated with altered axon structure and function. Consistent with these previous reports, here we observed compromised transverse band integrity and a significant increase in the variability of node of Ranvier length. The loss of the transverse band complex is further evidenced by the mislocalization of caspr, an essential component of the transverse band complex(Bhat et al., 2001; Boyle et al., 2001; Sun et al., 2009). Mis-localization of caspr suggests that the caspr-contactin complex is not appropriately binding to its glial ligand neurofascin 155. Consequently, disruption of these complexes results in a loss of proper myelin-axon anchoring (Bhat et al., 2001; Boyle et al., 2001) and could result in lateral movement of the sheath along the axon providing a viable explanation for the observed variation in node of Ranvier length that we observe in the conditional CST KO mice (Figure 4). Additionally, these paranodal junctions also maintain the proper clustering of the axonal proteins that constitute the node, paranode and juxtaparanode by providing a physical barrier that restricts lateral movement into the adjacent domains (Mierzwa et al., 2010b). Interestingly, we have previously shown, using the constitutive CST KO mice, that the loss of sulfatide results in destabilization of the paranodal axo-glial complexes and a loss of the respective protein clusters (Ishibashi et al., 2002b; A. Pomicter et al., 2013). Since this previous work was based on studies using the constitutive CST KO mice, the possibility remained that this sulfatide-dependent regulation was limited to developmental conditions with limited relevance to an adult-onset disease. Here, we show that adult-onset sulfatide depletion is sufficient to drive protein domain disruption at both the ultrastructural and molecular levels.
4.3. Adult-onset sulfatide depletion disrupts the function of myelinated axons
In addition to possibly facilitating the “sliding” of the myelin sheath along the axon, altered transverse band integrity may result in compromised axonal domain organization. Our IHC data showed that at 6- and 11-months post injection, the highly conserved spatial organization of the sodium and potassium channel domains was significantly compromised. Although myelin structure was spared, mis-localization of the ion channels translated functionally into loss of the myelinated (N1) amplitude. Similar abnormal protein domain localization pathologies have been observed using the experimental autoimmune encephalomyelitis (EAE) mouse model (Recks et al., 2013) as well as clinical studies showing substantial pathology in the NAWM, where sodium and potassium channel overlap(Gallego-Delgado et al., 2020b; Howell et al., 2010) and axonal degeneration preceding demyelination (Luchicchi et al., 2021a) have been observed.
Disruption of ion channel localization not only leads to mis-firing and inefficient functioning of the axon, but also leads to channelopathy. Craner et al. (2004) demonstrated that aberrant localization of Nav1.6 along extensive regions of a demyelinated fiber co-localize with sodium/calcium exchangers, causing a reversion of the exchangers’ function, resulting in increased intracellular calcium resulting in abnormal axonal function and ultimately axonal degeneration. (Craner et al., 2004; Stys et al., 1992). Our data show, at 6 months post injection, an increase in protein domain mislocalization and a functional ratio shift from N1 to N2; however, there is no significant difference in axonal degeneration. Consistent with limited axonal pathology in the sulfatide conditional KO mice is the retention of the myelin sheath. In contrast to demyelinated axons, dysmyelinated axons express Nav1.2 (Boiko et al., 2001; Westenbroek et al., 1992) and are less sensitive to this type of injury. Presently, it is unknown if the myelinated axons in the conditional KO mice present with a re-expression of the immature sodium channel isoform. Ultrastructural axonal degeneration reaches significance at 11 months post injection coinciding with an increase in the frequency of elongated Nav1.6 channel domains. It will be of interest to determine if the myelinated axons in the conditional KO mice present with an upregulation of Nav1.2, which has been reported following myelin disruption (Dupree et al., 2004). An upregulation of Nav1.2 expression is consistent with the observed compromised axonal function and might provide protection from degeneration expanding the window for myelin repair and ultimately axonal function.
Technology has greatly improved the resolution of magnetic resonance imaging (MRI) allowing the detection of lesions in what otherwise was considered NAWM(Inglese et al., 2018). While our studies suggest that a significant reduction of sulfatide leads to axonal degeneration, the dysmyelination that occurs consequential of sulfatide depletion may act to spare axonal integrity at least in the acute, or perhaps prodromic, stage of the disease. It remains to be determined if sulfatide depletion alone is sufficient to induce demyelination and to drive the transition of NAWM into lesion development. By the time lesions are detected, axonal pathology, which is at least in part consequential of sulfatide deletion and sufficient to result in functional deficits, has already developed. Our findings combined with the studies that have shown a loss of sulfatide in brain regions that reveal no myelin abnormalities are consistent with sulfatide depletion defining the prodromic stage of MS, the period of months or years before classic MS symptom onset (Makhani & Tremlett, 2021). It is unknown how the initial loss of sulfatide occurs, and how long sulfatide is reduced in NAWM prior to the onset of demyelination and plaque formation; however, detection of sulfatide levels should be considered as a possible diagnostic for MS in the future and may provide an indicator for initiation of therapeutic repair prior to irreversible axonal loss.
Supplementary Material
FIGURE 7. Decrease of N1 peak following adult-onset sulfatide depletion.

(ai) Compound action potential (CAP) recordings demonstrate comparable and robust N1 and N2 peaks between the CTL and CST-cKO mice 3-months PI. (aii) Quantification of the ratio of N1/N2 from ascending stimulus currents in the CTL and CSTcKO mouse at 3-months PI shows no significant difference between genotypes. (bi) By 6-months PI CSTcKO mice present with a reduced N1 peak and (bii) a significant reduction in the N1/N2 ratio between cKO and CTL mice. (ci) By 11-months PI, N1 peaks were not observed in the CST cKO mice but N2 peaks were spared. (cii) The loss of the N1 peak resulted in further separation of N1/N2 ratios between CTL and CSTcKO mice at 11-months PI with the ratios being zero due to loss of N1 peak. Analyzed using a 2-way ANOVA. ****p < .0001.
Acknowledgements:
All microscopy was performed at the VCU Microscopy Facility, supported, in part, by funding from NIH-NCI Cancer Center Support Grant P30 CA016059. The authors wish to acknowledge Dr. Tytus Bernas, Director of the VCU Microscopy Facility, and Terry Smith and Judy Williamson, electron microscopy technicians, for their invaluable support and technical expertise with tissue and section preparation. The authors also wish to thank Fran White, light microscopy technician, for her support and expertise with confocal microscopy imaging and Dr. Leroy Thacker of the VCU Biostatics Department for his assistance with statistical analyses. The authors acknowledge their funding from NIH R21 NS122152 (JLD) and NIA RF1 AG061729 (XH).
Literature Cited
- Yaghootfam Afshin, Sorkalla Thomas, Hanns Häberlein Volkmar Gieselmann, Kappler Joachim, and, & Eckhardt M. (2007, July 21). Cerebroside Sulfotransferase Forms Homodimers in Living Cells† (world) [Research-article]. American Chemical Society. 10.1021/bi700014q [DOI] [PubMed] [Google Scholar]
- Bagchi B, Al-Sabi A, Kaza S, Scholz D, O’Leary VB, Dolly JO, & Ovsepian SV (2014). Disruption of myelin leads to ectopic expression of K(V)1.1 channels with abnormal conductivity of optic nerve axons in a cuprizone-induced model of demyelination. PloS One, 9(2), e87736. 10.1371/journal.pone.0087736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker AJ, Phan N, Moulton RJ, Fehlings MG, Yucel Y, Zhao M, Liu E, & Tian GF (2002). Attenuation of the Electrophysiological Function of the Corpus Callosum after Fluid Percussion Injury in the Rat. Journal of Neurotrauma, 19(5), 587–599. 10.1089/089771502753754064 [DOI] [PubMed] [Google Scholar]
- Belachew S, Yuan X, & Gallo V. (2001). Unraveling Oligodendrocyte Origin and Function by Cell-Specific Transgenesis. Developmental Neuroscience, 23(4–5), 287–298. 10.1159/000048712 [DOI] [PubMed] [Google Scholar]
- Bell KA, Delong R, Goswamee P, & McQuiston AR (2021). The Entorhinal Cortical Alvear Pathway Differentially Excites Pyramidal Cells and Interneuron Subtypes in Hippocampal CA1. Cerebral Cortex, 31(5), 2382–2401. 10.1093/cercor/bhaa359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benusa SD, George NM, Sword BA, DeVries GH, & Dupree JL (2017). Acute neuroinflammation induces AIS structural plasticity in a NOX2-dependent manner. Journal of Neuroinflammation, 14(1), 116. 10.1186/s12974-017-0889-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, St Martin M, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JL, & Bellen HJ (2001). Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron, 30(2), 369–383. 10.1016/s0896-6273(01)00294-x [DOI] [PubMed] [Google Scholar]
- Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, Elledge SJ, Niebuhr DW, Scher AI, Munger KL, & Ascherio A. (2022). Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science (New York, N.Y.), 375(6578), 296–301. 10.1126/science.abj8222 [DOI] [PubMed] [Google Scholar]
- Boggs JM, Gao W, & Hirahara Y. (2008). Myelin glycosphingolipids, galactosylceramide and sulfatide, participate in carbohydrate–carbohydrate interactions between apposed membranes and may form glycosynapses between oligodendrocyte and/or myelin membranes. Biochimica et Biophysica Acta (BBA) - General Subjects, 1780(3), 445–455. 10.1016/j.bbagen.2007.10.015 [DOI] [PubMed] [Google Scholar]
- Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS, & Matthews G. (2001). Compact Myelin Dictates the Differential Targeting of Two Sodium Channel Isoforms in the Same Axon. Neuron, 30(1), 91–104. 10.1016/S0896-6273(01)00265-3 [DOI] [PubMed] [Google Scholar]
- Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, & Ranscht B. (2001). Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron, 30(2), 385–397. 10.1016/s0896-6273(01)00296-3 [DOI] [PubMed] [Google Scholar]
- Buscham TJ, Eichel-Vogel MA, Steyer AM, Jahn O, Strenzke N, Dardawal R, Memhave TR, Siems SB, Müller C, Meschkat M, Sun T, Ruhwedel T, Möbius W, Krämer-Albers E-M, Boretius S, Nave K-A, & Werner HB (2022). Progressive axonopathy when oligodendrocytes lack the myelin protein CMTM5. ELife, 11, e75523. 10.7554/eLife.75523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell JH, Schaller KL, Lasher RS, Peles E, & Levinson SR (2000). Sodium channel Nav1.6 is localized at nodes of Ranvier, dendrites, and synapses. Proceedings of the National Academy of Sciences, 97(10), 5616–5620. 10.1073/pnas.090034797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles P, Tait S, Faivre-Sarrailh C, Barbin G, Gunn-Moore F, Denisenko-Nehrbass N, Guennoc A-M, Girault J-A, Brophy PJ, & Lubetzki C. (2002). Neurofascin is a glial receptor for the paranodin/Caspr-contactin axonal complex at the axoglial junction. Current Biology: CB, 12(3), 217–220. 10.1016/s0960-9822(01)00680-7 [DOI] [PubMed] [Google Scholar]
- Clark KC, Josephson A, Benusa SD, Hartley RK, Baer M, Thummala S, Joslyn M, Sword BA, Elford H, Oh U, Dilsizoglu-Senol A, Lubetzki C, Davenne M, DeVries GH, & Dupree JL (2016). Compromised axon initial segment integrity in EAE is preceded by microglial reactivity and contact. Glia, 64(7), 1190–1209. 10.1002/glia.22991 [DOI] [PubMed] [Google Scholar]
- Coman I, Aigrot MS, Seilhean D, Reynolds R, Girault JA, Zalc B, & Lubetzki C. (2006). Nodal, paranodal and juxtaparanodal axonal proteins during demyelination and remyelination in multiple sclerosis. Brain: A Journal of Neurology, 129(Pt 12), 3186–3195. 10.1093/brain/awl144 [DOI] [PubMed] [Google Scholar]
- Craner MJ, Hains BC, Lo AC, Black JA, & Waxman SG (2004). Co-localization of sodium channel Nav1.6 and the sodium-calcium exchanger at sites of axonal injury in the spinal cord in EAE. Brain: A Journal of Neurology, 127(Pt 2), 294–303. 10.1093/brain/awh032 [DOI] [PubMed] [Google Scholar]
- Dupree JL, Coetzee T, Blight A, Suzuki K, & Popko B. (1998a). Myelin Galactolipids Are Essential for Proper Node of Ranvier Formation in the CNS. Journal of Neuroscience, 18(5), 1642–1649. 10.1523/JNEUROSCI.18-05-01642.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree JL, Coetzee T, Blight A, Suzuki K, & Popko B. (1998b). Myelin Galactolipids Are Essential for Proper Node of Ranvier Formation in the CNS. Journal of Neuroscience, 18(5), 1642–1649. 10.1523/JNEUROSCI.18-05-01642.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree JL, Coetzee T, Suzuki K, & Popko B. (1998). Myelin abnormalities in mice deficient in galactocerebroside and sulfatide. Journal of Neurocytology, 27(9), 649–659. 10.1023/A:1006908013972 [DOI] [PubMed] [Google Scholar]
- Dupree JL, Girault JA, & Popko B. (1999). Axo-glial interactions regulate the localization of axonal paranodal proteins. The Journal of Cell Biology, 147(6), 1145–1152. 10.1083/jcb.147.6.1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree JL, Polak PE, Hensley K, Pelligrino D, & Feinstein DL (2015). Lanthionine ketimine ester provides benefit in a mouse model of multiple sclerosis. Journal of Neurochemistry, 134(2), 302–314. 10.1111/jnc.13114 [DOI] [PubMed] [Google Scholar]
- Einheber S, Bhat MA, & Salzer JL (2006). Disrupted axo-glial junctions result in accumulation of abnormal mitochondria at nodes of ranvier. Neuron Glia Biology, 2(3), 165–174. 10.1017/S1740925X06000275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foran DR, & Peterson AC (1992). Paper for timeline of myelination: Myelin acquisition in the central nervous system of the mouse revealed by an MBP-Lac Z transgene. Journal of Neuroscience, 12(12), 4890–4897. 10.1523/JNEUROSCI.12-12-04890.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Möbius W, Diaz F, Meijer D, Suter U, Hamprecht B, Sereda MW, Moraes CT, Frahm J, Goebbels S, & Nave K-A (2012). Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature, 485(7399), 517–521. 10.1038/nature11007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego-Delgado P, James R, Browne E, Meng J, Umashankar S, Tan L, Picon C, Mazarakis ND, Faisal AA, Howell OW, & Reynolds R. (2020a). Neuroinflammation in the normal-appearing white matter (NAWM) of the multiple sclerosis brain causes abnormalities at the nodes of Ranvier. PLoS Biology, 18(12), e3001008. 10.1371/journal.pbio.3001008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego-Delgado P, James R, Browne E, Meng J, Umashankar S, Tan L, Picon C, Mazarakis ND, Faisal AA, Howell OW, & Reynolds R. (2020b). Neuroinflammation in the normal-appearing white matter (NAWM) of the multiple sclerosis brain causes abnormalities at the nodes of Ranvier. PLoS Biology, 18(12), e3001008. 10.1371/journal.pbio.3001008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Fresco GP, Sousa AD, Pillai AM, Moy SS, Crawley JN, Tessarollo L, Dupree JL, & Bhat MA (2006). Disruption of axo-glial junctions causes cytoskeletal disorganization and degeneration of Purkinje neuron axons. Proceedings of the National Academy of Sciences of the United States of America, 103(13), 5137–5142. 10.1073/pnas.0601082103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gbaguidi B, Guillemin F, Soudant M, Debouverie M, Mathey G, & Epstein J. (2022). Age-period-cohort analysis of the incidence of multiple sclerosis over twenty years in Lorraine, France. Scientific Reports, 12(1), Article 1. 10.1038/s41598-022-04836-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, & Nave KA (1998). Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science (New York, N.Y.), 280(5369), 1610–1613. 10.1126/science.280.5369.1610 [DOI] [PubMed] [Google Scholar]
- Hayashi A, Kaneko N, Tomihira C, & Baba H. (2013a). Sulfatide decrease in myelin influences formation of the paranodal axo-glial junction and conduction velocity in the sciatic nerve. Glia, 61(4), 466–474. 10.1002/glia.22447 [DOI] [PubMed] [Google Scholar]
- Hayashi A, Kaneko N, Tomihira C, & Baba H. (2013b). Sulfatide decrease in myelin influences formation of the paranodal axo-glial junction and conduction velocity in the sciatic nerve. Glia, 61(4), 466–474. 10.1002/glia.22447 [DOI] [PubMed] [Google Scholar]
- Hayes LW, & Jungawala FB (1976). Synthesis and Turnover of Cerebrosides and Phosphatidylserine of Myelin and Mcrosomal Fractions of Adult and Developing Rat Brain. 160, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirahara Y, Tsuda M, Wada Y, & Honke K. (2000). CDNA cloning, genomic cloning, and tissue-specific regulation of mouse cerebroside sulfotransferase. European Journal of Biochemistry, 267(7), 1909–1917. 10.1046/j.1432-1327.2000.01139.x [DOI] [PubMed] [Google Scholar]
- Honke K, Hirahara Y, Dupree J, Suzuki K, Popko B, Fukushima K, Fukushima J, Nagasawa T, Yoshida N, Wada Y, & Taniguchi N. (2002). Paranodal junction formation and spermatogenesis require sulfoglycolipids. Proceedings of the National Academy of Sciences of the United States of America, 99(7), 4227–4232. 10.1073/pnas.032068299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honke K, Tsuda M, Hirahara Y, Ishii A, Makita A, & Wada Y. (1997). Molecular cloning and expression of cDNA encoding human 3’-phosphoadenylylsulfate:galactosylceramide 3’-sulfotransferase. The Journal of Biological Chemistry, 272(8), 4864–4868. 10.1074/jbc.272.8.4864 [DOI] [PubMed] [Google Scholar]
- Howell Owain. W., Rundle Jon. L., Garg A, Komada M, Brophy Peter. J., & Reynolds R. (2010). Activated microglia mediate axo-glial disruption that contributes to axonal injury in multiple sclerosis. Journal of Neuropathology and Experimental Neurology, 69(10), 1017–1033. 10.1097/NEN.0b013e3181f3a5b1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley AF, & Stämpeli R. (1949). Evidence for saltatory conduction in peripheral myelinated nerve fibres. The Journal of Physiology, 108(3), 315–339. 10.1113/jphysiol.1949.sp004335 [DOI] [PubMed] [Google Scholar]
- Inglese M, Fleysher L, Oesingmann N, & Petracca M. (2018). Clinical applications of ultra-high field magnetic resonance imaging in multiple sclerosis. Expert Review of Neurotherapeutics, 18(3), 221–230. 10.1080/14737175.2018.1433033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi T, Dupree JL, Ikenaka K, Hirahara Y, Honke K, Peles E, Popko B, Suzuki K, Nishino H, & Baba H. (2002a). A Myelin Galactolipid, Sulfatide, Is Essential for Maintenance of Ion Channels on Myelinated Axon But Not Essential for Initial Cluster Formation. The Journal of Neuroscience, 22(15), 6507–6514. 10.1523/JNEUROSCI.22-15-06507.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi T, Dupree JL, Ikenaka K, Hirahara Y, Honke K, Peles E, Popko B, Suzuki K, Nishino H, & Baba H. (2002b). A myelin galactolipid, sulfatide, is essential for maintenance of ion channels on myelinated axon but not essential for initial cluster formation. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 22(15), 6507–6514. https://doi.org/20026705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch-Henriksen N, & Sørensen PS (2010). The changing demographic pattern of multiple sclerosis epidemiology. The Lancet. Neurology, 9(5), 520–532. 10.1016/S1474-4422(10)70064-8 [DOI] [PubMed] [Google Scholar]
- Kojima W, & Hayashi K. (2018). Changes in the axo-glial junctions of the optic nerves of cuprizone-treated mice. Histochemistry and Cell Biology, 149(5), 529–536. 10.1007/s00418-018-1654-0 [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, & Nave K-A (2003). Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nature Genetics, 33(3), 366–374. 10.1038/ng1095 [DOI] [PubMed] [Google Scholar]
- Lassetter AP, Corty MM, Barria R, Sheehan AE, Hill JQ, Aicher SA, Fox AN, & Freeman MR (2023). Glial TGFβ activity promotes neuron survival in peripheral nerves. The Journal of Cell Biology, 222(1), e202111053. 10.1083/jcb.202111053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang P-W, Pellerin L, Magistretti PJ, & Rothstein JD (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature, 487(7408), 443–448. 10.1038/nature11314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchicchi A, Hart B, Frigerio I, van Dam A, Perna L, Offerhaus HL, Stys PK, Schenk GJ, & Geurts JJG (2021a). Axon-Myelin Unit Blistering as Early Event in MS Normal Appearing White Matter. Annals of Neurology, 89(4), 711–725. 10.1002/ana.26014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchicchi A, Hart B, Frigerio I, van Dam A-M, Perna L, Offerhaus HL, Stys PK, Schenk GJ, & Geurts JJG (2021b). Axon-Myelin Unit Blistering as Early Event in MS Normal Appearing White Matter. Annals of Neurology, 89(4), 711–725. 10.1002/ana.26014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhani N, & Tremlett H. (2021). The multiple sclerosis prodrome. Nature Reviews. Neurology, 17(8), 515–521. 10.1038/s41582-021-00519-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallon BS, Shick HE, Kidd GJ, & Macklin WB (2002). Proteolipid Promoter Activity Distinguishes Two Populations of NG2-Positive Cells throughout Neonatal Cortical Development. The Journal of Neuroscience, 22(3), 876–885. 10.1523/JNEUROSCI.22-03-00876.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbois BN, Faull KF, Fluharty AL, Raval-Fernandes S, & Rome LH (2000). Analysis of sulfatide from rat cerebellum and multiple sclerosis white matter by negative ion electrospray mass spectrometry. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, 1484(1), 59–70. 10.1016/S1388-1981(99)00201-2 [DOI] [PubMed] [Google Scholar]
- Marcus J, Honigbaum S, Shroff S, Honke K, Rosenbluth J, & Dupree JL (2006). Sulfatide is essential for the maintenance of CNS myelin and axon structure. Glia, 53(4), 372–381. 10.1002/glia.20292 [DOI] [PubMed] [Google Scholar]
- Mierzwa AJ, Arevalo J-C, Schiff R, Chao MV, & Rosenbluth J. (2010a). Role of transverse bands in maintaining paranodal structure and axolemmal domain organization in myelinated nerve fibers: Effect on longevity in dysmyelinated mutant mice. The Journal of Comparative Neurology, 518(14), 2841–2853. 10.1002/cne.22367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierzwa AJ, Arevalo J-C, Schiff R, Chao MV, & Rosenbluth J. (2010b). Role of transverse bands in maintaining paranodal structure and axolemmal domain organization in myelinated nerve fibers: Effect on longevity in dysmyelinated mutant mice. The Journal of Comparative Neurology, 518(14), 2841–2853. 10.1002/cne.22367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morell P, & Radin NS (1969). Synthesis of cerebroside by brain from uridine diphosphate galactose and ceramide containing hydroxy fatty acid. Biochemistry, 8(2), 506–512. 10.1021/bi00830a008 [DOI] [PubMed] [Google Scholar]
- Morrison BM, Lee Y, & Rothstein JD (2013). Oligodendroglia metabolically support axons and maintain structural integrity. Trends in Cell Biology, 23(12). 10.1016/j.tcb.2013.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscatelli EA, & Isaacson E. (1969). Gas liquid chromatographic analysis of sphingosine bases in sphingolipids of human normal and multiple sclerosis cerebral white matter. Lipids, 4(6), 550–555. 10.1007/BF02531040 [DOI] [PubMed] [Google Scholar]
- Mukherjee C, Kling T, Russo B, Miebach K, Kess E, Schifferer M, Pedro LD, Weikert U, Fard MK, Kannaiyan N, Rossner M, Aicher M-L, Goebbels S, Nave K-A, Krämer-Albers E-M, Schneider A, & Simons M. (2020). Oligodendrocytes Provide Antioxidant Defense Function for Neurons by Secreting Ferritin Heavy Chain. Cell Metabolism, 32(2), 259–272.e10. 10.1016/j.cmet.2020.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton WT, & Cammer W. (1984). Isolation and Characterization of Myelin. In Morell P(Ed.), Myelin (pp. 147–195). Springer US. 10.1007/978-1-4757-1830-0_5 [DOI] [Google Scholar]
- O’Connor KC, Bar-Or A, & Hafler DA (2001). The Neuroimmunology of Multiple Sclerosis: Possible Roles of T and B Lymphocytes in Immunopathogenesis. Journal of Clinical Immunology, 21(2), 12. [DOI] [PubMed] [Google Scholar]
- Olsson T, Barcellos LF, & Alfredsson L. (2017). Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nature Reviews Neurology, 13(1), Article 1. 10.1038/nrneurol.2016.187 [DOI] [PubMed] [Google Scholar]
- Palavicini JP, Wang C, Chen L, Ahmar S, Higuera JD, Dupree JL, & Han X. (2016). Novel molecular insights into the critical role of sulfatide in myelin maintenance/function. Journal of Neurochemistry, 139(1), 40–54. 10.1111/jnc.13738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A. (1966). The Node of Ranvier in the Central Nervous System. Quarterly Journal of Experimental Physiology and Cognate Medical Sciences, 51(3), 229–236. 10.1113/expphysiol.1966.sp001852 [DOI] [PubMed] [Google Scholar]
- Peters A, Palay Sanford, & Webster Henry. (1992). The Fine Structure of the Nervous System: Neurons and Their Supporting Cells. The Quarterly Review of Biology, 67(1), 80. [Google Scholar]
- Piehl F. (2021). Current and emerging disease-modulatory therapies and treatment targets for multiple sclerosis. Journal of Internal Medicine, 289(6), 771–791. 10.1111/joim.13215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai AM, Thaxton C, Pribisko AL, Cheng J-G, Dupree JL, & Bhat MA (2009). Spatiotemporal Ablation of Myelinating Glia-Specific Neurofascin (NfascNF155) in Mice Reveals Gradual Loss of Paranodal Axoglial Junctions and Concomitant Disorganization of Axonal Domains. Journal of Neuroscience Research, 87(8), 1773–1793. 10.1002/jnr.22015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podbielska M, Ariga T, & Pokryszko-Dragan A. (2022). Sphingolipid Players in Multiple Sclerosis: Their Influence on the Initiation and Course of the Disease. International Journal of Molecular Sciences, 23(10), 5330. 10.3390/ijms23105330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, & Colonna M. (2015). TREM2 sustains microglial expansion during aging and response to demyelination. The Journal of Clinical Investigation, 125(5), 2161–2170. 10.1172/JCI77983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomicter AD, Shroff SM, Fuss B, Sato-Bigbee C, Brophy PJ, Rasband MN, Bhat MA, & Dupree JL (2010). Novel forms of neurofascin 155 in the central nervous system: Alterations in paranodal disruption models and multiple sclerosis. Brain, 133(2), 389–405. 10.1093/brain/awp341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomicter A, DeLoyht J, Hackett A, Purdie N, Sato-Bigbee C, Henderson S, & Dupree J. (2013). Nfasc155H and MAG are specifically susceptible to detergent extraction in the absence of the myelin sphingolipid sulfatide. Neurochemical Research, 38(12), 2490–2502. 10.1007/s11064-013-1162-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Palavicini JP, Wang J, Gonzalez NS, He S, Dustin E, Zou C, Ding L, Bhattacharjee A, Van Skike CE, Galvan V, Dupree JL, & Han X. (2021). Adult-onset CNS myelin sulfatide deficiency is sufficient to cause Alzheimer’s disease-like neuroinflammation and cognitive impairment. Molecular Neurodegeneration, 16, 64. 10.1186/s13024-021-00488-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Peles E, Trimmer JS, Levinson SR, Lux SE, & Shrager P. (1999). Dependence of Nodal Sodium Channel Clustering on Paranodal Axoglial Contact in the Developing CNS. Journal of Neuroscience, 19(17), 7516–7528. 10.1523/JNEUROSCI.19-17-07516.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recks MS, Stormanns ER, Bader J, Arnhold S, Addicks K, & Kuerten S. (2013). Early axonal damage and progressive myelin pathology define the kinetics of CNS histopathology in a mouse model of multiple sclerosis. Clinical Immunology (Orlando, Fla.), 149(1), 32–45. 10.1016/j.clim.2013.06.004 [DOI] [PubMed] [Google Scholar]
- Reeves TM, Phillips LL, & Povlishock JT (2005). Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Experimental Neurology, 196(1), 126–137. 10.1016/j.expneurol.2005.07.014 [DOI] [PubMed] [Google Scholar]
- Rodríguez Murúa S, Farez MF, & Quintana FJ (2022). The Immune Response in Multiple Sclerosis. Annual Review of Pathology, 17, 121–139. 10.1146/annurev-pathol-052920-040318 [DOI] [PubMed] [Google Scholar]
- Rosenbluth J, Dupree JL, & Popko B. (2003). Nodal sodium channel domain integrity depends on the conformation of the paranodal junction, not on the presence of transverse bands. Glia, 41(3), 318–325. 10.1002/glia.10179 [DOI] [PubMed] [Google Scholar]
- Schnapp B, peracchia, camillo, & Mugnaini E. (1976). THE PARANODAL AXO-GLIAL JUNCTION IN THE CENTRAL NERVOUS SYSTEM STUDIED WITH THIN SECTIONS AND FREEZE-FRACTURE. Neuroscience, 1, 181–190. [DOI] [PubMed] [Google Scholar]
- Shepherd MN, Pomicter AD, Velazco CS, Henderson SC, & Dupree JL (2012). Paranodal reorganization results in the depletion of transverse bands in the aged central nervous system. Neurobiology of Aging, 33(1), 203.e13–203.e24. 10.1016/j.neurobiolaging.2010.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff SM, Pomicter AD, Chow WN, Fox MA, Colello RJ, Henderson SC, & Dupree JL (2009). Adult CST-null mice maintain an increased number of oligodendrocytes. Journal of Neuroscience Research, 87(15), 3403–3414. 10.1002/jnr.22003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha K, Karimi-Abdolrezaee S, Velumian AA, & Fehlings MG (2006). Functional changes in genetically dysmyelinated spinal cord axons of shiverer mice: Role of juxtaparanodal Kv1 family K+ channels. Journal of Neurophysiology, 95(3), 1683–1695. 10.1152/jn.00899.2005 [DOI] [PubMed] [Google Scholar]
- Sturrock RR (1980). Myelination of the mouse corpus callosum. Neuropathology and Applied Neurobiology, 6(6), 415–420. 10.1111/j.1365-2990.1980.tb00219.x [DOI] [PubMed] [Google Scholar]
- Stys PK, Waxman SG, & Ransom BR (1992). Ionic mechanisms of anoxic injury in mammalian CNS white matter: Role of Na + channels and Na + -Ca 2+ exchanger. Journal of Neuroscience, 430–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Takagishi Y, Okabe E, Chishima Y, Kanou Y, Murase S, Mizumura K, Inaba M, Komatsu Y, Hayashi Y, Peles E, Oda S, & Murata Y. (2009). A Novel Caspr Mutation Causes the Shambling Mouse Phenotype by Disrupting Axoglial Interactions of Myelinated Nerves. Journal of Neuropathology & Experimental Neurology, 68(11), 1207–1218. 10.1097/NEN.0b013e3181be2e96 [DOI] [PubMed] [Google Scholar]
- Susuki K, Zollinger DR, Chang K-J, Zhang C, Huang CY-M, Tsai C-R, Galiano MR, Liu Y, Benusa SD, Yermakov LM, Griggs RB, Dupree JL, & Rasband MN (2018). Glial βII Spectrin Contributes to Paranode Formation and Maintenance. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 38(27), 6063–6075. 10.1523/JNEUROSCI.3647-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Andrews J, Waltz J, & Terry R. (1969). Ultrastructural studies of multiple sclerosis. International Academy of Pathology, 20(5), 444–454. [PubMed] [Google Scholar]
- Thomason EJ, Suárez-Pozos E, Afshari FS, Rosenberg PA, Dupree JL, & Fuss B. (2022). Deletion of the Sodium-Dependent Glutamate Transporter GLT-1 in Maturing Oligodendrocytes Attenuates Myelination of Callosal Axons During a Postnatal Phase of Central Nervous System Development. Frontiers in Cellular Neuroscience, 16. 10.3389/fncel.2022.905299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, & Bö L. (1998). Axonal Transection in the Lesions of Multiple Sclerosis. New England Journal of Medicine, 338(5), 278–285. 10.1056/NEJM199801293380502 [DOI] [PubMed] [Google Scholar]
- Wallin MT, Culpepper WJ, Campbell JD, Nelson LM, Langer-Gould A, Marrie RA, Cutter GR, Kaye WE, Wagner L, Tremlett H, Buka SL, Dilokthornsakul P, Topol B, Chen LH, & LaRocca NG (2019). The prevalence of MS in the United States: A population-based estimate using health claims data. Neurology, 92(10), e1029–e1040. 10.1212/WNL.0000000000007035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton C, King R, Rechtman L, Kaye W, Leray E, Marrie RA, Robertson N, La Rocca N, Uitdehaag B, van der Mei I, Wallin M, Helme A, Angood Napier C, Rijke N, & Baneke P. (2020). Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Multiple Sclerosis (Houndmills, Basingstoke, England), 26(14), 1816–1821. 10.1177/1352458520970841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Kunkel DD, Schwartzkroin PA, & Tempel BL (1994). Localization of Kv1.1 and Kv1.2, two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. Journal of Neuroscience, 14(8), 4588–4599. 10.1523/JNEUROSCI.14-08-04588.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, & Colonna M. (2015). TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer’s Disease Model. Cell, 160(6), 1061–1071. 10.1016/j.cell.2015.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Noebels JL, & Catterall WA (1992). Elevated expression of type II Na+ channels in hypomyelinated axons of shiverer mouse brain. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 12(6), 2259–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler D, Bandaru VVR, Calabresi PA, Nath A, & Haughey NJ (2008a). A defect of sphingolipid metabolism modifies the properties of normal appearing white matter in multiple sclerosis. Brain, 131(11), 3092–3102. 10.1093/brain/awn190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler D, Bandaru VVR, Calabresi PA, Nath A, & Haughey NJ (2008b). A defect of sphingolipid metabolism modifies the properties of normal appearing white matter in multiple sclerosis. Brain, 131(11), 3092–3102. 10.1093/brain/awn190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wight PA, & Dobretsova A. (2004). Where, when and how much: Regulation of myelin proteolipid protein gene expression. Cellular and Molecular Life Sciences CMLS, 61(7), 810–821. 10.1007/s00018-003-3309-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahara S, Kawamura N, Kishimoto Y, Saida T, & Tourtellotte WW (1982). A change in the cerebrosides and sulfatides in a demyelinating nervous system. Development of the methodology and study of multiple sclerosis and Wallerian degeneration. Journal of the Neurological Sciences, 54(2), 303–315. 10.1016/0022-510x(82)90191-5 [DOI] [PubMed] [Google Scholar]
- Yamate-Morgan H, Lauderdale K, Horeczko J, Merchant U, & Tiwari-Woodruff SK (2019). Functional Effects of Cuprizone-Induced Demyelination in the Presence of the mTOR-Inhibitor Rapamycin,. Neuroscience, 406, 667–683. 10.1016/j.neuroscience.2019.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- York EN, Martin S-J, Meijboom R, Thrippleton MJ, Bastin ME, Carter E, Overell J, Connick P, Chandran S, Waldman AD, & Hunt DPJ (2021). MRI-derived g-ratio and lesion severity in newly diagnosed multiple sclerosis. Brain Communications, 3(4), fcab249. 10.1093/braincomms/fcab249 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.