

Abstract
Jacobsen syndrome (MIM #147791) is a rare multisystem genomic disorder involving craniofacial abnormalities, intellectual disability, other neurodevelopmental defects and terminal truncation of chromosome 11q, typically deleting ~170 to >340 genes. We describe the first case of Jacobsen syndrome caused by congenital chromoanasynthesis, an extreme form of complex chromosomal rearrangement. Six duplications and five deletions occurred on one copy of chromosome 11q with microhomology signatures in the breakpoint junctions, indicating an all-at-once replication-based rearrangement mechanism in a gametocyte or early post-zygotic cell. Eighteen genes were deleted from the Jacobsen region, including KIRREL3, which is associated with intellectual disability.
Keywords: chromoanagenesis, chromothripsis, congenital, genome, cytogenetics
1. INTRODUCTION
Chromoanagenesis, or ‘chromosome rebirth’, refers collectively to three extreme forms of complex balanced or unbalanced chromosomal rearrangements--chromothripsis, chromoplexy and chromoanasynthesis—each of which appears to occur all at once in one or a few chromosomes in a single stressed cell (Pellestor, 2019). There is increasing evidence that sequestration in micronuclei precedes the chromosomal damage followed by reintegration with the primary nucleus (Ly et al., 2019; Zhang et al., 2015). The most common form of chromoanagenesis is chromothripsis (‘chromosome shattering’), which typically involves large numbers of clustered deletions with random reconnection of the remaining fragments by non-homologous end joining (NHEJ) (Stephens et al., 2011). Chromoplexy (or ‘sudden incapacity of a chromosome’) refers to balanced translocations among multiple chromosomes. Like chromothripsis, chromoanasynthesis (or ‘chromosome resynthesis’) involves oscillating copy number states, which occur in clusters in one or a few chromosomes with random orientation of rearranged fragments. However, in contrast to chromothripsis, copy number gains in chromoanasynthesis typically exceed losses, and the mechanism appears to involve aberrant DNA replication pathways, not NHEJ (Liu et al., 2011). One favored model is microhomology-mediated break-induced replication (MMBIR), which involves switching of the replicating single strand of DNA from stalled replication forks to available nearby active replication forks (Hastings, Lupski, Rosenberg, & Ira, 2009).
Approximately 5% of all cancers are thought to be caused by chromothripsis, with much higher frequencies in specific blood, brain, pancreatic and bone cancers (Li et al., 2020; Stephens et al., 2011). Chromothripsis has also been reported in children with developmental disorders, (Kloosterman et al., 2011; Kloosterman et al., 2012) in a few healthy individuals and in one extraordinary case of a patient with the primary immunodeficiency disorder warts-hypogammaglobulinemia-infections-myelokathexis (WHIM) syndrome, who was cured as an adult by acquired chromothriptic deletion of the hyperactive disease allele CXCR4R334X in a single advantaged hematopoietic stem cell (McDermott et al., 2015). Only 19 cases of congenital chromoanasynthesis have been reported, all associated with neurodevelopmental abnormalities (Collins et al., 2017; Kloosterman et al., 2012; Liu et al., 2011; Plaisancie et al., 2014; Redin et al., 2017; Zepeda-Mendoza & Morton, 2019).
Here we report a 6-year-old male with neuromotor and global developmental delays, intellectual disability, mild craniofacial abnormalities and 18 gene deletions on the terminal portion of one copy of chromosome 11q, diagnostic of Jacobsen syndrome (or ‘11q terminal deletion disorder’) (Favier, Akshoomoff, Mattson, & Grossfeld, 2015; Jacobsen et al., 1973). The 11q deletions are part of a more complex genomic rearrangement caused by congenital chromoanasynthesis.
2. MATERIALS AND METHODS
The family was enrolled in a screening protocol entitled ‘Evaluation of Patients with Immune Function Abnormalities’ (NCT00128973), approved by the NIAID Institutional Review Board. The parents provided written informed consent. Clinical laboratory assessments were in accordance with Clinical Laboratory Improvement Amendments standards.
For cytogenetic analysis, peripheral blood mononuclear cells were isolated from heparinized blood by Ficoll-Paque centrifugation and stimulated with IL-2 (Peprotech, Rocky Hill, NJ) and PB-Max Medium (Gibco, Carlsbad, CA) to create cultured lymphoblastoid cells in metaphase. GTW (G-banding by Trypsin treatment followed by Wright stain)-banded metaphases were obtained using established harvesting and banding techniques. Spectral and mBand karyotyping and Fluorescence in situ hybridization (FISH) were performed as detailed in the Supplemental Appendix. DNA microarray was performed by Quest Diagnostics (Chantilly, VA) using the CytoScan® HD SNP-oligo microarray (Affymetrix, Santa Clara, CA) following the manufacturer’s protocol.
Whole genome sequencing was performed as described in detail in the Supplementary Appendix. Briefly, genomic DNA extracted from whole blood cells was fragmented, ligated to bar-coded adapters and sequenced on an Illumina HiSeq 2500 instrument as paired-end 2 × 100 bp reads. Sequencing produced a total of 1.32 billion read pairs, resulting in an average coverage of 80X, and are available at Genbank accession number pending. Adapter-trimmed and quality filtered paired-end sequence reads were aligned to the reference human genome sequence GRCh37 (hg19) using Bowtie2 and TopHat (Trapnell, Pachter, & Salzberg, 2009). Candidate fusion junctions were validated by Sanger sequencing of junction specific amplicons generated by PCR. Whole genome and Sanger sequence data were interrogated manually to determine the position and junctional features including micro-homologous regions and small deletions and non-templated insertions. Rearrangements were visualized using Circos software (Krzywinski et al., 2009).
3. CASE REPORT AND RESULTS
The patient is a 6-year-old male born after 41 weeks gestation to a G1/P0 Asian mother and Caucasian father. There were dysmorphic craniofacial features including trigonocephaly, a flattened nasal bridge, mild hypertelorism, increased epicanthal folds, mild ptosis, low set ears, a thin upper lip, a small lower jaw and a slightly high-arched palate (Figure 1A and Table S1). A large café-au-lait spot was present on the left leg, and a Mongolian spot was present over the sacrum. Both feet were hyperpronated and the right testicle was undescended. Coloboma was present in the inferior iris and retina of the left eye and both optic nerves showed inferior sloping and elongation, consistent with coloboma. There was also chronic constipation.
Figure 1.
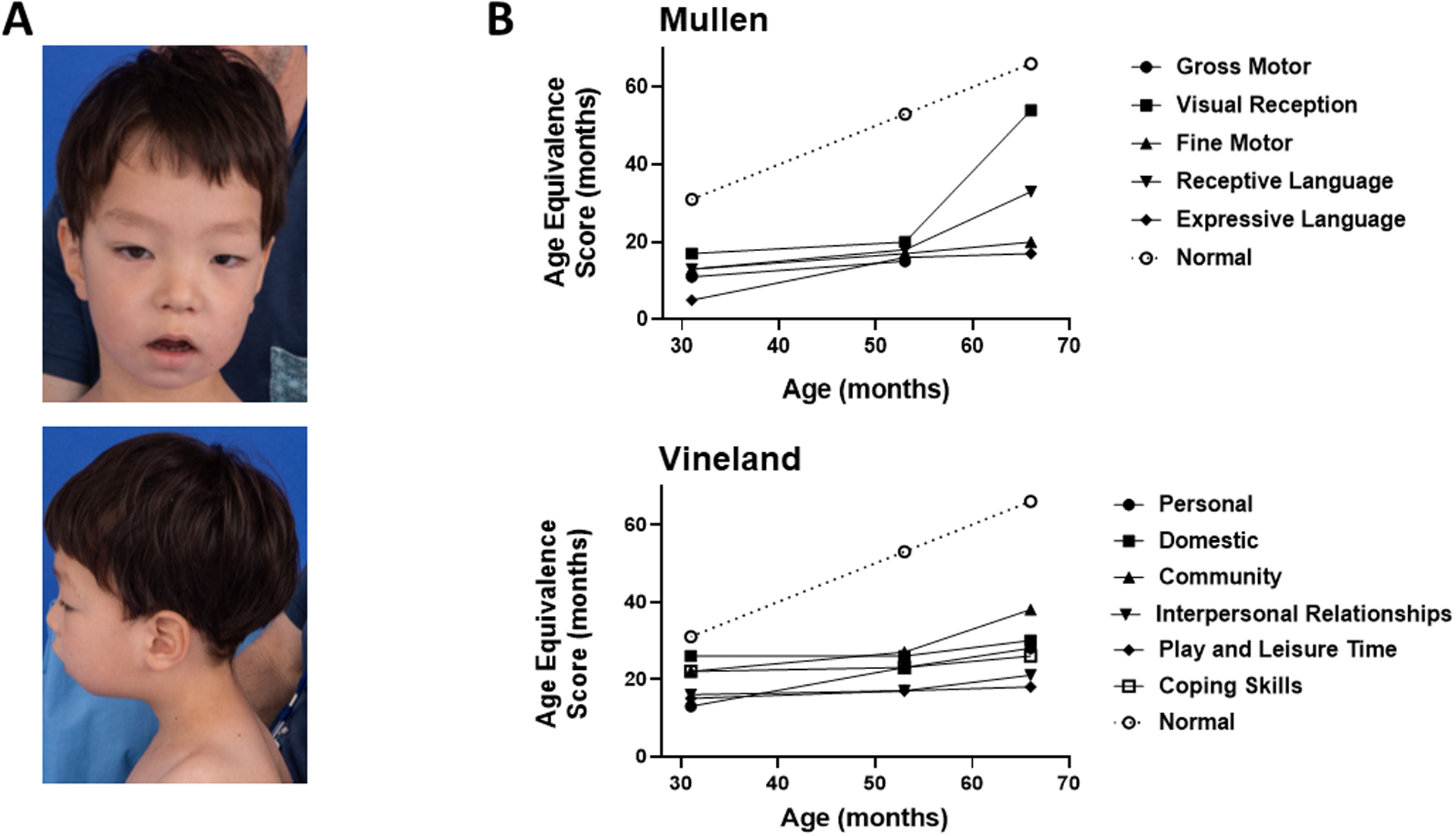
Developmental phenotypes of a patient with congenital chromoanasynthesis of chromosome 11q. (A) Dysmorphic craniofacial features. Clinical photography was obtained at age 5. (B) Neurodevelopmental delay. Top, language and motor delay were quantitated using the Mullen Scales of Early Learning tool; bottom, socialization delay was quantitated using the Vineland Adaptive Behavior Scales tool (Vineland-II). ‘Control’ depicts age-equivalent scores equal to the corresponding chronological age. Detailed developmental descriptions are in the Results section of the Supplementary Appendix. Results from the Communication and Motor Skills portions of the Vineland evaluation are in Table S1 and were consistent with results from the Mullen evaluation.
The growth trajectory was within normal limits, but truncal and lower extremity hypotonia and developmental delays were present. Beginning at age 2, research-based serial quantitative assessments of development were performed using the Mullen Scales of Early Learning and Vineland Adaptive Behavior Scales, Second Edition (Vineland-II) evaluation tools, which revealed marked delay of gross and fine motor skills, language and socialization (Figure 1B, Table S1, Supplemental Appendix). An emerging relative strength was visual reception, which includes basic nonverbal conceptual skills, but also requires visual and spatial skills; whereas, expressive language (and speech in particular) was consistently the weakest area.
At age 5 years, 3 months, the patient suddenly transitioned from crawling to walking independently with a slightly ataxic gait. He attended kindergarten, played well with other children, made good eye contact and was affectionate and curious, although he remained minimally verbal and met criteria for intellectual disability. Computerized tomography studies performed at age 4 years, 5 months revealed mild colpocephaly in the brain and no congenital malformations of internal organs in the abdomen or thorax.
G band and spectral karyotyping of metaphase chromosome spreads from cultured patient peripheral blood mononuclear cells consistently revealed a deletion of one copy of chromosome 11q24 (Figure 2A). Spectral karyotyping did not reveal interchromosomal translocations and mBand FISH identified no gross chromosome 11 rearrangements (Figure S1A–C). Microarray analysis of leukocyte DNA revealed 7 areas of copy number variation, (3 gains and 4 losses) on chromosome 11q. In aggregate, 4.894 Mb were duplicated, and 6.8 Mb were deleted (Figure 2B) for a net DNA loss of ~2 Mb. DNA isolated from a cheek swab sample gave the same result, making mosaicism unlikely. Microarray analyses of leukocyte DNA from both parents were normal. The presence of clustered copy number gains and losses was consistent with chromoanasynthesis.
Figure 2.
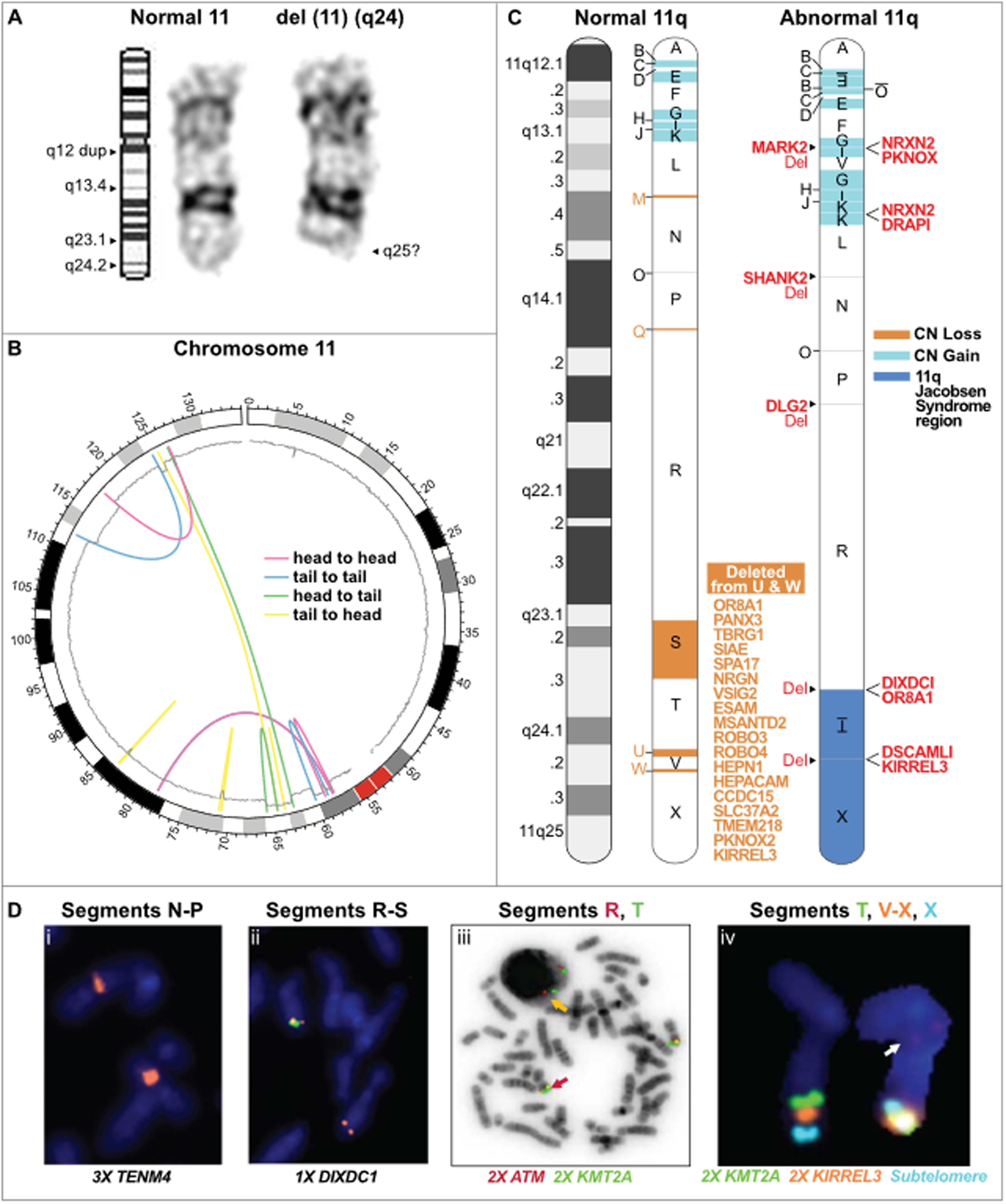
Chromoanasynthesis of chromosome 11q as a mechanism for terminal 11q deletions in a patient with Jacobsen syndrome. (A) G band cytogenetic analysis of chromosome 11 in a Jacobsen Syndrome patient. Left, ideogram of normal chromosome 11 with labeled q arm bands including location of a q12 duplication (dup); middle, normal copy of patient chromosome 11; right, abnormal copy of patient chromosome 11 with deletion of band q24. Results are representative of 10 individual metaphases. See Figure S1 for the complete G-banded karyotype and spectral karyotyping. (B) Circos plot summarizing the rearrangements of the derivative chromosome 11. The chromosome is arranged circularly end-to-end. The outermost ring represents the chromosome ideogram with the centromere indicated in red. The inner ring represents the copy number based on smoothed copy number values from the chromosome microarray data. Centrifugal and centripetal deviations of the copy number tracing mark regions of gain and loss, respectively. The colored lines indicate the position and orientation of the breakpoint junctions based on the paired-end read data. The orientation of the rearrangements are head to head (red); tail to tail (blue); head to tail (green); or tail to head (yellow), as indicated in the inset.
(C) Linear model of the derivative chromosome 11q arm. Left, normal chromosome 11q depicted as a G-banded ideogram (left) and as a segmented chromosome (right) with letter-named segments (A-X) whose boundaries are the breakpoints defined by whole genome sequencing of patient DNA. Right, model of derivative chromosome 11q. Deleted segments are in salmon; duplicated segments are in blue; inverted segments are underlined and written upside down. Segment O possibly had 3 copies based on reads in the Integrative Genomics Viewer. Genes that are partially deleted (Del) or fused are listed in red adjacent to their position on the derivative chromosome. The 18 genes deleted from segments U and W in the Jacobsen region are listed in salmon.
(D) Fluorescence in-situ hybridization of patient metaphase spreads using specific probes for the genes indicated at the bottom of each panel located in the segments indicated at the top of each panel. The copy number state is indicated by #X, next to the corresponding gene. Panels iii and iv show results of combined FISH for the indicated probes which are color-aligned with the segment names. In panel iii, the orange arrow points to the wild type arrangement of KMT2A/MLL (green signal) and ATM (red signal). In panel iv, the wild type signal arrangement of chromosome 11q is on the left and the abnormal arrangement is on the right. Evidence of segment V translocation is visible (arrow).
We next performed whole genome sequencing of patient leukocyte DNA with ~80X coverage. The data are openly available in dbGaP at https://dbgap.ncbi.nlm.nih.gov, accession number phs002036. Mate-pair analysis confirmed all 4 copy number losses and 3 copy number gains and revealed 4 additional copy number changes (3 duplications and 1 deletion) (Table S2). The gains were all clustered towards the centromere in the 11q12-q13 region and were segregated from most of the losses which were clustered towards the telomere mainly in the 11q23 region (Figures 2B and 2C). The breakpoints divided 11q into 24 segments; the gains and the remaining segments in the region of loss were rearranged into a derivative chromosome with 26 segments (Figure 2C). These included a tandem duplication of segment K, in situ inversion of segment T, which was not duplicated, and inverted and non-inverted rearrangements of multiple other segments. Nevertheless, the rearrangement was not completely random, since duplicated segments all remained in 11q12-q13 and surviving segments from the telomeric region remained in approximately the same sub-telomeric order in 11q23-q24. Segment V was the sole outlier in this regard, moving from 11q24.2 to 11q13.2. The full list of 183 duplicated and 73 deleted genes is provided in Table S2. One fusion gene was predicted, NRXN2/DRAP1 (Figure S2). Segment-specific FISH probes alone and in combination were used to confirm duplications and deletions, as well as the order of the rearrangements (Figure 2D and Figure S3).
All but one of the breakpoint junctions were confirmed by PCR and Sanger sequencing (Figures 3A and S4A) and all contained microhomology with or without small indels (Figure 3B and S4B). The last breakpoint junction (BE) could not be amplified by PCR because it was flanked by inverted long interspersed nuclear element (LINE) repeats. Four other breakpoint junctions occurred in LINE sequences. Microhomology at breakpoint junctions indicates a replication-based mechanism of rearrangement such as MMBIR,(Hastings et al., 2009; Liu et al., 2011) and a replication model composed of multiple fork switches compatible with the data is proposed in Figure 3C.
Figure 3.
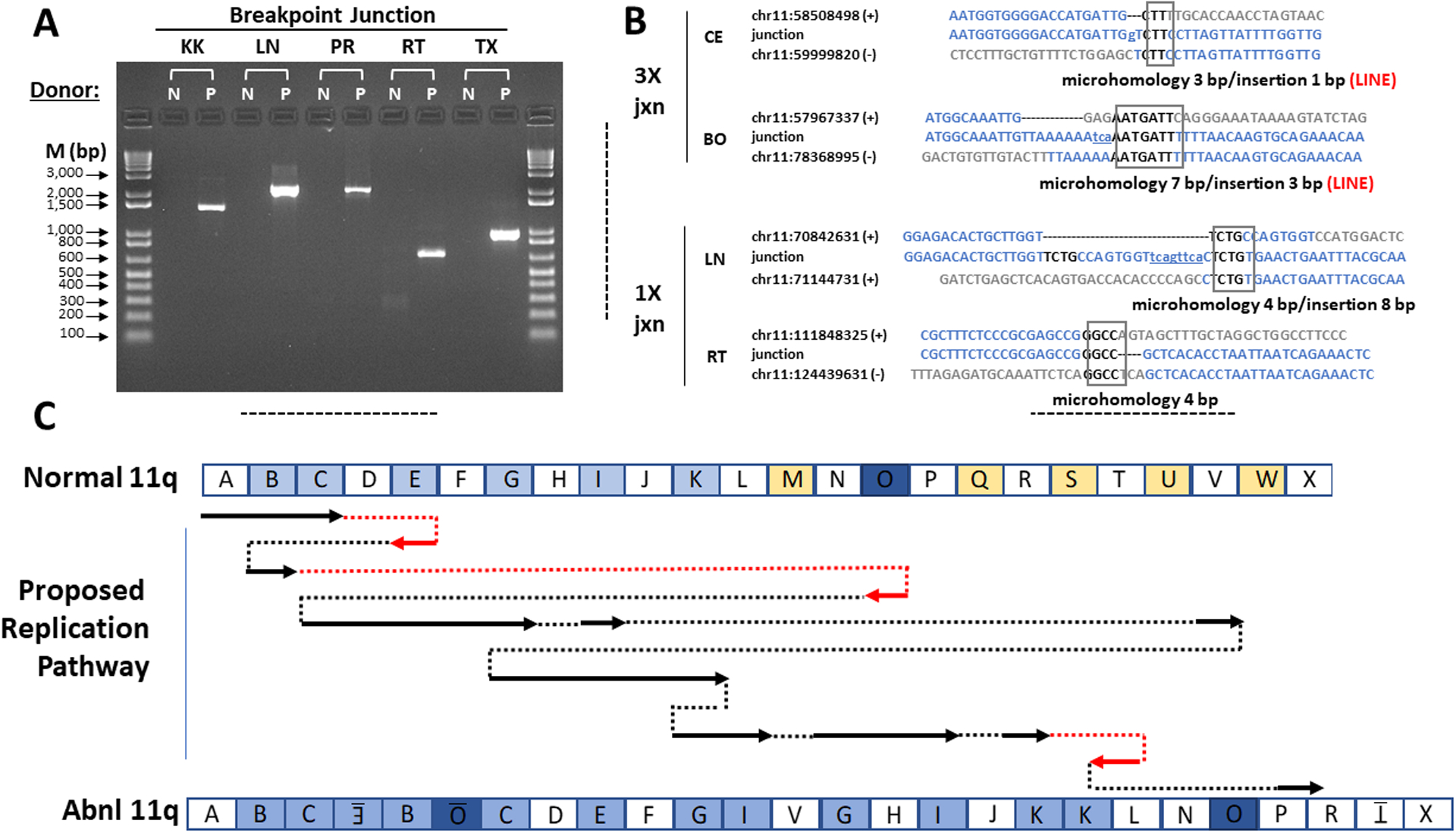
Microhomology-Mediated Break-Induced Replication as a potential mechanism for chromoanasynthesis. (A) Confirmation of patient chromosome 11 breakpoint junctions by PCR. Donors: N, normal control; P, patient. PCR primers were designed to amplify DNA only from the breakpoint junctions identified from patient DNA by whole genome sequencing. The names of the breakpoint junctions are composed of the two flanking segment names as defined in Figures 2C and 3C. M, markers (base pairs). (B) Breakpoint junction sequence analysis. Four of the 12 breakpoint junction sequences determined by the Sanger method are shown. The other junction sequences are in Figure S4B. At left is indicated the type of copy number variation at the indicated junction. The name of each junction is formed by the adjacent segments as defined in Figures 2C and 3C. For each junction, the top and bottom sequences shown in blue are those of the reference sequence + and - strands that contribute to the junction. The number is the chromosome 11 position from the hg19 reference sequence of the first nucleotide shown. The middle sequences are those determined experimentally by the Sanger method for the patient using primers designed to flank each breakpoint junction. The boxed sequence denotes regions of microhomology in the junction, and the lower-case and underlined nucleotides denote insertions, as summarized below each alignment. Both 3X junctions occurred at a LINE sequence as indicated by the word LINE in red. The BE breakpoint could not be validated with Sanger sequencing due to surrounding inverted LINE sequences. (C) Proposed chromoanasynthesis replication pathway on chromosome 11q in Jacobsen Syndrome patient. The top and bottom models of chromosome 11q are segmented according to the experimentally determined breakpoints as defined in Figure 2. In the proposed replication pathway, arrows denote regions of normal chromosome 11q that appear in the derivative patient chromosome 11q, either in the normal orientation (black) or inverted (red). Dotted lines indicate movement from one replication fork to another, either in the normal orientation (black) or the opposite orientation (red).
The clinical phenotypes in this patient occur in multiple developmental disorders. However, coexistence of terminal 11q deletions establishes the diagnosis of Jacobsen Syndrome. Importantly, only 18 genes were deleted from the Jacobsen region. Deleted segment U in 11q24.2 contains 17 of these (Figure 2C, Table S2), one of which, HEPACAM, is associated with megalencephalic leukoencephalopathy with cortical cysts (MIM# 604004) (Lopez-Hernandez et al., 2011). The 154 kb deleted segment W in the Jacobsen region includes only 5 exons of KIRREL3, which is associated with autosomal dominant immunodeficiency and intellectual defects (Talkowski et al., 2012). Of these pathologies, the patient only has evidence of intellectual impairments. Several other diseases have been associated with mutations in the 11q areas affected by copy number variation in the patient, as detailed in the Supplemental Appendix; however, none is manifest in the patient to date.
4. DISCUSSION
Approximately 90% of Jacobsen syndrome cases arise from de novo deletions on one copy of chromosome 11q with breakpoints occurring within or centromeric to 11q23.3, deleting 5–20 Mb of DNA and extending to the telomere (Favier et al., 2015). The remaining ~10% involve unbalanced segregation of a familial balanced translocation, de novo unbalanced translocations, or other chromosomal rearrangements of 11q, including deletions nested in the Jacobsen region. Most cases have deletions of ~170 to >340 terminal 11q genes. The smallest terminal deletion previously reported is 4.1 Mb in 11q25 from 130.3–134.5 Mb. This region includes only 21 genes, all of which lie distal to the 0.8 Mb deletions in the Jacobsen region in our patient, and this other patient had developmental delay, microcephaly and facial dysmorphisms (Ji, Wu, Wang, Wang, & Jiang, 2010).
The ~200 Jacobsen syndrome cases have presented with diverse phenotypes. The most penetrant (>90%) are neuromotor and language delay, cognitive impairment, learning disabilities and dysmorphic craniofacial and ocular features, including the ones in our patient, as well as lifelong bleeding and easy bruisability (Paris-Trousseau syndrome, MIM # 188025), which was absent. Importantly, Paris-Trousseau syndrome is caused by thrombocytopenia and platelet α granule dysfunction that has been linked to deletion of FLI1, which was intact in our patient (Stevenson et al., 2015). Behavioral problems are common in Jacobsen syndrome, including autism spectrum disorder (Akshoomoff, Mattson, & Grossfeld, 2015). Our patient was young, and significantly globally delayed, preventing comprehensive assessment of behavior. Other common Jacobsen phenotypes (~50%) include short stature and congenital heart, kidney and stomach defects, which were absent in our patient, and chronic constipation, which was present. Less common phenotypes include cryptorchidism, which was present, and sleep disturbances, primary immunodeficiency, osteopenia and sensorineural hearing loss, which were absent.
The large number of 11q genes typically deleted in Jacobsen syndrome complicates genotype-phenotype considerations. On one hand, the 18 gene deletion within the Jacobsen region in our patient represents a simplification of the problem. On the other hand, 184 gene duplications and 55 gene deletions occurred outside the Jacobsen region, and must also be considered. In this regard, several patients with speech delay have been reported who have smaller duplications within the boundaries of the patient’s 11q12.1 duplication (Collins et al., 2017). However, motor developmental delay, dysmorphic features and coloboma have not been reported for any of the 11q regions outside the Jacobsen region that were affected in the patient.
Previous analyses of Jacobsen patients with small deletions in the Jacobsen region as well as on mouse genetic models have focused on genes expressed in human brain as candidates to explain cognitive impairment, including B3GAT1, BSX and NRGN. Mice lacking B3GAT1 have impaired synaptic plasticity and learning (Yabuno et al., 2015). Mice lacking NRGN, which encodes neurogranin, have major defects in synaptic plasticity and long-term potentiation, which is important for learning and memory (Coldren et al., 2009). However, mice that are haploinsufficient for these genes are neurologically normal. Of these three candidate genes, our patient was hemizygous only for NRGN.
Every chromoanagenesis event results in a unique genotype. Future studies will be needed to address the contributions of each of the individual affected genes to our patient’s phenotypes; however, clearly the 18 in the Jacobsen region take priority. Our case re-emphasizes the general point that cytogenetics does not necessarily reveal the full complexity of chromosomal damage in genomic disorders.
Supplementary Material
Acknowledgements
We thank the patient and his parents for participating in this research, Kent Barbian for technical assistance, Austin Athman and Ryan Kissinger for assistance with graphical illustrations, and Harry Malech for creating the screening protocol and helpful discussions. This work was supported by the Divisions of Intramural Research of the National Institute of Allergy and Infectious Diseases, the National Cancer Institute, the National Institute of Mental Health, and the National Eye Institute, as well as the Clinical Center, all from the National Institutes of Health, USA.
Footnotes
Supplemental Data
Supplemental data include four figures and two tables
Declaration of Interests
The authors declare no competing interests.”
Data Sharing Statement
The data that support the findings of this study are openly available in dbGaP at https://dbgap.ncbi.nlm.nih.gov, accession number phs002036.
References/Bibliography
- Akshoomoff N, Mattson SN, & Grossfeld PD (2015). Evidence for autism spectrum disorder in Jacobsen syndrome: identification of a candidate gene in distal 11q. Genet Med, 17(2), 143–148. doi: 10.1038/gim.2014.86 [DOI] [PubMed] [Google Scholar]
- Coldren CD, Lai Z, Shragg P, Rossi E, Glidewell SC, Zuffardi O, … Grossfeld PD (2009). Chromosomal microarray mapping suggests a role for BSX and Neurogranin in neurocognitive and behavioral defects in the 11q terminal deletion disorder (Jacobsen syndrome). Neurogenetics, 10(2), 89–95. doi: 10.1007/s10048-008-0157-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RL, Brand H, Redin CE, Hanscom C, Antolik C, Stone MR, … Talkowski ME (2017). Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome. Genome Biol, 18(1), 36. doi: 10.1186/s13059-017-1158-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favier R, Akshoomoff N, Mattson S, & Grossfeld P (2015). Jacobsen syndrome: Advances in our knowledge of phenotype and genotype. Am J Med Genet C Semin Med Genet, 169(3), 239–250. doi: 10.1002/ajmg.c.31448 [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Lupski JR, Rosenberg SM, & Ira G (2009). Mechanisms of change in gene copy number. Nat Rev Genet, 10(8), 551–564. doi: 10.1038/nrg2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen P, Hauge M, Henningsen K, Hobolth N, Mikkelsen M, & Philip J (1973). An (11;21) translocation in four generations with chromosome 11 abnormalities in the offspring. A clinical, cytogenetical, and gene marker study. Hum Hered, 23(6), 568–585. doi: 10.1159/000152624 [DOI] [PubMed] [Google Scholar]
- Ji T, Wu Y, Wang H, Wang J, & Jiang Y (2010). Diagnosis and fine mapping of a deletion in distal 11q in two Chinese patients with developmental delay. J Hum Genet, 55(8), 486–489. doi: 10.1038/jhg.2010.51 [DOI] [PubMed] [Google Scholar]
- Kloosterman WP, Guryev V, van Roosmalen M, Duran KJ, de Bruijn E, Bakker SC, … Cuppen E (2011). Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum Mol Genet, 20(10), 1916–1924. doi: 10.1093/hmg/ddr073 [DOI] [PubMed] [Google Scholar]
- Kloosterman WP, Tavakoli-Yaraki M, van Roosmalen MJ, van Binsbergen E, Renkens I, Duran K, … Cuppen E (2012). Constitutional chromothripsis rearrangements involve clustered double-stranded DNA breaks and nonhomologous repair mechanisms. Cell Rep, 1(6), 648–655. doi: 10.1016/j.celrep.2012.05.009 [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, … Marra MA (2009). Circos: an information aesthetic for comparative genomics. Genome Res, 19(9), 1639–1645. doi: 10.1101/gr.092759.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, … Consortium P (2020). Patterns of somatic structural variation in human cancer genomes. Nature, 578(7793), 112–121. doi: 10.1038/s41586-019-1913-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Erez A, Nagamani SC, Dhar SU, Kolodziejska KE, Dharmadhikari AV, … Bi W (2011). Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell, 146(6), 889–903. doi: 10.1016/j.cell.2011.07.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Hernandez T, Ridder MC, Montolio M, Capdevila-Nortes X, Polder E, Sirisi S, … van der Knaap MS (2011). Mutant GlialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am J Hum Genet, 88(4), 422–432. doi: 10.1016/j.ajhg.2011.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly P, Brunner SF, Shoshani O, Kim DH, Lan W, Pyntikova T, … Cleveland DW (2019). Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat Genet, 51(4), 705–715. doi: 10.1038/s41588-019-0360-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott DH, Gao JL, Liu Q, Siwicki M, Martens C, Jacobs P, … Murphy PM (2015). Chromothriptic cure of WHIM syndrome. Cell, 160(4), 686–699. doi: 10.1016/j.cell.2015.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellestor F (2019). Chromoanagenesis: cataclysms behind complex chromosomal rearrangements. Mol Cytogenet, 12, 6. doi: 10.1186/s13039-019-0415-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaisancie J, Kleinfinger P, Cances C, Bazin A, Julia S, Trost D, … Vigouroux A (2014). Constitutional chromoanasynthesis: description of a rare chromosomal event in a patient. Eur J Med Genet, 57(10), 567–570. doi: 10.1016/j.ejmg.2014.07.004 [DOI] [PubMed] [Google Scholar]
- Redin C, Brand H, Collins RL, Kammin T, Mitchell E, Hodge JC, … Talkowski ME (2017). The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat Genet, 49(1), 36–45. doi: 10.1038/ng.3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, … Campbell PJ (2011). Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell, 144(1), 27–40. doi: 10.1016/j.cell.2010.11.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson WS, Rabbolini DJ, Beutler L, Chen Q, Gabrielli S, Mackay JP, … Morel-Kopp MC (2015). Paris-Trousseau thrombocytopenia is phenocopied by the autosomal recessive inheritance of a DNA-binding domain mutation in FLI1. Blood, 126(17), 2027–2030. doi: 10.1182/blood-2015-06-650887 [DOI] [PubMed] [Google Scholar]
- Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A, … Gusella JF (2012). Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell, 149(3), 525–537. doi: 10.1016/j.cell.2012.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, & Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics, 25(9), 1105–1111. doi: 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabuno K, Morise J, Kizuka Y, Hashii N, Kawasaki N, Takahashi S, … Oka S (2015). A Sulfated Glycosaminoglycan Linkage Region is a Novel Type of Human Natural Killer-1 (HNK-1) Epitope Expressed on Aggrecan in Perineuronal Nets. PLoS One, 10(12), e0144560. doi: 10.1371/journal.pone.0144560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zepeda-Mendoza CJ, & Morton CC (2019). The Iceberg under Water: Unexplored Complexity of Chromoanagenesis in Congenital Disorders. Am J Hum Genet, 104(4), 565–577. doi: 10.1016/j.ajhg.2019.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, … Pellman D (2015). Chromothripsis from DNA damage in micronuclei. Nature, 522(7555), 179–184. doi: 10.1038/nature14493 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are openly available in dbGaP at https://dbgap.ncbi.nlm.nih.gov, accession number phs002036.