

Abstract
The methodology of ab initio molecular dynamics, wherein finite-temperature dynamical trajectories are generated by using forces computed “on the fly” from electronic structure calculations, has had a profound influence in modern theoretical research. Ab initio molecular dynamics allows chemical processes in condensed phases to be studied in an accurate and unbiased manner, leading to new paradigms in the elucidation of microscopic mechanisms, rationalization of experimental data, and testable predictions of new phenomena. The purpose of this work is to give a brief introduction to the technique and to review several important recent developments in the field. Several illustrative examples showing the power of the technique have been chosen. Perspectives on future directions in the field also will be given.
Modern theoretical methodology, aided by the advent of high speed and massively parallel computing, has advanced to a level that the microscopic details of chemical processes in condensed phases can now be treated on a relatively routine basis. One of the most commonly used theoretical approaches for such studies is the molecular dynamics (MD) method, in which the classical Newtonian equations of motion for a system are solved numerically starting from a prespecified initial state and subject to a set of boundary conditions appropriate to the problem. MD methodology allows both equilibrium thermodynamic and dynamical properties of a system at finite temperature to be computed. The quality of a MD calculation rests largely on the method by which the forces are specified. In many applications, these forces are computed from an empirical model or “force field,” an approach that has enjoyed tremendous success in the treatment of systems ranging from simple liquids and solids to polymers and biological systems including proteins, membranes, and nucleic acids. Since most force fields do not include electronic polarization effects (see, however, ref. 1) and can treat chemical reactivity only through specialized techniques (see, e.g., ref. 2), it is often necessary to turn to the methodology of ab initio MD (AIMD).
AIMD is a rapidly evolving and growing technique that constitutes one of the most important theoretical tools developed in the last decades. In an AIMD calculation, finite-temperature dynamical trajectories are generated by using forces obtained directly from electronic structure calculations performed “on the fly” as the simulation proceeds. Thus, AIMD permits chemical bond breaking and forming events to occur and accounts for electronic polarization effects (3, 4). AIMD has been successfully applied to a wide variety of important problems in physics and chemistry and is now beginning to influence biology as well. In numerous studies, new physical phenomena have been revealed and microscopic mechanisms elucidated that could not have been uncovered by using empirical methods, often leading to new interpretations of experimental data and even suggesting new experiments to perform.
In its most ideal form, an AIMD calculation assumes only that the system is composed of N nuclei and Ne electrons, that the Born–Oppenheimer approximation is valid, and that the dynamics of the nuclei can be treated classically on the ground-state electronic surface. The total Hamiltonian is H = Te + Vee + VeN + TN + VNN ≡ Helec + TN + VNN, where the terms are the electronic kinetic energy, the electron–electron Coulomb repulsion, the electron–nuclear Coulomb attraction, the nuclear kinetic energy and the nuclear–nuclear Coulomb repulsion, respectively. If R1,..., RN ≡ R denote the nuclear positions, then the classical dynamics of the nuclei is given by an equation of motion
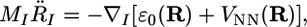 |
[1] |
where ε0(R) is the corresponding ground-state energy eigenvalue at the nuclear configuration R.
Since the ground-state electronic problem Helec(R)Ψ0(R) = ε0(R)Ψ0(R) cannot be solved exactly, approximate electronic structure methods are needed. In modern AIMD, the electronic structure method most commonly used is the Kohn–Sham (KS) formulation of density functional theory, wherein the total energy is expressed as a functional of n mutually orthonormal single-particle electron orbitals ψi(r), i = 1,..., n. The orbitals are related to the electronic density according to n(r) = Σi fi|ψi(r)|2, where fi is the occupation of the ith orbital. For spin-unrestricted calculations, n = Ne and fi = 1, whereas for the spin-restricted case, n = Ne/2 and fi = 2. The total energy is given by
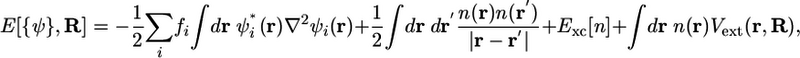 |
[2] |
where Vext(r, R) = –ΣIZIe2/|r – RI| is the total Coulomb attraction between one electron and the nuclei. Minimization of this functional over orbitals subject to an orthonormality condition 〈ψi|ψj〉 = δij yields the ground state energy ε0(R) appearing in Eq. 1. Although Eq. 2 is formally exact, the form of the exchange–correlation energy, Exc[n], is unknown and, therefore, must be approximated. Therefore, the quality of the electronic structure calculation rests on the quality of the approximation used for Exc. The most common classes of approximations are the local-density, generalized-gradient, and meta-generalized-gradient approximations. Such approximations generally cannot treat dispersion accurately; however, a recent class of nonlocal functionals (9–11) seems to improve significantly on this shortcoming. In the most straightforward type of AIMD calculation, Eq. 1 is integrated numerically by using a symplectic integrator, and, at each time step, the forces are computed by minimizing the KS energy functional in Eq. 2 at the present nuclear configuration. This type of AIMD is called Born–Oppenheimer dynamics. However, as was pointed out in ref. 3, the minimization must be carried out to a high level of accuracy to ensure that the classical nuclear Hamiltonian is properly conserved and does not drift. Alternatively, AIMD can be carried out by using the Car–Parrinello (CP) extended Lagrangian approach (12). In that approach, the KS orbitals are imbued with a fictitious time dependence, i.e., ψi(r) → ψi(r,t), and a dynamics for the orbitals is introduced that propagates an initially fully minimized set of orbitals to subsequent minima corresponding to each new nuclear configuration. This task is accomplished by designing the orbital dynamics in such a way that the orbitals are maintained at a “temperature” Te that is much smaller than the real nuclear temperature T but are still allowed to relax quickly in response to the nuclear motion. The complete motion of the system, i.e., the fictitious orbital dynamics and real nuclear dynamics, is specified by the CP Lagrangian (12),
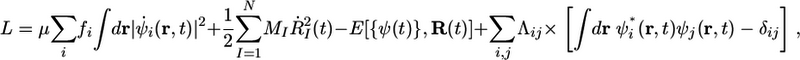 |
[3] |
where the last term in Eq. 3 involves a set of Lagrange multipliers Λij for maintaining the orthonormality of the orbitals as a constraint condition. The aforementioned conditions on the orbital dynamics are controlled through the first term in Eq. 3, a fictitious kinetic energy term involving a mass-like parameter μ, which has units of energy × time2. The fictitious temperature Te of the orbitals is controlled by the initial choice of their “velocities” 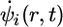 , and the adiabatic decoupling from the nuclear dynamics is controlled by the choice of μ, specifically, by choosing μ to be as small as is computationally feasible. As Tuckerman and Parrinello (13) showed, the value of μ can be increased if thermostatting techniques on the orbitals are used. Issues related to increasing μ were recently rediscovered by Galli and coworkers (14). This Lagrangian yields the CP equations of motion
, and the adiabatic decoupling from the nuclear dynamics is controlled by the choice of μ, specifically, by choosing μ to be as small as is computationally feasible. As Tuckerman and Parrinello (13) showed, the value of μ can be increased if thermostatting techniques on the orbitals are used. Issues related to increasing μ were recently rediscovered by Galli and coworkers (14). This Lagrangian yields the CP equations of motion
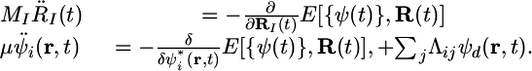 |
[4] |
Below, several recent, significant developments in AIMD methodology will be discussed, and selected examples illustrating the power of the approach will be presented.
System-Size Scaling and Orbital Localization
AIMD calculations typically are performed by using a plane-wave expansion of the KS orbitals. Because MD calculations of bulk systems employ periodic boundary conditions on the simulation cell, the orbitals should be treated as Bloch functions and include a proper sampling of the Brioullin zone. However, for many applications in chemistry, it is sufficient to treat only the Γ-point and expand the orbitals as
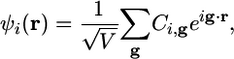 |
[5] |
where Cg,i is an expansion coefficient, V is the volume of the cell, and g = 2π h–1ĝ, with h as the cell matrix (V = deth) and ĝ as a vector of integers. In practice, the expansion is truncated by including only those g-vectors that satisfy |g|2/2 < Ecut, where Ecut is a chosen plane-wave kinetic energy cutoff. Plane waves are a natural choice of basis set for treating periodic systems and can be extended to treat clusters, surfaces, and wires as well (15–17). Although plane waves have the advantage of simplicity, they nevertheless lead to n2M scaling behavior in computational overhead with system size, where M is the number of plane waves. Nevertheless, the properties of nonmetallic matter are local, and plane waves, being fully delocalized functions, are not well suited for describing an electron density comprised of localized contributions. Indeed, formulations of AIMD in terms of localized basis functions such as Gaussians (18, 19) and discrete variable representations (4, 20) have been proposed.
Using localized basis sets potentially leads to linear scaling methodology; however, advantage of the local character of the basis functions cannot be taken unless the orbitals themselves also are spatially localized. Unfortunately, the orbitals that result from a minimization or the KS energy functional or are generated during the CP fictitious dynamics are not guaranteed to be localized because the total electronic energy is invariant with respect to a unitary transformation among the orbitals. If 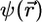 denotes a column vector of the n orbitals, and if a new column
denotes a column vector of the n orbitals, and if a new column 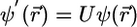 is defined, where U is a constant unitary matrix, then the total energy expressed in terms of
is defined, where U is a constant unitary matrix, then the total energy expressed in terms of 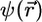 or
or 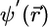 will have the same form. This unitary invariance property means that the output orbitals of a minimization calculation or step of CP evolution is highly likely to be an unknown linear combination of spatially localized orbitals and, hence, spatially “delocalized.” The problem of localizing the orbitals thus amounts to introducing a definition of spatial locality to find the appropriate inverse unitary transformation. A common approach for periodic systems is to define a functional Ω[{ψ}] by (22–25)
will have the same form. This unitary invariance property means that the output orbitals of a minimization calculation or step of CP evolution is highly likely to be an unknown linear combination of spatially localized orbitals and, hence, spatially “delocalized.” The problem of localizing the orbitals thus amounts to introducing a definition of spatial locality to find the appropriate inverse unitary transformation. A common approach for periodic systems is to define a functional Ω[{ψ}] by (22–25)
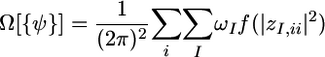 |
[6] |
where σ and L denote the typical spatial extent of a localized orbital and box length, respectively, and
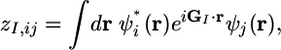 |
[7] |
is a complex number with GI = 2π (h–1)ĝI. Here, ĝI = (lI, mI, nI) is the Ith Miller index. The weights ωI have the dimension of (length)2.
Eq. 6 is analogous to the “spatial spread” functional originally introduced by Boys and Foster (21). Here, f(x) = 1 – x. Orbital localization is achieved by substituting ψ′(r) = Uψ (r) into Eq. 6 to give Ω[{ψ′}] and then minimizing with respect to Uij. The solution will be a unitary matrix U that generates a set of orbitals with minimal spatial spread. The orbitals generated in this way are known as “Wannier orbitals” (26). If zI,ii is evaluated with respect to these orbitals, then the orbital centers are known as “Wannier centers.” Fig. 1 illustrates the phenomenon of orbital localization in a configuration of an aqueous solution subject to periodic boundary conditions.
Fig. 1.

Illustration of orbital localization in a system of 64 water molecules with a single dissociated HCl molecule. (A) Shown in yellow is an isosurface of the square of a typical delocalized orbital. B shows a localized Wannier function.
The need to minimize the spread functional explicitly at each CP or minimization step is both cumbersome and computationally costly (27). It would be far better if the electronic structure problem could be reformulated in such a way that the spatial locality of the orbitals could be maintained implicitly as the AIMD calculation proceeds. It is in the solution of this problem that the CP approach has a distinct advantage over the Born–Oppenheimer method.
It is not widely appreciated that electronic structure theories based on single-particle orbitals, such as Hartree–Fock and KS density functional theory are mathematically equivalent to SU(n) quantum field theories. The fact that the CP scheme attributes a time variable to each orbital means that the orbitals can be regarded as evolving in a “space-time” similar to that used in relativistic quantum field theories (28). The extra time dimension in the CP scheme can be exploited to solve the problem of implicit orbital localization. The invariance of the total energy with respect to the unitary transformation is also known as “gauge invariance.” A quick check shows that not only the total energy but also the CP Lagrangian in Eq. 3 possesses gauge invariance. However, for the CP Lagrangian to yield an orbital dynamics that preserves the spatial locality of the orbitals implicitly, the Lagrangian must be invariant under time-dependent unitary transformations, i.e., ψ′(r, t) = U(t)ψ(r, t), and, unfortunately, Eq. 3 does not exhibit this property. The invariance can be achieved, however, by the introduction of an additional set of variables in the form of a traceless Hermitian matrix A(t). The elements of A(t) are known as gauge variables. The form of the fictitious orbital kinetic energy then is modified by replacing the time derivative ∂/∂t in the fictitious orbital kinetic energy by Dt = ∂/∂t – iA(t). Thus, if A(t) is required to transform according to A′(t) = U(t)A(t)U†(t) – i[U̇(t)]U†(t), under a time-dependent unitary transformation of the orbitals, then the resulting Lagrangian will possesses the desired invariance. The new Lagrangian takes the form (28, 29)
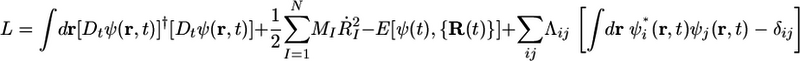 |
[8] |
In fact, the replacement in Eq. 8 is permissible because the form of the fictitious orbital kinetic energy is arbitrary. The equations of motion derived from the new Lagrangian contain A(t) but do not specify how A(t) should evolve. In fact, A(t) remains undetermined unless an additional condition is imposed on it. Choosing this condition is known as choosing a gauge or unitary representation of the orbitals. The phenomenon of choosing a gauge is well known in electrodynamics. Here, the gauge is fixed by imposing the condition that the spread functional in Eq. 6 be minimized along the orbital trajectory. It can be shown that this condition uniquely fixes A(t), and the resulting equations of motion, first reported in refs. 28 and 29, constitute a set of modified CP equations that implicitly preserve the spatial locality of the orbitals. We call the gauge chosen in this manner the “Wannier gauge.” It can, therefore, be expected that when such a scheme is combined with a localized basis set, the resulting method will be a linear scaling method.
Calculation of Observables: Example of IR Spectra
In the calculation of observables, the standard techniques used to analyze the positions and velocities in any MD simulation can be applied to AIMD trajectories as well. However, a unique feature of AIMD calculations over standard force-field-based MD calculations is the fact that the former permits direct access to the electronic properties of the system. As a result, a wide variety of spectroscopic observables (22–34) can be computed directly with a high degree of accuracy. The calculation of these quantities has been enabled by important developments in variational density functional perturbation theory (31–34) and make use of maximally localized Wannier functions.
IR spectroscopy represents one of the most sensitive methods for the study of hydrogen bonds (36). Indeed, until a few decades ago, the oversensitivity of the traditional IR transmission spectroscopy methods to the presence of hydrogen bonds often led to catastrophic saturation effects (37) that limited dramatically the utility of the approach for the study of aqueous systems. However, recent progress in the attenuated total reflection technique in linear spectroscopy (38) provides experimentalists with a convenient and accurate method to obtain optical and dielectric constants of liquids. Moreover, experimental advances in the field of nonlinear mid-IR spectroscopy (39) have opened the way for ultrafast time-resolved investigations of the dynamics of the hydrogen-bond network in aqueous solutions.
Theoretical calculations of the linear optical constants can be performed by using linear response theory from the Fourier transform of the autocorrelation function of the time derivative of the total dipole moment. Consequently, if total dipole moments could be unambiguously decomposed into a sum of local, molecule-based dipole moments, using orbital localization one could define the contribution of a molecular species to the IR spectrum by computing the time-dependent values of the cross-correlation between the local and total dipole moments.
IR absorptivity α(ν) is given by
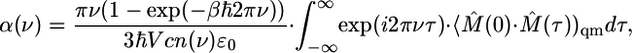 |
[9] |
where qm indicates a quantum mechanical ensemble average, α has Neperian units, an n(v) is the index of refraction and ε0 is the vacuum permittivity. In Eq. 9, M̂(0) and M̂ (t) represent the values of the quantum mechanical total dipole moment operator in the Heisenberg representation. By using an effective harmonic approximation (41) for the time correlation function, the absorptivity can be computed approximately by using a classical time correlation function according to
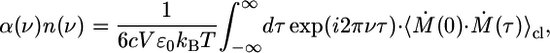 |
[10] |
where Ṁ = dM/dt. The calculation of the electronic contribution to the dipole moment M(τ) is slightly subtle in periodic systems because the electronic position operator r is not translationally invariant. However, as Resta (22–25) showed, the dipole moment can be computed in a translationally invariant way by (for a cubic box of length L)
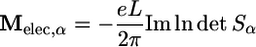 |
[11] |
where α = 1, 2, 3 and Sα,ij = zα,ij. Here, zα,ii is determined by the unit vectors along the axes of reciprocal space. Although Eq. 11 can be computed with respect to any orbital gauge, the Wannier gauge has the particular advantage that the matrix Sα is approximately diagonal, and, hence, the logarithm of the determinant reduces (approximately) to a simple sum, Mi = Σiμi. For molecular systems, the individual contributions could be grouped into contributions from each molecule, i.e., M = ΣAμA for each molecule A. If the system is composed of Nm molecules, then each molecular dipole moment could be expressed as
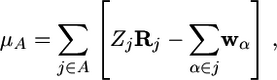 |
[12] |
where j indexes the atoms in the Ath molecule, A = 1,..., Nm, and α indexes all of the Wannier centers close to atom j with wα the position of the corresponding Wannier center. Such a decomposition allows contributions from different solute and solvent components to be isolated and identified in an IR spectrum. To account for both direct and cross-correlation terms in an effective manner, the correlation function 〈Ṁ (0)·Ṁ(τ)〉 is expressed as
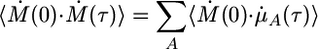 |
[13] |
Eq. 13 differs substantively from a method recently introduced by Gaigeot and Sprik (42). The utility of the Wannier gauge approach to the calculation of molecular IR spectra was tested in a computer experiment consisting of the calculation of the IR difference spectra of the excess proton in water. More precisely, the difference Δ(ν) between the absorption spectrum [α(ν)n(ν)]c of an aqueous solution of a strong acid of concentration “c” mol/liter and the absorption spectrum [α(ν)n(ν)]0 of pure water or water plus the conjugate base, i.e., Δ(ν) = [α(ν)n(ν)]c – [α(ν)n(ν)]0 was computed from a single simulation of the aqueous solution by using the Wannier orbital approach. The simulations performed include two systems as follows: a 0.85 M aqueous solution of HCl and a 0.85 M aqueous solution of H2F2 that were completely dissociated at 300 K. The generalized gradient approximation to density functional theory (45–46) and atomic pseudopotentials (47) was used on a system of 64 water molecules plus solute. All simulations were performed with the piny_md code (54) using a plane-wave basis set with a cutoff of 80 rydberg. Fig. 2 shows the theoretical difference spectra Δ(ν) defined above with c = 0.85 M. The figure shows that by using the maximally localized orbitals-based IR decomposition approach, it is possible to separate the spectrum of the excess proton from that of FHF– ion, even though both ions absorb strongly between 1,200 and 2,000 cm–1.
Fig. 2.

Theoretical difference spectra Δ(ν) (see text) scaled to a concentration c = 0.85 M. The red solid line is the spectrum of the HCl solution minus that of bulk water and that of the Cl. The red dashed line is the spectrum of HCl solution minus that of bulk water. The blue solid line is the spectrum of the H2F2 solution minus that of bulk water and that of the bifluoride ion. The blue dashed line is the spectrum of H2F2 solution minus that of bulk water.
Application: Addition of cis-1,3-butadiene to the Si(100)-2×1 Surface
The chemistry of hybrid structures composed of organic molecules and semiconductor surfaces (48, 49) is opening up exciting new avenues of development in molecular electronics, nanoscale devices, and surface lithography. The possibility of engineering such novel structures requires a detailed understanding of how organic molecules react with the semiconductor substrate. To realize the covalent attachment of nanoscale objects to the surface, controlled functionalization of the latter is required. Although the chemical vapor deposition technique is the currently used tool in this atomic level fabrication process, novel “carrier” scanning tunneling microscopic tips capable of holding reactive organic agents could open up an entirely new field of mechanistically selective surface functionalization.
Joint theoretical and experimental studies of conjugated dienes on a Si(100)-2×1 surface (48, 49) identified an analog version of one of the most important products of organic reactions, the Diels–Alder (DA) [4+2] adduct. The latter is a six-membered ring containing two Si atoms of a dimer at the (100)-2×1 surface and four C atoms of the reactant diene. Very recent scanning tunneling microscopic experiments (50) of 1,3-cyclohexadiene addition to the surface show that, in addition to the DA [4+2] adduct, other products are possible, specifically, [4+2]-like adducts that bridge two dimers within a row or across adjacent rows, and two intradimer [2+2] adducts. The later examples demonstrate that complex chemistry of conjugated dienes at semiconductor surfaces has exciting potential as a method for controlled synthesis of organic/semiconductor interfaces. Additionally, this chemistry can be used as a starting point for lithographic patterning schemes if appropriate methods can be found to remove unmodified adducts from the surface; however, it was shown for the [4+2] adduct that the retro-DA process was not observed on Si(100)-2×1. Instead, thermal decomposition was found to be the major reaction pathway upon heating (51). Because experiments and static ab initio calculations cannot identify specific mechanisms by which these addition products form, this system is ideal to study by using AIMD (52, 53). Moreover, the maximally localized orbital dynamics can be used to follow the progress of the reactions that occur on the surface. For the DA [4+2] adduct, an outstanding and controversial question of whether the reaction mechanism involves a concerted (symmetric or asymmetric) or stepwise formation of the two Si–C bonds remains unanswered; one might expect that similar questions arise for the other adducts. Furthermore, investigation of the energetic and mechanistic details of the DA [4+2] adduct formation on Si(100) could explain its heat-induced thermal decomposition. One explanation for this experimental finding might be the presence of large activation barriers along the retro-DA pathway. Assuming that energetic considerations play a major role, one might ask if there is any derivative of chemically substituted dienes that would favor a retro-DA reaction over thermal decomposition.
Our AIMD study focused on the underlying mechanistic aspects of the chemical adsorption of cis-1,3-butadiene to the Si(100)-2×1 surface. The calculations were performed the piny_md code (54) on a system of four silicon layers composed of 32 atoms (four surface dimers), a passivating bottom layer of hydrogens, and one cis-1,3-butadiene at a temperature of 300 K. Exchange and correlation were approximated by using a generalization gradient functional (45, 46) and a 35-rydberg plane-wave cutoff was used with atomic pseudopotentials, which is sufficient to converge the geometry of the butadiene and reproduce the change in energy per surface dimer upon reconstruction (55). Forty trajectories starting from a distribution of initial conditions above the surface were performed. A monofluorinated butadiene, was also considered, for which a cutoff of 80 rydberg was used. Rigorous treatment of the surface boundary conditions (13) allowed a box with periodic dimensions 15.34 × 7.67 Å and nonperiodic dimension 22.53 Å to be used.
This protocol predicts, in agreement with recent experiments (56), indicates that the room-temperature structure of the next surface is the c(4×2) buckled dimer structure. The average geometry of the dimers was found to be in good agreement with static ab initio calculations. Consequently, the charge distribution within each dimer is asymmetric, with an excess positive and negative charge on the lower and upper atoms, respectively. The distribution of final products agrees well (52, 53) with the experiments performed on 1,3-cyclohexadiene (50). In particular, the products obtained are shown in Fig. 5, which is published as supporting information on the PNAS web site, which include a DA [4+2] adduct (Fig. 3A), a [4+2]-like intrarow, interdimer adduct (Fig. 3B), a [4+2]-like interrow, interdimer adduct (Fig. 3C), and a [2+2] adduct (Fig. 3D). Moreover, if the progress of the C–C and C–Si bonds along representative trajectories in the ensemble is followed, it becomes clear that the Si–C bonds form asymmetrically and that there is a time interval in which only one C–C bond has the bond length of a single bond, whereas the remaining two have bond lengths between those of single and double bonds, suggesting a stable intermediate resonant structure (52, 53). Putting these results together, we have posited a mechanistic picture capable of rationalizing the product distribution.
Fig. 3.

Proposed mechanism of the addition product formation. Reprinted with permission from ref. 53 (Copyright 2005, American Chemical Society).
The mechanism is illustrated in Fig. 3. Each product begins with a nucleophilic attack of the C C double bond on the positive Si of a surface dimer. As Fig. 3 shows, the next step in the mechanism depends on the subsequent migration of the positive charge into the butadiene. This charge migration results in an intermediate carbocation (see Fig. 5A, which is published as supporting information on the PNAS web site) that can exist as a resonant structure (Fig. 3R). As a consequence of the resonance, the DA and other products form either by a second nucleophilic attack (Figs. 3 C and F–B and 5 C and D) or by attraction of a local positive charge in the carbocation to a negative Si on the surface (Figs. 3 A and D and 5 B and E). The snapshots in Fig. 5 also display electron distributions and Wannier center locations, which further support this mechanistic picture.
C double bond on the positive Si of a surface dimer. As Fig. 3 shows, the next step in the mechanism depends on the subsequent migration of the positive charge into the butadiene. This charge migration results in an intermediate carbocation (see Fig. 5A, which is published as supporting information on the PNAS web site) that can exist as a resonant structure (Fig. 3R). As a consequence of the resonance, the DA and other products form either by a second nucleophilic attack (Figs. 3 C and F–B and 5 C and D) or by attraction of a local positive charge in the carbocation to a negative Si on the surface (Figs. 3 A and D and 5 B and E). The snapshots in Fig. 5 also display electron distributions and Wannier center locations, which further support this mechanistic picture.
It is known in organic chemistry that DA reactions increase their rates with increasing strength of the electron-donating groups on the diene. Therefore, it would seem natural to assume that electron-withdrawing groups on the diene have an opposite effect. Furthermore, it can be expected that such groups would destabilize the DA [4+2] adduct, so it is more likely to undergo a heating induced retro-DA reaction. Through these concepts, we show that it is possible to “reverse engineer” a butadiene derivative that leads to a lower free energy barrier for the retro-DA reaction. To be considered useful for lithographic purposes, a substituted diene candidate has the following properties: (i) it participates in a spontaneous DA [4+2] reaction on the room temperature surface; and (ii) the [4+2] adduct undergoes a retro-DA reaction upon heating. To accommodate both requirements, the candidate substituent must have moderate electron-withdrawing ability to favor the retro-DA pathway, while it also should spontaneously react producing the DA [4+2] adduct at a sufficient rate. Based on qualitative arguments, fluorine was chosen as a test substituent, because it is electronegative yet is not considered to be a highly effective electron-withdrawing substituent such as CF3 or SO3H. Specifically, one of the hydrogens on the CC double bond of the isolated 1,3-butadiene was replaced by F.
Next, the thermodynamic free energy profile ΔG(ξ) along the DA [4+2] reaction path was determined for 1,3-butadiene and the monofluorinated derivative. Here, ξ is taken to be the relative coordinate between the mass centers of the two outer carbons of the 1,3-butadiene and the two Si atoms in the dimer forming the adduct ξ(r) = (1/2)|(rSi1 + rSi2) – (rC1 + rC4)|. ξ decreases from 3.90 to 1.96 Å as the butadiene approaches the surface and forms the DA adduct. The free energy profile was computed by using the blue moon ensemble method (8, 30) using 13 separate systems with values equidistantly distributed along the [1.97, 3.90 Å] interval. Eight-picosecond production runs were carried out after a 1.0-ps equilibration period at each point, for a total of 117 ps. The free energy profiles thus obtained for 1,3-butadiene and the fluorinated derivative are shown in Fig. 4. Both profiles show a shallow minimum at the intermediate carbocation state and, therefore, further support the mechanistic picture of Fig. 3. The figure also shows that, like 1,3-butadiene, F-butadiene has negligible activation barrier (4–5 kcal/mol) toward the DA [4+2] adduct, so that spontaneous reaction is expected at room temperature. Although similar in many qualitative aspects, there is a 6–8 kcal/mol decrease in the activation free energy for the retro-DA pathway. The activation energy level is still too high for a spontaneous retro-DA reaction upon heating, but this result serves as a proof of concept and illustrates the power of a potential computational engineering principle toward designing functionalizing agents with required chemical characteristics.
Fig. 4.

Free energy profiles ΔG(ξ) (See text) for 1,3-butadiene to form the DA [4 + 2] adduct on the Si (100)-2 × 1 surface (solid line) and for the fluorinated derivative to form the same adduct (dashed line). Inset illustrates the reaction.
Future Directions
Key issues that need to be addressed in the field of AIMD are the accuracy of the electronic structure method and the high computational overhead associated with the calculations. It is clear that progress on the former question will come from new breakthroughs in the development of density functionals and/or novel algorithms that allow higher-level electronic structure methods to be used in place of KS density functional theory (see, e.g., ref. 40). For the latter problem, questions concerning the extension of both time and length scales arise. Although linear scaling methods such as those alluded to here hold promise for extending length scales, they are still too computationally intensive and premature enough that alternative ideas are needed. In particular, extensions of AIMD to biological systems have been possible by combining AIMD with force-field-based MD (see ref. 35 for a perspective on this approach).
As large-scale computational resources become more powerful and more readily accessible, issues of software design and massively parallel algorithms will become an integral part of the solution. To meet this challenge, we have begun to pursue an approach based on new software design paradigms for performing AIMD calculations on large supercomputers (43). The concept is based on the charm++ runtime system (44), wherein the basic computational problem is divided arbitrarily into individual tasks independent of the number of processors. These “virtual processors” are then mapped onto physical processors by the charm++ system at runtime. In this way, charm++ is able to drastically minimize processor idle time and, thereby, give rise to highly efficient scaling at a very fine-grained level (43). With considerable effort proceeding on both the algorithmic and software design fronts, it is expected that AIMD calculations will continue to increase in their importance and potential to impact challenging problems in theoretical chemistry, physics, and biology.
Supplementary Material
Acknowledgments
This work was supported by National Science Foundation Grants CHE-0121375 and CHE-0310107. R.I. was supported by the Natural Science and Engineering Research Council of Canada.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Rick, S. W. & Stuart, S. J. (2002) Rev. Comp. Chem. 18, 89–146. [Google Scholar]
- 2.Warshel, A. & Weiss, R. M. (1980) J. Am. Chem. Soc. 102, 6218–6226. [Google Scholar]
- 3.Marx, D. & Hutter, J. (2000) in Modern Methods and Algorithms of Quantum Chemistry, ed. Grotendorst, J. (Forschungszentrum, Jülich, Germany) John von Neumann-Institut für Computing, Vol. 1, pp. 301–449. [Google Scholar]
- 4.Tuckerman, M. E. (2002) J. Phys. B Condens. Matter 14, R1297–R1355. [Google Scholar]
- 5.Ono, A., Kamoshida, N., Matsuura, E., Ishukawa, E., Eguchi, T. & Hasegawa, Y. (2003) Phys. Rev. B 67, 201306. [Google Scholar]
- 6.Yoshinobu, J., Yamashita, Y., Yasui, F., Mukai, K., Tsuneyuki, K., Akagi, K., Hamaguchi, K., Machida, S., Nagao, M., Sato, T. & Iwatsuki, M. (2001) J. Electron. Spectrosc. Related Phenomema 114, 383–387. [Google Scholar]
- 7.Becke, A. D. & Edgecombe, K. E. (1990) J. Chem. Phys. 92, 5397–5403. [Google Scholar]
- 8.Hynes, J. T., Kapral, R., Carter, E. A. & Ciccotti, G. (1989) Chem. Phys. Lett. 156, 472–477. [Google Scholar]
- 9.Rydberg, H., Dion, M., Jacobson, N., Schröder, E., Hyldgaard, P., Simak, S. I., Langreth, D. C. & Lundqvist, B. I. (2003) Phys. Rev. Lett. 91, 126402. [DOI] [PubMed] [Google Scholar]
- 10.Dion, M., Rydberg, H., Schröder, E., Langreth, D. C. & Lundqvist, B. I. (2004) Phys. Rev. Lett. 92, 246401. [DOI] [PubMed] [Google Scholar]
- 11.Langreth, D. C., Dion, M., Rydberg, H., Schröder, E., Hyldgaard, P. & Lundqvist, B. I. (2005) Int. J. Quant. Chem. 101, 599–610. [Google Scholar]
- 12.Car, R. & Parrinello, M. (1985) Phys. Rev. Lett. 55, 2471–2474. [DOI] [PubMed] [Google Scholar]
- 13.Tuckerman, M. E. & Parrinello, M. (1994) J. Chem. Phys. 101, 1302–1315. [Google Scholar]
- 14.Grossmann, J. C., Schwegler, E., Draeger, E. W., Gygi, F. & Galli, G. (2004) J. Chem. Phys. 120, 300–311. [DOI] [PubMed] [Google Scholar]
- 15.Martyna, G. J. & Tuckerman, M. E. (1999) 110, 2810–2821.
- 16.Minary, P., Tuckerman, M. E., Pihakari, K. A. & Martyna, G. J. (2002) J. Chem. Phys. 116, 5351–5362. [Google Scholar]
- 17.Minary, P., Morrone, J.A., Martyna, G. J. & Tuckerman, M. E. (2004) J. Chem. Phys. 121, 11949–11956. [DOI] [PubMed] [Google Scholar]
- 18.Lippert, G., Hutter, J. & Parrinello, M. (1997) Mol. Phys. 92, 477–487. [Google Scholar]
- 19.Lippert, G., Hutter, J. & Parrinello, M. (1999) Theor. Chem. Acc. 103, 124–140. [Google Scholar]
- 20.Liu, Y., Yarne, D. & Tuckerman, M. E. (2003) Phys. Rev. B 68, 125110. [Google Scholar]
- 21.Foster, J. M. & Boys, S. F. (1960) Rev. Mod. Phys. 32, 300–302. [Google Scholar]
- 22.Marzari, N. & Vanderbilt, D. (1997) Phys. Rev. B 56, 12847–12865. [Google Scholar]
- 23.Resta, R. (1998) Phys. Rev. Lett. 80, 1800–1803. [Google Scholar]
- 24.Silvestrelli, P. L. (1999) Phys. Rev. B 59, 9703–9706. [Google Scholar]
- 25.Berghold, G., Mundy, C. J., Romero, A. H., Hutter, J. & Parrinello, M. (2000) Phys. Rev. B 61, 10040–10048. [Google Scholar]
- 26.Wannier, G. H. (1937) Phys. Rev. 52, 191–197. [Google Scholar]
- 27.Sharma, M. & Car, R. (2003) Int. J. Quant. Chem. 95, 821–829. [Google Scholar]
- 28.Thomas, J. W., Iftimie, R. & Tuckerman, M. E. (2004) Phys. Rev. B 69, 125105. [Google Scholar]
- 29.Iftimie, R., Thomas, J. W. & Tuckerman, M. E. (2004) J. Chem. Phys. 120, 2169–2181. [DOI] [PubMed] [Google Scholar]
- 30.Sprik, M. & Ciccotti, G. (1998) J. Chem. Phys. 109, 7737–7744. [Google Scholar]
- 31.Putrino, A., Sebastiani, D. & Parrinello, M. (2000) J. Chem. Phys. 113, 7102–7109. [Google Scholar]
- 32.Baroni, S., Gianozzi, P. & Testa, A. (1987) Phys. Rev. Lett. 58, 1861–1864. [DOI] [PubMed] [Google Scholar]
- 33.Gonze, X. & Vigneron, J. P. (1989) Phys. Rev. B Condens. Matter 39, 13120–13128. [DOI] [PubMed] [Google Scholar]
- 34.Gonze, X. (1995) Phys. Rev. A At. Mol. Opt. Phys. 52, 1096–1114. [DOI] [PubMed] [Google Scholar]
- 35.Carloni, P., Rothlisberger, U. & Parrinello, M. (2002) Acc. Chem. Res. 35, 455–464. [DOI] [PubMed] [Google Scholar]
- 36.Pimentel, G. C. & McClellan, A. L. (1960) The Hydrogen Bond (Freeman, San Francisco).
- 37.Maréchal, Y. & Chamel, A. (1996) J. Phys. Chem. 100, 8551–8555. [Google Scholar]
- 38.Bertie, J. E. & Eysel, H. H. (1985) Appl. Spectrosc. 39, 392–401. [Google Scholar]
- 39.Elsaesser, T. & Bakker, H. J. (2002) Ultrafast Hydrogen Bonding Dynamics and Proton Transfer Processes in the Condensed Phase (Kluwer Academic, Dordrecht, The Netherlands).
- 40.Grossman, J. C. & Mitas, L. (2005) Phys. Rev. Lett. 94, 045403. [DOI] [PubMed] [Google Scholar]
- 41.Bader, J. S. & Berne, B. J. (1994) J. Chem. Phys. 100, 8359–8366. [Google Scholar]
- 42.Gaigeot, M. P. & Sprik, M. (2003) J. Phys. Chem. B 107, 10344–10358. [Google Scholar]
- 43.Vadali, R. V., Shi Y., Kumar, S., Kale, L. V., Tuckerman, M. E. & Martyna, G. J. (2004) J. Comp. Chem. 25, 2006–2022. [DOI] [PubMed] [Google Scholar]
- 44.Kale, L. V. & Krishnan, S. (1993) Sigplan Notices 28, 91–108. [Google Scholar]
- 45.Becke, A. D. (1998) Phys. Rev. A 38, 3098–3100. [DOI] [PubMed] [Google Scholar]
- 46.Lee, C., Yang, W. & Parr, R. C. (1998) Phys. Rev. B Condens. Matter 37, 785–789. [DOI] [PubMed] [Google Scholar]
- 47.Troullier, N. & Martins, J. L. (1991) Phys. Rev. B Condens. Matter 43, 1993–2006. [DOI] [PubMed] [Google Scholar]
- 48.Teplyakov, A. V., Kong, M. J. & Bent, S. F. (1997) J. Am. Chem. Soc. 119, 11100–11101. [Google Scholar]
- 49.Konecny, R. & Doren, D. J. (1997) J. Am. Chem. Soc. 119, 11098–11099. [Google Scholar]
- 50.Teage, L. C. & Boland, J. (2003) J. Phys. Chem. B 107, 3820–3823. [Google Scholar]
- 51.Teplyakov, A. V., Kong, M. J. & Bent, S. F. (1998) J. Chem. Phys. 108, 4599–4606. [Google Scholar]
- 52.Minary, P. & Tuckerman, M. E. (2004) J. Am. Chem. Soc. 126, 13920–13921. [DOI] [PubMed] [Google Scholar]
- 53.Minary, P. & Tuckerman, M. E. (2005) J. Am. Chem. Soc. 127, 1110–1111. [DOI] [PubMed] [Google Scholar]
- 54.Tuckerman, M. E., Yarne, D. A., Samuelson, S. O., Hughes, A. L. & Martyna, G. J. (2000) Comp. Phys. Commun. 128, 333–376. [Google Scholar]
- 55.Minary, P., Martyna, G. J. & Tuckerman, M. E. (2003) J. Chem. Phys. 118, 2527–2538. [Google Scholar]
- 56.Landemark, E., Karlsson, C. J., Chao, Y. C. & Uhrberg, R. I. G. (1992) Phys. Rev. Lett. 69, 1588–1591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}