

Abstract
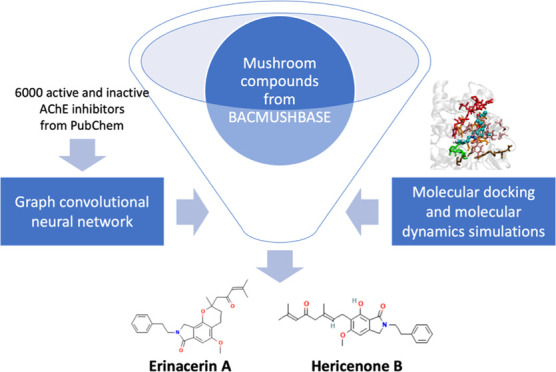
Alzheimer’s disease (AD) is the most common type of dementia, affecting over 50 million people worldwide. Currently, most approved medications for AD inhibit the activity of acetylcholinesterase (AChE), but these treatments often come with harmful side effects. There is growing interest in the use of natural compounds for disease prevention, alleviation, and treatment. This trend is driven by the anticipation that these substances may incur fewer side effects than existing medications. This research presents a computational approach combining machine learning with structural modeling to discover compounds from medicinal mushrooms with a high potential to inhibit the activity of AChE. First, we developed a deep neural network capable of rapidly screening a vast number of compounds to indicate their potential to inhibit AChE activity. Subsequently, we applied deep learning models to screen the compounds in the BACMUSHBASE database, which catalogs the bioactive compounds from cultivated and wild mushroom varieties local to Thailand, resulting in the identification of five promising compounds. Next, the five identified compounds underwent molecular docking techniques to calculate the binding energy between the compounds and AChE. This allowed us to refine the selection to two compounds, erinacerin A and hericenone B. Further analysis of the binding energy patterns between these compounds and the target protein revealed that both compounds displayed binding energy profiles similar to the combined characteristics of donepezil and galanthamine, the prescription drugs for AD. We propose that these two compounds, derived from Hericium erinaceus (also known as lion’s mane mushroom), are suitable candidates for further research and development into symptom-alleviating AD medications.
1. Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder that progressively worsens over time. Early symptoms of AD often manifest as memory, thinking, or reasoning deficits since the disease typically impairs the cortical brain regions implicated in critical thinking, learning, and memory. Common pathogenic indications of AD include significant loss of synaptic and neuronal tissues, astrogliosis, and the buildup of proteinaceous deposits.1 The underlying causes of clinical onset are likely connected to age-related factors and disease-promoting variables.
Although the exact cause of AD remains elusive, numerous hypotheses have been proposed to explain the development and progression of the symptoms. One hypothesis is the presence of extracellular amyloid plaques and neurofibrillary tangles.1,2 Amyloid plaques are made up of extracellular aggregates of amyloid-β (Aβ),3 a product from the cleavage of the amyloid precursor protein by β- and γ-secretases.4 Neurofibrillary tangles are abnormal, twisted aggregates of the tau proteins found within the neurons.5 However, treatments directed to counter Aβ accumulation and tau protein aggregation have so far failed to yield satisfactory results in clinical trials.6
Another hypothesis, the cholinergic hypothesis, posits that impaired cognitive function in AD and adult-onset dementia disorders is closely linked to deficiencies in cholinergic neurotransmission. Individuals with AD exhibit reduced levels of acetylcholine, a neurotransmitter that is crucial for transmitting messages between specific nerve cells. The decline in acetylcholine levels and the progressive loss of these nerve cells are linked to worsening symptoms. Acetylcholine is hydrolytically degraded by two cholinesterases, acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE).7−9 AChE is more abundant than BuChE in AD brains, contributing to acetylcholine degradation in the hippocampus and cerebral cortex.7 Cholinesterase inhibitors assist in alleviating AD symptoms and might even reduce the disease’s course. The first medication to treat AD was tacrine, an acridine derivative, which was approved by the Food and Drug Administration (FDA) in 1993.10 The drug is an effective, noncompetitive, reversible AChE/BuChE inhibitor; however, tacrine use has been discontinued owing to its high hepatotoxicity.10−12 In the following years, galanthamine, rivastigmine, donepezil, and memantine have been approved as symptomatic treatments for AD to manage the patients’ memory loss and cognitive impairment.12,13 The drugs donepezil, rivastigmine, and galanthamine block the acetylcholinesterase enzyme from degrading acetylcholine in the brain. Maintaining appropriate levels of acetylcholine enhances communication between nerve cells, which stabilizes or temporarily relieves some AD symptoms.
However, the effectiveness of these drugs is constrained due to their unfavorable side effects in the form of a loss of appetite, nausea, vomiting, and diarrhea, to name but a few.14 Notably, the available drugs are nowhere near the sought-after “magic bullet” mode of action as they have yet to put a stop to the disease progression. As such, many attempts have been made to develop new drugs with different structures and mechanisms of action. An emerging area of research focuses on naturally derived AChE inhibitors.15−19 As a case in point, huperzine A from Huperzia serrata is a licensed drug for AD in China.20
In the preclinical drug design phase, computer-aided development of novel chemical entities has proven indispensable and significantly contributed to a decrease in the time and resources required in the drug discovery process.21−25 The quantitative structure–activity relationship (QSAR) is a frequently utilized chemometrics method in computational modeling and drug development.26,27 Several computational methods have already been proposed for predicting potential cholinesterase inhibitors.28−33 However, predicting the biological activity of compounds is still challenging and a subject of research due to the complexity of representing molecular structures to capture relevant information for the task. Capable of addressing this complexity, deep learning techniques have become more prominent in the field of drug discovery. The growing availability of data from bioactivity experiments has allowed for the requisite integration of deep learning into the workflow. Its ability to capture nonlinearity in the data also allows it to tackle complex biochemistry problems effectively while minimizing the preprocessing steps.34,35 As a result, deep learning methods have demonstrated superior accuracy compared to traditional machine learning approaches.36,37 The techniques have found applications in myriad aspects of drug discovery, including the prediction of drug–target interactions.37,38 In addition, the application of convolutional neural networks (CNNs) to extract information from molecular structures as graphs has enabled the learning of molecular features and fingerprints.39 The approach demonstrates success in predicting molecular properties and exhibiting a superior predictive performance across a range of tasks.
With these capabilities, this work uses a deep graph convolutional neural network to identify AChE inhibitor candidates from an available database containing natural compounds from mushrooms (BACMUSHBASE; http://bacmushbase.sci.ku.ac.th/). Afterward, docking simulations were carried out to validate the predicted compounds and scope the choices to two compounds, erinacerin A and hericenone B from Hericium erinaceus. Upon a detailed examination of the binding energy patterns between these compounds and the target protein by molecular dynamics simulations, it was observed that both substances exhibited binding energy profiles similar to the combined characteristics of the two AD drugs donepezil and galanthamine. We propose that these two compounds demonstrate significant potential for future development as medications for AD.
2. Results and Discussion
The overall workflow of this study is presented in Figure 1. Briefly, a graph convolutional neural network (GCN) was trained on the structural data of compounds tested for their inhibitory effects on AChE. The trained model was employed to assign scores to the mushroom compounds from the BACMUSHBASE database. Based on the scores, 38 compounds were selected for subsequent molecular docking studies. By considering both deep learning and molecular docking scores, two promising candidates were identified and subjected to molecular dynamics simulations. The analysis of the interaction patterns between the potential compounds and the target protein combined with the investigation of the atomistic features extracted from the selected compounds provided insight into the mechanisms that underlie the inhibition of AChE by the compound candidates.
Figure 1.

Overall workflow of this study. Multiple in silico techniques, (i.e., deep learning, molecular docking, and molecular dynamics simulations) were successively applied to identify potential AChE inhibitors and elucidate the mechanisms underlying the inhibition exerted by the compound candidates.
2.1. Building a Graph Convolutional Neural Network for AChE Inhibitor Screening
A list of compounds tested against acetylcholinesterase (AChE) activity was retrieved from PubChem (https://pubchem.ncbi.nlm.nih.gov/) (NCBI protein accession: P22303). The data (accessed in January 2021) contained 51,505 data points. After redundancy was removed, 3001 compounds with IC50 ≤ 10 μM in inhibiting the AChE activity were defined as “active” compounds. 7825 compounds were “inactive” compounds as labeled by the database. Then, 3000 active and 3000 inactive compounds were randomly chosen and combined to form a workable data set for this study. We implemented the Latin hypercube algorithm to create random sets of seven to-be-tuned hyperparameters for a graph convolutional neural network (Table 1), resulting in 44 random sets chosen out of 432 possible combinations. For each hyperparameter set, we implemented the graph convolutional model with 5-fold cross-validation (CV) using atomic features extracted by the command MolGraphConvFeaturizer() in the DeepChem package.40 The hyperparameter set with the highest validating AUCs averaged among the 5-fold was selected (Table 1; averaged AUC = 0.9858 ± 0.0022). The model was then trained with the selected hyperparameter set on the whole data set (6000 data points) to yield a final model. The AUC scores for the rest of the hyperparameter sets are reported in Supporting Table S1.
Table 1. Hyperparameter Tuning.
| the hyperparameter varied in the model | varied values | value in the optimal hyperparameter set |
|---|---|---|
| number of nodes in the graph convolutional layer | 256, 512, 1024 | 1024 |
| activation function in the graph convolutional layer | ReLU, tanh | ReLU |
| number of nodes in the dense layer | 256, 512, 1024 | 256 |
| activation function in the dense layer | ReLU, tanh | ReLU |
| activation function in the graph gather layer | ReLU, tanh | tanh |
| dropout rate | 0.0, 0.1, 0.2 | 0.0 |
| learning rate | 10–4, 10–3 | 10–4 |
The final model consisted of a 1024-node graph convolutional layer with the ReLU activation function, a batch normalization layer, a graph pool layer, a 256-node dense layer with the ReLU activation function, another batch normalization layer, a graph gather layer with the tanh activation function, and an output node that assigns a probability of the compound being “active” (has inhibitory effect on AChE) using the SoftMax function. Considering the stochastic nature of the learning algorithm, whereby the models trained with the same data and hyperparameters may not necessarily yield identical results, we utilized the entire data set (3000 active and 3000 inactive compounds) to train five instances of the models adopting the optimal hyperparameter set in Table 1.
Subsequently, the trained models were used to predict the probability score of all substances in BACMUSHBASE (http://bacmushbase.sci.ku.ac.th/), a database of bioactive compounds from the mushroom species found in Thailand. The top five compounds with an average active probability score of more than 0.9, derived from predictions made by the five models, are shown in Table 2. The predicted scores of the complete list of the compounds are listed in Supporting File S1.
Table 2. Top Five Compounds from BACMUSHBASE as Potential AChE Inhibitors.
| compound name | mushroom source | averaged “active” probability ± sd |
|---|---|---|
| erinacerin A | H. erinaceus | 0.9622 ± 0.0416 |
| flavidulol C | Lactarius flavidulus S. Imai | 0.9609 ± 0.0353 |
| lucidimine B | Ganoderma lucidum | 0.9607 ± 0.0216 |
| lucidimine C | G. lucidum | 0.9424 ± 0.0242 |
| hericenone B | H. erinaceus | 0.9283 ± 0.0876 |
2.2. Validating Potential AChE Inhibitors by Molecular Docking
Next, molecular docking calculations by AutoDock Vina 1.241 were performed on a group of 40 compounds to further validate the deep learning model in terms of molecular interactions between the atomistic structure of each compound and the protein target (acetylcholinesterase; PDB ID: 4EY7). The 40 compounds consisted of two known inhibitors, donepezil and galanthamine, the top five compounds with a predicted probability score >0.9, 23 randomly sampled compounds with a predicted probability score between 0.1 and 0.9, and 10 compounds with a predicted probability score <0.1.
Figure 2a displays the active probability score predicted by the deep learning model along with the binding energy score estimated through molecular docking calculations for each of the 40 selected compounds. Nine of the 10 compounds with a probability score <0.1 had a relatively weak binding affinity (binding energy score above −6.13 kcal/mol). The compounds with a predicted probability score between 0.1 and 0.9 exhibited a binding energy in the range of −7.91 and −10.41 kcal/mol. Four out of five compounds with an active probability score >0.9 possessed relatively strong binding affinity (binding energy scores between −9.19 and −10.54). Donepezil displayed the strongest binding energy of −10.66 kcal/mol, while galanthamine had the binding energy of −9.90 kcal/mol. Supporting Table S2 lists the binding energy scores of all 40 selected compounds.
Figure 2.

Binding energy scores estimated from molecular docking plotted against (a) the active probability score predicted by the deep learning models and (b) the logarithm of the active probability score (log P) for each of the 40 selected compounds.
Plotting the binding energy score against the logarithm of the active probability score (log P) shown in Figure 2b displayed a roughly linear relationship with an R-squared value of 0.6, signifying a substantial agreement between the two methods. Therefore, among the bioactive compounds from BACMUSHBASE with a probability score exceeding 0.9, erinacerin A and hericenone B, with the strongest binding affinity, were proposed as candidates for new AChE inhibitors and were subsequently subject to further analysis.
2.3. Binding Interaction Networks between AChE and Candidate Compounds Compared to First-Line Prescription Drugs
In this section, molecular interactions between the AChE protein and each of the two proposed inhibitors, erinacerin A and hericenone B, were analyzed in comparison with the two AD drugs, donepezil and galanthamine. 50 ns molecular dynamics (MD) simulations in explicit solvents were performed for all four AChE complexes. After the simulations, MM/PBSA calculations were performed on the last 25 ns of each equilibrated trajectory, and the contributions of all of the energy terms and entropy are plotted in Figure 3. In the case of donepezil binding to AChE with the free energy of −166 ± 11 kJ/mol, the contribution of the electrostatic term to the binding free energy was dominant as the protonated donepezil became positively charged. However, the interfacial area between the highly soluble donepezil also became lost upon AChE binding, so the binding energy was penalized by the polar solvation term. On the other hand, a significantly lower energy penalty from the polar solvation term was observed for the uncharged galanthamine with a smaller AChE binding interface. Despite the lower contribution of both van der Waals and electrostatic terms to the AChE binding, the strength of the total binding free energy of −178 ± 25 kJ/mol for galanthamine to AChE became slightly stronger than donepezil.
Figure 3.
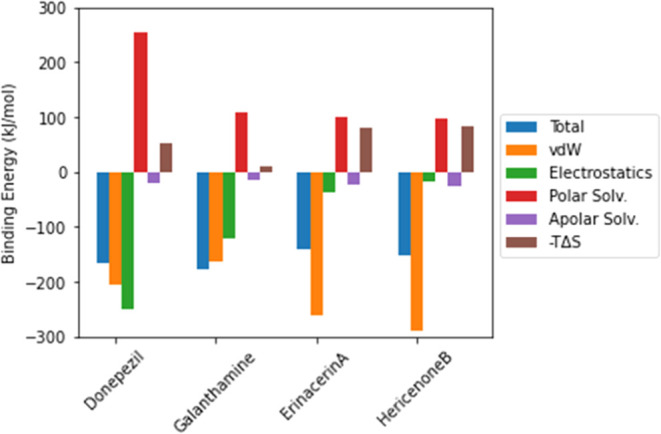
Relative contributions of the van der Waals, electrostatics, polar solvation, apolar solvation, and entropy (−TΔS) terms to the total binding energy of two confirmed AChE inhibitors and two predicted AChE inhibitors.
The binding free energy of the proposed compounds, erinacerin A and hericenone B, was dominated by the van der Waals term. The structures of both proposed compounds contained more hydrophobic functional groups than the structure of donepezil, so the contribution of the van der Waals term to AChE binding became stronger. However, the entropic penalty from the additional flexible chains caused the total AChE binding free energy of erinacerin A (−141 ± 16 kJ/mol) and hericenone B (−151 ± 16 kJ/mol) to be slightly weaker than that of donepezil and galanthamine, which was consistent with the active probability predicted by the deep learning model. The interaction networks of AChE amino acid residues to inhibitor binding were visualized in Figure 4, in which the active site “gorge” was divided into the top region (subsite I), side regions (subsites II and III), and the bottom region near the catalytic sites (subsite IV). Donepezil interacted with the top region of the AChE gorge through its methoxyl groups connected to the indene ring and with the bottom region through its phenyl ring (Figure 4a). Galanthamine utilized its phenyl ring part to bind with only one residue in the top region but utilized its adjacent methoxyl group to bind with all of the catalytic residues of AChE, which could explain its clinical use for treating mild to moderate dementia (Figure 4b). Similar to donepezil, erinacerin A employed the phenyl ring part to bind with the bottom region (Figure 4c), while hericenone B employed its branched aliphatic tail to form an extensive hydrophobic interaction network with the bottom region in concurrence with its strongest enthalpy contribution of AChE binding energy (Figure 4d). One of the common characteristics shared by all four inhibitors was the tertiary amine groups found near the side regions of AChE gorge. Moreover, the oxygen functional group, e.g., carbonyl, ether, and methoxyl, were also found binding to the side regions of the AChE gorge and contributed to the binding energy.
Figure 4.

Interaction analysis of the amino acid residues of AChE and the enzyme inhibitors (a) donepezil, (b) galanthamine, (c) erinacerin A, and (d) hericenone B generated by LigPlot 2.2.42 Red, pink, orange, and yellow dashed circles represent Subsite I (top region), Subsite II (outside region), Subsite III (inside region), and Subsite IV (bottom region), respectively.
The contributions of the AChE amino acid residues to inhibitor binding were estimated through the decomposition of the MM/PBSA energy averaged over the last 25 ns of each trajectory of the AChE–inhibitor complex. Figure 5 depicts per-residue MM/PBSA energy for inhibitor binding of the 33 selected residues with the highest degree of contribution, which resided in the top region (subsite I), the side regions (subsites II and III), the bottom region (subsite IV), and the catalytic residues Ser203, Glu334, and His447.
Figure 5.

(Left) Binding configurations of the inhibitors into different subsites of the active site gorge of AChE: subsite I (top region; red), subsite II (outside region; pink), subsite III (inside region; orange), subsite IV (bottom region; brown), and catalytic residues (green). (Right) Per-residue binding energy decomposition between the important AChE residues and (a) donepezil, (b) galanthamine, (c) erinacerin A, and (d) hericenone B.
Figure 5a shows that four glutamic acid residues within subsite I and subsite IV, as well as the catalytic residue Glu334 with a negatively charged side chain, largely contributed to the donepezil binding via Coulombic interactions with the protonated donepezil. For the case of galanthamine binding in Figure 5b, a very high per-residue energy contribution of approximately −15 kJ/mol was observed for catalytic residue His447 at the bottom of the AChE active site gorge. In contrast, a relatively small energy contribution was observed in the side regions, and almost no energy contribution was observed in the top region. Similar to donepezil, the energy decomposition profiles of erinacerin A (Figure 5c) and hericenone B (Figure 5d) showed that residue Tyr341 within subsite II of both proposed candidates was one of the important binding residues and was in common with that of donepezil. However, erinacerin A and hericenone B displayed a relatively lower contribution of the top region to binding energy but a significantly higher contribution of the bottom region, which was similar to galanthamine. The similarity between the binding mechanisms of the predicted inhibitors and the combined characteristics of two known inhibitors indeed makes a compelling case that erinacerin A and hericenone B may have the potential as an alternative AChE-targeting drug.
2.4. Exploring the Molecular Features Influencing AChE Binding
To further elucidate the featural patterns of the screened compounds from which the classification of active and inactive compounds against AChE was learned, the values of all 75 atomic features were extracted from the 40 sampled compounds (see the Methods section). Pearson correlation coefficients were calculated for each feature: one between the feature and the “active” probability score and another between said feature and the binding energy score. Three features with a Pearson correlation coefficient ≤−0.4 or ≥0.4 from both calculations were identified. Figure 6 plots the three feature values against the binding energy score from docking. Remarkably, the two known AChE inhibitors donepezil and galanthamine, along with the two candidate compounds erinacerin A and hericenone B, were among the compounds that possessed the highest values for all three selected features. The feature “symbol = C” (Figure 6a) possesses the highest correlation with the binding energy score, indicating that both known AChE inhibitors and predicted compounds contained a high C content ratio, which also reflected the lower ratio of finding hydrogens and functional groups that contained N or O atoms. Moreover, the “degree = 3” feature (Figure 6b) reflected the ring structures and the amino groups observed in all four molecules. Lastly, the “#H = 0” feature (Figure 6c) reflected the special functional groups containing N or O atoms with no covalent bonding with H, e.g., methoxyl, carbonyl, and tertiary amine groups. These commonly observed functional groups in the four compounds contributed to AChE binding: the ring structures interacted with the hydrophobic side chains, while the polar atoms with no adjacent hydrogen interacted with either the polar side chain or the backbone of the amino acids within the narrow gorge containing AChE active sites.
Figure 6.
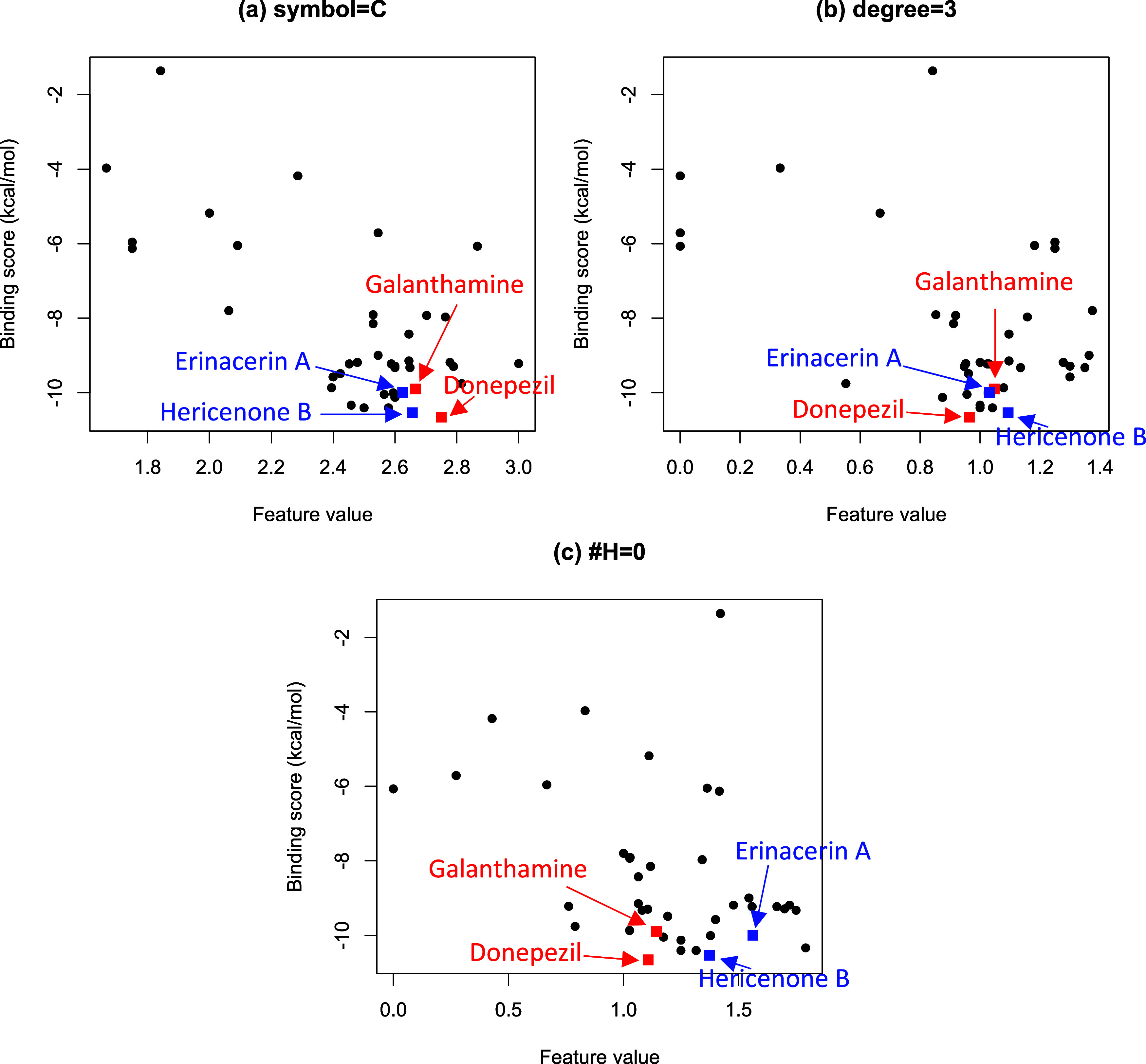
Extracted atomic features plotted against the binding energy estimated by molecular docking calculations with the highest correlations: (a) symbol = C, (b) degree = 3, and (c) total number of hydrogens = 0.
3. Conclusions
One of the major features of Alzheimer’s disease (AD) is an abnormally low amount of acetylcholine, which is induced by excessive acetylcholinesterase (AChE) activity. Several AChE inhibitors have been approved for their ability to alleviate AD symptoms, albeit with certain side effects. The search for new AChE inhibitors is ongoing and has proven to be a challenging endeavor. Compounds from herbal extracts, which are often associated with milder effects, could prove useful as an alternative treatment option.
This study utilized a combined deep learning and molecular modeling approach to rapidly screen candidate compounds from mushroom extracts as alternative AChE inhibitors. The deep learning neural network was trained on the available data set of the enzyme inhibitory assays indicating whether each compound was “active” or “inactive” against AChE. After optimizing hyperparameters for the best AUC score, the ensemble model was used to predict the probability of each compound being classified as an “active” compound from the mushroom extract database. Molecular docking calculations were then performed on a group of selected compounds with the highest, lowest, and moderate “active” probability and displayed a substantial correlation between the logarithm of the “active” probability score and the binding energy score. Subsequently, erinacerin A and hericenone B were selected as the candidates for AChE inhibitors based on the analysis of both methods. Afterward, we conducted molecular dynamics (MD) simulations and MM/PBSA free energy calculations to compare the two candidates with the two approved drug molecules. Our aim was to determine the AChE binding mechanisms involving functional groups and chemical features captured by our deep learning model. According to the per-residue MM/PBSA energy decomposition, galanthamine was bound more directly to the active residues at the active site gorge, while donepezil possessed a stronger binding energy through binding with the outer (top) regions of the active site gorge. Erinacerin A and hericenone B demonstrated the combined binding mechanisms of both donepezil and galanthamine while maintaining their strong binding energy. The potentials of erinacerin A and hericenone B as alternative AChE inhibitors were in good agreement with another report (https://patents.google.com/patent/WO2014098306A1/en), which validated the screening results.
The consensus derived from our deep learning and molecular modeling calculations led to the identification of erinacerin A and hericenone B as potential AChE inhibitors in this study. To prove the effectiveness of the predicted compounds, it is necessary to carry out in vitro tests, followed by dose–response studies, to determine the concentration-dependent effects on AChE inhibition. Furthermore, the chemical structures of erinacerin A and hericenone B can be optimized to improve their druglike properties and synthetic feasibility.
Our approach was endorsed by the fact that the screened compounds were considerably similar to donepezil. However, some compounds with reasonably high active probability, e.g., flavidulols A and C, had chemical structures similar to galanthamine. Therefore, different groups of compounds with different AChE binding modes could exist. In fact, the conformation of the AChE binding pocket has been shown to be diverse, and an ensemble of binding pocket conformations was suggested to improve the in silico screening.43 Improving the current approach in future work will be possible by addressing this fact.
Our drug discovery process involves identifying substances consistently highly ranked by deep learning and molecular modeling approaches. We believe that this approach can significantly improve the accuracy and efficiency of drug discovery. A similar strategy has been used to identify potential new mTOR inhibitors from Ganoderma lucidum and Lentinus polychrous Lev. before.44 Therefore, the approach should be efficiently applied to similar drug discovery tasks involving single enzyme targets, provided a sufficiently large data set is available.
4. Methods
4.1. AChE Inhibitor Data Set
A list of compounds with their inhibitory activity against AChE was retrieved from PubChem (Protein accession ID P22303) originally contained 51,505 data points. Using IC50 ≤ 10 μM as a cutoff, 3001 nonredundant compounds were defined as “active” compounds (having an inhibitory effect against AChE). 7825 compounds were “inactive” compounds (do not have an inhibitory effect) as labeled by the database. To create our data set, 3000 active and 3000 inactive compounds were randomly selected and combined.
4.2. Deep Learning
A graph convolutional neural network was implemented using the DeepChem package.40 The model comprised the following layers: a graph convolutional layer, a batch normalization layer, a graph pool layer, a dense layer, another batch normalization layer, and a graph gather layer. Seven hyperparameters listed in Table 1 were tuned to identify the best combination. The varied values, as listed in Table 1, could be combined into 432 combinations. To reduce the task, the Latin hypercube algorithm was implemented to select only 44 combinations to train the model and identify the best combination using the 5-fold cross-validation (CV) method. The combination with the highest ROC score averaged over the 5-fold was selected to train the final models.
Finally, due to the stochastic nature of the learning algorithm (models trained with the same data and hyperparameters do not necessarily produce identical results), the whole data set (3000 positive and 3000 negative data points) was used to train five instances of the models with the hyperparameter identified from the previous section. Then, the five independently trained models were employed to predict “active” probability scores for compounds from the BACMUSHBASE database (http://bacmushbase.sci.ku.ac.th/). The average predicted scores across the five models represent the ensemble predictions that were used to screen for top-ranking compounds for further investigation by structural methods.
4.3. Molecular Modeling
AutoDock Vina 1.241 was used to perform all molecular docking of 40 selected compounds, including two currently approved drugs, donepezil and galanthamine. The crystallographic structural data of acetylcholinesterase (PDB ID: 4EY7) was obtained and set as the receptor after stripping water, along with other ligand molecules and Gasteiger charge parametrization by AutoDockTools.45 Then, SMILES data of each selected compound was obtained from the BACMUCHBASE database (http://bacmushbase.sci.ku.ac.th/) before converting into 3D coordinates by Openbabel46 and Gasteiger charge parametrization by AutoDockTools.45 After that, a search space was defined by a 40 × 40 × 40 Å3 cubic box centered between the three catalytic triads of AChE. Finally, the molecular docking calculation was performed for each ligand with the exhaustiveness set to 20, and the binding energy of the best docking mode was output along with the docked configuration of the ligand on the receptor.
After selecting two candidate compounds based on the deep learning prediction and the molecular docking binding energy score, all-atom molecular dynamics (MD) simulations were performed for the AChE complexes with donepezil, galanthamine, and the two candidate compounds using the best binding mode from the molecular docking calculation as the starting structure. All protein and ligand molecules were parametrized through the GROMOS54A7 force field,47 in which the partial charge of each atom within the ligand molecules was obtained through semiempirical QM calculations by the MOPAC software48,49 implemented within the Automated Topology Builder (ATB) Web server tool50 along with other force field terms. The protonation states of all systems were set at pH 7 by default, with all of the Asp and Glu amino acid residues deprotonated and donepezil in their protonated form. Each system was explicitly solvated within a cubic simulation box filled with simple point-charge (SPC) water models and either Na+ or Cl– counterions to neutralize the total charge with an 8 Å minimum buffer distance between the protein surface and the periodic boundary, treated by particle-mesh Ewald (PME) summation.51 After performing an energy minimization of the whole system, a 1 ns simulated annealing was carried out to linearly increase the temperature from 100 to 300 K. After that, a 50 ns productive MD run under an NPT ensemble with a constant temperature of 300 K regulated by the velocity-rescale algorithm52 and a constant pressure of 1 atm regulated by the Parrinello–Rahman barostat was carried out.53 The 10 Å cutoff distance for short-ranged interactions was employed, and holonomic constraints by the P-LINC algorithm54 were applied for all covalent bonds with hydrogen so that a 2 fs time step was allowed. All MD simulations were performed by the GROMACS 2019 package55 before the calculations of root-mean-square deviation (RMSD) for both global conformational change and ligand shifting to ensure equilibration. Finally, the MM/PBSA calculation by the g_mmpbsa package56 was performed for each MD trajectory to obtain the time-averaged binding free energy between the protein complex and the docked compound contributed by electrostatics, van der Waals, along with both polar and apolar solvation terms. The contribution of entropy to the binding energy was calculated by Schlitter’s method based on the covariance matrix analysis implemented through the “gmx covar” and “gmx anaeig” modules, for which the change in entropy was determined between the bound protein/ligand complex and the unbound systems. For each calculation, the dielectric constants of proteins and water were set to 4 and 80, respectively, and the surface tension of the solvent was set to 0.0226778 kJ/(mol Å2). Salt concentration was set to 0.03 M, equivalent to the ratio between the numbers of counterions and water molecules in the simulation boxes. The contribution of each amino acid residue from the energy decomposition analysis was performed to elucidate the AChE binding mechanisms, which was illustrated through Ligplot 2.242 for the last simulated snapshot of each complex.
4.4. Molecular Feature Extraction and Exploration
To calculate the molecular feature, as shown in Figure 6, the ConvMolFeaturizer() command from the DeepChem package was used to extract 75 per-atom attributes from each compound. The featurization resulted in a per-atom feature matrix, denoted as F, with dimensions N × 75, where N represents the number of atoms in the respective compound. Each atom is represented by a 75-bit vector, encompassing atomistic features, such as atom type, degree, implicit valence, formal charges, hybridization type, aromaticity, and total number of hydrogens.
The structural information was then incorporated into matrix F through the computation of H = (A + I) × F, where A is the N × N adjacency matrix of the molecule with N atoms, I is the identity matrix, and H is the resulting N × 75 matrix. This matrix encapsulates the representation of each atom as a sum of its neighboring atoms’ features. Subsequently, atom-level features in matrix H were aggregated by averaging values across all atoms in the molecule, resulting in a 1 × 75 vector containing a molecule-level fingerprint for each compound. Next, a correlation between each of the 75 molecule-level features and the binding energy score was calculated to identify the features influencing the AChE binding affinity.
Acknowledgments
This research project was supported by Thailand Science Research and Innovation (TSRI) Basic Research Fund: The fiscal year 2022 under project number FRB650048/0164. G.P. was supported by the Program Management Unit for Human Resources & Institutional Development, Research and Innovation [grant number B01F630003].
Data Availability Statement
The codes and data sets can be found at the following link: https://github.com/pribnowbox/virtual-screening-of-AChE-inhibitors.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c10459.
The authors declare no competing financial interest.
Supplementary Material
References
- Hyman B. T.; Phelps C. H.; Beach T. G.; Bigio E. H.; Cairns N. J.; Carrillo M. C.; Dickson D. W.; Duyckaerts C.; Frosch M. P.; Masliah E.; et al. National institute on aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dementia 2012, 8 (1), 1–13. 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow K.; de Leon M. J.; Zetterberg H. Alzheimer’s disease. Lancet 2006, 368 (9533), 387–403. 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- Masters C. L.; Simms G.; Weinman N. A.; Multhaup G.; McDonald B. L.; Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 1985, 82 (12), 4245–4249. 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J.; Selkoe D. J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002, 297 (5580), 353–356. 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Medeiros R.; Baglietto-Vargas D.; LaFerla F. M. The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci. Ther. 2011, 17 (5), 514–524. 10.1111/j.1755-5949.2010.00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madav Y.; Wairkar S.; Prabhakar B. Recent therapeutic strategies targeting beta amyloid and tauopathies in Alzheimer’s disease. Brain Res. Bull. 2019, 146, 171–184. 10.1016/j.brainresbull.2019.01.004. [DOI] [PubMed] [Google Scholar]
- Nordberg A.; Ballard C.; Bullock R.; Darreh-Shori T.; Somogyi M. A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s disease. Primary Care Companion CNS Disord. 2013, 15 (2), 26731 10.4088/PCC.12r01412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard C. G.; Greig N. H.; Guillozet-Bongaarts A. L.; Enz A.; Darvesh S. Cholinesterases: roles in the brain during health and disease. Curr. Alzheimer Res. 2005, 2 (3), 307–318. 10.2174/1567205054367838. [DOI] [PubMed] [Google Scholar]
- Anand P.; Singh B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch. Pharm. Res. 2013, 36 (4), 375–399. 10.1007/s12272-013-0036-3. [DOI] [PubMed] [Google Scholar]
- Przybyłowska M.; Kowalski S.; Dzierzbicka K.; Inkielewicz-Stepniak I. Therapeutic potential of multifunctional tacrine analogues. Curr. Neuropharmacol. 2019, 17 (5), 472–490. 10.2174/1570159X16666180412091908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak M.; Holubova K.; Nepovimova E.; Krusek J.; Kaniakova M.; Korabecny J.; Vyklicky L.; Kuca K.; Stuchlik A.; Ricny J.; et al. The pharmacology of tacrine at N-methyl-d-aspartate receptors. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 75, 54–62. 10.1016/j.pnpbp.2017.01.003. [DOI] [PubMed] [Google Scholar]
- Korabecny J.; Zemek F.; Soukup O.; Spilovska K.; Musilek K.; Jun D.; Nepovimova E.; Kuca K.. Pharmacotherapy of Alzheimer’s Disease: Current State and Future Perspectives. In Drug Design and Discovery in Alzheimer’s Disease; Atta ur R.; Choudhary M. I., Eds.; Elsevier, 2014; pp 3–39. [Google Scholar]
- Tan C. C.; Yu J. T.; Wang H. F.; Tan M. S.; Meng X. F.; Wang C.; Jiang T.; Zhu X. C.; Tan L. Efficacy and safety of donepezil, galantamine, rivastigmine, and memantine for the treatment of Alzheimer’s disease: a systematic review and meta-analysis. J. Alzheimer’s Dis. 2014, 41 (2), 615–631. 10.3233/JAD-132690. [DOI] [PubMed] [Google Scholar]
- Ruangritchankul S.; Chantharit P.; Srisuma S.; Gray L. C. Adverse drug reactions of acetylcholinesterase inhibitors in older people living with dementia: A comprehensive literature review. Ther. Clin. Risk Manage. 2021, 17, 927–949. 10.2147/TCRM.S323387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos T. C.; Gomes T. M.; Pinto B. A. S.; Camara A. L.; de Andrade Paes A. M. Naturally occurring acetylcholinesterase inhibitors and their potential use for Alzheimer’s disease therapy. Front. Pharmacol. 2018, 9, 1192 10.3389/fphar.2018.01192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H. A.; Min B.-S.; Yokozawa T.; Lee J.-H.; Kim Y. S.; Choi J. S. Anti-Alzheimer and antioxidant activities of coptidis rhizoma alkaloids. Biol. Pharm. Bull. 2009, 32 (8), 1433–1438. 10.1248/bpb.32.1433. [DOI] [PubMed] [Google Scholar]
- Kamal Z.; Ullah F.; Ayaz M.; Sadiq A.; Ahmad S.; Zeb A.; Hussain A.; Imran M. Anticholinesterse and antioxidant investigations of crude extracts, subsequent fractions, saponins and flavonoids of Atriplex laciniata L.: potential effectiveness in Alzheimer’s and other neurological disorders. Biol. Res. 2015, 48 (1), 21 10.1186/s40659-015-0011-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S. Y.; Lee K. Y.; Sung S. H.; Park M. J.; Kim Y. C. Coumarins isolated from Angelica gigas inhibit acetylcholinesterase: structure–activity relationships. J. Nat. Prod. 2001, 64 (5), 683–685. 10.1021/np000441w. [DOI] [PubMed] [Google Scholar]
- Ingkaninan K.; Temkitthawon P.; Chuenchom K.; Yuyaem T.; Thongnoi W. Screening for acetylcholinesterase inhibitory activity in plants used in Thai traditional rejuvenating and neurotonic remedies. J. Ethnopharmacol. 2003, 89 (2), 261–264. 10.1016/j.jep.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Qian Z. M.; Ke Y. Huperzine A: is it an effective disease-modifying drug for Alzheimer’s disease?. Front. Aging Neurosci. 2014, 6, 216 10.3389/fnagi.2014.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge H.; Wang Y.; Li C.; Chen N.; Xie Y.; Xu M.; He Y.; Gu X.; Wu R.; Gu Q.; et al. Molecular dynamics-based virtual screening: Accelerating the drug discovery process by high-performance computing. J. Chem. Inf. Model. 2013, 53 (10), 2757–2764. 10.1021/ci400391s. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Wong S. E.; Lightstone F. C. Toward fully automated high performance computing drug discovery: a massively parallel virtual screening pipeline for docking and molecular mechanics/generalized Born surface area rescoring to improve enrichment. J. Chem. Inf. Model. 2014, 54 (1), 324–337. 10.1021/ci4005145. [DOI] [PubMed] [Google Scholar]
- Sliwoski G.; Kothiwale S.; Meiler J.; Lowe E. W. Jr. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66 (1), 334–395. 10.1124/pr.112.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L. The many roles of computation in drug discovery. Science 2004, 303 (5665), 1813–1818. 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- Okimoto N.; Futatsugi N.; Fuji H.; Suenaga A.; Morimoto G.; Yanai R.; Ohno Y.; Narumi T.; Taiji M. High-performance drug discovery: Computational screening by combining docking and molecular dynamics simulations. PLoS Comput. Biol. 2009, 5 (10), e1000528 10.1371/journal.pcbi.1000528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon S.; Bae H.; Jo J.; Yoon S. Comprehensive ensemble in QSAR prediction for drug discovery. BMC Bioinf. 2019, 20 (1), 521 10.1186/s12859-019-3135-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves B. J.; Braga R. C.; Melo-Filho C. C.; Moreira-Filho J. T.; Muratov E. N.; Andrade C. H. QSAR-based virtual screening: Advances and applications in drug discovery. Front. Pharmacol. 2018, 9, 418940 10.3389/fphar.2018.01275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J.; Yang R.; Gao L.; Zhou D.; Yang S.; Liu A. L.; Du G. H. Predictions of BuChE inhibitors using support vector machine and naive Bayesian classification techniques in drug discovery. J. Chem. Inf. Model. 2013, 53 (11), 3009–3020. 10.1021/ci400331p. [DOI] [PubMed] [Google Scholar]
- Fang J.; Li Y.; Liu R.; Pang X.; Li C.; Yang R.; He Y.; Lian W.; Liu A. L.; Du G. H. Discovery of multitarget-directed ligands against Alzheimer’s disease through systematic prediction of chemical-protein interactions. J. Chem. Inf. Model. 2015, 55 (1), 149–164. 10.1021/ci500574n. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Zhou G.; Zhang C.; Zhang M.; Chen Q.; Zheng L.; Niu B. Identify compounds’ target against Alzheimer’s disease based on in-silico approach. Curr. Alzheimer Res. 2019, 16 (3), 193–208. 10.2174/1567205016666190103154855. [DOI] [PubMed] [Google Scholar]
- Nguyen T. H.; Tran P.-T.; Pham N. Q. A.; Hoang V.-H.; Hiep D. M.; Ngo S. T. Identifying possible AChE inhibitors from drug-like molecules via machine learning and experimental studies. ACS Omega 2022, 7 (24), 20673–20682. 10.1021/acsomega.2c00908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu H.; Kumar R. N.; Garg P. Machine learning-based modeling to predict inhibitors of acetylcholinesterase. Mol. Diversity 2022, 26 (1), 331–340. 10.1007/s11030-021-10223-5. [DOI] [PubMed] [Google Scholar]
- Khan M. I.; Taehwan P.; Cho Y.; Scotti M.; de Menezes R. P. B.; Husain F. M.; Alomar S. Y.; Baig M. H.; Dong J. J. Discovery of novel acetylcholinesterase inhibitors through integration of machine learning with genetic algorithm based in silico screening approaches. Front Neurosci. 2023, 16, 1007389 10.3389/fnins.2022.1007389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Park S.; Min D.; Kim W. Comprehensive survey of recent drug discovery using deep learning. Int. J. Mol. Sci. 2021, 22 (18), 9983 10.3390/ijms22189983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Tan J.; Han D.; Zhu H. From machine learning to deep learning: progress in machine intelligence for rational drug discovery. Drug Discovery Today 2017, 22 (11), 1680–1685. 10.1016/j.drudis.2017.08.010. [DOI] [PubMed] [Google Scholar]
- Chen H.; Engkvist O.; Wang Y.; Olivecrona M.; Blaschke T. The rise of deep learning in drug discovery. Drug Discovery Today 2018, 23 (6), 1241–1250. 10.1016/j.drudis.2018.01.039. [DOI] [PubMed] [Google Scholar]
- Nag S.; Baidya A. T. K.; Mandal A.; Mathew A. T.; Das B.; Devi B.; Kumar R. Deep learning tools for advancing drug discovery and development. 3 Biotech 2022, 12 (5), 110 10.1007/s13205-022-03165-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimber T. B.; Chen Y.; Volkamer A. Deep learning in virtual screening: recent applications and developments. Int. J. Mol. Sci. 2021, 22 (9), 4435 10.3390/ijms22094435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvenaud D. K.; Maclaurin D.; Iparraguirre J.; Bombarell R.; Hirzel T.; Aspuru-Guzik A.; Adams R. P.. Convolutional networks on graphs for learning molecular fingerprints Advances in neural information processing systems 2015; Vol. 28.
- Ramsundar B.; Eastman P.; Walters P.; Pande V.. Deep Learning for the Life Sciences: Applying Deep Learning to Genomics, Microscopy, Drug Discovery, and More; O’Reilly Media, 2019. [Google Scholar]
- Eberhardt J.; Santos-Martins D.; Tillack A. F.; Forli S. AutoDock Vina 1.2.0: new docking methods, expanded force field, and python bindings. J. Chem. Inf. Model. 2021, 61 (8), 3891–3898. 10.1021/acs.jcim.1c00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; Swindells M. B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51 (10), 2778–2786. 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- van der Westhuizen C. J.; Stander A.; Riley D. L.; Panayides J. L. Discovery of novel acetylcholinesterase inhibitors by virtual screening, in vitro screening, and molecular dynamics simulations. J. Chem. Inf Model. 2022, 62 (6), 1550–1572. 10.1021/acs.jcim.1c01443. [DOI] [PubMed] [Google Scholar]
- Posansee K.; Liangruksa M.; Termsaithong T.; Saparpakorn P.; Hannongbua S.; Laomettachit T.; Sutthibutpong T. Combined deep learning and molecular modeling techniques on the virtual screening of new mTOR inhibitors from the Thai mushroom database. ACS Omega 2023, 8 (41), 38373–38385. 10.1021/acsomega.3c04827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30 (16), 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: an open chemical toolbox. J. Cheminf. 2011, 3, 33 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid N.; Eichenberger A. P.; Choutko A.; Riniker S.; Winger M.; Mark A. E.; van Gunsteren W. F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys J. 2011, 40 (7), 843–856. 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- Dewar M. J. S.; Thiel W. Ground states of molecules. 38. The MNDO method. Approximations and parameters. J. Am. Chem. Soc. 1977, 99 (15), 4899–4907. 10.1021/ja00457a004. [DOI] [Google Scholar]
- Stewart J. J. P. Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19 (1), 1–32. 10.1007/s00894-012-1667-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malde A. K.; Zuo L.; Breeze M.; Stroet M.; Poger D.; Nair P. C.; Oostenbrink C.; Mark A. E. An automated force field topology builder (ATB) and repository: version 1.0. J. Chem. Theory Comput. 2011, 7 (12), 4026–4037. 10.1021/ct200196m. [DOI] [PubMed] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98 (12), 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126 (1), 014101 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Parrinello M.; Rahman A. Crystal structure and pair potentials: A molecular-dynamics study. Phys. Rev. Lett. 1980, 45 (14), 1196–1199. 10.1103/PhysRevLett.45.1196. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J.; Fraaije J. G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18 (12), 1463–1472. 10.1002/(SICI)1096-987X(199709)18:12%3C1463::AID-JCC4%3E3.0.CO;2-H. [DOI] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Pall S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Kumari R.; Kumar R.; Lynn A.; g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54 (7), 1951–1962. 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The codes and data sets can be found at the following link: https://github.com/pribnowbox/virtual-screening-of-AChE-inhibitors.