

Abstract
Aims
Increased miR-375 levels has been implicated in rodent models of myocardial infarction (MI) and with patients with heart failure. However, no prior study had established a therapeutic role of miR-375 in ischemic myocardium. Therefore, we assessed whether inhibition of MI-induced miR-375 by LNA anti-miR-375 can improve recovery after acute MI.
Methods and results
Ten weeks old mice were treated with either control or LNA anti miR-375 after induction of MI by LAD ligation. The inflammatory response, cardiomyocyte apoptosis, capillary density and left ventricular (LV) functional, and structural remodelling changes were evaluated. Anti-miR-375 therapy significantly decreased inflammatory response and reduced cardiomyocyte apoptosis in the ischemic myocardium and significantly improved LV function and neovascularization and reduced infarct size. Repression of miR-375 led to the activation of 3-phosphoinositide-dependent protein kinase 1 (PDK-1) and increased AKT phosphorylation on Thr-308 in experimental hearts. In corroboration with our in vivo findings, our in vitro studies demonstrated that knockdown of miR-375 in macrophages modulated their phenotype, enhanced PDK-1 levels, and reduced pro-inflammatory cytokines expression following LPS challenge. Further, miR-375 levels were elevated in failing human heart tissue.
Conclusion
Taken together, our studies demonstrate that anti-miR-375 therapy reduced inflammatory response, decreased cardiomyocyte death, improved LV function, and enhanced angiogenesis by targeting multiple cell types mediated at least in part through PDK-1/AKT signalling mechanisms.
Keywords: MiRNA, Inflammation, Myocardial infarction, Cardiac repair
1. Introduction
Ischemic heart disease is the major cause of morbidity and mortality in the USA. Potential therapeutic approaches target inhibition of cardiomyocyte apoptosis, increase coronary flow to facilitate neovascularization that collectively may improve cardiac function.1,2 Recent studies have highlighted the therapeutic potential of microRNAs (miRNA) in myocardial ischemia.3–5 We and others have previously shown that miRs regulates multiple cellular processes such as proliferation, differentiation, cell metabolism, apoptosis, and angiogenesis.6–12
Genome wide miRNA analysis revealed that miR-375 is expressed in various tissues and organs and is significantly reduced in different types of cancers,13–17 thus suggesting that miR-375 could be an important cancer-related miRNA.18
miR-375 has been recently implicated in patients with heart failure and diabetes.19,20 We and others have shown that miR-375 is associated with MI in mice and cardiac hypertrophy in rats.6,21 However, no prior study had established a therapeutic role of miR-375 in ischemic myocardium.
We report here that therapeutic silencing of miR-375 using LNA anti miR-375, attenuates MI-induced inflammation, cardiomyocyte apoptosis and restores LV function, and remodelling.
2. Methods
2.1. Animal model
This study conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. All experiments confirm to the protocols approved by the Institutional Animal Care and Use Committee of Temple University School of Medicine. Eight-weeks-old Wild-type (WT) male mice of C57BL/6J background were procured from Jackson Research Laboratory (Bar Harbor, ME).
2.2. Human heart tissues samples
Heart tissue samples were obtained from failing human hearts at the time of transplantation at the Houston Methodist De Bakey Heart and Vascular Center, Houston Methodist Hospital, Houston, Texas, and immediately frozen in liquid nitrogen and stored at − 80 °C until use. Normal hearts were obtained from donor hearts not used for transplantation and were collected and stored in the same manner. Study subjects’ details are provided in Table 1. The study was conducted in accordance with the Declaration of Helsinki. All tissues were collected with patients consent for research purposes and protocol was approved by the Houston Methodist Research Institutional Review Board.
Table 1.
Patients involved in this study
| Race | Sex | Age | Aetiology | Diabetes | |
|---|---|---|---|---|---|
| Caucasian | F | 63 | Ischemic | No | Ischemic |
| Caucasian | M | 67 | Ischemic | No | Ischemic |
| Caucasian | M | 63 | Ischemic | Yes | Ischemic |
| African American | M | 48 | Ischemic | Yes | Ischemic |
| Caucasian | M | 67 | Ischemic | No | Ischemic |
| Caucasian | F | 66 | Pulmonary hypertension | No | Control |
| Hispanic | F | 51 | Pulmonary hypertension | No | Control |
| African American | F | 47 | Pulmonary hypertension | No | Control |
| Hispanic | F | 31 | Pulmonary hypertension | No | Control |
2.3. Myocardial infarction and study design
Mice were anesthetized with 2% isoflurane inhalation with an isoflurane delivery system (Viking Medical, Medford, NJ) and were subjected to MI by ligation of left anterior descending coronary artery (LAD) as described previously.6,7 Immediately after LAD ligation, mice received a single sub-cutaneous (S.C.), injection of scrambled oligo control (catalogue number 4464076, Applied Biosystems; n = 20) or LNA anti miR-375 (5′CACGCGAGCCGAACGAACAA3′, Exiqon, Skelstedet, Denmark) (10 mg/kg, n = 20). Cardiac function was examined on 7, 14, and 28 days using by echocardiography and morphological analyses were performed on 28 days post-MI. At the end of the experiment, mice were euthanized by an overdose of tribromoethanol, 200 mg/kg BW, ip.
2.4. Isolation of adult mouse cardiomyocytes
Adult mouse cardiomyocytes were isolated as described previously.7,22,23 For detailed information, please see Supplementary material online.
2.5. Cardiac endothelial cells and macrophages isolation, and flow cytometry
Mice were euthanized by an overdose of tribromoethanol 200 mg/kg BW, ip. Cardiac endothelial cells were isolated by magnetic bead separation using CD31+ beads and further purity was confirmed by CD31+ (clone 390) cells using LSR-II flow cytometer. Cardiac macrophages were identified as CD45+ (clone 30F11), CD11bhigh (clone M1/70), F4/80high (clone BM8), Ly-6Cl°w (clone AL-21), and cell sorted using FACS Aria-II. Enriched populations of endothelial cells and macrophages were used for quantitative real time (RT)–PCR analyses.
2.6. Echocardiography
Mice were anesthetized with 2% isoflurane inhalation with an isoflurane delivery system (Viking Medical, Medford, NJ). Transthoracic two-dimensional M-mode echocardiogram was obtained using Vevo 770 (Visual Sonics, Toronto, Canada) equipped with 30 MHz transducer. Echocardiographic studies were performed before (baseline) and at 7, 14, and 28 day’s post-MI on mice anesthetized with a mixture of 1.5% isoflurane and oxygen (1 L/min). The internal diameter of the LV was measured in the short-axis view from M-mode recordings in end diastole, end systole and ejection fraction (EF), and fractional shortening (FS) were calculated using corresponding formulas as previously described.6,7
2.7. Morphometric studies
Mice were euthanized by an overdose of tribromoethanol, 200 mg/kg BW, ip. The hearts fixed by perfusion with 10% buffered formalin. Hearts were then cut into three slices (apex, mid-LV, and base) and paraffin embedded. The morphometric analysis including infarct size, wall thickness, and percent fibrosis was performed on Masson’s trichrome stained tissue sections using Image-J software (NIH, version 1.30, http://rsb.info.nih.gov/ij/). Fibrosis area was measured to determine percent fibrosis.6
2.8. Histology
Immunofluorescence staining for tissue sections were performed as described previously (9). In the mice received scrambled control oligo or LNA anti miR-375 the formation of new capillary network was assessed by Isolectin B4 as described before,6 positive staining in 10 randomly selected low-power visual fields (LPF) 28 days post-MI. Nuclei were counter-stained with 4′, 6-diamidino-2-phenylindole (DAPI, 1:10000, Sigma Aldrich, St Louis, MO), and sections were examined with a fluorescent microscope (Nikon, Japan).
2.9. Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labelling (TUNEL) staining for apoptosis in the myocardium was performed as previously described
Please see Supplementary material online for more details.6
2.10. RT–PCR
Expression levels of different genes (primer list included in Table 2) and miR-375 (mature miRNA sequence UUUGUUCGUU CGGCUCGC GUGA; Assay ID: 000564, Applied Biosystems) were measured using quantitative miRNA stem loop RT–PCR technology (TaqMan miRNA Assays; Applied Biosystems). This assay uses gene specific stem cell loop RT primers and TaqMan probes to detect mRNA or mature miRNA transcripts. Transcription was performed using 2 µg or 10 ng total RNA and the TaqMan miRNA RT kit (Applied Biosystems). RT–PCR was performed on an Applied Biosystems 770 apparatus using the TaqMan Universal PCR Master Mix, No AmpErase UNG (Applied Biosystems). The amplification steps consisted of initial denaturation at 95 °C, followed by 40 cycles of denaturation at 95 °C for 15 s and annealing at 60 °C for 1 min. The TaqMan specific primer 18S or U6 small nucleolar RNA was used for normalization with the threshold delta-delta cycle method (Gene Expression Macro; Bio-Rad, Hercules, CA, USA).
Table 2.
List of genes used in this study
| S.NO | Gene | Forward primer | Reverse primer | Probe |
|---|---|---|---|---|
| 1 | Mcp-1 | CTTCCTCCACCACCATGCA | CCAGCCGGCAACTGTGA | CCCTGTCATGCTTCTGGGCCTGC |
| 2 | TNF-α | GGCTGCCCCGACTACGT | AGGTTGACTTTCTCCTGGTATGAGA | CCTCACCCACACCGTCAGCCG |
| 3 | IL-1β | CTACAGGCTCCGAGATGAACAAC | TCCATTGAGGTGGAGAGCTTTC | AGCCTCGTGCTGTCGGACCCATATG |
| 4 | Chil3 | TTCTGAATGAAGGAGCCACTGA | ATTGTCATAACCAACCCACTCATTAC | ATGCCCCCCAGGAAGTACCCTATGC |
| 5. | Arg-1 | AAGCCAGGGACTGACTACCTTAAA | TGATGCCCCAGATGGTTTTC | CACCTAAGTGACTGTGAATGCGCCACA |
| 6 | IL-6 | TTCCATCCAGTTGCCTTCTTG | GGGAGTGGTATCCTCTGTGAAGTC | TGCTGGTGACAACCACGGCCTTC |
| 7. | iNOS | GGGCAGCCTGTGAGACCTT | GCATTGGAAGTGAAGCGTTTC | TGTCCGAAGCAAACATCACATTCAGATCC |
| 8. | PDK-1 | TGGCTTCATGCAGGTGTCAT | GGGCAGGCTGGTTTCCA | CTCCTCCTCTTCCCACTCCCTGTCTACG |
2.11. LPS induced miR-375 expression in RAW 264.7 and siRNA experiments (in vitro studies)
Cells (ATTC TIB-71), murine macrophage cell line was pre-treated with LPS (10 ng/mL, Sigma) for 24 h then anti miR-375 (50 nM, Applied Biosystems) or diluent were added at the end of 24 h incubation. Cells were harvested 24 h post-anti-miR-375 treatment and changes in the levels of miR-375, PDK-1, p-AKT, tAKT levels, (M1 markers) TNF-α, IL-1β, mcp-1, and arginase-1 (M2 marker) were quantified by quantitative RT–PCR or western blot. Results are represented as s.e.m for five independent experiments.
Small interfering RNA (siRNA) sequences targeting mouse PDK-1 were synthesized from Invitrogen (PDk-1 siRNA or a negative control [siRNA-NC]) was used at a final concentration of 100 nm according to the manufacturer’s instructions, and the cells were transfected for 24 h. Subsequently, the knockdown efficiency in RAW cells was determined by western blot. In addition, 50 nm anti-miR-375 (Applied Biosystems) was introduced alone or in combination with 100 nm PDK-1-1 SiRNA using Lipofectamine™ RNAiMAX (Invitrogen) in RAW cells. Cells were harvested 24 h post-anti-miR-375 treatment and changes in the levels of miR-375, PDK-1, p-AKT, tAKT levels, (M1 markers) TNF-α, IL-1β, mcp-1, and arginase-1 (M2 marker) were quantified by quantitative RT–PCR or western blot.
2.12. Statistics
Data are expressed as mean ± SEM. Analyses were performed using Prism 6.0a (GraphPad Software Inc.). The group means were compared using a t-test (for two groups) and ANOVA, followed by Bonferroni post-tests (for > 2 groups). P-values of <0.05 indicate statistical significance.
3. Results
3.1. miR-375 is up-regulated in failing human hearts
We have previously reported that miR-375 expression was significantly enhanced in post-MI mice hearts.6 In this study, we directly compared the expression of miR-375 between normal and ischemic human heart tissues. Our results demonstrate that miR-375 expression was significantly up-regulated in the failing human hearts compared to normal hearts (Figure 1). These findings suggest the clinical significance of miR-375.
Figure 1.
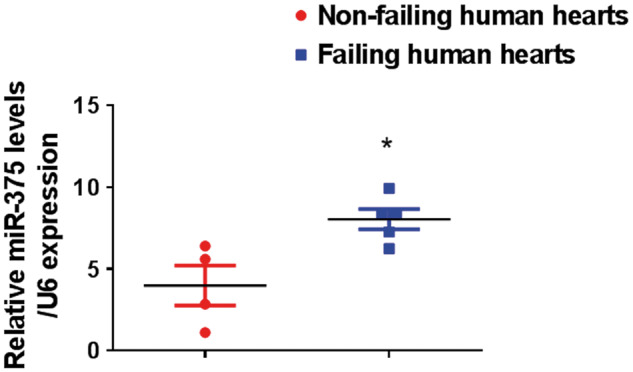
miR-375 expression is elevated in failing human hearts. Quantitative RT–PCR analysis of miR-375 expression in the LV tissue of the failing hearts (n = 5) and non-failing hearts (n = 4) normalized to control group U6 housekeeping gene. *P < 0.05 vs. non-failing hearts.
3.2. Myocardial miR-375 knockdown attenuates post-MI LV dysfunction
Increased levels of miR-375 in human failing ischemic hearts may suggest a significant role of this miRNA in ischemic recovery and regeneration. To test this hypothesis, we studied whether myocardial miR-375 knockdown may attenuate LV dysfunction. We determined the knockdown efficiency of a single dose s.c. administration of 10 mg/kg LNA anti miR-375 in vivo, and found significantly reduced miR-375 levels in the LV tissue, compared to controls post-MI at day 5 and at day 28 (Figure 2A). LV functions were determined by echocardiography in mice injected with either control or LNA anti miR-375 on days 7, 14, and 28 after the induction of MI. Percent EF and percent FS was similar at baseline. Compared to scrambled control miR, administration of LNA anti miR-375 injection a day after MI, significantly improved %EF and %FS (P < 0.001 vs. baseline; Figure 2B, C) 2 and 4 weeks after MI, suggesting that a single administration of LNA anti miR-375 post-MI attenuates ischemia-induced cardiac dysfunction.
Figure 2.

Myocardial miR-375 knockdown attenuates post-MI LV dysfunction. (A) Quantitative RT–PCR analysis of miR-375 expression at day 5 and day 28 in the scrambled ctrl or LNA-anti-miR-375 treated hearts (normalized to control group U6; n = 5). ***P < 0.001 vs. sham group; $< 0.05 vs. LNA anti-miR-375 treated group. (B, C) Quantification of %EF and %FS, in the hearts treated with scrambled or LNA anti-miR-375. %EF, percent ejection fraction; %FS, percent fractional shortening. n = 5–7/group. **P< 0.01 vs. scrambled ctrl group.
3.3. Myocardial miR-375 knockdown enhances neovascularization and reduces fibrosis after MI
To determine the effect of miR-375 knockdown on myocardial neovascularization, we assessed capillary density in the border zone of the infarct at 28 days post-MI. The number of Isolectin/B4 (+) capillaries were significantly higher in mice that received LNA anti miR-375 compared to those receiving scrambled ctrl (Figure 3A, B, P < 0.001 vs. scrambled ctrl groups). To determine whether improved capillary density after MI may improve cardiac remodelling we assessed infarct size at 28 days post-MI. Knockdown of miR-375 resulted in a significant reduction in the infarct size (Figure 3C, D, P < 0.001 vs. scrambled ctrl group).
Figure 3.

Myocardial miR-375 knockdown enhances neovascularization and reduces fibrosis after MI. (A, B) Capillary density (isolectin staining, green fluorescence) in border zone of LV infarct at 28 days post-MI in scrambled or LNA-anti-miR-375 treated hearts. Capillaries were stained with Isolectin-Alexa-555 (red) and nuclei were counterstained with DAPI (blue). (40×, Scale bar 100 μm). (C) Quantification of border zone capillary number across treatments presented as the number of Isolectin B4-positive capillaries (green) and DAPI-stained nuclei (blue) per high power field (HPF) (n = 5/each group). ***P< 0.001 vs. scrambled ctrl group. (D-E) Masson’s trichrome stained heart sections (28 days post-MI). (F) Quantitative analysis of infarct wall thickness at 28 days post-MI in scrambled or LNA-anti-miR-375 treated hearts. Scale = 1mm; n = 5/each group. ***P< 0.001 vs. scrambled ctrl group.
3.4. Myocardial miR-375 knockdown inhibits post-MI LV inflammation and promotes M2 macrophage polarization
We have previously reported that miR-375 is an independent downstream target of IL-10 in bone marrow progenitor angiogenic cells.6 We have previously shown that IL-10 controls miR-375 expression in bone marrow angiogenic progenitor cells.24 However, role of miR-375 in regulating macrophage polarization has never been studied. To test whether miR-375 knockdown may induce macrophage polarization, we evaluated the expression levels of M1 and M2-related genes. Compared to scrambled control treated group, 5 days post-MI quantitative RT–PCR analysis showed that miR-375 knockdown resulted in a significant repression of various M1 markers IL-1β (P < 0.01), TNF-α (P < 0.001), iNOS (P < 0.05), IL-6 (P < 0.01), and significantly increased M2 marker expression levels such as arginase-1(P < 0.01) and chitinase-like-3 (Chi313) (P < 0.01) (Figure 4A–F). We also found increased IL-10 levels (although not significant) in the LNA treated hearts compared to controls (Supplementary material online, Figure S1). The flow cytometric analysis revealed increased CD206+ (an M2 marker) cells in the LNA anti miR-375 treated hearts compared to controls (Figure 4G). Our mouse cytokine multiplex assay demonstrated significant reduction of at least six pro-inflammatory cytokines including IL-6 (P < 0.01), IP-10 (P < 0.01), MCP-1 (P < 0.06), MIP1-α (P < 0.07), MIP1-β (P < 0.01), and RANTES (P < 0.01) in LNA anti miR-375 group compared to scrambled ctrl (Supplementary material online, Figure S2). Reduction in pro-inflammatory cytokines was further corroborated by significant reduction in the infiltration of CD68+ cells (a common macrophage marker) in the infarct border zone in LNA anti miR-375 treated group compared to controls (Figure 4H–J, P < 0.001 vs. scrambled ctrl). Since LNA anti miR-375 therapy could reduce the inflammatory gene expression and also inflammatory cell response, we therefore examined cardiomyocyte apoptosis after LNA anti miR-375 administration in the ischemic myocardium (5 days after MI). The number of TUNEL (+) apoptotic cells were twice as high in scrambled ctrl vs. LNA anti miR-375 treated hearts (Figure 4K–M, P < 0.01 vs. scrambled ctrl), suggesting that miR-375 knockdown decreases cardiomyocyte apoptosis in the ischemic myocardium. Taken together with improved neovascularization and reduced infarct size these data suggest that miR-375 knockdown improves post-MI regeneration and remodelling processes in the ischemic myocardium with substantial preservation of LV function.
Figure 4.

Myocardial miR-375 knockdown inhibits post-MI LV inflammation. (A–F) Quantitative RT–PCR analysis of mRNA expression of M1 and M2 macrophage markers (TNF-α, IL-β, iNOS, IL-6, arginase-1, and Chi313) in the border zone of LV infarct at 5 days post-MI in control and LNA anti-miR-375 treated groups. mRNA expression normalized to 18S expression. n = 5/group, **P < 0.01, ***P< 0.001 vs. scrambled control. (G) Quantification of CD206+ cells by flow cytometry (M2 macrophages). n = 5/group, **P < 0.01 vs. scrambled control. (H, I) Immunofluorescent staining of macrophages (CD68+, red) in the border zone of infarct at 5 days post-MI. n = 5/group, ***P< 0.001 vs. scrambled control. (J) Quantitative analysis of infiltrating CD68+ cells per LPF at 5 days post-MI. 40×, Scale bar 100μm; n = 5/group, ***P< 0.001 vs. scrambled control. (K, L) Representative TUNEL staining image for cardiomyocyte apoptosis (red nuclei), alpha actinin (red), DAPI (blue) in border zone of LV infarct at 5 days post-MI. (M) Quantitative analysis of TUNEL+ cardiomyocytes in LNA-anti-miR-375 vs. scrambled ctrl-treated hearts. 20×, Scale bar 100μm; n = 5/group. ***P < 0.001 vs. scrambled control.
3.5. PDK-1 is a direct target of miR-375 in vivo
We have previously shown that PDK-1 is a predicted target of miR-375 in bone marrow progenitor cells.6 PDK-1 is also upstream kinase for AKT. Therefore, we assessed the levels of PDK-1 (Figure 5A, B) and total (tAKT) and phosphorylated (pAKT) (Figure 5A, C) in the border zone of infarct at 5 day’s post-MI in the scrambled ctrl and LNA anti miR-375 injected mice. There was a significant increase in PDK-1 levels (P < 0.001 vs. scrambled ctrl) and AKT phosphorylation (P < 0.05 vs. scrambled ctrl), suggesting that increase in pAKT levels may be mediated by PDK-1. To further corroborate increased capillary density in infarcted myocardium with activation of AKT, a well-known positive regulator of angiogenesis, we evaluated vascular endothelial growth factor (VEGF) expression in scrambled control and LNA anti miR-375 treated groups. VEGF expression levels were significantly increased in LNA anti miR-375 group compared to controls (Supplementary material online, Figure S3). Taken together, these data suggest that the miR-375 knockdown mediated increase in PDK-1 expression and activation of downstream survival and angiogenesis signalling may be responsible for improvement in ischemia-induced cardiac recovery.
Figure 5.

PDK-1 is a potential target of miR-375 in different cardiac cells. (A–C) Representative western blots and the quantification for PDK-1, pAKT/total AKT protein expression in LV tissue at 5 days post-MI. Equal loading of proteins in each lane is shown by β-actin. n = 5/group., **P < 0.01, ***P < 0.001 vs. scrambled ctrl group. Quantitative RT–PCR analysis of miR-375, normalized to control group U6 housekeeping gene (D–F) and its target PDK-1 expression normalized to 18S expression (G-I) in isolated cardiomyocytes, cardiac endothelial cells, and cardiac macrophages from sham and MI mice. n= 5/group. ***P < 0.001, **P < 0.01, *P < 0.05 vs. scrambled ctrl group.
LNA anti miR-375 injection may affect one or more cell types within the heart tissue. To address this question we studied the effects of LNA anti miR-375 on three types of cells in the heart—the isolated cardiomyocytes (rod shaped cells, Supplementary material online, Figure S4A), endothelial cells (CD31+ cells, Supplementary material online, Figure S4B, C), and macrophages (CD45+/F4/80High/CD11BHigh/LY6c l°w, Supplementary material online, Figure S4D) from control and LNA anti miR-375 treated groups. miR-375 levels were significantly repressed, whereas miR-375 target gene PDK-1 levels were significantly elevated in all three isolated cell types (Figure 5D–Ixs). These results suggest that, inhibition of miR-375 may provide cardiac protection post-MI, by acting on multiple cardiac cell types.
3.6. Modulation of inflammatory response by knockdown of miR-375 in macrophages
In order to understand the molecular mechanisms of LNA anti-miR-375 mediated inhibition of inflammation and M1 to M2 macrophage switch in vivo, we performed in vitro experiments in RAW 264.7 cells (ATTC TIB-71), murine macrophage cell line. RAW cells were treated with LPS (10 ng/mL) to induce inflammatory signal. LPS treatment for 24 h significantly increased expression of TNF-α, IL-β, MCP-1 (M1 markers) (Figure 6A–C). Treatment with LNA anti miR-375 markedly reduced LPS induced M1 markers expression and promoted arginase-1 expression (M2 marker) (Figure 6A–D). To investigate whether miR-375 mediated affects were mediated via PDK-1, we treated RAW cells with anti-miR-375 or scrambled oligo. PDK-1 expression levels were significantly increased in anti-miR-375-treated RAW cells (Figure 6E, P < 0.01 vs. scrambled ctrl). Further we assessed PDK-1 and pAKT levels in the RAW cells subjected to LPS treatment with or without anti-miR-375 (Figure 6F). LPS significantly reduced PDK-1 and pAKT levels, anti-miR-375-treatment markedly increased PDK-1 and pAKT levels (Figure 6G, H). Additionally, anti-miR-375 restored PDK-1 and pAKT levels in the LPS treated cells (Figure 6G, H). These in vitro studies confirm our in vivo findings and suggests that knockdown of miR-375 promote M2 polarization and enhance PDK-1-mediated cardiac recovery post-MI.
Figure 6.

Modulation of inflammatory response by knockdown of miR-375 in macrophages. RAW cells were treated with LPS (10 ng/mL), LPS (10 ng/mL) + anti miR-375 (50 nM), or anti miR-375 alone (50 nM) for 24 h. (A–D) Relative mRNA expression levels of TNF-α, IL-1β, mcp-1, and arginase-1 were measured by quantitative RT–PCR, normalized to 18S. n = 5/group. ***P < 0.001 vs. control group; ###P < 0.001 vs. LPS treated group. (E) PDK-1 mRNA levels were measured by quantitative RT–PCR, normalized to 18S. n = 5/group. **P < 0.001 vs. scrambled ctrl. (F–H) Representative western blots and the quantification for PDK-1, pAKT protein expression in LV tissue at 5 days post-MI. Equal loading of proteins in each lane is shown by total AKT and β-actin. n = 5/group. **P < 0.01, *P < 0.05 vs. control group; ##P < 0.01, #P < 0.05 vs. LPS treated group.
3.7. Effects of miR-375 on inflammatory response are PDK-1 dependent in macrophages
To further confirm whether miR-375 mediated effects were via PDK-1, we knocked down PDK-1 in RAW 264.7 cells using siRNA (Figure 7A, B) and further assessed their inflammatory response subjected to LPS stimulus. PDK-1 silencing exaggerated LPS induced proinflammatory cytokine gene expression compared to controls. Intriguingly in the co-transfection of PDK-1 siRNA and anti-miR-375 in RAW 264.7 cells, the effect of anti-miR-375 on anti-inflammatory gene expression ability was completely abolished by PDK-1 siRNA (Figure 7C–F). These data confirm that miR-375 regulates macrophage polarization in part via PDK-1 dependent manner.
Figure 7.

Effects of miR-375 on inflammatory response are PDK-1 dependent in macrophages. RAW cells were transfected with NC-siRNA or PDK-1 for 48 h. (A) Representative PDK-1 protein levels demonstrated by western blot. (B) Quantification of PDK-1 levels normalized to β-actin. RAW cells transfected with scrambled control (50 nM), anti-miR-375 (50 nM), combination of PDK-1 siRNA (100 nM), and anti-miR-375(50 nM), or PDK-1 siRNA (100 nM) alone for 48 h and stimulated with LPS (10 ng/mL) for 24 h. =5/group. ***P < 0.001 vs. scrambled ctrl. (C–F) Relative mRNA expression levels of TNF-α, IL-1β, mcp-1, and arginase-1 were measured by quantitative RT–PCR, normalized to 18S. n = 5/group. ***P < 0.001, **P < 0.01, *P < 0.05 vs. scrambled ctrl; ###P < 0.001, ##P < 0.01, #P < 0.05 vs. LPS treated group.
4. Discussion
It is well-documented that several miRNAs are dysregulated in the cardiovascular diseases. We have previously demonstrated that knockdown of miR-375 in the bone marrow angiogenic progenitor cells exhibited therapeutic potential post-MI.6 In the current study, we assessed the direct inhibition miR-375 in the ischemic heart. Emerging evidence suggests an association of decreased miR-375 expression in different types of cancer.13–17 Since this has been an important miR related to cancers and their levels were elevated in the human failing heart samples, we hypothesized that inhibition of miR-375 may play an important role in attenuating left ventricular dysfunction after MI in mice. Our conclusion is based on the following experimental findings: (i) miR-375 is elevated in the failing human hearts; (ii) direct inhibition of miR-375 in the myocardium by LNA-based miR-375 inhibitor therapy reduces inflammatory response after MI; (iii) knockdown of miR-375 enhances survival of cardiomyocytes; (iv) enhances neovascularization, reduces infarct size, and ultimately attenuates the LV dysfunction after MI; (v) mechanistically, miR-375 effects on observed benefits appear to be mediated at least in part through PDK-1/AKT signalling mechanisms.
The therapeutic applicability of systemically delivered LNA-modified anti-miRs has been reported.25–27 It is important to consider that LNA based miR inhibitors have made their way to clinical trials. MiR-122 inhibition has been tested in phase-II clinical trials in Hepatitis-C (HCV) patients with encouraging results.28 LNA-based miR inhibition has been thoroughly validated to be efficacious in inducing sustained and potent silencing of miR in the heart.25,29 In the current studies we obtained miR-375 silencing in isolated cardiomyocytes, endothelial cells, and macrophages upon single injection of 10 mg/kg LNA miR-375 inhibitor subcutaneously (s.c.). This single injection attenuated cardiac dysfunction post-MI. Corroborating with our studies a recent report suggested that two injections of 5 mg/kg of LNA-anti-miR had a sustained effect on silencing miR-24 in cardiac endothelial cells in vivo.30 One recent study reported that, LNA-anti-miR-1 at a dose of 1 mg/kg exhibited cardiac protective effects.31 Therefore, we propose that, low doses of LNA miR inhibitors might be sufficient to induce potential silencing in different cardiac cells in the injured heart.
We found that miR-375 knockdown increases the expression of PDK-1, suggesting PDK-1 as a potential target. Furthermore, we showed that knockdown of miR-375 increased the phosphorylation of Akt in post-MI heart by targeting PDK-1. This is consistent with our recent report showing miR-375 directly targets PDK-1 at the protein level in bone marrow angiogenic progenitor cells.24 Further ex vivo knockdown of miR-375 in BMPACs before transplantation into ischemic heart post-MI also increased PDK-1 and pAKT levels. We speculate this could be due to reduced miR-375 levels in the BMPAC exosomes leading to increased PDK-1 levels in the MI heart.24 Thus activation of the PDK-1/AKT signalling pathway may represent a potential protective mechanism contributing to the improved cardiac functional outcome observed in the LNA anti-miR-375 therapy MI mice.
It is well-known phenomenon that neovascularization plays an integral role in infarct healing process.2 In the current study, capillary density was increased in MI mice treated with LNA-anti miR-375 treated hearts compared to scrambled controls. In corroboration with this study, we have previously shown that, miR-375 knockdown enhances endothelial cell functions and angiogenic activity in vitro,24 it is likely that enhanced angiogenesis observed in this study reflects miR-375 knockdown in resident endothelial cells. Further, we have also demonstrated significant increase in VEGF levels in the LNA anti miR-375 treated hearts compared to scrambled controls. The observed increase in VEGF levels could directly be linked to the observed increase in the activation of AKT (upstream of VEGF) in the LNA miR-375 treated groups compared to controls.32 Furthermore, loss of PDK-1 has been implicated in endothelial cell apoptosis and impaired angiogenesis.33,34 Thus activation of the PDK-1/AKT signalling pathway represent a protective mechanism enabling neovascularization.
Pathological remodelling of the heart is accompanied by increased apoptosis.35 We have previously shown that, miR-375 knockdown in BMPACS reduces cardiomyocyte apoptosis via release of paracrine factors.24 In the present study, LNA-anti miR-375 treatment in MI was associated with a decreased cardiomyocyte apoptosis compared to scrambled controls, it is likely that reduced apoptosis observed in this study reflects miR-375 knockdown in resident cardiomyocytes and increased PDK-1 levels. Further, it is well-established that, PDK-1/AKT signalling plays a critical role in cardiovascular biology.34,36 Cardiac-specific disruption of PDK-1 in mice, resulted in lethal heart failure and reduced PDK-1 protein levels in end stage of failing human hearts reveals the possibility that functional alterations of PDK-1 could be implicated in the pathogenesis of heart failure.34,36 Thus increased expression of PDK-1 expression in hearts, isolated endothelial cells and cardiomyocytes of LNA anti miR-375-treated mice might be associated with enhanced angiogenesis and cardiomyocyte survival leading to the improved cardiac function.
It is suggested that sustained proinflammatory response in the myocardium could lead to LV dysfunction and remodelling.37,38 We have previously reported that, miR-375 is an independent downstream target of IL-10.5 However, role of miR-375 in regulating macrophage polarization has never been studied. In the present study, we demonstrated that LNA anti-miR-375 directly targets PDK-1 signalling in polarized macrophages residing in injured myocardial tissue. As MI has been linked with enhanced M1 macrophage polarization and suppressed M2 phenotype resulting in impaired tissue repair.39 Interestingly, in our study, M2-like macrophage markers arginase-1, Chi3l3, and CD206 levels were significantly increased. Also anti-inflammatory cytokine IL-10 levels were partially increased in the LNA anti-miR-375 treated hearts compared to controls. As IL-10 has been shown to activate M2 phenotype leading to abundance of regulatory macrophages with anti-inflammatory activity.40 Further pro-inflammatory markers IL-1β, TNF-α, iNOS, IL-6, IP-10, MCP-1, MIP1α, MIP1-β, and RANTES were significantly reduced in the LNA anti miR-375 treated hearts compared to the controls. Also, our findings corroborate with a previously published study showing exaggerated increase in pro-inflammatory gene expression in mice lacking PDK-1 in macrophages.41 This evidence indicated that PDK-1 might play a pivotal role in miR-375-mediated regulation of macrophage polarization. Our results demonstrated that miR-375 inhibition repressed the M1 macrophage switch and promoted M2 macrophage polarization via activating PDK-1. Our PDK-1 knockdown experiments in this study, further confirmed the crucial role of PDK-1 in modulating inflammatory response in macrophages. Thus increased expression of PDK-1 expression in hearts and isolated macrophages of LNA anti miR-375-treated mice, suggesting PDK-1 might serve as a molecular regulator in macrophage polarization and a potential therapeutic target for reducing inflammatory response post-MI.
We have in the past published various aspects of IL-10 signalling and function in cardiovascular injuries.2,24,35,42–45 First, IL-10 recombinant therapy is expensive and further IL-10 peptide is short lived. The present study is the logical extension of new mechanistic insights into IL-10, focusing on miR-375 as an independent downstream target of IL-10. As miRNA based therapies already made their way to clinical trials. We have shown PDK-1 as a potential target of miR-375, however, we cannot exclude the role of other miR-375-regulated target genes in the observed benefits, and future studies will be important to delineate other pathways that might mediate such effects.
In summary, our data demonstrate that inhibition of miR-375 mediate pleiotropic beneficial effects such as enhanced angiogenesis, reduced inflammatory response after MI, and direct cardiomyocyte protection all, are capable of limiting infarct size and preserving post-MI cardiac function and integrity. LNA anti miR-375 therapy inhibits miR-375 levels in several cells within heart tissue post-MI leading to activation of PDK-1/AKT signalling (Figure 8). Increased expression of PDK-1 (potential target of miR-375) enhances neovascularization, reduces cardiomyocyte death and mediates an M1 to M2 phenotype macrophage switch that collectively improve cardiac recovery post-MI. Thus, miR-375 knockdown appears to be a feasible approach to limit ischemic injury and might prove to be an attractive therapeutic strategy for patients with MI.
Figure 8.

Proposed mechanism of anti-miR-375 therapy mediated cardiac repair. LNA anti-miR-375 therapy regulated miR-375 levels in multiple cardiovascular cells (cardiomyocytes, endothelial cells, and macrophages) post-MI heart leading to activation of PDK-1/AKT signalling, PDK-1 (direct target of miR-375), thereby enhancing the neovascularization, reduced cardiomyocyte death, and macrophage phenotype switch (M1 to M2 phenotype) mediated cardiac repair. PDK-1, 3-phosphoinositide-dependent protein kinase 1.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
V.N.S.G.: Conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing; S.K.V., D.J.: Collection and analysis of data; S.K.V., Z.C., M.K.: Conception and design; C.B., D.A.V., E.G., W.K.: Conception and design; E.N., M.C., Y.T. and J.I.: Collection and/or assembly of data; R.K.: Conception and design, final editing, and approval of manuscript.
Supplementary Material
Acknowledgements
This work was supported in part by funding from the National Institute of Health grants HL091983, HL126186, HL053354, and HL108806. V.N.S.G is supported by American Heart Association postdoctoral grant 15POST22720022. S.K.V. is supported with American Heart Association—Scientist Development Grant 14SDG20480104. D.A.G. is supported with AHA American Heart Association—Grant in aid 17GRNT33400035. Y.Y. is supported with AHA pre-doctoral grant 17PRE33370001.
Conflict of interest: none declared.
References
- 1. Kocher AA, Schuster MD, Szabolcs MJ, Takuma S, Burkhoff D, Wang J, Homma S, Edwards NM, Itescu S.. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat Med 2001;7:430–436. [DOI] [PubMed] [Google Scholar]
- 2. Krishnamurthy P, Thal M, Verma S, Hoxha E, Lambers E, Ramirez V, Qin G, Losordo D, Kishore R.. Interleukin-10 deficiency impairs bone marrow-derived endothelial progenitor cell survival and function in ischemic myocardium. Circ Res 2011;109:1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bernardo BC, Nguyen SS, Winbanks CE, Gao XM, Boey EJ, Tham YK, Kiriazis H, Ooi JY, Porrello ER, Igoor S, Thomas CJ, Gregorevic P, Lin RC, Du XJ, McMullen JR.. Therapeutic silencing of miR-652 restores heart function and attenuates adverse remodeling in a setting of established pathological hypertrophy. FASEB J 2014;28:5097–5110. [DOI] [PubMed] [Google Scholar]
- 4. Bernardo BC, Gao XM, Tham YK, Kiriazis H, Winbanks CE, Ooi JY, Boey EJ, Obad S, Kauppinen S, Gregorevic P, Du XJ, Lin RC, McMullen JR.. Silencing of miR-34a attenuates cardiac dysfunction in a setting of moderate, but not severe, hypertrophic cardiomyopathy. PLoS One 2014;9:e90337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, Fasanaro P, Sun N, Wang X, Martelli F, Robbins RC, Wu JC.. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation 2010;122:S124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garikipati VN, Krishnamurthy P, Verma SK, Khan M, Abramova T, Mackie AR, Qin G, Benedict C, Nickoloff E, Johnson J, Gao E, Losordo DW, Houser SR, Koch WJ, Kishore R.. Negative regulation of miR-375 by interleukin-10 enhances bone marrow-derived progenitor cell-mediated myocardial repair and function after myocardial infarction. Stem Cells 2015;33:3519–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Joladarashi D, Srikanth Garikipati VN, Thandavarayan RA, Verma SK, Mackie AR, Khan M, Gumpert AM, Bhimaraj A, Youker KA, Uribe C, Suresh Babu S, Jeyabal P, Kishore R, Krishnamurthy P.. Enhanced cardiac regenerative ability of stem cells after ischemia-reperfusion injury: role of human CD34+ cells deficient in microRNA-377. J Am Coll Cardiol 2015;66:2214–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Khan M, Nickoloff E, Abramova T, Johnson J, Verma SK, Krishnamurthy P, Mackie AR, Vaughan E, Garikipati VN, Benedict C, Ramirez V, Lambers E, Ito A, Gao E, Misener S, Luongo T, Elrod J, Qin G, Houser SR, Koch WJ, Kishore R.. Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circ Res 2015;117:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sahoo S, Klychko E, Thorne T, Misener S, Schultz KM, Millay M, Ito A, Liu T, Kamide C, Agrawal H, Perlman H, Qin G, Kishore R, Losordo DW.. Exosomes from human CD34(+) stem cells mediate their proangiogenic paracrine activity. Circ Res 2011;109:724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009;136:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krol J, Loedige I, Filipowicz W.. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 2010;11:597–610. [DOI] [PubMed] [Google Scholar]
- 12. Almeida MI, Reis RM, Calin GA.. MicroRNA history: discovery, recent applications, and next frontiers. Mutat Res 2011;717:1–8. [DOI] [PubMed] [Google Scholar]
- 13. Mazar J, DeBlasio D, Govindarajan SS, Zhang S, Perera RJ.. Epigenetic regulation of microRNA-375 and its role in melanoma development in humans. FEBS Lett 2011;585:2467–2476. [DOI] [PubMed] [Google Scholar]
- 14. Avissar M, Christensen BC, Kelsey KT, Marsit CJ.. MicroRNA expression ratio is predictive of head and neck squamous cell carcinoma. Clin Cancer Res 2009;15:2850–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li X, Lin R, Li J.. Epigenetic silencing of microRNA-375 regulates PDK1 expression in esophageal cancer. Dig Dis Sci 2011;56:2849–2856. [DOI] [PubMed] [Google Scholar]
- 16. Ding L, Xu Y, Zhang W, Deng Y, Si M, Du Y, Yao H, Liu X, Ke Y, Si J, Zhou T.. MiR-375 frequently downregulated in gastric cancer inhibits cell proliferation by targeting JAK2. Cell Res 2010;20:784–793. [DOI] [PubMed] [Google Scholar]
- 17. Brase JC, Johannes M, Schlomm T, Falth M, Haese A, Steuber T, Beissbarth T, Kuner R, Sultmann H.. Circulating miRNAs are correlated with tumor progression in prostate cancer. Int J Cancer 2011;128:608–616. [DOI] [PubMed] [Google Scholar]
- 18. Tsukamoto Y, Nakada C, Noguchi T, Tanigawa M, Nguyen LT, Uchida T, Hijiya N, Matsuura K, Fujioka T, Seto M, Moriyama M.. MicroRNA-375 is downregulated in gastric carcinomas and regulates cell survival by targeting PDK1 and 14-3-3zeta. Cancer Res 2010;70:2339–2349. [DOI] [PubMed] [Google Scholar]
- 19. Akat KM, Moore-McGriff D, Morozov P, Brown M, Gogakos T, Correa Da Rosa J, Mihailovic A, Sauer M, Ji R, Ramarathnam A, Totary-Jain H, Williams Z, Tuschl T, Schulze PC.. Comparative RNA-sequencing analysis of myocardial and circulating small RNAs in human heart failure and their utility as biomarkers. Proc Natl Acad Sci U S A 2014;111:11151–11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao H, Guan J, Lee HM, Sui Y, He L, Siu JJ, Tse PP, Tong PC, Lai FM, Chan JC.. Up-regulated pancreatic tissue microRNA-375 associates with human type 2 diabetes through beta-cell deficit and islet amyloid deposition. Pancreas 2010;39:843–846. [DOI] [PubMed] [Google Scholar]
- 21. Feng HJ, Ouyang W, Liu JH, Sun YG, Hu R, Huang LH, Xian JL, Jing CF, Zhou MJ.. Global microRNA profiles and signaling pathways in the development of cardiac hypertrophy. Braz J Med Biol Res 2014;47:361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou YY, Wang SQ, Zhu WZ, Chruscinski A, Kobilka BK, Ziman B, Wang S, Lakatta EG, Cheng H, Xiao RP.. Culture and adenoviral infection of adult mouse cardiac myocytes: methods for cellular genetic physiology. Am J Physiol Heart Circ Physiol 2000;279:H429–436. [DOI] [PubMed] [Google Scholar]
- 23. Garcia-Prieto J, Garcia-Ruiz JM, Sanz-Rosa D, Pun A, Garcia-Alvarez A, Davidson SM, Fernandez-Friera L, Nuno-Ayala M, Fernandez-Jimenez R, Bernal JA, Izquierdo-Garcia JL, Jimenez-Borreguero J, Pizarro G, Ruiz-Cabello J, Macaya C, Fuster V, Yellon DM, Ibanez B.. beta3 adrenergic receptor selective stimulation during ischemia/reperfusion improves cardiac function in translational models through inhibition of mPTP opening in cardiomyocytes. Basic Res Cardiol 2014;109:422. [DOI] [PubMed] [Google Scholar]
- 24. Garikipati VN, Krishnamurthy P, Verma SK, Khan M, Abramova T, Mackie AR, Qin G, Benedict C, Nickoloff E, Johnson J, Gao E, Losordo DW, Houser SR, Koch WJ, Kishore R.. Negative regulation of miR-375 by interleukin-10 enhances bone marrow-derived progenitor cell-mediated myocardial repair and function after myocardial infarction. Stem Cells 2015;33:3519–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hullinger TG, Montgomery RL, Seto AG, Dickinson BA, Semus HM, Lynch JM, Dalby CM, Robinson K, Stack C, Latimer PA, Hare JM, Olson EN, van Rooij E.. Inhibition of miR-15 protects against cardiac ischemic injury. Circ Res 2012;110:71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Montgomery RL, Hullinger TG, Semus HM, Dickinson BA, Seto AG, Lynch JM, Stack C, Latimer PA, Olson EN, van Rooij E.. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation 2011;124:1537–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nagpal V, Rai R, Place AT, Murphy SB, Verma SK, Ghosh AK, Vaughan DE.. MiR-125b is critical for fibroblast-to-myofibroblast transition and cardiac fibrosis. Circulation 2016;133:291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, Persson R, King BD, Kauppinen S, Levin AA, Hodges MR.. Treatment of HCV infection by targeting microRNA. N Engl J Med 2013;368:1685–1694. [DOI] [PubMed] [Google Scholar]
- 29. Bernardo BC, Gao XM, Winbanks CE, Boey EJ, Tham YK, Kiriazis H, Gregorevic P, Obad S, Kauppinen S, Du XJ, Lin RC, McMullen JR.. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc Natl Acad Sci U S A 2012;109:17615–17620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fiedler J, Jazbutyte V, Kirchmaier BC, Gupta SK, Lorenzen J, Hartmann D, Galuppo P, Kneitz S, Pena JT, Sohn-Lee C, Loyer X, Soutschek J, Brand T, Tuschl T, Heineke J, Martin U, Schulte-Merker S, Ertl G, Engelhardt S, Bauersachs J, Thum T.. MicroRNA-24 regulates vascularity after myocardial infarction. Circulation 2011;124:720–730. [DOI] [PubMed] [Google Scholar]
- 31. Pan Z, Sun X, Ren J, Li X, Gao X, Lu C, Zhang Y, Sun H, Wang Y, Wang H, Wang J, Xie L, Lu Y, Yang B.. miR-1 exacerbates cardiac ischemia-reperfusion injury in mouse models. PLoS One 2012;7:e50515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K.. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest 2005;115:2108–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Park H, Lee S, Shrestha P, Kim J, Park JA, Ko Y, Ban YH, Park DY, Ha SJ, Koh GY, Hong VS, Mochizuki N, Kim YM, Lee W, Kwon YG.. AMIGO2, a novel membrane anchor of PDK1, controls cell survival and angiogenesis via Akt activation. J Cell Biol 2015;211:619–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Di RM, Feng QT, Chang Z, Luan Q, Zhang YY, Huang J, Li XL, Yang ZZ.. PDK1 plays a critical role in regulating cardiac function in mice and human. Chin Med J (Engl) 2010;123:2358–2363. [PubMed] [Google Scholar]
- 35. Verma SK, Krishnamurthy P, Barefield D, Singh N, Gupta R, Lambers E, Thal M, Mackie A, Hoxha E, Ramirez V, Qin G, Sadayappan S, Ghosh AK, Kishore R.. Interleukin-10 treatment attenuates pressure overload-induced hypertrophic remodeling and improves heart function via signal transducers and activators of transcription 3-dependent inhibition of nuclear factor-kappaB. Circulation 2012;126:418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Han Z, Jiang Y, Yang Y, Li X, Yang Z, Cao K, Wang DW.. Deletion of PDK1 causes cardiac sodium current reduction in mice. PLoS One 2015;10:e0122436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Christia P, Frangogiannis NG.. Targeting inflammatory pathways in myocardial infarction. Eur J Clin Invest 2013;43:986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun M, Dawood F, Wen WH, Chen M, Dixon I, Kirshenbaum LA, Liu PP.. Excessive tumor necrosis factor activation after infarction contributes to susceptibility of myocardial rupture and left ventricular dysfunction. Circulation 2004; 110: 3221–3228. [DOI] [PubMed] [Google Scholar]
- 39. Lambert JM, Lopez EF, Lindsey ML.. Macrophage roles following myocardial infarction. Int J Cardiol 2008;130:147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mosser DM, Edwards JP.. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008;8:958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chaurasia B, Mauer J, Koch L, Goldau J, Kock AS, Bruning JC.. Phosphoinositide-dependent kinase 1 provides negative feedback inhibition to Toll-like receptor-mediated NF-kappaB activation in macrophages. Mol Cell Biol 2010;30:4354–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krishnamurthy P, Lambers E, Verma S, Thorne T, Qin G, Losordo DW, Kishore R.. Myocardial knockdown of mRNA-stabilizing protein HuR attenuates post-MI inflammatory response and left ventricular dysfunction in IL-10-null mice. Faseb J. 2010;24:2484–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R.. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ Res 2009;104:e9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kishore R, Krishnamurthy P, Garikipati VN, Benedict C, Nickoloff E, Khan M, Johnson J, Gumpert AM, Koch WJ, Verma SK.. Interleukin-10 inhibits chronic angiotensin II-induced pathological autophagy. J Mol Cell Cardiol 2015;89:203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Verma SK, Garikipati VN, Krishnamurthy P, Khan M, Thorne T, Qin G, Losordo DW, Kishore R.. IL-10 accelerates re-endothelialization and inhibits post-injury intimal hyperplasia following carotid artery denudation. PLoS One 2016;11:e0147615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.