

Summary
Population genetics continues to identify genetic variants associated with diseases of the immune system and offers a unique opportunity to discover mechanisms of immune regulation. Multiple genetic variants linked to severe fungal infections and autoimmunity are associated with caspase recruitment domain-containing protein 9 (CARD9). We leverage the CARD9 R101C missense variant to uncover a biochemical mechanism of CARD9 activation essential for antifungal responses. We demonstrate that R101C disrupts a critical signaling switch whereby phosphorylation of S104 releases CARD9 from an autoinhibited state to promote inflammatory responses in myeloid cells. Furthermore, we show that CARD9 R101C exerts dynamic effects on the skin cellular contexture during fungal infection, corrupting inflammatory signaling and cell–cell communication circuits. Card9 R101C mice fail to control dermatophyte infection in the skin, resulting in high fungal burden, yet show minimal signs of inflammation. Together, we demonstrate how translational genetics reveals molecular and cellular mechanisms of innate immune regulation.
In brief
Brandt et al. employ translational genetics to show a missense variant linked to fungal immunodeficiency, CARD9 R101C, disrupts a signaling switch mediated by phosphorylation of S104. Disabling this switch impairs inflammatory cytokine programs in myeloid cells, that are amplified by chemokines from fibroblasts and keratinocytes, to promote recruitment of leukocytes.
Introduction
Insights from monogenic immunodeficiencies offer an unique opportunity to dissect mechanisms of protein function, signal transduction, and immune response regulation. Genetic variants associated with these phenotypes are rare, due to evolutionary selection, and typically result in loss of expression. In some cases, however, immunodeficiencies are associated with variants that do not affect protein expression but alter function instead. Uncovering the mechanistic basis for how these variants alter immune function can offer original strategies for therapeutic interventions that modulate immunity with greater precision. This general approach has proven to be successful in oncology, where detailed mechanistic studies of somatic mutations in oncogenes such as BRAF and KRAS have led to insights that fueled the development of targeted cancer therapeutics1. Here, we set out to uncover mechanisms of antifungal immunity through translational genetics, with the rationale that understanding germline encoded missense variants associated with immunodeficiency can reveal mechanisms of immunity that may have value for future development of anti-inflammatory therapeutics to treat autoimmunity.
Fungi are a natural part of the human commensal flora; however, invasive fungal infections are a growing concern worldwide as they are one of the major causes of infectious-disease related mortality, and the available treatments are limited. Most cases are driven by opportunistic fungi2, which can lead to life-threatening infections, especially in immunocompromised patients, causing fungal sepsis and damage to vital organs. The appearance of drug-resistant strains, such as Candida auris, is problematic in hospitals and residential care facilities and represents a major health burden3. Given the increase in the number of patients immunocompromised due to chronic illnesses, medication, or invasive medical procedures, as well as the emergence of drug-resistant fungal species, there is a need to develop effective treatments for co-emerging fungal infections4,5.
Studies of patients suffering from fungal infections without any known immunodeficiencies have led to the discovery of defects in specific immune genes and signaling pathways that can result in uncontrolled fungal invasion6,7. Additionally, GWAS studies have identified several common genetic variants that increase susceptibility to candidemia without full penetrance8. In particular, caspase recruitment domain-containing protein 9 (CARD9) has repeatedly been implicated in antifungal defense9, with at least 24 known loss-of-function mutations that lead to increased susceptibility to fungal infections. GWAS studies have also implicated CARD9 variants in the pathogenesis of inflammatory bowel disease10–12, ankylosing spondylitis13, primary sclerosing cholangitis14 and IgA nephropathy15. These GWAS studies highlight the importance of understanding how genetic variation in CARD9 is involved in fine-tuning the immune response, opening possibilities for therapeutic intervention.
CARD9 is an adaptor protein that signals downstream of ITAM-containing receptors in myeloid cells16–18. Recognition of fungal cell wall carbohydrates by C-type lectin receptors (CLRs) and subsequent downstream signaling through CARD9 is critical for activation of nuclear factor-κB (NFκB) and initiation of an antifungal immune response19. The CLR Dectin-1 engages fungal carbohydrates and, through a cascade of events, leads to CARD9 activation and the formation of a filamentous complex consisting of CARD9, BCL10, and MALT1 (CBM)20. Post-translational modifications (PTMs) such as phosphorylation and ubiquitination play an important role in regulating CARD9 activity and its ability to form the CBM complex. Despite significant progress, key questions remain regarding how CARD9 is activated and how understanding the structural dynamics of this process can be exploited for drug discovery.
In the current study, we sought further insight into the mechanistic regulation of CARD9 signaling by taking a translational genetics approach. The natural emergence of coding variants in CARD9 associated with fungal disease provides valuable genetic leads for structure-function studies aimed at deciphering how CARD9 activity is controlled. In particular, we focused on CARD9 R101C, linked to dermatophytosis in humans, to demonstrate that R101 regulates a phosphorylation switch that is essential for CARD9 activity and protection from fungal infection in mouse models. Thus, we demonstrate how genetics can be applied to uncover biochemical mechanisms of CARD9 signaling and contextualize the consequences of disease-associated variants in protective tissue immunity in vivo.
Results
CARD9 linker residues R101 and S104 are required to elicit cytokine responses
CARD9 adopts an autoinhibited conformation, yet it remains unclear how structural dynamics relieve autoinhibition to promote filamentation and subsequent assembly of the CBM complex21. The autoinhibited conformation of CARD9 is stabilized by interactions between the coiled-coil 1 (CC1) domain, key amino acid residues in the CARD domain, and the linker region between the CARD and CC1 domains21. This interface may be a key region that functions as an activating switch in response to PTMs. To gain further insight into the mechanisms regulating CARD9 activity, we evaluated genetic variants that predispose to severe fungal disease (Figure 1A). Only two CARD9 variants, R101C and R101L, are located in the linker region, which is notably conserved between CARD9 and CARD11 in higher vertebrates (Figure 1B)9,22,23. The R101 sidechain exhibits conformational dynamicity in published nuclear magnetic resonance (NMR) structural models21, leading us to hypothesize that it may function as an activating switch. Additionally, R101 is located in proximity to S104 (Figure 1C), which together comprise a common phosphorylation motif, RXXS, that is disrupted when arginine is replaced with another amino acid at position 101. To determine if R101 functions in a potential switch mechanism mediated by phosphorylation of the RXXS motif, we first set out to define the requirement for R101 and S104 in CARD9 signaling downstream of Dectin-1 in response to fungal stimuli. Accordingly, we ectopically expressed either full-length CARD9 (CARD FL) or CARD9 variants in Card9 knock-out (Card9−/−) bone marrow-derived dendritic cells (BMDCs). The risk variant for fungal infection, CARD9 R101C, completely ablated the cytokine response to heat killed Candida albicans (HKCA), heat killed dermatophyte Trichophyton rubrum (HKTR), and whole glucan particles (WGP) but not to LPS (Figures 1D, 1E, and S1A). Similarly, S104 was required for CARD9-dependent signaling, as S104N substitution, which is incapable of phosphorylation, ablated cytokine production downstream of Dectin-1 ligands (Figures 1D, 1E, and S1A). The same results were obtained using bone marrow-derived macrophages (BMDMs) (Figures S1B and S1C).
Figure 1. CARD9 linker residues R101 and S104 are required to elicit cytokine responses.
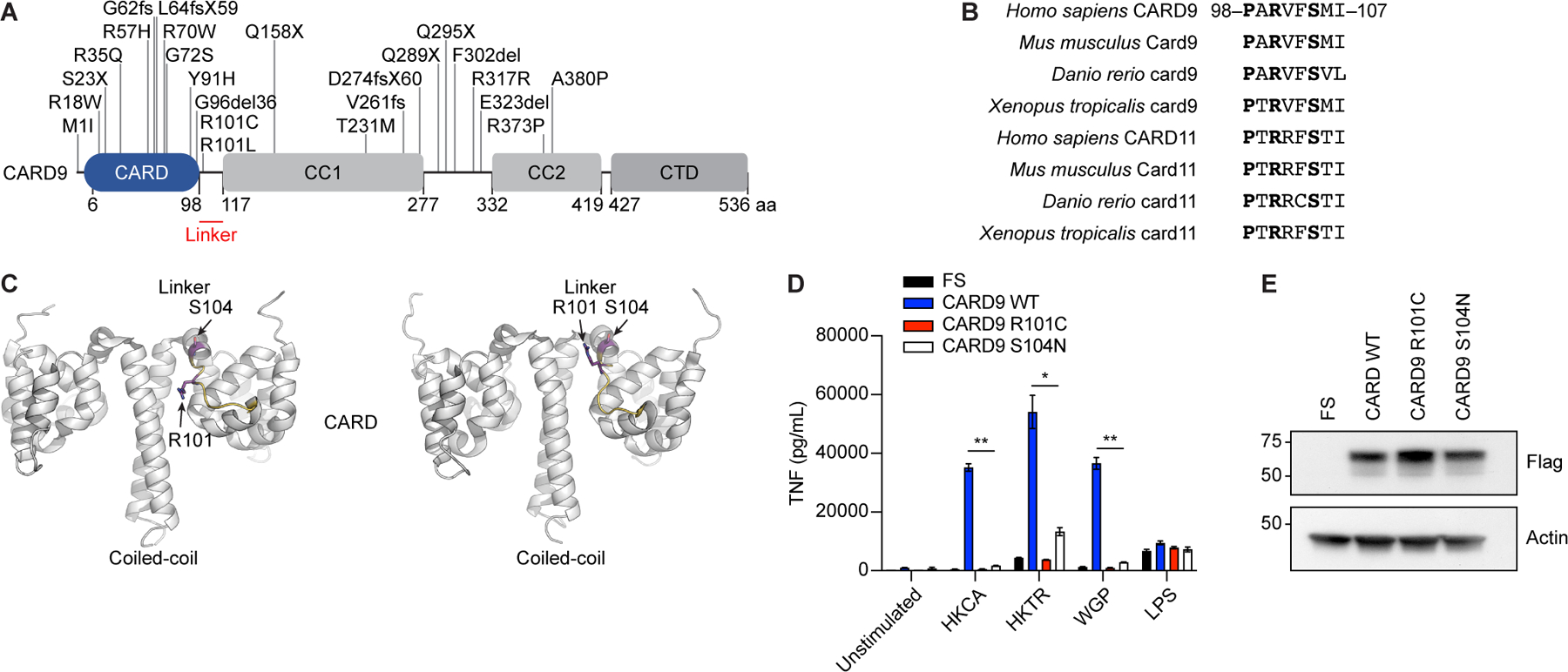
A, Schematic representation of known CARD9 genetic risk variants associated with susceptibility to fungal infection. Linker region is highlighted in red. CTD, C-terminal domain; aa, amino acids. B, Alignment of the linker region of CARD9 and CARD11 in higher vertebrates. C, Schematic representation of the dynamic CARD9 2–142 structure (PBD ID: 6N2M), projecting R101 and S104 locations in the linker region, visualized using PyMOL 2.4.0 (Schrödinger, LLC). D, Tnf concentration in the supernatant of Card9−/− murine BMDCs transduced with lentiviruses containing empty vector (FS), full-length CARD9 WT, CARD9 R101C, or CARD9 S104N and stimulated with HKCA (MOI 1:10), HKTR (MOI 1:10), WGP (50 μg/ml), or LPS (10 ng/ml) for 24 h. (n=3 mice per condition). E, Expression of CARD9 in the lysates from panel D determined by western blot with the indicated antibodies. Molecular weight markers (kDa) are shown to the left. Each experiment was repeated 3 times. Data represent mean +/− SEM. *p<0.05, **p<0.01 from paired t-tests. See also Figure S1.
Card9 activation through S104 phosphorylation is impaired by the R101C variant implicated in fungal disease
To directly assess the relationship between the CARD9 R101C risk variant and S104 phosphorylation, we generated a mouse model of Card9 R101C and a phospho-specific monoclonal antibody that recognizes phosphorylated S104. Analysis of BMDCs and splenic CD11c cells from wild-type (WT) and Card9 R101C mice confirmed unimpaired expression of Card9 protein (Figures S1D–S1G). Phospho-S104 (pS104) antibody specificity was verified by ELISA against different peptides, and we intentionally selected clones that recognize pS104 in the context of either R101 or C101 to determine if R101C substitution can impair phosphorylation of S104 (Figure S1H). We then used the phospho-S104 antibody to determine the phosphorylation state in primary cells upon stimulation with fungi by immunoprecipitating Card9 from either WT or Card9 R101C BMDCs. Following HKCA stimulation, we observed S104 phosphorylation in WT but not Card9 R101C BMDCs (Figure 2A). To determine the status of CBM complex formation following stimulation, we immunoprecipitated Bcl10 from either WT or Card9 R101C BMDCs. While we observed stimulation-dependent association of Bcl10 and Card9 in WT cells, this did not occur in Card9 R101C cells (Figure 2B). Consistent with these findings, we observed impaired Nfκb activation by evaluating phosphorylation and nuclear translocation of p65 in R101C BMDCs after stimulation with fungal ligands (Figures 2C and 2D). BMDCs from Card9 R101C mice also showed reduced production of cytokines following stimulation (Figures 2E and S1I). Both BMDMs and monocytes isolated from Card9 R101C mice also exhibited dampened responses to fungal stimulation (Figures S1J–S1M). Taken together, our results suggest that the R101C variant disrupts a critical kinase recognition motif, impairing S104 phosphorylation and subsequent activation of Card9 signaling, and results in a lack of immune response to fungal stimulation.
Figure 2. Card9 activation through S104 phosphorylation is impaired by the R101C variant implicated in fungal disease.
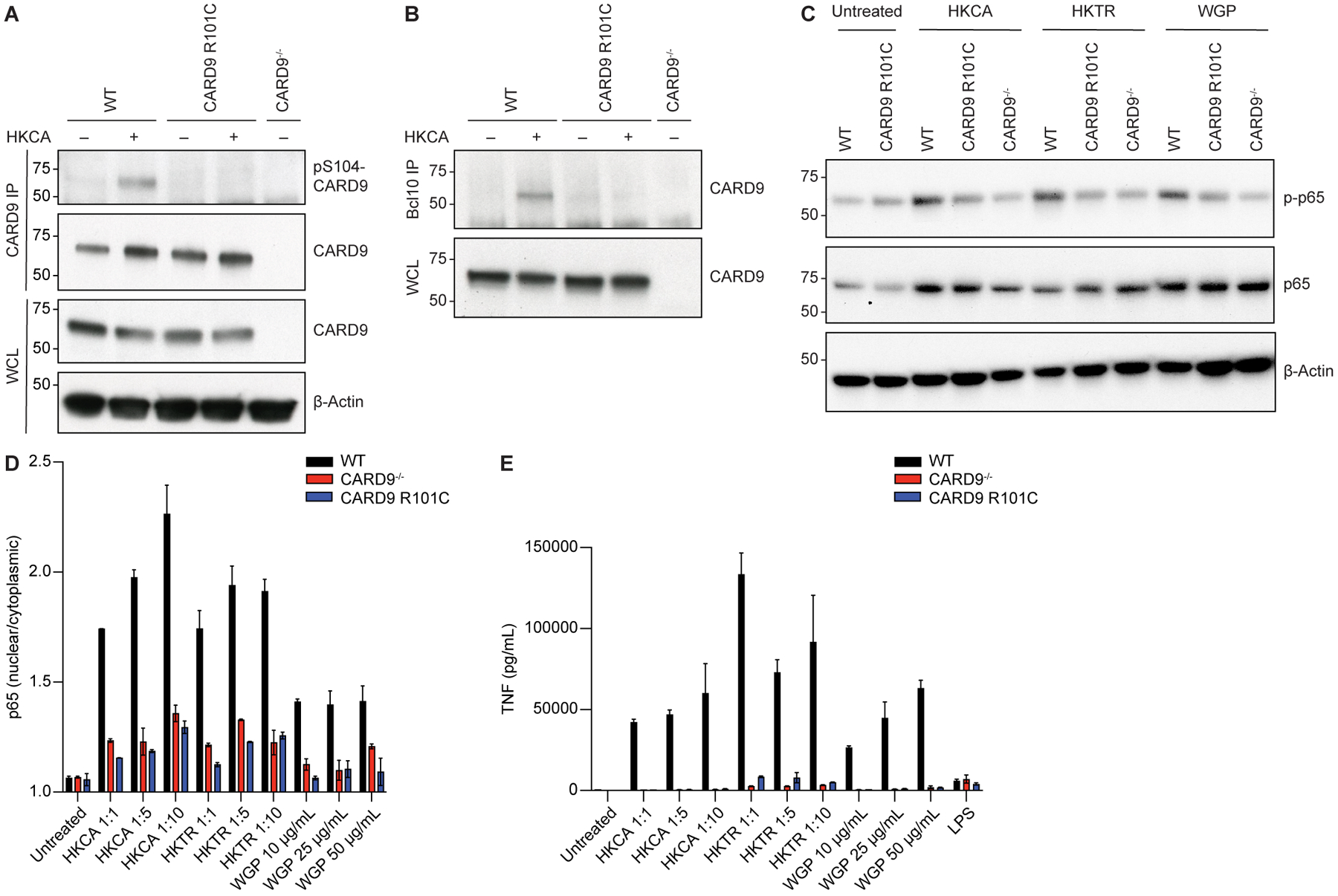
A, Card9 immunoprecipitate (IP) from WT, Card9 R101C, or Card9−/− BMDCs treated with HKCA (MOI 1:10) for 30 min. B, Bcl10 IP from WT, Card9 R101C, or Card9−/− BMDCs treated with HKCA (MOI 1:10) for 30 min. C, Representative western blot of WT, Card9 R101C, or Card9−/− BMDCs treated with HKCA (MOI 1:10), HKTR (MOI 1:10), or WGP (50 μg/ml) for 15 minutes. In A-C, molecular weight markers (kDa) are shown to the left; WCL, whole cell lysate. D, Nuclear p65 translocation in WT, Card9−/−, or Card9 R101C BMDCs treated with indicated doses of HKCA, HKTR, or WGP for 30 min. E, Tnf concentration in the supernatant from WT, Card9−/−, or Card9 R101C BMDCs treated with indicated doses of HKCA, HKTR, WGP, or LPS (10 ng/ml) for 24 h. (n=3 mice per condition). Each experiment was repeated 3 times. Data represent mean +/− SEM. See also Figure S1.
CARD9 R101 functions as a signaling switch activated by S104 phosphorylation
In order to identify the kinase that phosphorylates Card9 at S104, we performed a CRISPR screen by perturbing key kinases and then measuring cytokine production in response to Dectin-1 stimulation. We screened kinases that phosphorylate RXXS motifs24 and are expressed in BMDCs or were identified as Card9 interacting proteins12,25. We anticipated that this approach would identify kinases that phosphorylate Card9 in addition to any kinases acting upstream, downstream, or parallel to Card9. For example, we performed a knockout (KO) of the tyrosine kinase Syk as a positive control, because it is known to act upstream of Card9 and ablate cytokine production in response to Dectin-1 stimulation. Out of 18 additional RXXS kinases tested, CRISPR KO of Pak2 and Pkcδ resulted in IL-6 reduction in response to Dectin-1 stimulation (Figures 3A and S2A). To implicate the kinase that directly phosphorylates Card9 at S104, we performed secondary screening assays. We first evaluated p65 nuclear translocation, as this process is more proximal to Card9 activation, occurs on a time scale that correlates with S104 phosphorylation (Figures 2A and 2D), and is completely ablated by R101C mutation (Figure 2D). Only Pkcδ KO reduced p65 translocation to the nucleus, reflecting a reduction in Nfκb signaling following Dectin-1 stimulation (Figure 3B). Given that Pkcδ was previously shown to phosphorylate Card9 at T23120, we determined whether it can also phosphorylate S104. An in vitro phosphorylation assay demonstrated that Pkcδ does indeed phosphorylate S104, and that an intact RXXS motif in the linker domain is required for this phosphorylation but not for Card9 phosphorylation at TXR motifs like T231 (Figure 3C). Having shown that Pkcδ is sufficient to phosphorylate S104, we sought to formally demonstrate its requirement. Accordingly, both pharmacologic inhibition of Pkc with 10μM sotrastaurin and genetic KO of Pkcδ led to inhibition of Card9 S104 phosphorylation and activity in stimulated BMDCs (Figures 3D, 3E, S2B, and S2C).
Figure 3. CARD9 R101 functions as a signaling switch activated by S104 phosphorylation.
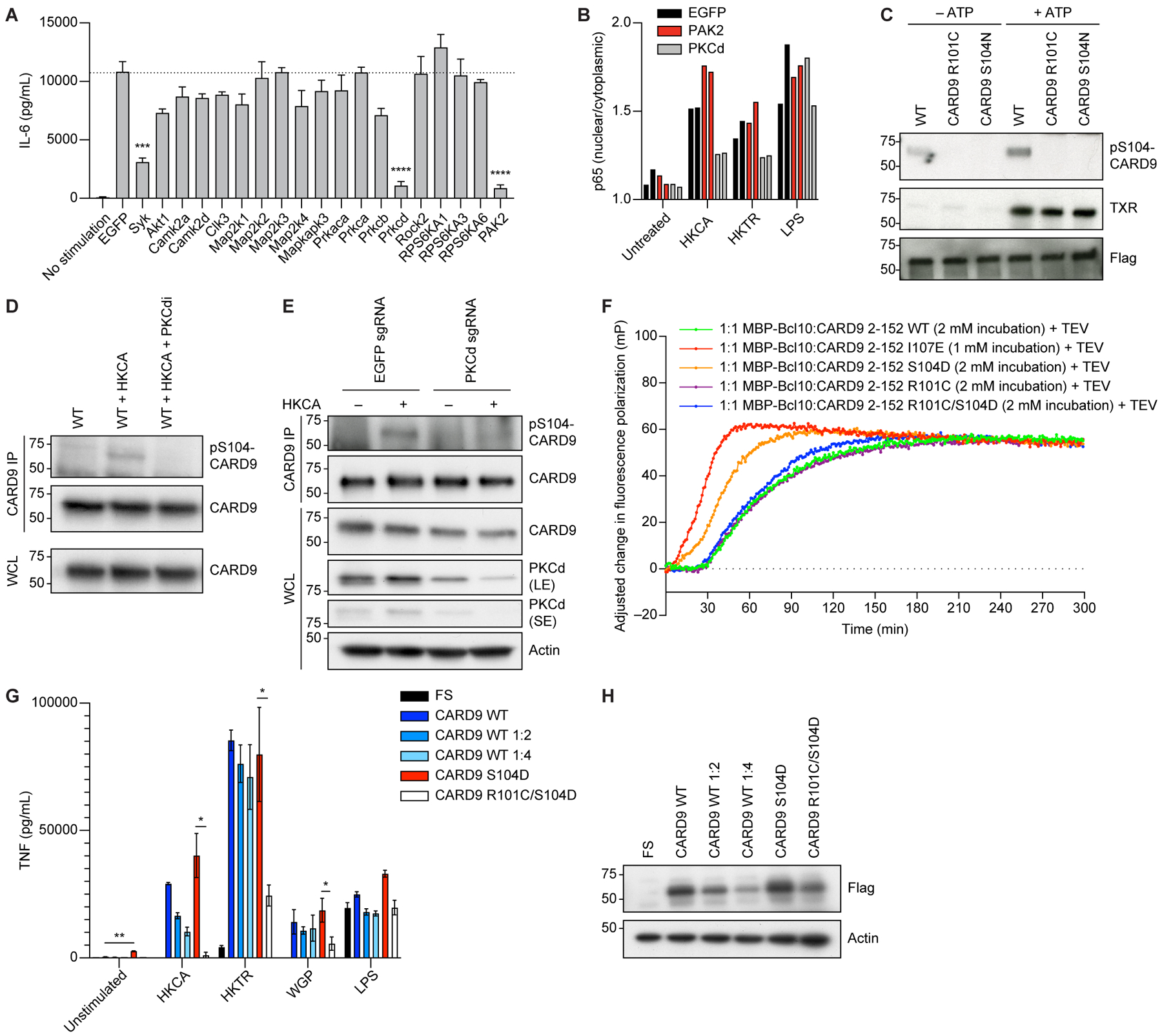
A, BMDCs expressing Cas9 were transduced with guides for the corresponding genes on day 2 and selected with puromycin. On day 9, cells were stimulated with 100 μg/ml WGP overnight, and Il-6 concentrations in the supernatant were measured with ELISA. B, BMDCs expressing Cas9 were transduced with guides for the corresponding genes on day 2 and selected with puromycin. On day 9, cells were stimulated with HKCA (MOI 1:10), HKTR (1:10), or LPS (10 ng/ml) for 30 min. p65 nuclear translocation was measured using fluorescence imaging. C, CARD9 WT, CARD9 R101C, or CARD9 S104N were overexpressed in HEK 293T cells. Following anti-flag pull-down, beads were incubated with purified PKCδ in the presence or absence of ATP and processed for western blotting with the indicated antibodies. D, WT BMDCs were treated with HKCA (MOI 1:10) or HKCA (MOI 1:10) plus the PKCδ inhibitor sotrastaurin (PKCdi) for 30 min and processed for immunoprecipitation with an anti-Card9 antibody. Samples were analyzed by western blot with the indicated antibodies. E, Control or Pkcδ KO BMDCs were treated with HKCA (MOI 1:10) for 30 min and processed for immunoprecipitation with an anti-Card9 antibody. Samples were analyzed by western blot with the indicated antibodies. LE, long exposure; SE, short exposure. F, Normalized fluorescence polarization representing filament formation between different CARD9 2–152 variants and maltose binding protein (MBP)-tagged BCL10 (average of quadruplicates). G, Tnf concentration in the supernatant from Card9−/− BMDCs transduced with lentiviruses containing either empty vector (FS), CARD9 WT, CARD9 WT diluted 1:2, CARD9 WT diluted 1:4, CARD9 S104D, or CARD9 R101C/S104D and stimulated with HKCA (MOI 1:10), HKTR (MOI 1:10), WGP (50 μg/ml), or LPS (10 ng/ml) for 24 h. H, Expression of CARD9 in the lysates from panel G determined by western blot with the indicated antibodies. In C-E and H, molecular weight markers (kDa) are shown to the left; IP, immunoprecipitate; WCL, whole cell lysate. Each experiment was repeated 3 times. Data represent mean +/− SEM. *p<0.05, **p<0.01, *** p < 0.001, **** p < 0.0001 from paired t-tests. See also Figure S2.
We next sought to further define how R101 and phosphorylation of S104 by PKCδ regulate CARD9 activation. Prior studies demonstrated that hydrophobic interactions between the CARD domain, linker region, and CC1 domain maintain CARD9 in an autoinhibited state21. Disruption of these interactions through mutagenesis (I107E) releases CARD9 from the autoinhibited state and induces NFκB activation as well as formation of BCL10-templating filaments21. We first tested if the phosphomimetic mutation S104D in CARD9 is sufficient to catalyze BCL10 filamentation. Employing a fluorescence polarization-based BCL10 polymerization assay using purified CARD9 and BCL10 proteins21,26,27, we demonstrated that WT CARD9 (amino acids 2–152) had no effect on the rate of BCL10 filamentation, whereas S104D accelerated the process (Figures 3F and S2D). Both variants were compared to CARD9 I107E that has been previously described to accelerate the filament formation21. Moreover, the CARD9 R101C/S104D double mutant had no effect on BCL10 polymerization, indicating that R101C overrides and prevents the activation of CARD9 by S104D (Figures 3F and S2D). However, CARD9 R101C/I107E was competent to accelerate filament formation, indicating that R101C does not completely incapacitate the protein, rather it can be activated by I107E-mediated release of the CARD domain (Figure S2E).
Having demonstrated the requirement for R101 and S104 phosphorylation in CARD9 signaling, we hypothesized that phosphomimetic mutation at the S104 site might promote or potentiate cytokine responses in a CARD9-dependent manner. Expression of CARD9 S104D in Card9−/− BMDCs led to increased baseline cytokine production in unstimulated cells without impairing Dectin-1 or LPS responses (Figures 3G, 3H, and S2F). To determine if S104 and R101 function cooperatively in cells, we generated an R101C/S104D double mutant and demonstrated that S104D was not sufficient to rescue the impaired cytokine production of R101C after fungal stimulation (Figures 3G, 3H, and S2F). Similar results were obtained when CARD9 S104D and R101C/S104D variants were expressed in Card9−/− BMDMs (Figures S1B and S1C). Overall, our data are consistent with a model in which phosphorylation at S104 causes a conformational switch allowing for the CARD domain to release from the coiled-coil domain and catalyze BCL10 polymerization for downstream cytokine production. This conformational switch in CARD9 requires arginine at position 101. Thus, the R101C mutation both disrupts the RXXS phosphorylation motif and disarms the switch mechanism that allows for the release of the CARD domain.
Card9 R101C mice are predisposed to systemic fungal infection
We next set out to determine how CARD9 R101C impacts global antifungal responses in vivo. Extensive immunophenotyping in Card9 R101C mice did not reveal any changes in immune cell subset distribution at steady state in the spleen, thymus, or bone marrow, and spontaneous development of fungal infection was not observed (Figures S3A–S3H). Next, we utilized a model of systemic candidiasis induced by intravenous infection. Mice harboring the R101C variant, similar to Card9−/− mice, showed severely reduced survival, with most of the mice succumbing to the C. albicans infection between days 2–4 post-infection (Figure 4A). Further analysis of the mice at day 2 (D2) post-infection revealed that Card9 R101C mice displayed 100 times higher fungal burden in the kidney and 10 times higher fungal burden in the brain than WT mice, which closely mimicked the phenotype observed in Card9−/− mice (Figure 4B). Histopathological analysis of both kidneys and brains obtained from infected mice showed significantly increased presence of fungal mats in Card9 R101C mice (Figures 4C–4F). Immunophenotyping of kidneys and brains demonstrated increased neutrophil infiltration in both organs from infected Card9 R101C mice and severe impairment of inflammatory monocyte infiltration in the brains of infected mutant mice (Figures 4G–4J, S3I–S3N). Additionally, brains from infected Card9 R101C mice showed decreases in the presence of dendritic cells and increases in eosinophil infiltration (Figures S3O and S3P). Spleens of infected Card9 R101C mice showed reductions in macrophage infiltration and in CD8a+ dendritic cells (Figures S3Q–S3W). Cytokine analysis demonstrated signs of increased systemic inflammation in infected Card9−/− and Card9 R101C mice as seen by Il-6 both in the serum and kidney (Figure 4K). Taken together, these data point to the importance of R101 in regulating the activity of Card9 and innate immunity in the context of systemic antifungal responses.
Figure 4. Card9 R101C mice are predisposed to systemic fungal infection.
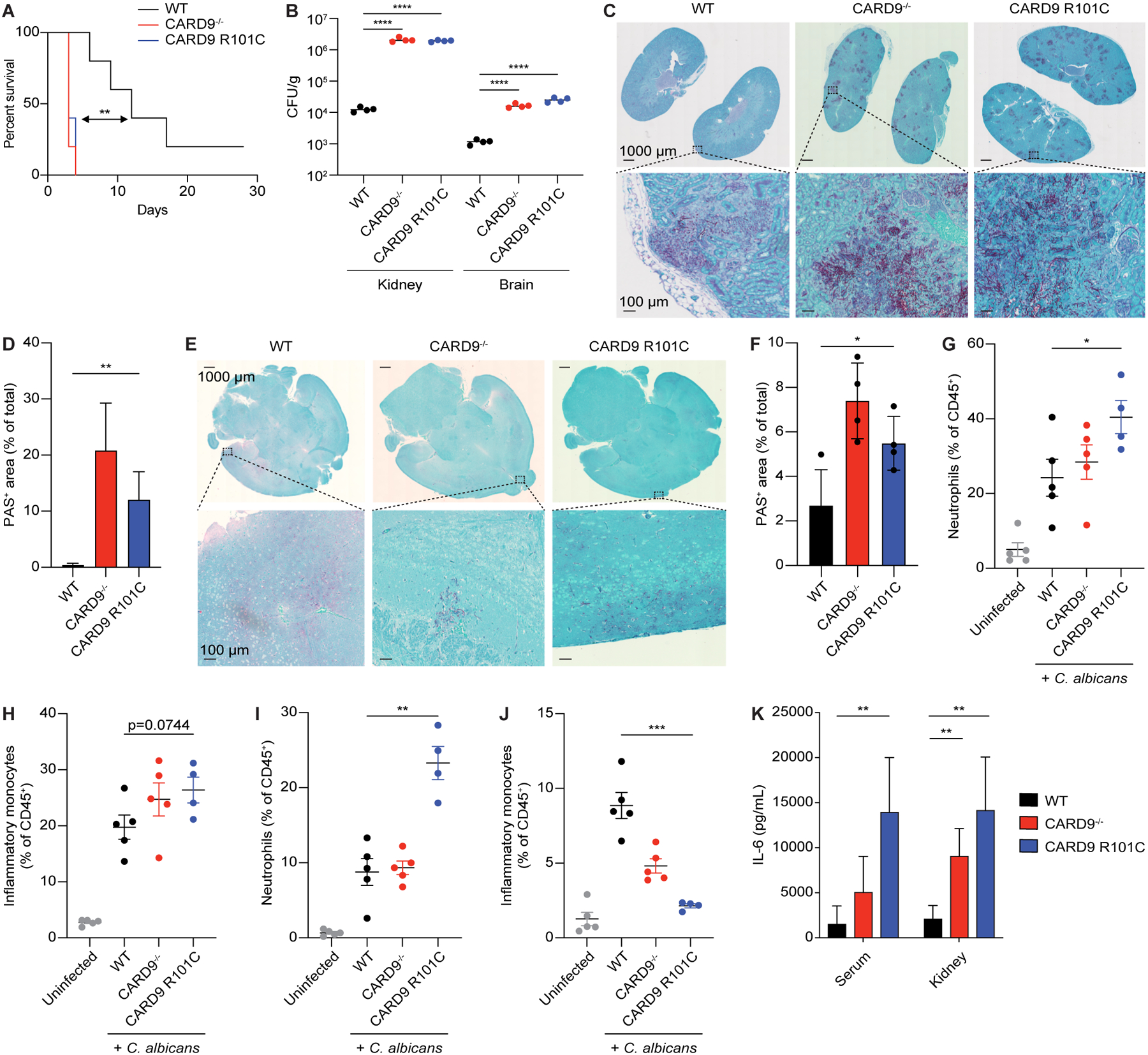
A, Survivorship over time of mice injected intravenously with live C. albicans (n=10 for each genotype). B, Colony forming units (CFUs) from the kidneys or brains of mice injected intravenously with live C. albicans analyzed on D2 post-infection (n=4 for each genotype). C, Representative PAS staining of kidneys from mice in panel B. D, Quantification of panel C. E, Representative PAS staining of brains from mice in panel B. F, Quantification of panel E. G-H, Flow cytometry analysis of G, neutrophil composition and H, inflammatory monocytes in the kidneys from mice in panel B. I-J, Flow cytometry analysis of I, neutrophil composition and J, inflammatory monocytes in the brains from mice in panel B. K, IL-6 concentrations in the serum and kidney from mice in panel B. Each experiment was repeated at least 2 times. Data represent mean +/− SEM. *p<0.05, **p<0.01,***p<0.001, ****p<0.0001 from paired t-tests. Scale bars represent 1000 μm or 100 μm. See also Figure S3.
Card9 R101C mutation impairs spore clearance in a mouse model of dermatophytosis
Given that CARD9 R101C has been linked to dermatophytosis from T. rubrum infection, we investigated the role of this mutation in the immune response to this pathogen using a mouse model of dermatophytosis23. Mice were injected intradermally with T. rubrum in the back skin and analyzed on D2 and day 9 (D9) post-infection. Card9 R101C mice showed a higher burden of fungal spores compared to WT mice, mimicking the phenotype observed in Card9−/− mice (Figure 5A). Histological analysis of harvested skin demonstrated a well-defined pyogranuloma containing mostly neutrophils, focal dermal fibrosis, and severe acanthosis in WT mice infected with T. rubrum, while formation of pyogranuloma and infiltration with immune cells was severely impaired in both Card9−/− and Card9 R101C mice (Figure 5B). After infection, focal accumulation of large vacuolated macrophages, neovascularization, and mild focal acanthosis with underlying focal dermal granulation and fibrosis sporadically presented in the skin of infected Card9−/− and Card9 R101C mice (Figure 5B). No differences in skin composition at steady state were observed between different genotypes. Analysis of the immune cell composition of infected skin revealed severe impairment in immune responses, with reduced infiltration of monocytes and neutrophils as well as decreased secretion of the neutrophil chemoattractant, Cxcl1 (Figures 5C–5F and S4A). Additionally, Card9 R101C mice demonstrated persistent infection at D9 post-infection, whereas live fungal organisms were not recoverable from WT mice (Figure 5G). Histological analysis of the skin revealed increased immune infiltration and presence of pyogranuloma in WT mice that was reduced in both Card9−/− and Card9 R101C mice (Figure 5H). Similar to what was observed for the early timepoint, at D9 post-infection, Card9 R101C showed impaired immune response demonstrated by lack of monocyte and neutrophil infiltration (Figures 5I–5L and S4A).
Figure 5. Card9 R101C mutation impairs spore clearance in a mouse model of dermatophytosis.
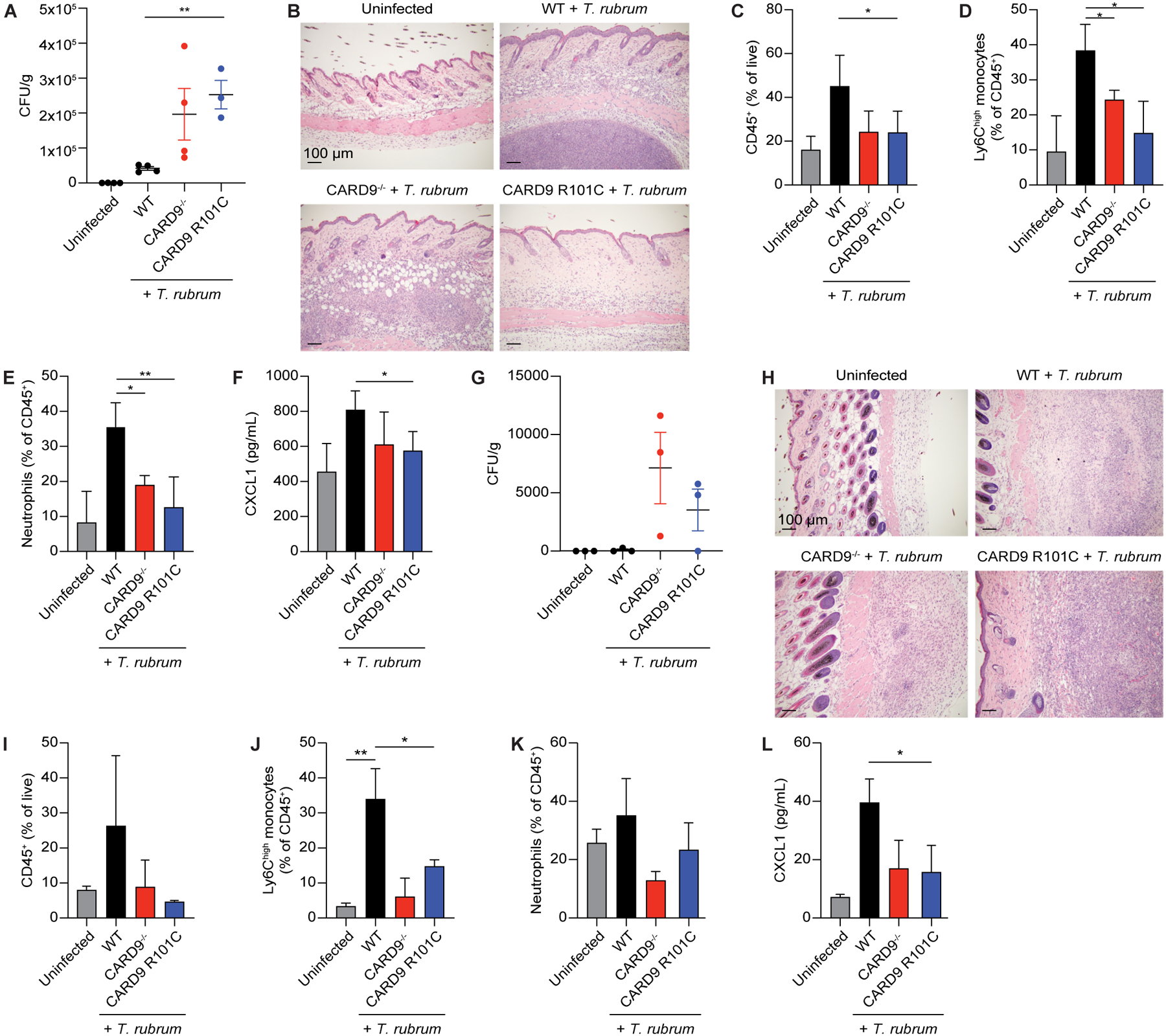
A, CFUs from back skin of mice injected intradermally with live T. rubrum analyzed on D2 post-infection (n=4 for each genotype). B, Representative H&E staining of back skin from mice in panel A. C-E, Flow cytometry analysis of C, total CD45+ cells, D, monocytes, and E, neutrophils in the back skin from mice in panel A. F, Cxcl1 concentration in the back skin from mice in panel A measured by ELISA. G, CFUs from the back skin of mice injected intradermally with live T. rubrum analyzed on D9 post-injection (n=4 for each genotype). H, Representative H&E staining of back skin from mice in panel G. I-K, Flow cytometry analysis of I, total CD45+ cells, J, monocytes, and K, neutrophils in the back skin from mice in panel G. L, Cxcl1 concentration in the back skin from mice in panel G measured by ELISA. Each experiment was repeated at least 2 times. Scale bars represent 100 μm. Data represent mean +/− SEM. *p<0.05, **p<0.01 from paired t-tests. See also Figure S4.
To further validate our findings, we implemented a foot pad model of dermatophytosis with transdermal injection of fungal spores into the foot pad. Similar to findings in the back skin infection model, Card9 R101C mice infected with T. rubrum showed impaired clearance of spores and lack of neutrophil infiltration into the injection site (Figures S4B–S4D). This recapitulated the response observed in Card9−/− mice. Together, these data indicate that Card9 R101 is essential for Card9-mediated activation of the immune response against fungal pathogens. Moreover, Card R101C mice exhibited a similar phenotype to Card9−/− mice, which accurately models the severe impairment in antifungal immunity observed in human patients with this rare variant.
Card9 R101C disrupts intercellular coordination through dynamic remodeling of immune, stromal, and keratinocyte cell states
Given the divergent inflammatory responses to fungi observed between WT and Card9 R101C mice, we sought to determine how Card9 R101C remodels skin immunity and cell states. In particular, we set out to explain the dichotomy between high fungal burden and reduced cellular infiltration and inflammation in Card9 R101C mice. We performed single-cell RNA-sequencing (scRNAseq) of skin from WT and Card9 R101C mice infected with T. rubrum at D2 and D9 post-infection. In total, we recovered 75,647 cells that yielded 17 clusters broadly partitioned into three compartments—immune cells, stromal cells, and keratinocytes, which broadly aligned with previously defined keratinocyte states28 (Figures 6A and S5A–S5G; Table S1). Together, these data comprise a comprehensive skin cell atlas. The prevalence of myeloid cells, in particular the Il1b-high monocyte cluster 1 and Gal3-high monocyte cluster 2, was altered in Card9 R101C skin (Figures 6B, S5A, and S5B; Table S2). Although Card9 expression was detected only in myeloid cells, and most highly in Langerhans cells (Figures S5C and S5H), we sought to investigate cell autonomous effects of Card9 on the skin immune response to fungi that may point to important crosstalk between immune cell types during infection. Differential expression analysis as well as pathway analysis between WT and R101C immune cells at D2 within each cluster revealed a broad reduction in expression of innate immune signaling genes in both myeloid cells and T cells (Figures 6C, 6D, and S6A–S6E; Tables S3 and S4). In contrast to R101C, WT cells exhibited upregulation of interferon-induced genes (Ifitm2/3 and Gbp2/5), Il1a/b, chemokines (Cxcl1/2/3), and Lcn2 across all myeloid populations and Th17 cells (Figures 6C, 6D, and S6A–S6E). In agreement with our flow cytometry data, we observed reduction in CD45+ cells expressing Ly6C markers, Tnf, Il6, and Cxcl1 in infected Card9 R101C skin (Figures S6F–S6N).
Figure 6. Card9 R101C impairs inflammatory signaling pathways and cell–cell communication circuitry in skin immune, stromal, and keratinocyte clusters.
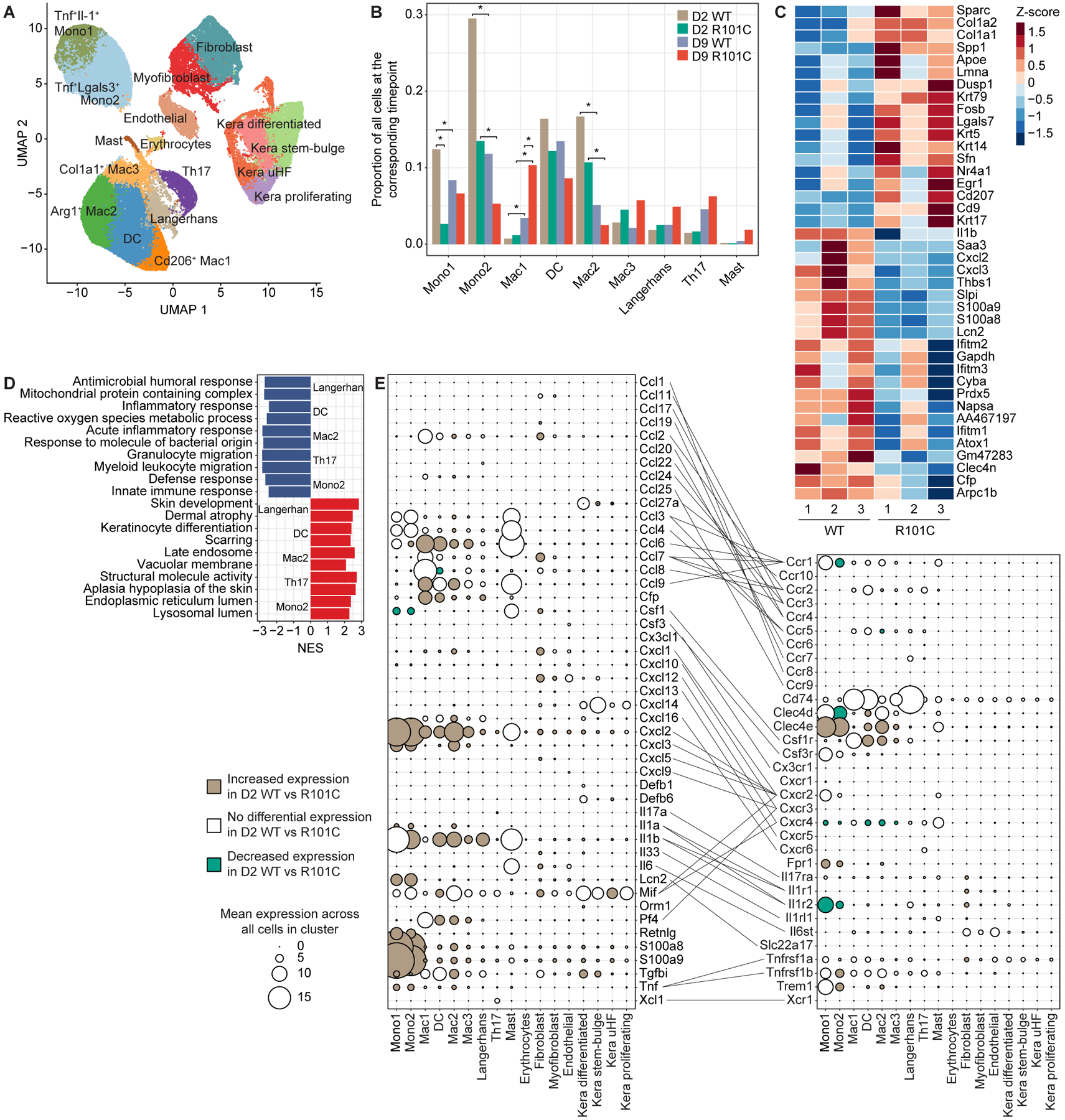
A, Clustering of cells based on expression obtained after scRNA-seq of Card9 WT and Card9 R101C skin analyzed on D2 and D9 post-infection with T. rubrum. n=6 for WT and n=6 for R101C (3 replicates of each WT and R101C at each timepoint). B, Prevalence of each immune cluster across Card9 genotypes and timepoints. Bar plots show the mean prevalence of each cluster across 3 replicates. For each cluster, 4 comparisons were conducted: D2 WT vs D2 R101C; D9 WT vs D9 R101C; D2 WT vs D9 WT; D2 R01C vs D9 R101C. Comparisons were conducted using Dirichlet multinomial regression and corrected for multiple testing using the Benjamini-Hochberg method. *FDR p < 0.05. C, Top 20 significant (FDR P < 0.05) DEGs between D2 WT and D2 R101C cells within the Langerhans cell cluster. D, Top pathways enriched in immune clusters based on DEGs between D2 WT cells and D2 R101C cells within each cluster. NES indicates normalized enrichment score from GSEA using DEGs between D2 WT cells and D2 R101C cells within each cluster; NES < 0 indicates upregulated in WT, NES > 0 indicates upregulated in R101C. E, Mean expression of cytokines, chemokines, effector molecules (left dot plot), and their corresponding receptors (right dot plot) in each cluster. Lines connect ligands to cognate receptors. For each molecule, the mean expression across all cells in each cluster regardless of Card9 genotype is shown. Increasing dot size indicates higher expression of the marker in the corresponding cluster. Colors indicate whether the molecule is significantly upregulated in D2 WT vs R101C cells in the corresponding cluster (beige), D2 R101C vs WT cells in the corresponding cluster (green), or not differentially expressed between D2 WT and R101C cells in the corresponding cluster (white). See also Figures S5 and S6 and Tables S1–S4.
To define the cell–cell communication circuits driving impaired monocyte recruitment and broad loss of inflammation in Card9 R101C mice, we first curated an extensive list of chemokines, cytokines, and their receptors. We then investigated their expression across all clusters, and whether their expression depends on Card9 genotype at D2 post-infection29. This analysis revealed a highly dynamic, Card9-dependent network of interactions within and between specific immune, stromal, and keratinocyte clusters at the early timepoint of infection (Figure 6E; Table S3). Upon infection, WT myeloid cells produced inflammatory mediators such as Il1, Tnf, Cxcl2, and Cxcl3 that both lead to recruitment of monocytes and neutrophils to the infected tissue as well as activation of stroma and keratinocytes. Activated fibroblasts and keratinocytes amplify the immune response by secreting chemokines such as Cxcl1, Cxcl2, Cxcl3, Cxcl12, and Cxcl14. In particular, Cxcl2, Cxcl3, and Il1b were expressed most highly in monocyte cluster 1 and were also upregulated in WT across most other clusters at D2, suggesting a key role in the antifungal immune response. Together, these data suggest that Card9 disrupts positive feedback loops comprised of myeloid cells that recruit monocytes and other immune cells primarily through the Il-1 and Cxcl2/Cxcr2 axes.
Despite the fact that stromal cells and keratinocytes do not express Card9 (Figure S5H), we observed dramatic differential expression of chemokines and receptors in these cell types in WT relative to R101C mice after infection, suggesting that Card9 exerts widespread cell non-autonomous effects on tissue immunity (Figure 6E). Consistent with our analysis of immune clusters, the top differentially expressed genes (DEGs) and pathways at D2 between WT and R101C cells in stromal and keratinocyte clusters were related to the innate immune response (Figures 7A–7D; Tables S3 and S4). Gene set enrichment analysis (GSEA) also indicated that oxidative stress and phosphorylation pathways were enriched in WT compared to Card9 R101C keratinocytes (Figures S7A–S7D; Tables S3–S5), which may reflect overall cell stress due to ongoing strong immune response. These data suggest that the primary cell non-autonomous consequence of Card9 R101C is the disruption of gene expression programs related to innate immunity in stromal cells and keratinocytes.
Figure 7. Cell non-autonomous effects of Card9 R101C.
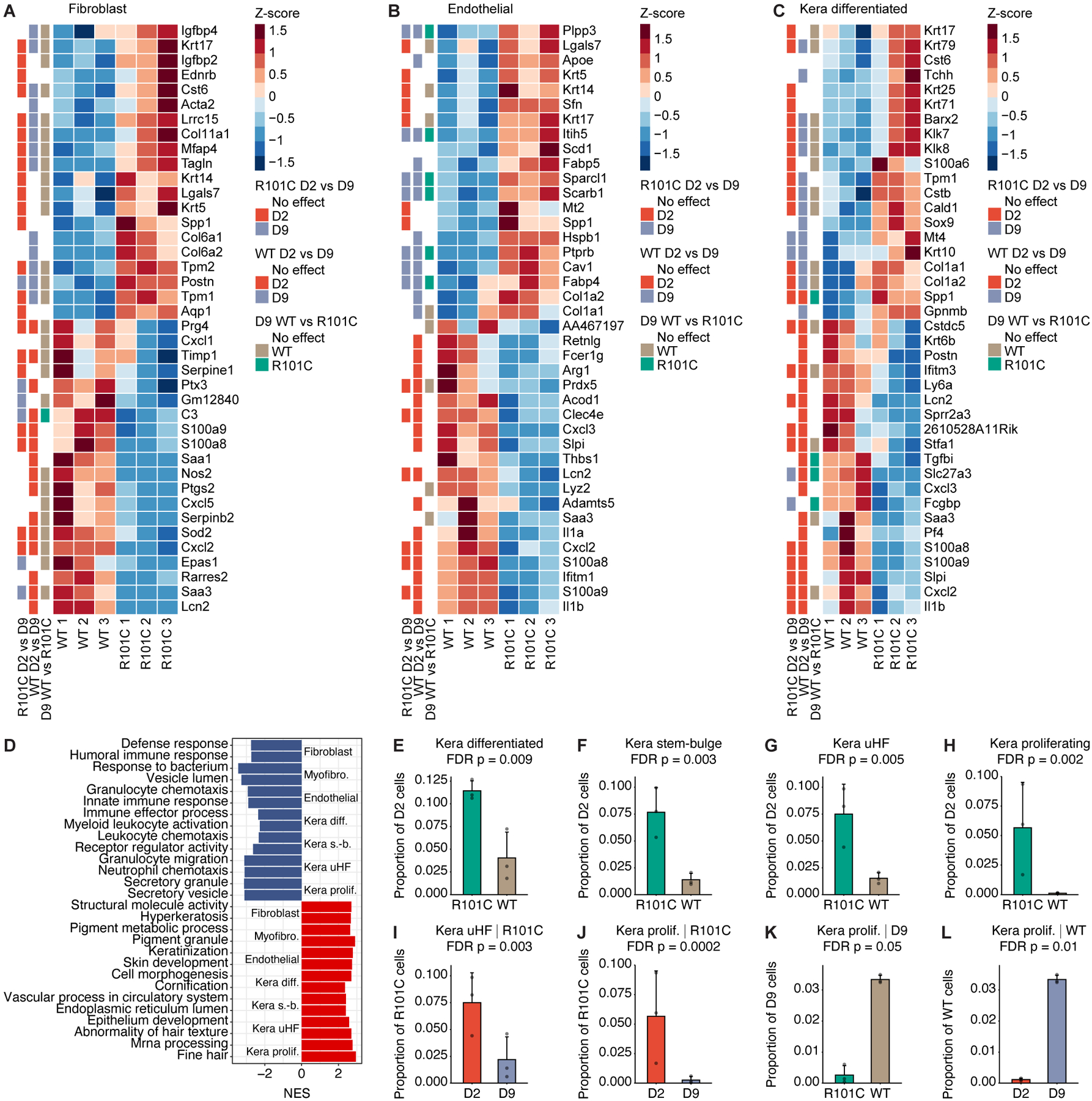
A-C, Heatmaps depict top 20 upregulated and downregulated genes between D2 WT and R101C cells in the A, fibroblast, B, endothelial, and C, kera differentiated clusters. Row annotations show whether the gene is significantly differentially expressed in the R101C D2 vs D9 comparison, WT D2 vs D9 comparison, and D9 WT vs R101C comparison. D, Top pathways enriched in stromal and keratinocyte clusters based on DEGs between D2 WT and R101C cells within each cluster. NES indicates normalized enrichment score from GSEA; NES < 0 indicates upregulated in WT, NES > 0 indicates upregulated in R101C. E-H, Changes in keratinocyte cluster frequency between D2 WT and R101C cells. Specifically, plots show proportion of all cells at D2 in each sample for keratinocyte clusters with significantly different (FDR P < 0.1; Dirichlet multinomial regression) prevalence in WT and R101C cells at D2. I-L, Changes in keratinocyte cluster frequencies between D2 and D9 and between Card9 genotypes. Specifically, plots show proportion of all cells at the indicated timepoint in each sample for keratinocyte clusters with significantly different (FDR P < 0.1; Dirichlet multinomial regression) prevalence in WT and R101C cells or between D2 and D9. Plots are shown for significant comparisons. See also Figure S7 and Tables S3–S6.
To further study the consequences of impaired immune response in keratinocytes, we first examined the prevalence of each keratinocyte cluster in our atlas across Card9 genotypes and timepoints. At D2 post-infection, all four keratinocyte clusters were expanded in skin from Card9 R101C mice relative to WT mice (Figures 7E–7H). Moreover, both the kera proliferating and kera uHF clusters were strongly enriched in Card9 R101C mice at D2 compared to D9 (Figures 7I and 7J; Table S6). In contrast, the proliferative keratinocyte response was delayed in WT mice (Figures 7K and 7L; Table S6), suggesting that high inflammation in WT cells may delay wound healing. Together, these data suggest that Card9 R101C promotes an early state-specific, proliferative keratinocyte response.
To identify changes in keratinocyte cell state independent of cluster prevalence, we also examined specific keratinocyte DEGs related to wound healing across timepoints. While some wound healing genes such as the collagen and keratin family members were upregulated both at D2 and D9 in keratinocytes (Figures S7E–S7I), others—such as the connexin Gja1 (Cx43), previously shown to regulate keratinocyte proliferation in skin wound healing—were induced early in R101C keratinocytes but later in WT keratinocytes. Thus, the cell non-autonomous effects of Card9 R101C include disruption of wound healing and inflammation in keratinocytes across multiple timepoints of infection.
Taken together, our analyses suggest that Card9 R101C disrupts fungal sensing, wound healing, and inflammatory responses, and thus may result in the perception of sterile tissue damage that elicits a distinct response in Card9 R101C relative to WT contexts. More broadly, we showed that through both cell autonomous and non-autonomous pathways, Card9 R101C disrupts an intercellular communication network that amplifies chemokine-driven recruitment of effector immune cells essential for resolution of the infection.
Discussion
Naturally occurring genetic variants provide opportunities to learn about protein functions and modes of regulation that would be challenging to uncover by directed mutagenesis. Immunodeficiencies associated with infectious diseases manifest in distinct phenotypes, making it easier to identify predisposing loss-of-function variants. CARD9 is a prime example demonstrating how rare variants can result in severe fungal infections30. To identify additional modes of regulation of anti-fungal responses, and in particular CARD9 signaling, we analyzed known CARD9 genetic variants. We focused specifically on the R101C mutation located in the linker region of the protein, hypothesizing based on structural modeling that the linker may be crucial for CARD9 activation and CBM complex formation. Indeed, we observed that introducing the R101C variant resulted in a loss-of-function phenotype in primary cells without affecting protein expression and stability. Additionally, we provided evidence that arginine to cysteine substitution at the 101 position disables an activating signaling switch mediated by phosphorylation of the neighboring S104 within a minimal kinase recognition motif RXXS, which is essential for CARD9 activation.
Structural models indicate that CARD9 adopts an autoinhibited multimeric conformation. Truncated CARD9 containing the CARD and CC1 domains adopts a dimeric form21, and we observed that full-length CARD9, with CARD, CC1, CC2, and C-terminal domains, exists as a tetrameric dimer of dimers. Still, several questions remain regarding how CARD9 is released from an autoinhibited state to form CARD9 filaments that template BCL10 polymerization and assembly of the signaling-competent CBM complex to activate NFκB. The interface of the CARD domain with the linker and CC1 domain is mediated through hydrophobic interactions and aromatic packing, including multiple amino acids of both the CARD and CC1 regions, with the linker acting as a key switch region21. Disruption of the interface frees the CARD domain, providing a platform for subsequent filament formation21. We demonstrated that S104D phosphomimetic mutation accelerated BCL10 filamentation in vitro, presumably by freeing the CARD domain. Importantly, S104D only induced BCL10 filamentation when position 101 was arginine, suggesting that S104 phosphorylation and R101 operate together to activate CARD9.
Along with our results, previous research has identified point mutations that can disrupt the CARD-CC1 interaction, leading to robust activation of NFκB in an overexpression system21. Structural models of the autoinhibited CARD9 dimer derived from NMR depict multiple conformations of the R101 sidechain such that it is located pointing downwards between the CARD and CC1 domains or upwards aligned with the side chain of S104 in the linker domain21. In this context, our results suggest a model for how phosphorylation at S104 may cooperate with R101 to actuate a switch mechanism. We propose that S104 phosphorylation may initiate engagement of the negatively charged phosphate on S104 with the positively charged R101 sidechain, thus rotating R101 out of the CARD-CC1 interface and disrupting the autoinhibited conformation to allow for templating BCL10 polymerization. This model suggests the possibility of developing inhibitors that function as intermolecular glues to lock the autoinhibited CARD9 complex in the off state, thus disabling the R101/S104 switch mechanism.
Our work demonstrating CARD9 regulation by S104 phosphorylation complements prior studies functionally characterizing polymorphisms in CARD9 implicated in immunodeficiency or autoimmunity30–32. While most variants create hypomorphic or null alleles, several others provided key insights into the modes of CARD9 regulation. One example is the R70W variant that disrupts assembly of CARD filaments21,33. Our work suggests that the R101C variant disrupts the R101 switch mechanism triggered by S104 phosphorylation, which may be an initiating event that promotes filamentation, and the R70W variant acts at a later stage in the process preventing proper assembly of filaments. Another variant associated with fungal infection disrupts a critical phosphorylation site in CARD9 at position T231, which links CARD9 activation through PKCδ9,20. In the context of our work, this suggests that PKCδ may phosphorylate S104 to activate the R101 switch and T231 to promote dissociation of coiled-coil domains within the autoinhibited multimer, together allowing for the assembly of CARD9 into filaments. Additional PTMs on CARD9, such as ubiquitination on K125, may further promote templating BCL10 polymerization by disruption of the autoinhibited CARD9 multimer or perhaps more likely by stabilizing the assembled filaments and promoting recruitment of other signaling proteins necessary for NFκB activation12.
Efforts to decipher mechanisms of CARD9 variants have been fruitful, yet many questions remain. For example, the missense variant CARD9 S12N was modeled in mice and implicated in regulating type 2 immune responses and fungal immunity34. Although prior GWAS implicated S12N (rs4077515) in IBD and other autoimmune diseases14,15,35,36, this variant has been shown to be in linkage disequilibrium with additional SNPs upstream of Card9 that are associated with more significant risk of IBD37,38 and upregulated CARD9 expression15 (GTEx Portal on 10/03/2023 and/or dbGaP accession number phs000424.vN.pN on 10/02/2023). Deeper functional dissection of SNPs in this locus is warranted in order to fully understand the mechanisms responsible for IBD risk in this complex Card9 haplotype. Future work delineating the sequence of events, such as PTMs and conformational changes leading to CARD9 activation and CBM complex formation, holds promise for guiding therapeutic development in autoimmune diseases associated with Card9.
Functional genetics can reveal mechanisms of CARD9 regulation and also provide an opportunity to connect those mechanisms to clinical phenotypes through deeper characterization of immune regulation in vivo. In order to better mimic the clinical phenotypes linked to CARD9 deficiencies, we applied two distinct models to study the R101C variant. We observed extreme susceptibility to systemic C. albicans infection with fungal overgrowth in the brain and kidney as well as systemic inflammation, a phenotype that reflects clinical disease associated with multiple Card9 variants. As the CARD9 R101C missense variant has been described in connection with deep dermatophytosis from T. rubrum infection, we also implemented a mouse model of dermatophytosis23. In this model, Card9 R101C was associated with a lack of monocyte infiltration in the skin as well as reduced innate immune activation of Card9-expressing myeloid cells. Furthermore, we demonstrate that cell-intrinsic expression of Card9 is not required for it to exert broad effects on skin cell states. The cell non-autonomous impact of Card9 was exemplified by pronounced cell state changes (i.e. DEGs) and corruption of wound healing and resolution of inflammation.
We observed Card9-dependent cell non-autonomous effects on stromal cells and keratinocytes, which express chemoattractants and cytokines essential for recruitment of immune cells into inflamed tissue sites. Indeed, recent studies have highlighted the importance of nonimmune cells in the regulation of all phases of the immune response39. In the case of ulcerative colitis, oncostatin M from myeloid cells acts on stromal cells to potentiate chemokine secretion in a manner that contributes to anti-TNF treatment resistance40. Along similar lines, abnormal activation of IL-1 signaling circuits in the inflamed intestine drives neutrophil recruitment through a cellular network associated with resistance to anti-TNF therapy41. Both mechanisms provide insights into unique disease pathotypes that disrupt tissue homeostasis. Similarly, we identified a complex cellular network in the skin that provides protective tissue immunity against fungal infection. Applying this translational genetics approach to other innate immune signaling pathways may deepen our understanding of the crosstalk between immune and non-immune cells and inform the design of targeted treatments for immune-mediated diseases.
Limitations of the Study
Here, we identify mechanisms by which the CARD9 R101C variant disrupts signaling downstream of pathogen recognition receptors, and more broadly, how this variant impairs tissue-level immunity to dermatophyte infection. Specifically, we uncovered a cell autonomous role for CARD9 in fungal detection by skin resident myeloid cells and a cell non-autonomous role for CARD9 in coordinating a chemokine amplification loop mediated by stromal cells. Further work will be required to dissect the ligand–receptor pairs that mediate this complex cellular network controlled by CARD9. Tissue- and cell type-specific models will also be informative to further define the cascade of events during the immune response. Nevertheless, key insights were gained from studying a mouse model of the CARD9 R101C variant. Mechanistically, our results support a model in which R101 functions as a switch that is stabilized in the open configuration when S104 is phosphorylated by PKCδ. Future structural biology efforts will be revealing in determining precisely how S104 phosphorylation changes the conformational dynamics of CARD9, and how these events promote assembly of a competent signalosome. Understanding the biochemical mechanisms controlling CARD9 activation has potential value in drug discovery, as a dominant negative CARD9 variant has been linked to protection from autoimmunity, suggesting a rationale for developing inhibitors.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ramnik J. Xavier (rxavier@broadinstitute.org).
Materials availability
Card9 R101C knock-in mice and the phospho-S104 Card9 monoclonal antibody are available from the lead contact upon request.
Data and code availability
Raw fastq files for the scRNAseq experiment and a processed Seurat object with cell metadata have been deposited at GEO and are publicly available as of the date of publication under accession GSE210224. Accession numbers are additionally listed in the Key Resources Table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| AF488-CD19 | BioLegend | Cat. # 115521 |
| PE/DZ594-CD23 | BioLegend | Cat. # 101634 |
| BV421-CD21/35 | BioLegend | Cat. # 123421 |
| BUV395-CD93 | BD Biosciences | Cat. # 745595 |
| BV785-CD11b | BioLegend | Cat. # 101243 |
| APC Cy7-F4/80 | BioLegend | Cat. # 123118 |
| AF647-Ly6G | BioLegend | Cat. # 127609 |
| BV650-CD11c | BioLegend | Cat. # 117339 |
| PE Cy7-CD3e | BioLegend | Cat. # 100320 |
| PE Cy5-CD122 | BioLegend | Cat. # 123219 |
| BV510-CD8α | BioLegend | Cat. # 100752 |
| PE-CD4 | BioLegend | Cat. # 100512 |
| AF700-CD45 | BioLegend | Cat. # 103128 |
| BV605-IA/IE | BD Horizon | Cat. # 563413 |
| BUV661-CD5 | BD Biosciences | Cat. # 741459 |
| BUV395-CD4 | BD Biosciences | Cat. # 563790 |
| PacBlu-CD25 | BioLegend | Cat. # 102022 |
| PE-CD44 | BioLegend | Cat. # 103008 |
| APC Cy7-CD117 | BioLegend | Cat. # 105826 |
| PE Cy7-γδTCR | BioLegend | Cat. # 118124 |
| APC-TCRβ | BioLegend | Cat. # 109212 |
| FITC-NK1.1 | BioLegend | Cat. # 108717 |
| FITC-TER119 | BioLegend | Cat. # 116206 |
| BV605-B220 | BioLegend | Cat. # 103244 |
| PE Cy7-CD43 | BioLegend | Cat. # 143210 |
| PE-CD24 | BioLegend | Cat. # 138503 |
| APC-BP1 | eBioscence | Cat. # 17-5891-82 |
| BV421-IgM | BioLegend | Cat. # 406517 |
| BUV395-IgD | BD Horizon | Cat. # 564274 |
| BV650-CD150 | BioLegend | Cat. # 115932 |
| AF700-CD48 | BioLegend | Cat. # 103426 |
| PE Cy5-CD34 | BioLegend | Cat. # 119312 |
| FITC-CD16/32 | BioLegend | Cat. # 101305 |
| BV785-SCA1 | BioLegend | Cat. # 108139 |
| BV711-F4/80 | BioLegend | Cat. # 123147 |
| BV480-CD117 | BD Biosciences | Cat. # 566074 |
| BUV496-CD123 | BD Biosciences | Cat. # 750813 |
| BV570- CD90.2 | BioLegend | Cat. # 105329 |
| PE CF594-Siglec-F | BD Biosciences | Cat. # 562757 |
| PE Cy5 -CD3 | BioLegend | Cat. # 100310 |
| PE-CD103 | BioLegend | Cat. # 121406 |
| eFluor450-Ly6C | Thermo Fisher Scientific | Cat. # 48-5932-82 |
| BV605-Ly6G | BioLegend | Cat. # 127639 |
| BUV805-CD45 | BD Biosciences | Cat. # 748370 |
| PE Cy7-CD64 | BioLegend | Cat. # 139314 |
| BUV737-CD19 | BD Biosciences | Cat. # 612781 |
| AF700-CD8b | BioLegend | Cat. # 126618 |
| AF647-IgA | SouthernBiotech | Cat. # 1040–31 |
| BUV563-IA/IE | BD Biosciences | Cat. # 748846 |
| FITC-EpCAM | BioLegend | Cat. # 118207 |
| BV785-CD45 | BioLegend | Cat. # 103149 |
| AF647-CD64 | BioLegend | Cat. # 139322 |
| BV650-Ly6G | BioLegend | Cat. # 127641 |
| PerCP-Cy5.5-Ly6C | BioLegend | Cat. # 128011 |
| BV421-CD11b | BioLegend | Cat. # 101235 |
| PE-Cy7-CD11c | BioLegend | Cat. # 117339 |
| AF700-IA/IE | BioLegend | Cat. # 107621 |
| Biotin-CD11c | BioLegend | Cat. # 117304 |
| CARD9 | Cell Signaling Technology | Cat. # 12283S |
| Bcl10 | Santa Cruz Biotechnology | Cat. # sc-5273 |
| CARD9 | Santa Cruz Biotechnology | Cat. # sc-374569 |
| p-p65 | Cell Signaling Technology | Cat. # 3033S |
| p65 | Cell Signaling Technology | Cat. # 8242S |
| Flag M2 | Sigma Millipore | Cat. # F1894 |
| pTXR | Cell Signaling Technology | Cat. # 2351S |
| PKCd | Cell Signaling Technology | Cat. # 9616 |
| pS104-Card9 | This study | This study |
| p65 | Santa Cruz Biotechnology | Cat. # sc-8008 |
| Anti-rabbit HRP | Daco | Cat. # P0448 |
| Anti-mouse HRP | Daco | Cat. # P0447 |
| AF488 anti-mouse | Invitrogen | Cat. # A-21202 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Sabouraud dextrose agar | Thermo Fisher Scientific | Cat. # BD210950 |
| Remel Dermatophyte test medium | Thermo Fisher Scientific | Cat. # R01365 |
| 1X RBC Lysis Buffer | Thermo Fisher Scientific | Cat. # 00433357 |
| Liberase | Roche | Cat. # 385040 |
| DNAse I | Roche | Cat. # 10104159001 |
| ViaKrome Live/Dead dye | Beckman Coulter | Cat. # C36628 |
| Zombie UV Live/Dead | BioLegend | Cat. # 423108 |
| DMEM | Thermo Fisher Scientific | Cat. # 10569044 |
| RPMI with glutamax | Life Technologies | Cat. # 72400–120 |
| GM-CSF | PeproTech | Cat. # 315–03 |
| M-CSF | PeproTech | Cat. # 315–02 |
| IL-3 | PeproTech | Cat. # 213–13 |
| Lipofectamine 3000 reagent | Thermo Fisher Scientific | Cat. # L3000–015 |
| Cas9 | IDT | Cat. # 1081059 |
| YPD broth | Thermo Fisher Scientific | Cat. # DF0428-07-7 |
| WGP | Invivogen | Cat. # tltl-wgp |
| LPS | Invivogen | Cat. # tlrl-peklps |
| Sotrastaurin | Selleckchem | Cat. # S2791 |
| 1x Halt Protease & Phosphatase Inhibitor Single-Use Cocktail | Thermo Fisher Scientific | Cat. # 78442 |
| Pierce Universal Nuclease | Thermo Fisher Scientific | Cat. # 88701 |
| Dynabeads Protein G beads | Life Technologies | Cat. # 10004D |
| Laemmli buffer | BioRad | Cat. # 1610737 |
| Western Lightning Chemiluminescence Reagent | Perkin Elmer Life Sciences | Cat. # NEL104001EA |
| 4% PFA | Thermo Scientific | Cat. # J19943-K2 |
| 1x Hoechst 33342 | Life Technologies | Cat. # H3570 |
| RIPA buffer | Boston BioProducts | Cat. # BP-115 |
| MagStrep XT beads | Fisher Scientific | Cat. # NC0776437 |
| Sulfo-SMCC | Thermo/Fisher | Cat. # 22322 |
| activated KLH | Thermo/Fisher | Cat. # 77600 |
| activated BSA | Thermo/Fisher | Cat. # 77110 |
| Protein G | Cytiva Life Sciences | Cat. # P-00067 |
| Immobilon-P membranes | Millipore Sigma | Cat. # IPVH08130 |
| Tris-Glycince-SDS-Running buffer (10x) | Boston BioProducts | Cat. # BP-150 |
| Transfer buffer (10x) | Boston BioProducts | Cat. # BP-190 |
| Tris Buffered Saline-Tween (20x, with 1% Tween-20, pH 7.4) | Boston BioProducts | Cat. # IBB-181X |
| 4–20% Bio-Rad Mini-PROTEAN TGX Stain-Free Gels | BioRad | Cat. # 4568095 |
| β-mercaptoethanol | Sigma | Cat. # M3148–25ML |
| EDTA | Invitrogen | Cat. # 15575–038 |
| LS columns | Miltenyi Biotech | Cat. # 130-042-401 |
| Brilliant Stain Buffer | BD Biosciences | Cat. # 566349 |
| Critical commercial assays | ||
| CXCL1 ELISA | Thermo Fisher Scientific | Cat. # EMCXCL1 |
| Monocyte isolation kit | Miltenyi Biotech | Cat. # 130-100-629 |
| P3 Primary Cell 4D-Nucleofactor X kit S | Lonza | Cat. # V4XP-3032 |
| BD Cytometric Bead Array (CBA) Mouse IL-6 Flex Set | BD Biosciences | Cat. # 558301 |
| BD Cytometric Bead Array (CBA) Mouse TNFa Flex Set | BD Biosciences | Cat. # 558299 |
| PKCd Kinase Enzyme System | Promega | Cat. # V3401 |
| Chromium Single Cell 3’ Gene Expression kit | 10x Genomics | Cat. # PN-1000120 |
| Deposited data | ||
| Single-cell RNA-sequencing data | This study | GSE210224; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE210224 |
| Experimental models: Cell lines | ||
| HEK293T | Lab strain, mycoplasma free | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: Card9−/− | Hara et al.17 | N/A |
| Mouse: Card9R101C | This study | N/A |
| Fungi: Candida albicans strain SC5314 | Jatin Vyas Lab (Massachusetts General Hospital) | N/A |
| Fungi: Trichophyton rubrum | Paul Verweij (Radboud University Medical Centre, Netherlands) | Clinical isolate |
| Oligonucleotides | ||
| sgRNA sequences are provided in Table S7 | IDT | N/A |
| Recombinant DNA | ||
| pMD601-CARD92–152/WT | This study | N/A |
| pMD602-CARD92–152/I107E | This study | N/A |
| pMD604- CARD92–152/S104D | This study | N/A |
| pMD605-CARD92–152/R101C | This study | N/A |
| pMD606-CARD92–152/R101C/S104D | This study | N/A |
| pMD613-Bcl10Fl/C29A/C57A | This study | N/A |
| pCDH-CMV | Addgene | Cat. # 72265 |
| pCDH-CMV-CARD9 | This study | N/A |
| pCDH-CMV-CARD9R101C | This study | N/A |
| pCDH-CMV-CARD9S104N | This study | N/A |
| pCDH-CMV-CARD9S104D | This study | N/A |
| Software and algorithms | ||
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com/ |
| FlowJo | FlowJo | https://www.flowjo.com/ |
| Harmony | Perkin Elmer | N/A |
| Cell Ranger v.6.0.0 | 10x Genomics | N/A |
| Seurat v4.0.1 | Hao et al.44 | N/A |
| Other | ||
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mice
All animal studies complied with relevant ethical regulations and were conducted according to protocols #2003N000158 and #2008N000078 approved by the Institutional Animal Care and Use Committee (IACUC) at Massachusetts General Hospital (MGH). Card9−/− mice were described previously17. Card9R101C CRISPR knock-in mice were generated on the C57BL/6 background for the purpose of this study. Mice were maintained in specific-pathogen free conditions and co-housed in the same room in the animal facility at MGH with a 12-hour light:dark cycle, room temperature of 21°C, and relative humidity of 30–70%. All experiments were performed using male and female mice aged 12–18 weeks. Mice were age- and sex-matched in each experiment, and littermates were used. The number of mice used in each experiment is provided in the figure legends.
Candida albicans culture and infection
C. albicans (strain ATCC SC5314) was grown on yeast-peptone-dextrose (YPD; 1% yeast extract, 2% bactopeptone, 2% glucose, 2% agar) at 30°C. For each infection, a fresh colony was picked or inoculated directly from frozen stocks and grown in YPD broth overnight at 30°C with agitation. Prior to infection, fungi were washed three times with PBS, counted using a LUNA™ Automated Cell Counter (Logos Biosystems, Annandale, VA), and diluted in PBS to 7.5 × 105 yeast/ml. Each mouse was infected with 200 μl of the suspension (150,000 yeast) via lateral tail vein injection. Control mice were infected with PBS alone. Mice were monitored daily for the duration of the experiment as per MGH IACUC protocol #2008N000078. Mice were assessed based upon a 12-point scale to determine when an individual animal should be euthanized due to illness and removed from the study. This was developed in conjunction with the MGH IACUC and includes the following symptoms and points assigned to each in parentheses: hunched posture (3), ruffled and/or matted fur (3), shivering (3), abnormal breathing (increased respiratory rate) (12), 75% reduction in activity compared to controls (3), inactivity leading to inability to acquire food or water (12), and barrel rolling (12). If any animal reaches 12 points, they are humanely euthanized and counted as a death for the purposes of survival analysis.
Heat-killed Candida albicans preparation
For each preparation, a fresh colony was picked and grown in BD Difco YPD broth (Thermo Fisher Scientific #DF0428-07-7) overnight at 37°C with agitation. Fungi were centrifuged for 10 min at 2000 rpm, washed once with PBS, and counted. Concentration was adjusted to 1 × 109/ml in PBS, and fungi were heat-killed by incubation at 95°C for 30 min.
Trichophyton rubrum culture and infection
T. rubrum (clinical isolate was provided by Paul Verweij at Radboud University Medical Centre in the Netherlands) was plated on BD Difco Sabouraud dextrose agar (SDA) (Fisher Scientific #BD210950) and kept at room temperature (RT) in a humid chamber for up to 1 month to allow sporulation. For spore isolation, a culture plate was incubated for 5 min with PBS with 0.05% Triton X-100, scraped, and passed through a 100-μm cell strainer. Suspension was centrifuged for 10 min at 2000 rpm, washed once with PBS, manually counted using a Neubauer counting chamber, and resuspended in PBS to the desired concentration. For the back skin infection, back skin was shaved and 50 μl of spore suspension was injected intradermally into 4 separate spots. Skin was also abrased with a pumice stone, and 100 μl of spore suspension was applied topically. For footpad injections, 50 μl of spore suspension was injected into the left back foot pad. Experiments were performed according to MGH IACUC protocol #2003N000158. For preparation of heat killed T. rubrum (HKTR), spore suspension was incubated at 95°C for 30 min.
Quantification of fungal burden
For determination of fungal burdens after infection, mice were euthanized with CO2, and kidneys and brains were harvested and placed into conical tubes containing sterile PBS. Tubes were weighed before and after addition of the organ to determine the organ weight. Organs were homogenized using an immersion homogenizer (Omni TH, Omni International, Kennesaw, GA), and serial dilutions of the homogenate were spread onto YPD plates. Plates were incubated 24–48 hours at 30°C until visible colonies formed. Colony forming units (CFUs) were counted, and CFU/g of tissue was calculated for each organ. For T. rubrum, back or foot pad skin was harvested with sterile tools and placed in 1 ml of PBS. Tissue was dissociated with a mechanical homogenizer. Serial dilutions were performed and plated on Remel Dermatophyte test medium (Thermo Fisher Scientific #R01365). Plates were incubated over 5 days at RT in a humid chamber, and CFUs were counted. Data is represented as CFU/g of harvested tissue.
Immunophenotyping
Single-cell suspension for flow cytometry analysis from the spleen and thymus was prepared by disrupting tissue between the frosted ends of two microscope slides using Staining Buffer. Bone marrow single-cell suspension was prepared by flushing femurs and tibia. Red blood cell (RBC) lysis was performed using 1X RBC Lysis Buffer. Kidney tissue was cut into small pieces with scissors and incubated for 20 min in digestion solution containing Liberase (Roche #385040) and DNAse I (Roche #10104159001) at 37°C with agitation. After the digestion, samples were placed on ice, and the reaction was stopped using a stop buffer containing 5% FBS and 1 mM EDTA. Cells were centrifuged for 5 min at 1500 rpm and 4°C and washed once with PBS. Cells were passed through a 100 μM cell strainer. Brain tissue was processed as previously described42. Briefly, tissue was homogenized using a syringe plunger and 100 mM filter on ice. Suspension was separated using a Percoll gradient, and the leukocyte interphase was collected after centrifugation at 2450 rpm for 20 min at 4°C. Cells were stained using an appropriate antibody cocktail described in the following section for 1 h on ice, washed, and run on a CytoFLEX LX flow cytometer (Beckman Coulter) or Cytek Aurora (Cytek Biosciences). Isolated single cells were filtered through a 40-μM filter and re-suspended in FACS buffer followed by flow cytometry analysis.
Three multi-color flow cytometry panels for CytoFLEX FL were developed to assess major myeloid and lymphoid subsets in the spleen, thymus, and bone marrow, comparing controls with the genetically modified mouse models. The following antibodies were used for a 16-color flow cytometry spleen panel: AF488-CD19 (clone 6D5), PE/DZ594-CD23 (clone B3B4), BV421-CD21/35 (clone 7E9), BUV395-CD93 (clone 493), BV785-CD11b (clone M1/70), APC Cy7-F4.80 (clone BM8), AF647-Ly6G (clone 1A8), BV650-CD11c (clone HL3), PE Cy7-CD3e (clone 145–2C11), PE Cy5-CD122 (clone 5H4), BV510-CD8α (clone 53–6.7), PE-CD4 (clone H129.19), AF700-CD45 (clone 30-F11), BV605-IA/IE (clone M5/114.15.2), BUV661-CD5 (clone 53–7.3), and ViaKrome Live/Dead dye (Beckman Coulter, #C36628). The following antibodies were used for a 10-color flow cytometry thymus panel: BUV395-CD4 (clone GK1.5), BV510-CD8α (clone 53–6.7), AF700-CD45 (clone 30-F11), PacBlu-CD25 (clone PC61), PE-CD44 (clone IM7), APC Cy7-CD117 (clone 2B8), PE Cy7-γδTCR (clone GL3), APC-TCRβ (clone H57–597), lineage cocktail includes: FITC-NK1.1 (clone PK136) & FITC-TER119 (clone TER-119) & AF488-CD19 (clone 6D5) and ViaKrome Live/Dead dye (Beckman Coulter, #C36628). The following antibodies were used for a 13-color flow cytometry bone marrow panel: BV605-B220 (clone RA3–6B2), PE Cy7-CD43 (clone S11), PE-CD24 (clone 30-F1), APC-BP1 (clone 6C3), BV421-IgM (clone RMM-1), BUV395-IgD (clone 11–26), BV650-CD150 (clone TC15–12F12.2), AF700-CD48 (clone HM48–1), PE Cy5-CD34 (clone MEC14.7), FITC-CD16/32 (clone 93), BV785-SCA1 (clone Ly-6A/E), APC Cy7-CD117 (clone 2B8), and ViaKrome Live/Dead dye (Beckman Coulter, #C36628).
One multi-color flow cytometry panel for Cytek Aurora was developed to assess major myeloid and lymphoid subsets in the spleen, kidney, and brain, comparing controls with the genetically modified mouse models. The following antibodies were used for a 22-color flow cytometry panel: Zombie UV Live/Dead (BioLegend, #423108), BV711-F4/80 (clone BM8), BUV395-CD4 (clone GK1.5), BV480-CD117 (clone 2B8), BUV496-CD123 (clone 5B11), APC Cy7-TCRB (clone H57–597), BV570-CD90.2 (clone 30-H12), PE CF594-Siglec-F (clone E50–2440), BV650-CD11c (clone N418), PE Cy5-CD3 (clone 145–2C11), PE-CD103 (clone 2E7), eFluor450-Ly6C (clone HK1.4), BV605-Ly6G (clone 1A8), BUV805-CD45 (clone 30-F11), BV510-CD8a (clone 53–6.7), PE Cy7-CD64 (clone X54–5/7.1), BUV737-CD19 (clone 1D3), AF700-CD8b (clone YTS156.7.7), BV785-CD11b (cloneM1/70), AF647-IgA, BUV563-IA/IE (clone M5/114.15.12), and FITC-EpCAM (clone G8.8).
Histology
Tissue was harvested into Tissue-Tec cassettes (#4118–01) and fixed in 10% neutral buffered formalin, trimmed, processed routinely, embedded in paraffin, and stained with hematoxylin and eosin (H&E) and Periodic Acid-Schiff (PAS). Histology was performed by Tufts Comparative Medicine Services at Tufts University, Boston, MA. Histopathology was evaluated by a board certified veterinary pathologist.
Isolation of splenic CD11c+ cells
Single-cell splenic suspension prepared as described above were incubated with biotin anti-mouse-CD11c antibody (BioLegend #117304, clone # N418) for 20 min on ice. Cells were washed once with MACS buffer (PBS pH 7.2, 0.5% BSA, 2mM EDTA) and incubated with anti-biotin MicroBeads (Miltenyi Biotec #130-090-485) according to the product instructions. Cells were positively selected using MACS cell separation MS columns (Miltenyi Biotec #130-042-201) according to the manufacturer’s instructions. Selected cells were centrifuged for 5 min at 1500 rpm, washed once with PBS, and processed for western blotting.
Preparation of single-cell suspension from back skin
Skin was harvested with sterile tools, cut into small pieces with scissors, and incubated for 1.5 h in digestion solution containing Liberase (Roche #385040) and DNAse I (Roche #10104159001) at 37°C with agitation. After the digestion, samples were placed on ice, and the reaction was stopped using a stop buffer containing 5% FBS and 1 mM EDTA. Cells were centrifuged for 5 min at 1500 rpm and 4°C and washed once with PBS. Cells were passed through a 100-μM cell strainer and counted. For the flow cytometry analysis, the following 9-color panel was used: BV785-CD45 (clone 30-F11), AF488-F4/80 (cloneBM8), AF647-CD64 (clone X54–5/7.1), BV650-Ly-6G (clone 1 A8), PerCP-Cy5.5-Ly-6C (clone HK1.4), BV421-CD11b (cloneM1/70), PE-Cy7-CD11c (clone N418), AF700-I-A/I-E (clone M5/114.15.2), and ViaKrome Live/Dead dye (Beckman Coulter, #C36628). Samples were analyzed on a CytoFLEX LX flow cytometer (Beckman Coulter) or Cytek Aurora flow cytometer (Cytek Biosciences).
CXCL1 ELISA
Back or foot pad skin was harvested with sterile tools and placed in 1 ml of PBS. Tissue was homogenized by mechanical disruption and centrifuged for 5 min at 1500 rpm. Supernatant was processed for CXCL1 ELISA (Thermo Fisher Scientific #EMCXCL1) according to manufacturer’s protocol.
Cell culture
HEK293T cells were maintained at 37°C and 5% CO2 in Gibco DMEM (Thermo Fisher Scientific #10569044) supplemented with 10% fetal calf serum and 15 μg/ml gentamycin sulfate.
Mouse bone marrow-derived dendritic and macrophage cell cultures
To generate BMDCs and BMDMs, murine bone marrow cells obtained from murine femurs and tibia were cultured for 7 days in complete RPMI with glutamax (Life Technologies #72400–120) supplemented with 10% FBS, 15 μg/ml gentamycin sulfate and 37.5 ng/ml recombinant murine GM-CSF (PeproTech #315–03) (for BMDCs) or 25 ng/ml M-CSF (PeproTech #315–02) and 5 ng/ml IL-3(PeproTech #213–13) (for BMDMs) at 37°C and 5% CO2. Media was changed at day 3, and fresh media was added at day 5 of the culture. Cells were used for functional assays on day 8.
Bone marrow monocyte isolation
Monocytes from bone marrow were isolated using a monocyte isolation kit (Miltenyi Biotech #130-100-629) and LS columns (Miltenyi Biotech #130-042-401) according to the manufacturer’s instructions. Isolated cells were plated in 96-well plates and stimulated overnight at 37°C and 5% CO2.
METHOD DETAILS
Lentivirus production and BMDC and BMDM transduction
All constructs used were cloned into a pCDH-CMV backbone (Addgene #72265). For virus preparation and BMDC or BMDM infection, protocols from the Broad Institute’s RNAi Consortium shRNA Library were used (http://www.broadinstitute.org/rnai/trc/lib). In brief, 70% confluent HEK293T cells were transfected using Lipofectamine 3000 reagent (Thermo Fisher Scientific #L3000–015) to generate viruses with desired constructs. Media containing viruses were harvested 48 h post transfection, filtered through 22-μM cell filters, and supplemented with 8 μg/ml of polybrene. BMDCs or BMDMs used for infection were grown in 12-well plates. On day 2 post isolation, media was aspirated, and 2 ml of media containing virus was added per well. Plates were centrifuged for 1.5 h at 2250 rpm and 30°C. After centrifugation, the virus-containing media was removed and replaced with fresh RPMI supplemented with GM-CSF (for BMDCs) or M-CFS and IL-3 (for BMDMs). Two days post-transduction, fresh RPMI supplemented with 5 μg/ml puromycin was added to the wells. Functional assays were performed 3–4 days later. In experiments involving reconstitution of CARD9 in Card9−/− BMDCs or BMDMs, puromycin selection was omitted. Sequences of all guides used for CRISPR KO are listed in Table S7.
CRISPR using RNP
BMDCs were nucleofected as previously described43. Briefly, sgRNA and Cas9 (IDT #1081059) were mixed and incubated for 20 min at RT. Following that, isolated bone marrow cells (2e6/reaction) were mixed with the sgRNA/Cas9 complexes and nucleofected using P3 Primary Cell 4D-Nucleofactor X kit S (Lonza #V4XP-3032) and program CM 137 in Lonza 4D-Nucleofector System. Following nucleofection, cells were plated in 12-well plates in RPMI media supplemented with GM-CSF and processed for analysis on day 8.
BMDC or BMDM stimulation
Mature BMDCs or BMDMs were harvested and plated in 96-well plates at a concentration of 1 × 105/cells per well. The following day, media was replaced with media containing either HKCA (MOI 1:10), HKTR (MOI 1:10), WGP (50 μg/ml) (Invivogen #tltl-wgp), or LPS (10 ng/ml) (Invivogen #tlrl-peklps) and incubated for 24 h. For p-p65 detection, cells were stimulated for 15 min.
Cytometric bead array
Cytokine concentration in serum or cell culture supernatants was assessed using BD Cytometric Bead Array (CBA) Mouse/Rat Soluble Protein Kit according to the manufacturer’s instruction.
Immunoprecipitation and western blotting
Mature BMDCs were harvested, and 3 × 107 cells were plated per 15-cm dish. The following day, HKCA (MOI 1:10) was added to the media for 30 min at 37°C. In experiments with PKCδ inhibitor, 10 μM sotrastaurin (Selleckchem #S2791) was added 1 h before stimulation with HKCA. Cells were harvested and washed once with PBS. For CARD9 immunoprecipitation to detect pS104-CARD9, the cell pellet was resuspended in 100 μl of lysis buffer (50mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 1% NP-40) supplemented with 1x Halt Protease & Phosphatase Inhibitor Single-Use Cocktail (Thermo Fisher Scientific #78442) and Pierce Universal Nuclease (Thermo Fisher Scientific #88701) and kept on ice for 30 min. SDS was then added to the tube to a final concentration of 1%, and lysates were heated at 95°C for 10 min. Denatured cell lysate was placed on ice and diluted with standard lysis buffer to a final SDS concentration of 0.01%. Lysate was incubated for 30 min and centrifuged at 2000 rpm for 10 min at 4°C. 50 μl of the supernatant was collected as a whole cell lysate (WCL) fraction. The remaining supernatant was collected and incubated overnight with 10 μl/sample of CARD9 antibody (Cell Signaling Technology #12283S) at 4°C while rotating. The next day, 50 μl of Dynabeads Protein G beads (Life Technologies #10004D) were added and incubated for 1 h rotating at 4°C. Immunoprecipitates were washed 3 times with standard lysis buffer and resuspended in 25 μl of 2x Laemmli buffer (BioRad #1610737) supplemented with 5% β-mercaptoethanol followed by boiling for 10 min at 75°C. For Bcl10 immunoprecipitation to detect CBM complex formation, cell pellets were resuspended in lysis buffer (PBS, 0.5% NP-40) supplemented with 1x Halt Protease & Phosphatase Inhibitor Single-Use Cocktail and Pierce Universal Nuclease and kept on ice for 10 min. Lysate was centrifuged for 10 min at 2000 rpm and 4°C. 50 μl of the supernatant was collected as a WCL fraction. The remaining supernatant was incubated overnight with 200 μl/sample of Bcl10 antibody (Santa Cruz Biotechnology #sc-5273) at 4°C rotating. The next day, 50 μl of Dynabeads Protein G beads were added and incubated for 1 h rotating at 4°C. Immunoprecipitates were washed 3 times with standard lysis buffer and resuspended in 25 μl of 2x Laemmli buffer supplemented with 5% β-mercaptoethanol followed by boiling for 10 min at 75°C. Immunoprecipitates and WCL samples were resolved by SDS-PAGE using Bio-Rad Mini-PROTEAN TGX Stain-Free Gels and Boston Bioproducts SDS-Running buffer and transferred to Immobilon-P membranes (Millipore Sigma #IPVH08130) in Boston Bioproducts Transfer buffer. Membranes were blocked for 1 h in 5% milk in TBS-T and immunoblotted with the indicated antibodies overnight in TBS-T supplemented with 5% BSA. Detection was performed by enhanced chemiluminescence with the Western Lightning Chemiluminescence Reagent (Perkin Elmer Life Sciences #NEL104001EA) followed by blotting with the secondary HRP-conjugated antibodies (Daco). The following antibodies were used for western blotting: CARD9 (clone A-8 Santa Cruz Biotechnology #sc-374569), p-p65 (Cell Signaling #3033S), p65 (Cell Signaling #8242S), Bactin-HRP (Cell Signaling #5125S), Flag M2 (Sigma Milipore #F1894), PKCδ (Cell Signaling #9616) and pTXR (Cell Signaling #2351S).
p65 nuclear translocation assay
Cells were plated in a 96-well plate suitable for a fluorescence microscope (PerkinElmer #6055302). The following day cells were stimulated for 30 min and processed for fluorescence staining. Cells were washed 3 times with PBS and fixed for 20 min in 4% PFA (Thermo Scientific #J19943-K2) at RT. Following 3 washes with PBS, cells were permeabilized using 0.5% Triton X-100 and 2% BSA in PBS for 1 h at RT. Cells were washed 3 times with PBS and incubated overnight at 4°C with anti-p65 antibody (1:100, Santa Cruz Biotechnology clone F-6 #sc-8008) in 1% BSA. The following day, cells were washed 3 times with PBS and incubated for 1 h at RT with a secondary 488-conjugated anti-mouse antibody and 1x Hoechst 33342 (Life Technologies #H3570) in 1% BSA. Cells were imaged using a Perkin Elmer Opera Phenix Imaging System and analyzed using Harmony Software.
In vitro phosphorylation assay
70% confluent HEK293T cells were transfected using Lipofectamine 3000 reagent (Thermo Fisher Scientific #L3000–015) to overexpress CARD9 variants. The following day, cells were lysed using RIPA buffer (Boston BioProducts #BP-115) supplemented with 1x Halt Protease & Phosphatase Inhibitor Single-Use Cocktail and Pierce Universal Nuclease and kept on ice for 10 min. Lysate was centrifuged for 10 min at 14000 rpm and 4°C. Supernatant was incubated for 1 h with MagStrep XT beads (Fisher Scientific #NC0776437) at 4°C, rotating. Following 2 washes with RIPA buffer and 2 washes with PBS, samples were split in half and processed for in vitro phosphorylation using PKCd Kinase Enzyme System (Promega #V3401) following the manufacturer’s instructions. ATP was used to control the reaction. After 1 h, the reaction was stopped by adding 2x Laemmli buffer containing 5% β-mercaptoethanol and boiling for 10 min at 75°C.
pS104-Card9 antibody generation
Peptides and antibodies were developed by Vivitide (formerly known as New England Peptide).
Peptide synthesis:
Peptides were made using standard Fmoc solid-state peptide synthesis and purified using high-performance liquid chromatography (HPLC) to >95% purity as determined by HPLC and/or LC/MS.
| Name | Lot # | Sequence |
|---|---|---|
| Card9 pS104 | 3702–2 | Ac-CEPARVF(pS)MIIDAS-amide |
| Card9 S104 NP | LP01491 | Ac-CEPARVFSMIIDAS-amide |
| Card9 pS104 R101C | LP01490 | H2N-EPACVF(pS)MIIDAS-amide |
Preparation of immunogen (maleimide):
Peptide 3702–2 above was conjugated to Sulfo-SMCC (Thermo/Fisher # 22322) activated KLH (Thermo/Fisher # 77600) through the sulfhydryl side chain of the added terminal cysteine, then emulsified 1:1 (volume) with complete Freund’s adjuvant (CFA)/incomplete Freund’s adjuvant (IFA).
Immunization protocol:
50–100 μg of immunogen was administered via intraperitoneal injection using the Freund’s adjuvant system. The immunizations were scheduled as follows:
Day 0 Pre-immune + Boost (100 μg immunogen with CFA)
Day 14 Boost (50 μg immunogen with IFA)
Day 28 Boost (50 μg immunogen with IFA)
Day 42 Boost (50 μg immunogen with IFA)
Day 49 Tail Bleed
Animals:
3 Balb/c mice (8–12 weeks old) were immunized, with animal protocols approved by Vivitide’s Animal Care and Use committee.
Preparation of screening materials (maleimide):
Peptides were conjugated to Sulfo-EMCS (Thermo/Fisher # 22307) activated BSA (Thermo/Fisher # 77110) through the sulfhydryl side chain of the added terminal cysteine.
Preparation of screening materials (amine):
The peptide was conjugated to BSA (Thermo/Fisher # 77110) through the N-terminal free amine with bi-functional amine linker glutaraldehyde.
Fusion of spleen cells:
Mouse 2 was euthanized, and the spleen was excised. The spleen cells were dissociated and fused with a NS1 (murine myloma) cell line via typical PEG protocols, then plated into 16 plates.
Screening for reactive clones:
Culture supernatants from all plates were screened by ELISA via peptides conjugated to EMCS-activated BSA. Ninety-four positive wells are selected from this initial test for expansion and a subsequent re-confirmation assay. Twenty positive wells from this test were selected to have one vial of cells frozen down, with supernatant saved for subsequent additional testing.
Sub-cloning:
Two positive parental cell lines were chosen to be further expanded and screened again by ELISA. The two best daughter clones from each were further expanded and isotyped (all clones were IgG1).
Production:
Supernatant from one daughter clone (4F9.H5) was scaled up to 250 mL by roller-bottle production. The resultant material was purified by Protein G (Cytiva Life Sciences # P-00067) chromatography.
CARD9 and BCL10 expression and protein purification
The expressions and protein purifications of CARD9 2–152 and MBP-BCL10 constructs were done according to previously described methods21 with slight modifications. Modifications include lysing 72 h CARD9 2–152 pellets using an Avestin Microfluidizer under denaturing conditions. After the final Superdex S200 gel filtration step, CARD9 2–152 protein was concentrated to 2 mM. 1 h MBP-BCL10 pellet was lysed in nickel buffer using an Avestin Microfluidizer, and post-Ni-NTA affinity column elutions were frozen down and stored at −80°C in 1 mL aliquots for subsequent fluorescence polarization assays.
BCL10 fluorescence polarization assay
The fluorescence polarization assay preparations and assays were performed according to previously described methods21 with slight modifications. 2 μM Alexa Fluor 488 C5 maleimide was added to one thawed aliquot of BCL10 and purified over a Superdex 200 Increase gel filtration column. The monomeric BCL10 peak was collected, stored at 4°C in the dark, and used in the fluorescence polarization assay within 1 h of the purification. 1–2 mM of CARD9 2–152 were incubated with 4 mM EDTA and shook at 500 rpm and 37°C for 60 minutes to allow for filament formation. The assay was performed in 20 mM Tris, 150 mM NaCl, and 0.5 mM TCEP (pH 7.5) in a final volume of 20 μL in a black 384-well plate. 2 μM MBP-BCL10 and incubated CARD9 were added to the plate initially, and 0.05 mg/mL tobacco etch virus (TEV) protease was added right before the fluorescence polarization readout. Fluorescence polarization was measured by exciting at 495 nm and monitoring at 520 nm on a PHERAstar FSX plate reader at 25°C.
Single-cell RNA-sequencing
Single-cell suspensions prepared as described above were processed using the Chromium Single Cell 3’ Gene Expression kit (v3.1, 10x Genomics) per the manufacturer’s instructions. Libraries were sequenced on the Illumina NovaSeq SP per the manufacturer’s instructions.
Pre-processing of single-cell RNA-sequencing data
Pre-processing of raw fastq files (6 WT, 6 Card9 R101C) was conducted using Cell Ranger v.6.0.0 (10x genomics). Raw reads were aligned to the mm10 reference genome, and the Cell Ranger count function was used to generate UMI count matrices for each sample. Count matrices for each sample were aggregated prior to downstream processing. Cells with greater than 20% mitochondrial gene (“mt-“) expression were removed from the count matrix, followed by removal of all mitochondrial genes from the matrix. We also removed ribosomal genes and the non-coding RNAs Neat1 and Malat1. We examined the distribution of log10(mean gene UMI count) across all genes, and log10(library size) across all genes, and filtered the matrix at −4.0 and 2.6 for each of these values, respectively, to remove genes with low read count and cells with abnormally low library size. 75,647 cells and 20,715 genes remained after these filters. The mean library size was 3,948.41, and the mean number of genes captured per cell was 1,455.63.
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of single-cell RNA-sequencing data
The final count matrix of 75,647 cells and 20,715 genes was input into Seurat v4.0.144 for downstream analysis. The top 10 principal components of the gene expression data were used as input to the FindNeighbors() function, after which we used RunUMAP() to generate a final dimensionality reduction of the data. This analysis yielded 17 clusters. Cluster enrichments in WT or R101C mice were calculated as proportion of all cells in each replicate, and statistics were evaluated using Dirichlet multinomial regression, as previously described45. Clusters were considered significantly differentially enriched in a condition at FDR P < 0.1. We used the FindMarkers() function in Seurat to find DEGs. Genes were considered significantly differentially expressed at FDR P < 0.05. We performed DEG analysis within the three broad lineages described in Figure 6A (immune cells, stromal cells, and keratinocytes) and between WT and R101C within each cluster separately at D2 or D9, and/or between D2 and D9 for WT and R101C cells. Pathway analyses for WT vs R101C DEGs were conducted using the C5 pathway sets from MsigDB; pathways and DEGs were evaluated using gene set enrichment analysis (GSEA) as implemented in the fgsea() function in R. To align keratinocyte clusters identified in the present study with previously defined clusters28, we obtained DEGs for all previously defined clusters28 and evaluated enrichment of our cluster DEGs in those clusters using fgsea(). High normalized enrichment score (NES) indicates positively enriched; low NES indicates negatively enriched. We applied the MitoCarta oxidative phosphorylation signature to scRNA-seq data using the AddModuleScore() function in Seurat. For analyses in Figures S7H and S7I, we examined the Card9 DEGs overlapping with the wound healing (http://www.informatics.jax.org/go/term/GO:0042060) and inflammatory response (http://www.informatics.jax.org/go/term/GO:0006954) gene sets obtained from gene ontology.
Statistical analysis
Mouse experiments included 4–10 mice per group and were repeated at least 2 times, with data being reproducible between the repeats. Mice were randomly allocated into age- and sex-matched experimental groups. When possible, littermates were used. No outliers were excluded. Differences between experimental groups were assessed using paired t-tests unless otherwise indicated. All statistical analysis was performed using GraphPad Prism v.9 unless otherwise specified. P value below 0.05 was considered statistically significant.
Supplementary Material
Table S7. Sequences of CRISPR guides, Related to Methods.
Table S6. Cluster enrichment for WT and R101C, Related to Figure 7. Cluster enrichments calculated via Dirichlet multinomial regression between WT cells at D2 and WT cells at D9 as well as R101C cells at D2 and R101C cells at D9.
Table S2. Cluster enrichments at D2, Related to Figure 6. Cluster enrichments calculated via Dirichlet multinomial regression between WT and R101C cells at D2.
Table S1. DEGs defining clusters, Related to Figure 6. DEGs defining all clusters phenotyped via scRNA-seq analysis.
Table S3. DEGs for each cluster, Related to Figures 6 and 7. DEGs between WT and R101C cells at D2 and D9 for each cluster.
Table S4. Pathway GSEAs for each cluster, Related to Figures 6 and 7. Pathway GSEAs for DEGs between WT and R101C cells at D2 and D9 for each cluster.
Table S5. Cluster enrichments at D9, Related to Figure 7. Cluster enrichments calculated via Dirichlet multinomial regression between WT and R101C cells at D9.
Highlights.
CARD9 R101C missense variant is associated with recurrent fungal infections.
R101C disrupts a previously undescribed PKCδ phosphorylation site at S104.
pS104 actuates a signaling switch to activate CARD9 and allow Bcl10 polymerization.
CARD9 R101C mouse model reveals cellular antifungal response networks in skin.
Acknowledgements
We thank Paul Verweij at Radboud University Medical Centre in the Netherlands for providing the T. rubrum strain, Theresa Reimels and Elizabeth Heppenheimer for editing the manuscript and figures, and all members of the Xavier lab for the constructive discussions and suggestions. This work was supported by funding from The Leona M. and Harry B. Helmsley Charitable Trust and the National Institutes of Health (grants AI137325, DK043351, and DK135492 to R.J.X., grants AI15081 and AI152499 to J.M.V.). J.L.R. was supported by NIH/NIAID grant 1K08AI14755. The graphical abstract was created with BioRender.com.
Footnotes
Declaration of Interests
R.J.X. is a co-founder of Celsius Therapeutics and Jnana Therapeutics, scientific advisory board member at Nestlé, and board director at MoonLake Immunotherapeutics; these organizations had no roles in this study.
References
- 1.Huang L, Guo Z, Wang F, and Fu L (2021). KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther 6, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bongomin F, Gago S, Oladele RO, and Denning DW (2017). Global and Multi-National Prevalence of Fungal Diseases-Estimate Precision. J Fungi (Basel) 3. 10.3390/jof3040057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Desoubeaux G, Coste AT, Imbert C, and Hennequin C (2022). Overview about Candida auris: What’s up 12 years after its first description? J. Mycol. Med 32, 101248. [DOI] [PubMed] [Google Scholar]
- 4.Firacative C (2020). Invasive fungal disease in humans: are we aware of the real impact? Mem. Inst. Oswaldo Cruz 115, e200430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodrigues ML, and Nosanchuk JD (2020). Fungal diseases as neglected pathogens: A wake-up call to public health officials. PLoS Negl. Trop. Dis 14, e0007964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fodil N, Langlais D, and Gros P (2016). Primary Immunodeficiencies and Inflammatory Disease: A Growing Genetic Intersection. Trends Immunol. 37, 126–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pathakumari B, Liang G, and Liu W (2020). Immune defence to invasive fungal infections: A comprehensive review. Biomed. Pharmacother 130, 110550. [DOI] [PubMed] [Google Scholar]
- 8.Kumar V, Cheng S-C, Johnson MD, Smeekens SP, Wojtowicz A, Giamarellos-Bourboulis E, Karjalainen J, Franke L, Withoff S, Plantinga TS, et al. (2014). Immunochip SNP array identifies novel genetic variants conferring susceptibility to candidaemia. Nat. Commun 5, 4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vaezi A, Fakhim H, Abtahian Z, Khodavaisy S, Geramishoar M, Alizadeh A, Meis JF, and Badali H (2018). Frequency and Geographic Distribution of CARD9 Mutations in Patients With Severe Fungal Infections. Front. Microbiol 9, 2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beaudoin M, Goyette P, Boucher G, Lo KS, Rivas MA, Stevens C, Alikashani A, Ladouceur M, Ellinghaus D, Törkvist L, et al. (2013). Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet. 9, e1003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, Boucher G, Ripke S, Ellinghaus D, Burtt N, et al. (2011). Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet 43, 1066–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao Z, Conway KL, Heath RJ, Rush JS, Leshchiner ES, Ramirez-Ortiz ZG, Nedelsky NB, Huang H, Ng A, Gardet A, et al. (2015). Ubiquitin Ligase TRIM62 Regulates CARD9-Mediated Anti-fungal Immunity and Intestinal Inflammation. Immunity 43, 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pointon JJ, Harvey D, Karaderi T, Appleton LH, Farrar C, Stone MA, Sturrock RD, Brown MA, and Wordsworth BP (2010). Elucidating the chromosome 9 association with AS; CARD9 is a candidate gene. Genes Immun. 11, 490–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janse M, Lamberts LE, Franke L, Raychaudhuri S, Ellinghaus E, Muri Boberg K, Melum E, Folseraas T, Schrumpf E, Bergquist A, et al. (2011). Three ulcerative colitis susceptibility loci are associated with primary sclerosing cholangitis and indicate a role for IL2, REL, and CARD9. Hepatology 53, 1977–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, Fasel D, Lata S, Prakash S, Shapiro S, et al. (2014). Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat. Genet 46, 1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gross O, Gewies A, Finger K, Schäfer M, Sparwasser T, Peschel C, Förster I, and Ruland J (2006). Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 442, 651–656. [DOI] [PubMed] [Google Scholar]
- 17.Hara H, Ishihara C, Takeuchi A, Imanishi T, Xue L, Morris SW, Inui M, Takai T, Shibuya A, Saijo S, et al. (2007). The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat. Immunol 8, 619–629. [DOI] [PubMed] [Google Scholar]
- 18.Hara H, Ishihara C, Takeuchi A, Xue L, Morris SW, Penninger JM, Yoshida H, and Saito T (2008). Cell type-specific regulation of ITAM-mediated NF-kappaB activation by the adaptors, CARMA1 and CARD9. J. Immunol 181, 918–930. [DOI] [PubMed] [Google Scholar]
- 19.Drummond RA, Franco LM, and Lionakis MS (2018). Human CARD9: A Critical Molecule of Fungal Immune Surveillance. Front. Immunol 9, 1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strasser D, Neumann K, Bergmann H, Marakalala MJ, Guler R, Rojowska A, Hopfner K-P, Brombacher F, Urlaub H, Baier G, et al. (2012). Syk kinase-coupled C-type lectin receptors engage protein kinase C-δ to elicit Card9 adaptor-mediated innate immunity. Immunity 36, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holliday MJ, Witt A, Rodríguez Gama A, Walters BT, Arthur CP, Halfmann R, Rohou A, Dueber EC, and Fairbrother WJ (2019). Structures of autoinhibited and polymerized forms of CARD9 reveal mechanisms of CARD9 and CARD11 activation. Nat. Commun 10, 3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grumach AS, de Queiroz-Telles F, Migaud M, Lanternier F, Filho NR, Palma SMU, Constantino-Silva RN, Casanova JL, and Puel A (2015). A homozygous CARD9 mutation in a Brazilian patient with deep dermatophytosis. J. Clin. Immunol 35, 486–490. [DOI] [PubMed] [Google Scholar]
- 23.Lanternier F, Pathan S, Vincent QB, Liu L, Cypowyj S, Prando C, Migaud M, Taibi L, Ammar-Khodja A, Stambouli OB, et al. (2013). Deep dermatophytosis and inherited CARD9 deficiency. N. Engl. J. Med 369, 1704–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson JL, Yaron TM, Huntsman EM, Kerelsky A, Song J, Regev A, Lin T-Y, Liberatore K, Cizin DM, Cohen BM, et al. (2023). An atlas of substrate specificities for the human serine/threonine kinome. Nature 613, 759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu W, Rush JS, Graham DB, Cao Z, and Xavier RJ (2020). USP15 Deubiquitinates CARD9 to Downregulate C-Type Lectin Receptor-Mediated Signaling. Immunohorizons 4, 670–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qiao Q, Yang C, Zheng C, Fontán L, David L, Yu X, Bracken C, Rosen M, Melnick A, Egelman EH, et al. (2013). Structural architecture of the CARMA1/Bcl10/MALT1 signalosome: nucleation-induced filamentous assembly. Mol. Cell 51, 766–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holliday MJ, Ferrao R, de Leon Boenig G, Estevez A, Helgason E, Rohou A, Dueber EC, and Fairbrother WJ (2018). Picomolar zinc binding modulates formation of Bcl10-nucleating assemblies of the caspase recruitment domain (CARD) of CARD9. J. Biol. Chem 293, 16803–16817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joost S, Zeisel A, Jacob T, Sun X, La Manno G, Lönnerberg P, Linnarsson S, and Kasper M (2016). Single-Cell Transcriptomics Reveals that Differentiation and Spatial Signatures Shape Epidermal and Hair Follicle Heterogeneity. Cell Syst 3, 221–237.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, and Locati M (2004). The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677–686. [DOI] [PubMed] [Google Scholar]
- 30.Ji C, Yang Z, Zhong X, and Xia J (2021). The role and mechanism of CARD9 gene polymorphism in diseases. Biomed. J 44, 560–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drummond RA, and Lionakis MS (2016). Mechanistic Insights into the Role of C-Type Lectin Receptor/CARD9 Signaling in Human Antifungal Immunity. Front. Cell. Infect. Microbiol 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gavino C, Hamel N, Zeng JB, Legault C, Guiot M-C, Chankowsky J, Lejtenyi D, Lemire M, Alarie I, Dufresne S, et al. (2016). Impaired RASGRF1/ERK-mediated GM-CSF response characterizes CARD9 deficiency in French-Canadians. J. Allergy Clin. Immunol 137, 1178–1188.e7. [DOI] [PubMed] [Google Scholar]
- 33.De Bruyne M, Hoste L, Bogaert DJ, Van den Bossche L, Tavernier SJ, Parthoens E, Migaud M, Konopnicki D, Yombi JC, Lambrecht BN, et al. (2018). A CARD9 Founder Mutation Disrupts NF-κB Signaling by Inhibiting BCL10 and MALT1 Recruitment and Signalosome Formation. Front. Immunol 9, 2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu X, Xu J-F, Zheng G, Lu H-W, Duan J-L, Rui W, Guan J-H, Cheng L-Q, Yang D-D, Wang M-C, et al. (2018). CARD9S12N facilitates the production of IL-5 by alveolar macrophages for the induction of type 2 immune responses. Nat. Immunol 19, 547–560. [DOI] [PubMed] [Google Scholar]
- 35.McGovern DPB, Gardet A, Törkvist L, Goyette P, Essers J, Taylor KD, Neale BM, Ong RTH, Lagacé C, Li C, et al. (2010). Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat. Genet 42, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Franke A, McGovern DPB, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al. (2010). Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet 42, 1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Z, Liu R, Gao H, Jung S, Gao X, Sun R, Liu X, Kim Y, Lee H-S, Kawai Y, et al. (2023). Genetic architecture of the inflammatory bowel diseases across East Asian and European ancestries. Nat. Genet 55, 796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang H, Fang M, Jostins L, Umićević Mirkov M, Boucher G, Anderson CA, Andersen V, Cleynen I, Cortes A, Crins F, et al. (2017). Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 547, 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nowarski R, Jackson R, and Flavell RA (2017). The Stromal Intervention: Regulation of Immunity and Inflammation at the Epithelial-Mesenchymal Barrier. Cell 168, 362–375. [DOI] [PubMed] [Google Scholar]
- 40.West NR, Hegazy AN, Owens BMJ, Bullers SJ, Linggi B, Buonocore S, Coccia M, Görtz D, This S, Stockenhuber K, et al. (2017). Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med 23, 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Friedrich M, Pohin M, Jackson MA, Korsunsky I, Bullers SJ, Rue-Albrecht K, Christoforidou Z, Sathananthan D, Thomas T, Ravindran R, et al. (2021). IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat. Med 27, 1970–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lionakis MS, Lim JK, Lee C-CR, and Murphy PM (2011). Organ-specific innate immune responses in a mouse model of invasive candidiasis. J. Innate Immun 3, 180–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Freund EC, Lock JY, Oh J, Maculins T, Delamarre L, Bohlen CJ, Haley B, and Murthy A (2020). Efficient gene knockout in primary human and murine myeloid cells by non-viral delivery of CRISPR-Cas9. J. Exp. Med 217. 10.1084/jem.20191692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, Herbst RH, Rogel N, Slyper M, Waldman J, et al. (2019). Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 178, 714–730.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S7. Sequences of CRISPR guides, Related to Methods.
Table S6. Cluster enrichment for WT and R101C, Related to Figure 7. Cluster enrichments calculated via Dirichlet multinomial regression between WT cells at D2 and WT cells at D9 as well as R101C cells at D2 and R101C cells at D9.
Table S2. Cluster enrichments at D2, Related to Figure 6. Cluster enrichments calculated via Dirichlet multinomial regression between WT and R101C cells at D2.
Table S1. DEGs defining clusters, Related to Figure 6. DEGs defining all clusters phenotyped via scRNA-seq analysis.
Table S3. DEGs for each cluster, Related to Figures 6 and 7. DEGs between WT and R101C cells at D2 and D9 for each cluster.
Table S4. Pathway GSEAs for each cluster, Related to Figures 6 and 7. Pathway GSEAs for DEGs between WT and R101C cells at D2 and D9 for each cluster.
Table S5. Cluster enrichments at D9, Related to Figure 7. Cluster enrichments calculated via Dirichlet multinomial regression between WT and R101C cells at D9.
Data Availability Statement
Raw fastq files for the scRNAseq experiment and a processed Seurat object with cell metadata have been deposited at GEO and are publicly available as of the date of publication under accession GSE210224. Accession numbers are additionally listed in the Key Resources Table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| AF488-CD19 | BioLegend | Cat. # 115521 |
| PE/DZ594-CD23 | BioLegend | Cat. # 101634 |
| BV421-CD21/35 | BioLegend | Cat. # 123421 |
| BUV395-CD93 | BD Biosciences | Cat. # 745595 |
| BV785-CD11b | BioLegend | Cat. # 101243 |
| APC Cy7-F4/80 | BioLegend | Cat. # 123118 |
| AF647-Ly6G | BioLegend | Cat. # 127609 |
| BV650-CD11c | BioLegend | Cat. # 117339 |
| PE Cy7-CD3e | BioLegend | Cat. # 100320 |
| PE Cy5-CD122 | BioLegend | Cat. # 123219 |
| BV510-CD8α | BioLegend | Cat. # 100752 |
| PE-CD4 | BioLegend | Cat. # 100512 |
| AF700-CD45 | BioLegend | Cat. # 103128 |
| BV605-IA/IE | BD Horizon | Cat. # 563413 |
| BUV661-CD5 | BD Biosciences | Cat. # 741459 |
| BUV395-CD4 | BD Biosciences | Cat. # 563790 |
| PacBlu-CD25 | BioLegend | Cat. # 102022 |
| PE-CD44 | BioLegend | Cat. # 103008 |
| APC Cy7-CD117 | BioLegend | Cat. # 105826 |
| PE Cy7-γδTCR | BioLegend | Cat. # 118124 |
| APC-TCRβ | BioLegend | Cat. # 109212 |
| FITC-NK1.1 | BioLegend | Cat. # 108717 |
| FITC-TER119 | BioLegend | Cat. # 116206 |
| BV605-B220 | BioLegend | Cat. # 103244 |
| PE Cy7-CD43 | BioLegend | Cat. # 143210 |
| PE-CD24 | BioLegend | Cat. # 138503 |
| APC-BP1 | eBioscence | Cat. # 17-5891-82 |
| BV421-IgM | BioLegend | Cat. # 406517 |
| BUV395-IgD | BD Horizon | Cat. # 564274 |
| BV650-CD150 | BioLegend | Cat. # 115932 |
| AF700-CD48 | BioLegend | Cat. # 103426 |
| PE Cy5-CD34 | BioLegend | Cat. # 119312 |
| FITC-CD16/32 | BioLegend | Cat. # 101305 |
| BV785-SCA1 | BioLegend | Cat. # 108139 |
| BV711-F4/80 | BioLegend | Cat. # 123147 |
| BV480-CD117 | BD Biosciences | Cat. # 566074 |
| BUV496-CD123 | BD Biosciences | Cat. # 750813 |
| BV570- CD90.2 | BioLegend | Cat. # 105329 |
| PE CF594-Siglec-F | BD Biosciences | Cat. # 562757 |
| PE Cy5 -CD3 | BioLegend | Cat. # 100310 |
| PE-CD103 | BioLegend | Cat. # 121406 |
| eFluor450-Ly6C | Thermo Fisher Scientific | Cat. # 48-5932-82 |
| BV605-Ly6G | BioLegend | Cat. # 127639 |
| BUV805-CD45 | BD Biosciences | Cat. # 748370 |
| PE Cy7-CD64 | BioLegend | Cat. # 139314 |
| BUV737-CD19 | BD Biosciences | Cat. # 612781 |
| AF700-CD8b | BioLegend | Cat. # 126618 |
| AF647-IgA | SouthernBiotech | Cat. # 1040–31 |
| BUV563-IA/IE | BD Biosciences | Cat. # 748846 |
| FITC-EpCAM | BioLegend | Cat. # 118207 |
| BV785-CD45 | BioLegend | Cat. # 103149 |
| AF647-CD64 | BioLegend | Cat. # 139322 |
| BV650-Ly6G | BioLegend | Cat. # 127641 |
| PerCP-Cy5.5-Ly6C | BioLegend | Cat. # 128011 |
| BV421-CD11b | BioLegend | Cat. # 101235 |
| PE-Cy7-CD11c | BioLegend | Cat. # 117339 |
| AF700-IA/IE | BioLegend | Cat. # 107621 |
| Biotin-CD11c | BioLegend | Cat. # 117304 |
| CARD9 | Cell Signaling Technology | Cat. # 12283S |
| Bcl10 | Santa Cruz Biotechnology | Cat. # sc-5273 |
| CARD9 | Santa Cruz Biotechnology | Cat. # sc-374569 |
| p-p65 | Cell Signaling Technology | Cat. # 3033S |
| p65 | Cell Signaling Technology | Cat. # 8242S |
| Flag M2 | Sigma Millipore | Cat. # F1894 |
| pTXR | Cell Signaling Technology | Cat. # 2351S |
| PKCd | Cell Signaling Technology | Cat. # 9616 |
| pS104-Card9 | This study | This study |
| p65 | Santa Cruz Biotechnology | Cat. # sc-8008 |
| Anti-rabbit HRP | Daco | Cat. # P0448 |
| Anti-mouse HRP | Daco | Cat. # P0447 |
| AF488 anti-mouse | Invitrogen | Cat. # A-21202 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Sabouraud dextrose agar | Thermo Fisher Scientific | Cat. # BD210950 |
| Remel Dermatophyte test medium | Thermo Fisher Scientific | Cat. # R01365 |
| 1X RBC Lysis Buffer | Thermo Fisher Scientific | Cat. # 00433357 |
| Liberase | Roche | Cat. # 385040 |
| DNAse I | Roche | Cat. # 10104159001 |
| ViaKrome Live/Dead dye | Beckman Coulter | Cat. # C36628 |
| Zombie UV Live/Dead | BioLegend | Cat. # 423108 |
| DMEM | Thermo Fisher Scientific | Cat. # 10569044 |
| RPMI with glutamax | Life Technologies | Cat. # 72400–120 |
| GM-CSF | PeproTech | Cat. # 315–03 |
| M-CSF | PeproTech | Cat. # 315–02 |
| IL-3 | PeproTech | Cat. # 213–13 |
| Lipofectamine 3000 reagent | Thermo Fisher Scientific | Cat. # L3000–015 |
| Cas9 | IDT | Cat. # 1081059 |
| YPD broth | Thermo Fisher Scientific | Cat. # DF0428-07-7 |
| WGP | Invivogen | Cat. # tltl-wgp |
| LPS | Invivogen | Cat. # tlrl-peklps |
| Sotrastaurin | Selleckchem | Cat. # S2791 |
| 1x Halt Protease & Phosphatase Inhibitor Single-Use Cocktail | Thermo Fisher Scientific | Cat. # 78442 |
| Pierce Universal Nuclease | Thermo Fisher Scientific | Cat. # 88701 |
| Dynabeads Protein G beads | Life Technologies | Cat. # 10004D |
| Laemmli buffer | BioRad | Cat. # 1610737 |
| Western Lightning Chemiluminescence Reagent | Perkin Elmer Life Sciences | Cat. # NEL104001EA |
| 4% PFA | Thermo Scientific | Cat. # J19943-K2 |
| 1x Hoechst 33342 | Life Technologies | Cat. # H3570 |
| RIPA buffer | Boston BioProducts | Cat. # BP-115 |
| MagStrep XT beads | Fisher Scientific | Cat. # NC0776437 |
| Sulfo-SMCC | Thermo/Fisher | Cat. # 22322 |
| activated KLH | Thermo/Fisher | Cat. # 77600 |
| activated BSA | Thermo/Fisher | Cat. # 77110 |
| Protein G | Cytiva Life Sciences | Cat. # P-00067 |
| Immobilon-P membranes | Millipore Sigma | Cat. # IPVH08130 |
| Tris-Glycince-SDS-Running buffer (10x) | Boston BioProducts | Cat. # BP-150 |
| Transfer buffer (10x) | Boston BioProducts | Cat. # BP-190 |
| Tris Buffered Saline-Tween (20x, with 1% Tween-20, pH 7.4) | Boston BioProducts | Cat. # IBB-181X |
| 4–20% Bio-Rad Mini-PROTEAN TGX Stain-Free Gels | BioRad | Cat. # 4568095 |
| β-mercaptoethanol | Sigma | Cat. # M3148–25ML |
| EDTA | Invitrogen | Cat. # 15575–038 |
| LS columns | Miltenyi Biotech | Cat. # 130-042-401 |
| Brilliant Stain Buffer | BD Biosciences | Cat. # 566349 |
| Critical commercial assays | ||
| CXCL1 ELISA | Thermo Fisher Scientific | Cat. # EMCXCL1 |
| Monocyte isolation kit | Miltenyi Biotech | Cat. # 130-100-629 |
| P3 Primary Cell 4D-Nucleofactor X kit S | Lonza | Cat. # V4XP-3032 |
| BD Cytometric Bead Array (CBA) Mouse IL-6 Flex Set | BD Biosciences | Cat. # 558301 |
| BD Cytometric Bead Array (CBA) Mouse TNFa Flex Set | BD Biosciences | Cat. # 558299 |
| PKCd Kinase Enzyme System | Promega | Cat. # V3401 |
| Chromium Single Cell 3’ Gene Expression kit | 10x Genomics | Cat. # PN-1000120 |
| Deposited data | ||
| Single-cell RNA-sequencing data | This study | GSE210224; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE210224 |
| Experimental models: Cell lines | ||
| HEK293T | Lab strain, mycoplasma free | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: Card9−/− | Hara et al.17 | N/A |
| Mouse: Card9R101C | This study | N/A |
| Fungi: Candida albicans strain SC5314 | Jatin Vyas Lab (Massachusetts General Hospital) | N/A |
| Fungi: Trichophyton rubrum | Paul Verweij (Radboud University Medical Centre, Netherlands) | Clinical isolate |
| Oligonucleotides | ||
| sgRNA sequences are provided in Table S7 | IDT | N/A |
| Recombinant DNA | ||
| pMD601-CARD92–152/WT | This study | N/A |
| pMD602-CARD92–152/I107E | This study | N/A |
| pMD604- CARD92–152/S104D | This study | N/A |
| pMD605-CARD92–152/R101C | This study | N/A |
| pMD606-CARD92–152/R101C/S104D | This study | N/A |
| pMD613-Bcl10Fl/C29A/C57A | This study | N/A |
| pCDH-CMV | Addgene | Cat. # 72265 |
| pCDH-CMV-CARD9 | This study | N/A |
| pCDH-CMV-CARD9R101C | This study | N/A |
| pCDH-CMV-CARD9S104N | This study | N/A |
| pCDH-CMV-CARD9S104D | This study | N/A |
| Software and algorithms | ||
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com/ |
| FlowJo | FlowJo | https://www.flowjo.com/ |
| Harmony | Perkin Elmer | N/A |
| Cell Ranger v.6.0.0 | 10x Genomics | N/A |
| Seurat v4.0.1 | Hao et al.44 | N/A |
| Other | ||