

Abstract
Purpose of review:
A subset of patients with interstitial lung diseases (ILD), like rheumatoid arthritis (RA) associated ILD and chronic hypersensitivity pneumonitis (HP), will experience a disease course similar to patients with idiopathic pulmonary fibrosis (IPF). They also often have a usual interstitial pneumonia (UIP) pattern of fibrosis. While the standard of care for patients with RA-ILD and chronic HP is immunosuppression, the optimal treatment for patients with progressive disease and a UIP pattern remains unknown.
Recent findings:
Recent research has highlighted shared risk factors, disease behavior and pathobiology between RA-ILD, chronic HP and IPF. The presence of a UIP pattern, in both RA-ILD and chronic HP, is associated with a worse prognosis. Moreover, genetic risk factors, previously well characterized in IPF, are increasingly being linked to RA-ILD and chronic HP. The MUC5B promoter variant rs5705950, telomerase complex mutations and short telomere lengths are also linked to an increased susceptibility to pulmonary fibrosis in RA and chronic HP.
Summary:
IPF shares several clinical, genetic and biological features with other ILDs exhibiting the UIP pattern. The optimal pharmacologic management of these patients remains uncertain. Several on-going trials are evaluating the efficacy of antifibrotic medications in these other diagnoses and may change how we approach ILD treatment.
Keywords: interstitial lung disease, usual interstitial pneumonia, idiopathic pulmonary fibrosis, rheumatoid arthritis, hypersensitivity pneumonitis
Introduction
Interstitial lung diseases (ILD) form a heterogeneous group of diffuse parenchymal lung diseases of various etiologies, clinical course and prognosis1. The classical diagnostic approach to ILD heavily relies on the identification of the cause or trigger responsible for the development of ILD, including an underlying autoimmune condition (e.g. rheumatoid arthritis) and an environmental antigen (e.g. birds, mold). Patients are then classified, according to their clinical, radiological and pathological features, as having a diagnosis such as idiopathic pulmonary fibrosis (IPF), hypersensitivity pneumonitis (HP), connective tissue disease (CTD) related-ILD, or unclassifiable ILD1–6.
This diagnostic framework shapes the subsequent medical management of patients with ILD. While antifibrotic medications, like pirfenidone and nintedanib, are being used to slow disease progression in IPF, corticosteroids and/or other immunosuppressive agents are frequently used in patients with HP and CTD-ILD7–14. Further, immunosuppression is avoided in patients with IPF given the increased morbidity and mortality observed with the use of combination therapy with prednisone, azathioprine and N-acetylcysteine (NAC)15.
However, it is increasingly recognized that a proportion of patients with non-IPF ILD also progress in a manner very similar to that of IPF. Diseases in particular include rheumatoid arthritis associated ILD (RA-ILD) and chronic HP, specifically in those with the usual interstitial pneumonia (UIP) pattern of fibrosis. Recent work in these two disease states suggests that they share several clinical features and pathogenetic mechanisms with IPF16,17. While the optimal therapy for non-IPF ILDs such as RA-ILD and chronic HP remains unclear, the standard of care is immunosuppression. It has been hypothesized that these diseases could have more benefit and less risk from anti-fibrotic therapies, such as pirfenidone or nintedanib, rather than treatment with immunosuppression18–20.
This review will highlight our current understanding of the UIP lesion in idiopathic (IPF) and other non-idiopathic forms of UIP (RA-ILD and chronic HP), explore the evidence and knowledge gaps regarding the various therapeutic options available for patients with ILD and discuss how research progress may challenge our current paradigms and change how we approach treatment in ILD.
The Usual Interstitial Pneumonia pattern
The pattern of usual interstitial pneumonia (UIP) was first recognized and described on surgical lung biopsies and autopsies by Dr Averill Liebow. The UIP pattern was the most prevalent pathological pattern in patients with idiopathic interstitial pneumonias (IIP)21. Further characterization of the UIP pattern on pathology and imaging occurred over the next several decades, shaping our current understanding of UIP and ILD pathogenesis22.
Currently, the UIP pattern can be recognized in patients with ILD on high-resolution computed tomography (HRCT) and/or surgical lung biopsy. On HRCT, features of subpleural and basal predominance and honeycombing with or without peripheral traction bronchiectasis or bronchiolectasis are required to meet the UIP radiological pattern criteria6 (Figure 1). On lung pathology, UIP is defined by the presence of dense fibrosis with architectural distortion, a subpleural predominance and/or pareseptal distribution, fibroblast foci and patchy involvement of the lung6 (Figure 2). Although the UIP pattern is characteristic of IPF it is not pathognomonic and thus is also frequently encountered in patients with other forms of ILD, most commonly in RA-ILD and chronic HP23,24.
Figure 1:

High-resolution computed tomography image of usual interstitial pneumonia pattern - subpleural and basal predominance and honeycombing with or without peripheral traction bronchiectasis or bronchiolectasis.
Figure 2:
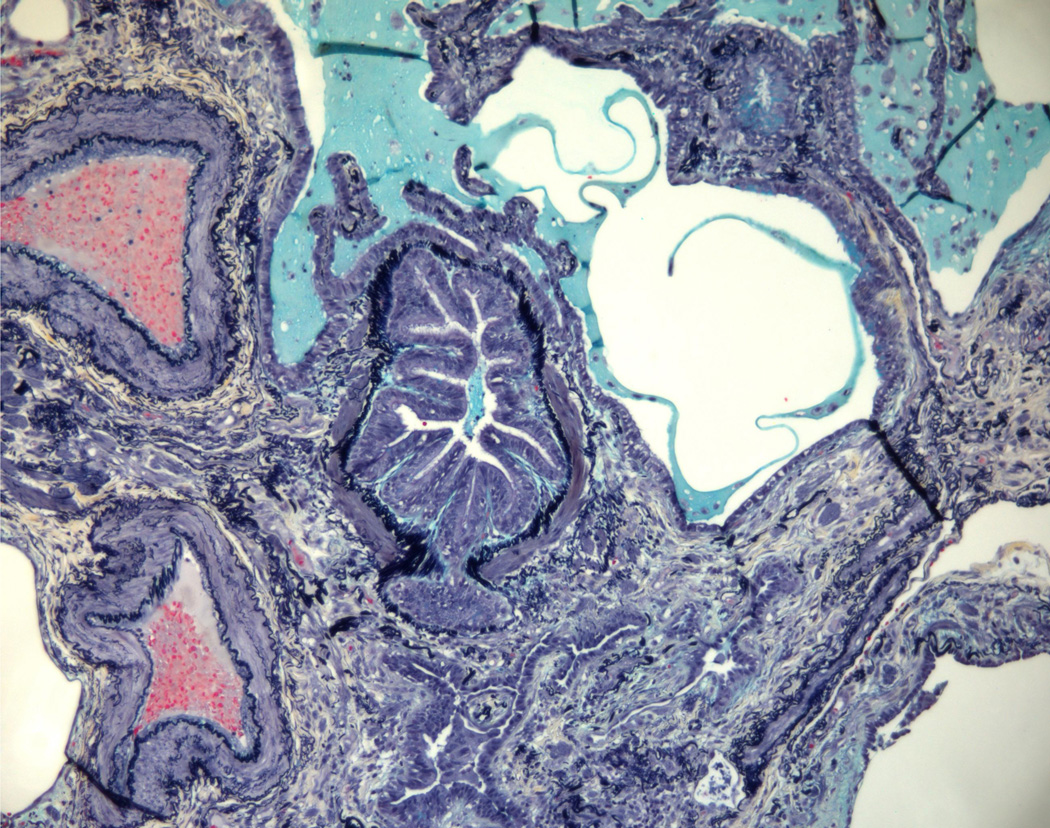
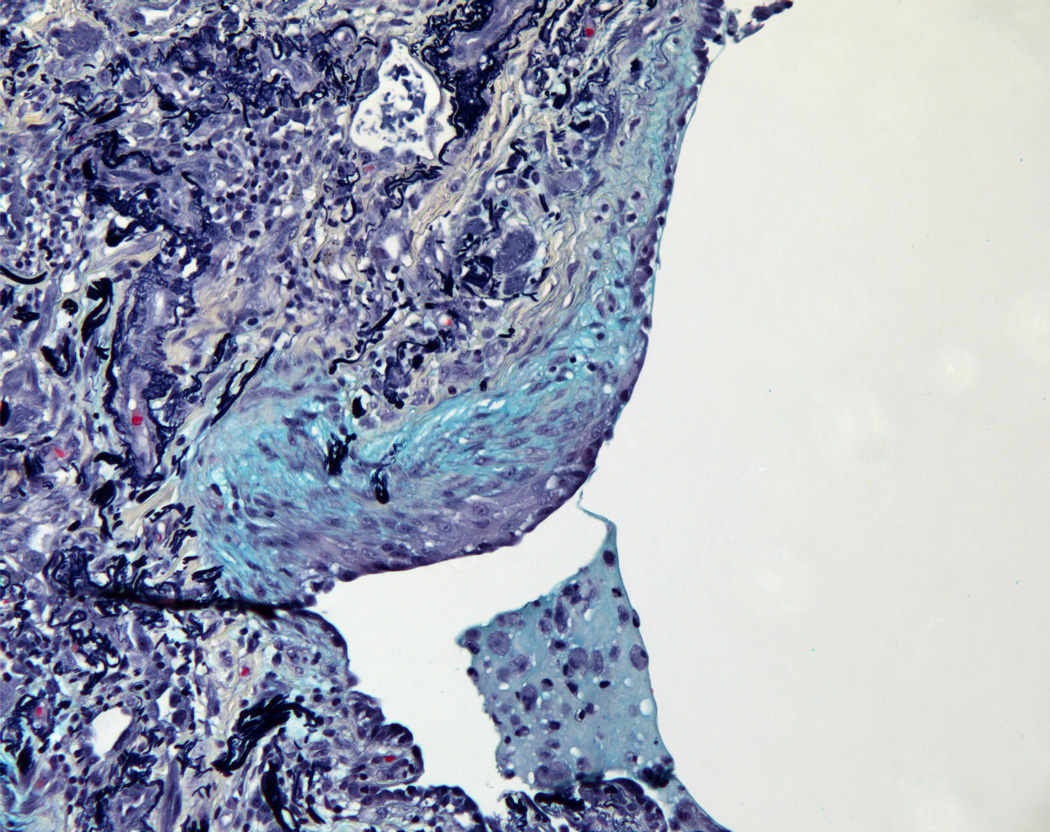
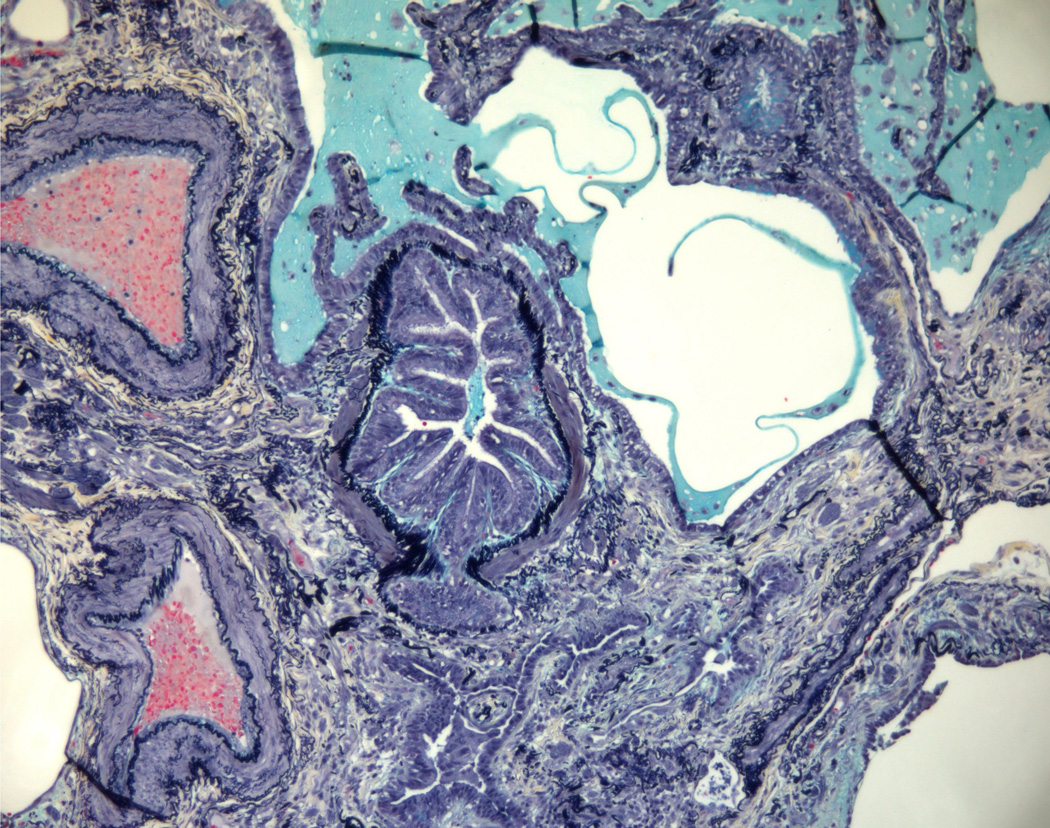
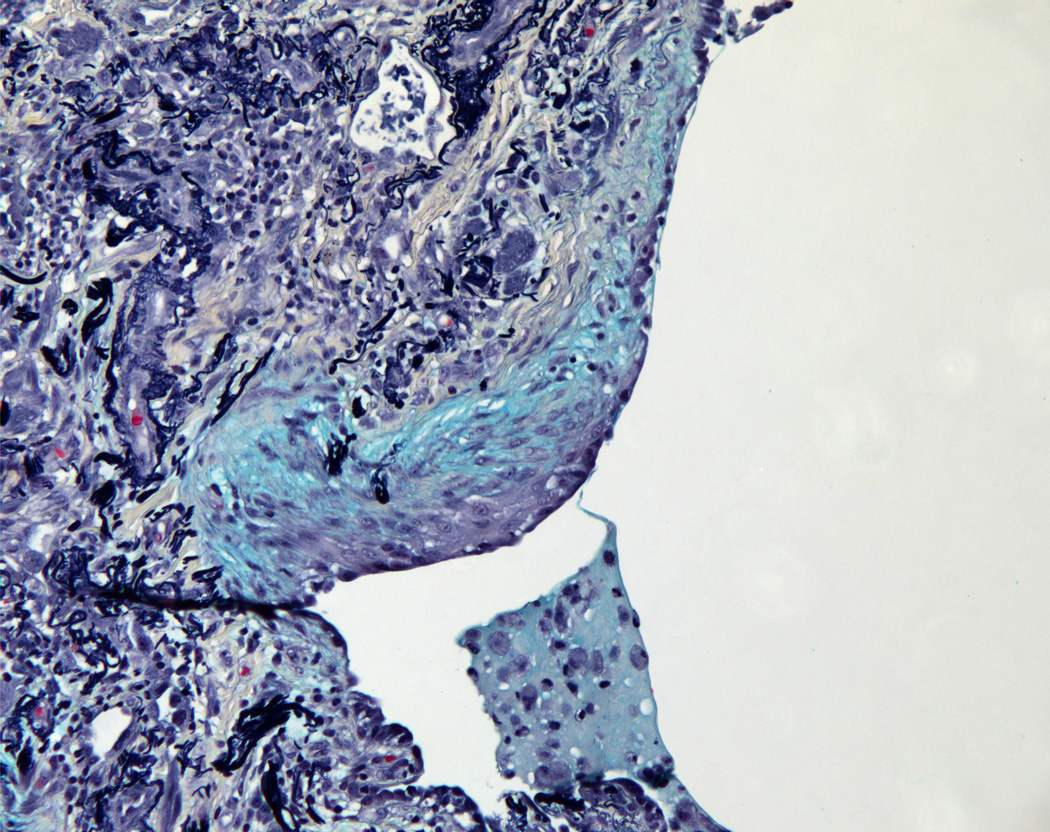
(2A) Pentachrome (Movat) stain of peripheral honeycombed lung in a patient with usual interstitial pneumonia (courtesy of Dr. Carlyne Cool). The honeycomb spaces are filled with mucus and lined by bronchiolar-type epithelium. The pleura is at the bottom right. Black = elastic tissue; yellow = mature fibrosis; blue = mucopolysaccharides (including mucus and myofibroblastic/fibroblastic tissue); red = fibrin blood; purple = nuclei. (2B) Pentachrome (Movat) stain of fibroblast focus (blue). The focus is capped by cuboidal epithelial cells and overlies an area of dense fibrosis/elastic tissue.
Idiopathic UIP
Idiopathic pulmonary fibrosis (IPF)
IPF is a progressive fibrotic ILD characterized by the presence of the usual interstitial pneumonia pattern on imaging or on lung pathology in the absence of any other known cause of ILD (e.g. CTD, occupational or environmental exposure or drug toxicity)6,25. IPF is a disease of the aging population and is more common in men and former smokers26. Although the disease course of IPF is heterogeneous and unpredictable in individual patients, it is associated with a limited survival time of 3 to 5 years after diagnosis25. While still considered to be a rare disease, IPF is the most common and most studied ILD. In the past years, our understanding of IPF has evolved and is now considered a disease of epithelial cell dysfunction resulting from a complex interaction between genetic and environmental risk factors27,28.
Several gene variants have been linked to an increased risk of IPF in sporadic and familial cases26. First, the presence of the MUC5B promoter minor-allele single nucleotide polymorphism (SNP) rs35705950 is strongly associated with an increase in IPF susceptibility29,30. When present, the variant allele rs35705950 SNP leads to an up-regulation of MUCB expression in the lungs, suggesting a role for airway mucins in the pathogenesis of IPF29,31. Paradoxically, this same MUC5B polymorphism has been found to be associated with better survival in patients with IPF32. Second, genome-wide association studies have identified the genetic variant rs5743890 in TOLLIP, an important regulator of innate immune responses. The TOLLIP mutation has also been associated with a higher risk of IPF33,34. Finally, telomere dysfunction and shortening is believed to play a role in the pathobiology of IPF with telomere attrition being one of the aging-associated processes playing a role in disease development35. Higher susceptibility to IPF was found in patients exhibiting mutations in the genes TERT, TERC, PARN and RTEL1, parts of the telomerase complex 35–39. Short telomere length (TL) has also been found to be predictive of mortality among patients with IPF40.
Disease behavior of progressive fibrotic ILD has also been observed in patients with other ILD diagnosis3,41. Most notably, this progressive disease phenotype has been observed and characterized in patients with rheumatoid arthritis-associated ILD (RA-ILD) and chronic HP 23,42,43.
Non-idiopathic Forms of UIP
Rheumatoid arthritis-associated interstitial lung disease (RA-ILD)
Rheumatoid arthritis (RA) is a connective tissue disease characterized by inflammatory arthritis and highly prevalent extra-articular and systemic manifestations44,45. ILD is one of the most common pulmonary manifestations in RA and the presence of ILD significantly impairs patient disease course, prognosis, and health-related quality of life45–48. Patients with RA-ILD often exhibit clinical characteristics similar to those of patients with IPF. Further, the prevalence of UIP pattern both on HRCT or surgical lung biopsy is higher in RA-ILD than in other types of CTD-ILD23,49. In RA-ILD, the UIP pattern of disease appears to be more frequent in older male patients with a positive history of smoking and is associated with a poorer prognosis and a natural history similar to IPF50–55. Additionally, RA-ILD shares several risk factors for mortality with IPF25,52,56. Patient variables like age, male gender, pulmonary function tests (forced vital capacity and diffusion capacity of the lung for carbon monoxide), extent of fibrosis and presence of the UIP pattern are known to be significant predictors of mortality in both diseases52,57–59.
RA-ILD also shares some genetic risk factors with IPF 55,60,61. Whole exome sequencing in a cohort of patients with RA-ILD identified heterozygous mutations in the TERT, RTEL1, PARN and SFTPC coding regions55,62. Those mutations, previously described to be prevalent in patients with IPF, are similarly linked to ILD susceptibility in RA. Further, the prevalence of patients with short telomere length (TL), below the tenth percentile, is similar in patients with RA-ILD and IPF61. Last, the presence of the MUC5B promoter variant rs5705950 is a strong risk factor for RA-ILD and is associated with the radiological pattern of UIP among RA-ILD patients60,61. The MUC5B promoter variant could represent a shared risk factor for the UIP pattern and may be a more generalizable genetic risk variant across various ILD diagnoses29,60,63.
Further, serum biomarkers that have been studied in IPF are being increasingly recognized in patients with RA-ILD64,65. For example, a peripheral biomarker signature combining matrix metalloproteinase (MMP7), pulmonary and activation-regulated chemokine (PARC) and surfactant protein D (SP-D) combined with clinical risk factors may enhance the detection of ILD in patients with RA64.
Chronic hypersensitivity pneumonitis (chronic HP)
HP is a complex disease thought to result from an repetitive exposure to one of the various causative organic antigens known to initiate an inflammatory response in the lungs of individuals with a genetic predisposition66. While some patients with HP may experience a complete resolution of their lung disease, others will go on to develop pulmonary fibrosis and present chronic symptoms similar to those of IPF43.
Historically, patients with HP were classified according to their clinical manifestations and symptom duration as having either acute, subacute or chronic HP67,68. More recently, it was proposed patients should instead be categorized into acute or chronic HP based on the absence or presence of fibrosis on radiology or lung biopsy42,43. The presence and extent of such fibrotic changes was found to represent a better surrogate of disease behavior in patients with HP since those who develop pulmonary fibrosis often experience chronic and progressive disease associated with a poorer prognosis24,69,70. Accordingly, presence of the UIP pattern on HRCT or surgical lung biopsy is known to be associated with an increased mortality among patients with chronic HP24,71.
Further, the presence of short TL and the MUC5B promoter variant rs35705950 are known to be associated with extent of fibrosis, the histopathologic pattern of UIP and reduced survival in patients with chronic HP63,72. These recent findings suggest that among patients with chronic HP, individuals with short TL or MUC5B rs35705950 may be at higher risk to develop pulmonary fibrosis63. These data support a shared genetic risk profile, and consequently suggest a shared pathobiology between chronic HP and IPF29,33.
Finally, on surgical lung biopsy and/or explanted lungs, patients with a UIP pattern of fibrosis and a diagnosis other than IPF (either chronic HP, CTD-ILD or unclassifiable ILD) also exhibit molecular markers of telomere dysfunction and senescence commonly found in IPF73. Interestingly, patients with non-IPF ILDs and a pathologic UIP pattern have shorter telomeres compared to age-matched healthy controls and show elevated levels of p16 and p21, 2 known molecular markers of senescence in alveolar type II cells73.
Treatment of ILD
Idiopathic pulmonary fibrosis
The most recent international guidelines recommend the use of either pirfenidone or nintedanib for the treatment of patients with IPF7. Both medications have been shown to reduce the decline of forced vital capacity (FCV) by about 50% after 1 year on therapy 8,9,26. Additionally, these drugs also improve other important outcomes: pirfenidone reduces the incidence of respiratory-related hospitalizations, nintedanib decreases the frequency of acute exacerbations and pooled analysis suggest they may also both reduce mortality 9,74–76.
Historically, prior to the era of antifibrotic medications, the recommended therapeutic approach for IPF was the combination of azathioprine, prednisone and N-acetylcysteine5. However, in the PANTHER-IPF trial, patients receiving this combination of drugs were found to be at increased risk of death and hospitalization compared to those on placebo15. A secondary data analysis of the PANTHER-IPF data has recently demonstrated that a high proportion of patients in the trial (62%) had a TL inferior to the tenth percentile. Further, in those receiving the combination of azathioprine, prednisone and N-acetylcysteine, having a short TL was associated with an increased risk of death, lung transplantation, hospitalization or lung function decline77. These results raise several questions relative to the safety of the use of immunosuppressive therapy in patients with other types of ILDs that exhibit shared clinical, genetic and biologic characteristics with IPF.
Rheumatoid arthritis-associated interstitial lung disease
Immunosuppressive therapy is considered a central part of the management of patients with RA-ILD78,79. Most of the evidence guiding the treatment of CTD-ILD and RA-ILD emerges from studies conducted in populations of patients with scleroderma-related ILD (Scl-ILD). Prospective randomized clinical trials have demonstrated the safety and efficacy of immunosuppressive therapies (i.e. cyclophosphamide and mycophenolate mofetil (MMF)) in Scl-ILD12,13,80. However, the improvement in lung function associated with both these medications remains modest. Additionally, several retrospective studies have shown lung function stabilization or improvement and reassuring tolerability in patients with various types of CTD-ILD receiving immunosuppressive medications81–83. Despite those favorable results, many patients with RA-ILD still experience disease progression associated with a significant impact on life expectancy49,84. The optimal medical management of these patients remains unknown and there is a pressing need for new therapeutic agents and a more comprehensive treatment algorithm. Recently, a phase 2 open-label study demonstrated pirfenidone can be well tolerated in patients with Scl-ILD and paved the way for further research evaluating the use and efficacy of antifibrotics in patients with RA-ILD and other CTD-ILD85.
Chronic hypersensitivity pneumonitis
Identification and removal of the causal antigen remains central to the medical management of chronic HP. However, many patients present with a high burden of symptoms or have disease progression despite antigen remediation and require additional therapy42,43. Unfortunately, few prospective studies have evaluated the different therapeutic approaches for HP. In patients with acute HP, corticosteroid use has been shown to alleviate symptoms, although it did not lead to better long-term outcomes when compared to antigen eradication alone86,87. The management of chronic HP remains even more challenging. Immunosuppressive agents like MMF and azathioprine (AZA) may be beneficial and help stabilize lung function, although further prospective studies are needed to validate their effectiveness in patients with chronic HP10,11.
The way forward
Several unanswered questions remain regarding the optimal management of patients with ILD. Patients with various ILD diagnoses exhibiting a UIP pattern represent a huge challenge for clinicians and create a pressing need for the medical community. Considering the several shared attributes between IPF and other progressive forms of ILD with a UIP pattern, the available IPF medications may also represent effective therapies in these other diseases. Currently, there are several on-going trials evaluating the effectiveness of nintedanib and pirfenidone in non-IPF ILDs, including Scl-ILD, unclassifiable ILD, progressive fibrosing ILD, RA-ILD, and chronic HP (Table 1). If these studies yield positive results, the landscape for the management of patients with ILD will likely be greatly modified and antifibrotic medications may have relevance across various ILD subtypes. Perhaps future research will lead to changes in the diagnostic approach and management of ILD. One possibility includes moving away from the classification of ILD by specific etiology, to a more personalized approach involving the identification of the underlying pattern, genetic risk profile and disease behavior of the ILD. Nonetheless, some elements of the management will always remain specific to the clinical diagnosis of ILD, such as antigen remediation in HP and treatment of extra-pulmonary manifestations in patients with CTD-ILD.
Table 1:
Randomized clinical trials evaluating antifibrotics in interstitial lung disease other than idiopathic pulmonary fibrosis
| Study name | Trial Registration number | Patient population | |
|---|---|---|---|
| SENSCIS | A randomized, placebo-controlled clinical trial of nintedanib in patients with systemic sclerosis-associated interstitial lung disease88 | NCT02597933 | SSc-ILD |
| SLS III | Scleroderma Lung Study III: combining the anti-fibrotic effects of pirfenidone with mycophenolate for treating scleroderma-related interstitial lung disease | NCT03221257 | SSc-ILD |
| PF-ILD | A double-blind, randomized, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease41 | NCT02999178 | Progressive fibrosing lung disease |
| Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: design of a double-blind, randomized, placebo-controlled phase II trial89 | NCT03099187 | Fibrosing unclassifiable interstitial lung disease | |
| TRAIL1 | Phase II Study of Pirfenidone in Patients With RAILD | NCT02808871 | RA-ILD |
| A randomized, double-blind, placebo-controlled, study of efficacy and safety of pirfenidone in patients with fibrotic hypersensitivity pneumonitis | NCT02958917 | Fibrotic hypersensitivity pneumonitis |
Abbreviations
SSc-ILD: systemic sclerosis-associated interstitial lung disease
RA-ILD: rheumatoid arthritis-associated interstitial lung disease
Conclusion
Several ILDs, particularly in those with the UIP pattern, share common epidemiologic, genetic and pathobiologic features with IPF. The optimal management of these patients with the UIP pattern remains unclear and more evidence is needed to clarify the best treatment approach. Areas of treatment uncertainty include the role of immunosuppression and antifibrotic therapy in this patient population. On-going clinical trials are currently evaluating the effectiveness of antifibrotic medications in various ILD diagnoses and will likely shape how we approach ILD in the future.
Key points:
IPF shares several clinical, genetic and biological features with other ILD diagnoses such as chronic RA-ILD and chronic HP
The UIP pattern of fibrosis is encountered in patients with RA-ILD and chronic HP and is associated with a worse prognosis.
Genetic risk factors well characterized in IPF (e.g.MUC5B promoter variant rs5705950, telomerase complex mutations and short telomere lengths) are also linked to an increase susceptibility to pulmonary fibrosis in RA and chronic HP.
Financial support and sponsorship
NIH/NHLBI K23 HL 138131
Conflicts of interest
JM has received consulting fees from Boehringer Ingelheim and Hoffman-La Roche unrelated to the current work, JSL has received consulting fees from Celgene, Boehringer Ingelheim, Genentech, and Galapagos, unrelated to the current work
REFERENCES
- 1.Schwarz MI, King TE. Interstitial Lung Disease - 5th ed.: People’s Medical Publishing House; USA; 2010. [Google Scholar]
- 2.American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med 2002;165:277–304. [DOI] [PubMed] [Google Scholar]
- 3.Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013;188:733–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradley B, Branley HM, Egan JJ, et al. Interstitial lung disease guideline: the British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax 2008;63 Suppl 5:v1–58. [DOI] [PubMed] [Google Scholar]
- 5.Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med 2018;198:e44–e68. * New international guidelines providing radiologic classification and diagnostic algorithm for idiopathic pulmonary fibrosis.
- 7.Raghu G, Rochwerg B, Zhang Y, et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med 2015;192:e3–19. [DOI] [PubMed] [Google Scholar]
- 8.King TE Jr., Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. The New England journal of medicine 2014;370:2083–92. [DOI] [PubMed] [Google Scholar]
- 9.Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. The New England journal of medicine 2014;370:2071–82. [DOI] [PubMed] [Google Scholar]
- 10.Morisset J, Johannson KA, Vittinghoff E, et al. Use of Mycophenolate Mofetil or Azathioprine for the Management of Chronic Hypersensitivity Pneumonitis. Chest 2017;151:619–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adegunsoye A, Oldham JM, Fernandez Perez ER, et al. Outcomes of immunosuppressive therapy in chronic hypersensitivity pneumonitis. ERJ open research 2017;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. The Lancet Respiratory medicine 2016;4:708–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. The New England journal of medicine 2006;354:2655–66. [DOI] [PubMed] [Google Scholar]
- 14.Lee SH, Park MS, Kim SY, et al. Factors affecting treatment outcome in patients with idiopathic nonspecific interstitial pneumonia: a nationwide cohort study. Respir Res 2017;18:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raghu G, Anstrom KJ, King TE Jr., Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. The New England journal of medicine 2012;366:1968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cottin V, Hirani NA, Hotchkin DL, et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. European respiratory review : an official journal of the European Respiratory Society 2018;27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wells AU, Brown KK, Flaherty KR, Kolb M, Thannickal VJ. What’s in a name? That which we call IPF, by any other name would act the same. The European respiratory journal 2018;51. [DOI] [PubMed] [Google Scholar]
- 18.Richeldi L, Varone F, Bergna M, et al. Pharmacological management of progressive-fibrosing interstitial lung diseases: a review of the current evidence. European respiratory review : an official journal of the European Respiratory Society 2018;27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cottin V, Wollin L, Fischer A, Quaresma M, Stowasser S, Harari S. Fibrosing interstitial lung diseases: knowns and unknowns. European respiratory review : an official journal of the European Respiratory Society 2019;28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olson AL, Gifford AH, Inase N, Fernandez Perez ER, Suda T. The epidemiology of idiopathic pulmonary fibrosis and interstitial lung diseases at risk of a progressive-fibrosing phenotype. European respiratory review : an official journal of the European Respiratory Society 2018;27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liebow AACC. The interstitial pneumonias, frontiers of pulmonary radiology. New York: Grune and Stratton; 1969. [Google Scholar]
- 22.Noble PW, Homer RJ. Back to the future: historical perspective on the pathogenesis of idiopathic pulmonary fibrosis. American journal of respiratory cell and molecular biology 2005;33:113–20. [DOI] [PubMed] [Google Scholar]
- 23.Kim EJ, Elicker BM, Maldonado F, et al. Usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J 2010;35:1322–8. [DOI] [PubMed] [Google Scholar]
- 24.Wang P, Jones KD, Urisman A, et al. Pathologic Findings and Prognosis in a Large Prospective Cohort of Chronic Hypersensitivity Pneumonitis. Chest 2017. [DOI] [PubMed]
- 25.Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;183:431–40. [DOI] [PubMed] [Google Scholar]
- 26.Lederer DJ, Martinez FJ. Idiopathic Pulmonary Fibrosis. The New England journal of medicine 2018;378:1811–23. [DOI] [PubMed] [Google Scholar]
- 27.Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet (London, England) 2017;389:1941–52. [DOI] [PubMed] [Google Scholar]
- 28.Wolters PJ, Blackwell TS, Eickelberg O, et al. Time for a change: is idiopathic pulmonary fibrosis still idiopathic and only fibrotic? Lancet Respir Med 2018;6:154–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. The New England journal of medicine 2011;364:1503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee MG, Lee YH. A meta-analysis examining the association between the MUC5B rs35705950 T/G polymorphism and susceptibility to idiopathic pulmonary fibrosis. Inflammation research : official journal of the European Histamine Research Society [et al] 2015;64:463–70. [DOI] [PubMed] [Google Scholar]
- 31.Nakano Y, Yang IV, Walts AD, et al. MUC5B Promoter Variant rs35705950 Affects MUC5B Expression in the Distal Airways in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med 2016;193:464–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peljto AL, Zhang Y, Fingerlin TE, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. Jama 2013;309:2232–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noth I, Zhang Y, Ma SF, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med 2013;1:309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allen RJ, Porte J, Braybrooke R, et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: a genome-wide association study. Lancet Respir Med 2017;5:869–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am J Respir Crit Care Med 2014;189:1161–72. [DOI] [PubMed] [Google Scholar]
- 36.Stuart BD, Choi J, Zaidi S, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nature genetics 2015;47:512–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fingerlin TE, Murphy E, Zhang W, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nature genetics 2013;45:613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. The New England journal of medicine 2007;356:1317–26. [DOI] [PubMed] [Google Scholar]
- 39.Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proceedings of the National Academy of Sciences of the United States of America 2007;104:7552–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stuart BD, Lee JS, Kozlitina J, et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation. Lancet Respir Med 2014;2:557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flaherty KR, Brown KK, Wells AU, et al. Design of the PF-ILD trial: a double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ open respiratory research 2017;4:e000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vasakova M, Morell F, Walsh S, Leslie K, Raghu G. Hypersensitivity Pneumonitis: Perspectives in Diagnosis and Management. Am J Respir Crit Care Med 2017. [DOI] [PubMed]
- 43.Salisbury ML, Myers JL, Belloli EA, Kazerooni EA, Martinez FJ, Flaherty KR. Diagnosis and Treatment of Fibrotic Hypersensitivity Pneumonia. Where We Stand and Where We Need to Go. Am J Respir Crit Care Med 2016. [DOI] [PMC free article] [PubMed]
- 44.Gabriel SE. The epidemiology of rheumatoid arthritis. Rheumatic diseases clinics of North America 2001;27:269–81. [DOI] [PubMed] [Google Scholar]
- 45.Turesson C, O’Fallon WM, Crowson CS, Gabriel SE, Matteson EL. Extra-articular disease manifestations in rheumatoid arthritis: incidence trends and risk factors over 46 years. Ann Rheum Dis 2003;62:722–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bongartz T, Nannini C, Medina-Velasquez YF, et al. Incidence and mortality of interstitial lung disease in rheumatoid arthritis: a population-based study. Arthritis Rheum 2010;62:1583–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olson AL, Swigris JJ, Sprunger DB, et al. Rheumatoid arthritis-interstitial lung disease-associated mortality. Am J Respir Crit Care Med 2011;183:372–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Natalini JG, Swigris JJ, Morisset J, et al. Understanding the determinants of health-related quality of life in rheumatoid arthritis-associated interstitial lung disease. Respiratory medicine 2017;127:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Assayag D, Elicker BM, Urbania TH, et al. Rheumatoid arthritis-associated interstitial lung disease: radiologic identification of usual interstitial pneumonia pattern. Radiology 2014;270:583–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee HK, Kim DS, Yoo B, et al. Histopathologic pattern and clinical features of rheumatoid arthritis-associated interstitial lung disease. Chest 2005;127:2019–27. [DOI] [PubMed] [Google Scholar]
- 51.Solomon JJ, Ryu JH, Tazelaar HD, et al. Fibrosing interstitial pneumonia predicts survival in patients with rheumatoid arthritis-associated interstitial lung disease (RA-ILD). Respir Med 2013;107:1247–52. [DOI] [PubMed] [Google Scholar]
- 52.Assayag D, Lubin M, Lee JS, King TE, Collard HR, Ryerson CJ. Predictors of mortality in rheumatoid arthritis-related interstitial lung disease. Respirology 2014;19:493–500. [DOI] [PubMed] [Google Scholar]
- 53.Solomon JJ, Chung JH, Cosgrove GP, et al. Predictors of mortality in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J 2015. [DOI] [PubMed]
- 54.Song JW, Lee HK, Lee CK, et al. Clinical course and outcome of rheumatoid arthritis-related usual interstitial pneumonia. Sarcoidosis, vasculitis, and diffuse lung diseases : official journal of WASOG / World Association of Sarcoidosis and Other Granulomatous Disorders 2013;30:103–12. [PubMed] [Google Scholar]
- 55.Juge PA, Borie R, Kannengiesser C, et al. Shared genetic predisposition in rheumatoid arthritis-interstitial lung disease and familial pulmonary fibrosis. Eur Respir J 2017;49. [DOI] [PubMed] [Google Scholar]
- 56.Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Annals of internal medicine 2012;156:684–91. [DOI] [PubMed] [Google Scholar]
- 57.Morisset J, Vittinghoff E, Lee BY, et al. The performance of the GAP model in patients with rheumatoid arthritis associated interstitial lung disease. Respir Med 2017;127:51–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yunt ZX, Chung JH, Hobbs S, et al. High resolution computed tomography pattern of usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease: Relationship to survival. Respir Med 2017;126:100–4. [DOI] [PubMed] [Google Scholar]
- 59.Solomon JJ, Chung JH, Cosgrove GP, et al. Predictors of mortality in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J 2016;47:588–96. [DOI] [PubMed] [Google Scholar]
- 60. Juge PA, Lee JS, Ebstein E, et al. MUC5B Promoter Variant and Rheumatoid Arthritis with Interstitial Lung Disease. The New England journal of medicine 2018;379:2209–19. ** Recent research exploring the genetics of rheumatoid arthritis related interstitial lung disease and the role of MUC5B promoter variant as a risk factor for interstitial lung disease
- 61.Newton CA, Oldham JM, Ley B, et al. Telomere Length and Genetic Variant Associations with Interstitial Lung Disease Progression and Survival. Eur Respir J 2019. [DOI] [PMC free article] [PubMed]
- 62.Wolters PJ. A recurring theme in pulmonary fibrosis genetics. The European respiratory journal 2017;49. [DOI] [PubMed] [Google Scholar]
- 63.Ley B, Newton CA, Arnould I, et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: an observational cohort-control study. Lancet Respir Med 2017. [DOI] [PMC free article] [PubMed]
- 64.Doyle TJ, Patel AS, Hatabu H, et al. Detection of Rheumatoid Arthritis-Interstitial Lung Disease Is Enhanced by Serum Biomarkers. Am J Respir Crit Care Med 2015;191:1403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen J, Doyle TJ, Liu Y, et al. Biomarkers of rheumatoid arthritis-associated interstitial lung disease. Arthritis & rheumatology (Hoboken, NJ) 2015;67:28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Selman M, Pardo A, King TE, Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 2012;186:314–24. [DOI] [PubMed] [Google Scholar]
- 67.Lacasse Y, Selman M, Costabel U, et al. Clinical diagnosis of hypersensitivity pneumonitis. Am J Respir Crit Care Med 2003;168:952–8. [DOI] [PubMed] [Google Scholar]
- 68.Schuyler M, Cormier Y. The diagnosis of hypersensitivity pneumonitis. Chest 1997;111:534–6. [DOI] [PubMed] [Google Scholar]
- 69.Mooney JJ, Elicker BM, Urbania TH, et al. Radiographic fibrosis score predicts survival in hypersensitivity pneumonitis. Chest 2013;144:586–92. [DOI] [PubMed] [Google Scholar]
- 70.Kern RM, Singer JP, Koth L, et al. Lung transplantation for hypersensitivity pneumonitis. Chest 2015;147:1558–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chung JH, Montner SM, Adegunsoye A, et al. CT findings associated with survival in chronic hypersensitivity pneumonitis. European radiology 2017;27:5127–35. [DOI] [PubMed] [Google Scholar]
- 72.Newton CA, Batra K, Torrealba J, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. The European respiratory journal 2016;48:1710–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee SJ, La J, Aziz S, et al. Molecular Markers of Telomere Dysfunction and Senescence are Common Findings in the Usual Interstitial Pneumonia Pattern of Lung Fibrosis. European Respiratory Journal 2018; 52: Suppl. 62, OA5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ley B, Swigris J, Day BM, et al. Pirfenidone Reduces Respiratory-related Hospitalizations in Idiopathic Pulmonary Fibrosis. American journal of respiratory and critical care medicine 2017;196:756–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. The Lancet Respiratory medicine 2017;5:33–41. [DOI] [PubMed] [Google Scholar]
- 76.Richeldi L, Cottin V, du Bois RM, et al. Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS((R)) trials. Respiratory medicine 2016;113:74–9. [DOI] [PubMed] [Google Scholar]
- 77. Newton CA, Zhang D, Oldham JM, et al. Telomere Length and Use of Immunosuppressive Medications in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med 2018. *** This recent publication explores the association between short telomere length and various outcomes of patients with idiopathic pulmonary fibrosis while receiving immunusopressive therapy
- 78.Volkmann ER, Tashkin DP. Treatment of Systemic Sclerosis-related Interstitial Lung Disease: A Review of Existing and Emerging Therapies. Annals of the American Thoracic Society 2016;13:2045–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee JS, Fischer A. Current and emerging treatment options for interstitial lung disease in patients with rheumatic disease. Expert review of clinical immunology 2016;12:509–20. [DOI] [PubMed] [Google Scholar]
- 80.Hoyles RK, Ellis RW, Wellsbury J, et al. A multicenter, prospective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis and rheumatism 2006;54:3962–70. [DOI] [PubMed] [Google Scholar]
- 81.Swigris JJ, Olson AL, Fischer A, et al. Mycophenolate mofetil is safe, well tolerated, and preserves lung function in patients with connective tissue disease-related interstitial lung disease. Chest 2006;130:30–6. [DOI] [PubMed] [Google Scholar]
- 82.Fischer A, Brown KK, Du Bois RM, et al. Mycophenolate mofetil improves lung function in connective tissue disease-associated interstitial lung disease. J Rheumatol 2013;40:640–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oldham JM, Lee C, Valenzi E, et al. Azathioprine response in patients with fibrotic connective tissue disease-associated interstitial lung disease. Respiratory medicine 2016;121:117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Winstone TA, Assayag D, Wilcox PG, et al. Predictors of mortality and progression in scleroderma-associated interstitial lung disease: a systematic review. Chest 2014;146:422–36. [DOI] [PubMed] [Google Scholar]
- 85.Khanna D, Albera C, Fischer A, et al. An Open-label, Phase II Study of the Safety and Tolerability of Pirfenidone in Patients with Scleroderma-associated Interstitial Lung Disease: the LOTUSS Trial. J Rheumatol 2016;43:1672–9. [DOI] [PubMed] [Google Scholar]
- 86.Monkare S. Influence of corticosteroid treatment on the course of farmer’s lung. Eur J Respir Dis 1983;64:283–93. [PubMed] [Google Scholar]
- 87.Kokkarinen JI, Tukiainen HO, Terho EO. Effect of corticosteroid treatment on the recovery of pulmonary function in farmer’s lung. Am Rev Respir Dis 1992;145:3–5. [DOI] [PubMed] [Google Scholar]
- 88.Distler O, Highland KB, Gahlemann M, et al. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. The New England journal of medicine 2019. [DOI] [PubMed]
- 89.Maher TM, Corte TJ, Fischer A, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: design of a double-blind, randomised, placebo-controlled phase II trial. BMJ open respiratory research 2018;5:e000289. [DOI] [PMC free article] [PubMed] [Google Scholar]