

Abstract
Ten subjects received 600 to 1,200 mg of the human immunodeficiency virus type 1 (HIV-1) protease inhibitor ritonavir per day. Following 2 weeks of therapy, plasma HIV RNA levels decreased by a mean of 1.57 (range, 0.89 to 1.96) log units. With continued therapy, HIV RNA levels began to rise in eight subjects. The initial rise in plasma RNA levels was temporally associated with the development and quantitative increase in the V82 resistance mutation. Doubling times of the V82A mutant virus were estimated to be 2.4 to 4.8 days. An L63P/A mutation was commonly present at baseline even in subjects with a durable virologic response. The concomitant acquisition of an L63P/A mutation with the V82A/F mutation at the time when plasma RNA levels rebounded suggests a role for the L63P/A mutation in improving the fitness of the V82A/F mutation. Subsequent additional genotypic changes at codons 54 and 84 were often associated with further increases in plasma RNA levels. Ongoing viral replication in the presence of drugs resulted in the appearance of additional genotypic changes, including the L90M saquinavir resistance mutation, and decreased phenotypic susceptibility. The relative fitness of the protease V82A ritonavir resistance mutation and reverse transcriptase T215Y/F zidovudine resistance mutation following drug withdrawal were estimated to be 96 to 98% that of the wild type. Durability of the virologic response was associated with plasma RNA levels at the nadir. A virologic response beyond 60 days was not observed unless plasma HIV RNA levels were suppressed below 2,000 copies/ml, consistent with estimates from V82A doubling times for selection of a single resistance mutation to dominate the replicating population.
Efforts to treat human immunodeficiency virus type 1 (HIV-1)-infected individuals have been directed towards the development of inhibitors of viral enzymes necessary for viral replication. The viral reverse transcriptase (RT) is the target of most drugs now in use. Virus variants with reduced susceptibility to several of these drugs have been selected in vitro (2, 11, 46) and have emerged with therapy in vivo (22, 42, 44). Genotypic changes in the RT gene following long-term zidovudine (ZDV) therapy have been associated with phenotypic losses in sensitivity to ZDV (21, 22). Establishing the in vivo relationship between the development of genotypic resistance and changes in plasma HIV RNA levels during ZDV monotherapy, however, required the development of methodologies to measure plasma HIV RNA levels and to determine the amounts of genotypic variants in a population. Recently correlations of the development of genotypic resistance in plasma virus RNA with the loss of viral suppression in vivo have been demonstrated for the K70R mutation with ZDV (9) and the M184V mutation with lamivudine (42). Interestingly, the K70R mutation is associated with only an eightfold decrease in susceptibility to ZDV in vitro (21) while the M184V mutation results in a >500-fold decrease in susceptibility to lamivudine (46). Similarly, with the nonnucleoside RT inhibitors, such as delavirdine and nevirapine, the Y181C mutation results in >100-fold reduction in drug susceptibility in vitro (37) and has been associated with loss of HIV suppression in patients (12, 37, 39).
A new class of drugs has been evaluated in phase I/II clinical trials (8, 26). The target of these drugs is the HIV protease which is required for cleavage of the Gag and Gag-Pol polyproteins and production of infectious virus (20, 35). As with the RT inhibitors, HIV variants with reduced susceptibility to protease inhibitors have been isolated in vitro (15, 18, 25, 34). Characterization of these drug-resistant variants has revealed several changes in the protease amino acid sequence which have been associated with low-level (<10-fold) decreases in phenotypic susceptibility to a number of inhibitors (6, 7, 28). Some of these mutations result in a reduced susceptibility to more than one protease inhibitor (6, 7, 33, 41, 47).
We have previously described a differential hybridization assay for determination of relative amounts of wild-type (WT) virus and resistant mutants (MUT) in the plasma HIV RNA of infected individuals (10). The assay is linear and reproducible over a wide dynamic range. Here we apply this technology to detect increases in the amount of plasma virus with specific mutations in association with increases in plasma HIV RNA levels following treatment with ritonavir (Norvir). Molla et al. (28) have described an ordered accumulation of mutations during ritonavir therapy based on the frequency of mutations observed. Here we describe an ordered pattern of accumulation based on the accumulation of mutations associated with changes in viral load during ritonavir therapy. Furthermore, differential hybridization allowed estimates of the doubling times of MUT populations during viral load rebounds and of the fitness of the expanding viral populations. Linear regression analysis of the rate of reversion of MUT populations following cessation of therapy provided an estimate of fitness of the replicating MUT population. These data indicate that the doubling time of the expanding resistant population is short, that the fitness of the resistant population is very close to that of the WT virus, and, that as a result, reversion of MUT populations in the absence of selective pressure is a slow process. Finally, the durability of the virologic response was correlated with the maximum suppression of viral replication, suggesting that the appearance of virus containing multiple mutations is a result of continued selection during ongoing viral replication.
(This study was presented in part at the 3rd Conference on Retroviruses and Related Infections, January 28 to February 1, 1996.)
MATERIALS AND METHODS
Patients and sample material.
The subjects represent a subset of the study subjects previously described (26). All subjects on previous antiretroviral therapy discontinued all medications except Pneumocystis carinii pneumonia prophylaxis for 2 weeks before therapy was initiated.
HIV-1 RNA quantification.
HIV-1 RNA in plasma was quantified by branched-DNA (bDNA) signal amplification-based hybridization with a Quantiplex HIV RNA assay kit (32). Plasma samples with values below the detection limit of the standard assay (10,000 HIV-1 RNA equivalents per ml, expressed as copies per milliliter) were tested by a modified bDNA assay with a detection limit of 500 HIV copies/ml (19).
RT-PCR of the HIV-1 protease gene.
Plasma virus RNA was prepared as previously described (16). PCR was performed as a modification of the procedure described by Eastman et al. (10). cDNA synthesis was performed on 10 μl of prepared RNA (100-μl equivalent of the starting plasma). The 100-μl reaction mixture included a buffer that contained 50 mM Tris (pH 8.3), 2.5 mM MgCl2, 10 mM KCl, 0.1 mg of bovine serum albumin/ml, 0.25 mM (each) deoxynucleoside triphosphate, and 30 pmol each of the primers PROT2083.UP (5′-GGAAGGACACCAAMTGAAAGA-3′, bp 1596 to 1616, HIV-1SF-2 [40]) and biotinylated PROT2662.DOWN (5′-ATTCCTGGCTTTAATTTTACTGG-3′, bp 2127 to 2149). Samples were heated to 65°C for 90 s, cooled to 42°C for 1 min before 200 U of Moloney murine leukemia virus RT (GIBCO/BRL) was added, and incubated at 42°C for 15 min. The samples were heated to 100°C for 2 min, then cooled to 55°C for 5 min, during which time 5 U of Taq polymerase (Perkin-Elmer, Norwalk, Conn.) was added. The temperature was raised to 72°C for 3 min, with 40 subsequent cycles of denaturation at 94°C for 1 min, annealing at 55°C for 1 min, and extension at 72°C for 3 min to generate a 554-bp fragment.
Analysis of PCR product.
For differential hybridization analysis, 10 μl of the PCR product was diluted in 90 μl of phosphate buffer, pH 5.5. Fifty microliters was dispensed to duplicate wells in which streptavidin (XENOPORE Corporation, Saddle Brook, N.J.) was covalently bound and incubated at 50°C for 30 min. The wells were washed, and the bound PCR product was denatured in 0.15 N NaOH for 5 min followed by washing. To the appropriate wells, 100 μl of hybridization solution (5× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 1% sodium dodecyl sulfate, 0.5% polyvinylpyrrolidone, 0.5% bovine serum albumin) containing either WT or MUT alkaline phosphatase-labeled probe (Table 1) was added and incubated for 1 h at the appropriate temperature. All wells were washed three times for 5 min each time at the appropriate temperature with prewarmed 1× SSC–0.1% sodium dodecyl sulfate, followed by three washes with prewarmed 1× SSC–0.1% Triton X-100, followed by four room-temperature washes in 1× SSC. For chemiluminescent detection, 50 μl of chemiluminescent substrate (LumiphosTM 530; Lumigen, Detroit, Mich.) was added to each well, incubated at 37°C for 30 min, and measured on a luminometer (Chiron, Emeryville, Calif.). In order to account for the amount of PCR product bound to each well, each well was stripped with 0.15 N NaOH and rehybridized with an alkaline phosphatase-labeled probe to a highly conserved region, the generic (GNR) probe, and washed as described above. The probes, probe sequences, map locations, hybridization temperatures, and wash temperatures are presented in Table 1. Controls for hybridizations with MUT probes were generated by site-directed mutagenesis (25). Alternatively, a biotinylated synthetic oligonucleotide containing both MUT probe and GNR probe target sequences was used as a hybridization control. HIV-1HXB2 was used as the WT control.
TABLE 1.
Description of hybridization probes
| Probe | Sequence | Locationa (nt) | Temp (°C)
|
|
|---|---|---|---|---|
| Hybridization | Wash | |||
| M46 WT | ACCAAAAATGATAGKRGG | 1934–1951 | 45 | 45 |
| M46I MUT | ACCAAAAATHATAGKRGG | 1934–1951 | 45 | 45 |
| M46L MUT | ACCAAAAYTGATAGKRGG | 1934–1951 | 45 | 45 |
| I54 WT | AGGTWTTATYAAAGTAA | 1958–1974 | 35 | 35 |
| I54V MUT | AGGTWTTGTYAAAGTAA | 1958–1974 | 35 | 35 |
| L63 WT | TCAGATACTYRTAGAAA | 1985–2001 | 45 | 40 |
| L63P MUT | TCAGATACCYRTAGAAA | 1985–2001 | 45 | 40 |
| L63A MUT | TCARATAGCYRTAGAAA | 1985–2001 | 45 | 40 |
| A71 WT | ACATAAAGCTRTAGGTA | 2009–2025 | 45 | 35 |
| A71V MUT | ACATAAAGTTRTAGGTA | 2009–2025 | 45 | 35 |
| V77 WT | GTATTAGTAGGRCCTA | 2028–2043 | 35 | 35 |
| V77I MUT | GTATTAATAGGRCCTA | 2028–2043 | 35 | 35 |
| V82 WT | ACMCCTGTCAACRTAAT | 2042–2057 | 50 | 35 |
| V82A MUT | TACMCCTGCCAACRTA | 2042–2057 | 50 | 35 |
| V82F MUT | TACMCCTTTCAACRTA | 2042–2057 | 35 | 35 |
| I84 WT | TDYCAACATAATTGG | 2048–2062 | 40 | 30 |
| I84V MUT | TDYCAACGTAATTGG | 2048–2062 | 40 | 30 |
| L90 WT | AGRAAYCTGTTRACTCA | 2064–2080 | 45 | 40 |
| L90M MUT | AGRAAYCTGATGACTCA | 2064–2080 | 45 | 40 |
| Protease GNR | CAGAGCCAACAGCCC | 1700–1714 | 45 | 40 |
HIV-1SF2 (40).
Estimation of MUT populations.
The relative proportions of MUT and WT populations for each probe set was initially determined. The relative light units (RLUs) for each sample hybridized with the WT- or MUT-specific probe were normalized to the respective RLUs hybridized with the GNR probe to account for the quantity of PCR product bound to each well (WT:GNRraw = WT RLUs/GNRWT RLUs; MUT:GNRraw = MUT RLUs/GNRMUT RLUs). Furthermore, these values were normalized to 100% WT and 100% MUT control values to control for the specific activity of the individual probes [WT:GNR = (WT:GNR)test/(WT:GNR)WTcontrol; MUT:GNR = (MUT:GNR)test/(MUT/GNR)MUTcontrol]. The portion of MUT species was estimated by multiplication of the plasma HIV RNA levels (HIV copies per milliliter) by the proportion of MUT [HIV copies per milliliter × MUT:GNR/(MUT:GNR + WT:GNR)].
Method for back calculating viral densities prior to initiation of therapy.
The method for back calculating viral densities makes use of CD4+ T-cell data to infer the growth rate of MUT viruses at previous time points. The model makes three main assumptions: (i) that viral growth can be described by means of the simple mathematical model presented by Perelson et al. (36), (ii) that peripheral blood CD4+ T-cell counts can be taken as a measure of the target cell densities, and (iii) that immune responses that may contribute to the death of infected cells and clearance of free virus do not change appreciably following the initiation of drug therapy. The model of Perelson et al. (36), when applied to MUT virus, consists of the following equations: dI/dt = βVT − δI and dV/dt = nδI − cV, where T is the density of target cells, I is the density of infected T cells, V is the density of free HIV, β is the infection rate, δ is the death rate, n is the number of virions released by an infected cell, and c is the clearance rate of free virus. The rate of change of a MUT virus, λ, under these equations can be shown to be
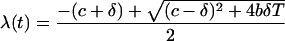 |
1 |
where b = nβ is a growth rate parameter characteristic of each virus.
The densities of MUT viruses at previous times were estimated through repeated application of the formula V(t − Δt) = V(t)e−λ(t)Δt, where V(t) is the viral density at time t and Δt is the amount of time between successive CD4+-T-cell counts. For purposes of back calculation, it was assumed that T is proportional to the density of CD4+ T cells (which generally increases as virus declines) but that b, c, and δ are constant. For the values of c and δ, published estimates were used (36). The growth rate parameter b was then estimated by evaluating the solution of equation (1) for b at time points where data existed on both CD4+-T-cell counts and the rate of change of MUT virus. In cases where there was more than one estimate for b, the average b was used.
Because the formula V(t − Δt) = V(t)e−λ(t)Δt is only an approximation (this becomes exact as Δt approaches zero), these numerical procedures were tested against simulated data sets in which the number of data points was similar to that in the actual trials. Baseline MUT virus densities estimated by this procedure generally came within about 10% of the true values, indicating that rounding errors in the back-calculation procedure do not seriously distort estimates for the densities of MUT viruses at baseline.
Exponential growth rate method for back calculating MUT densities.
MUT virus densities were also back calculated under the simplest assumption, that the rate of growth of MUT virus is constant from the time of drug initiation to viral rebound. This was accomplished by a linear regression of log MUT virus versus time through the first four to five MUT hybridization values.
This method almost certainly understates the amount of virus at early time points. Previous theoretical treatments, such as those presented by Nowak et al. (30), McLean and Nowak (27), and Stillianakis et al. (45), suggest that the rate of change in the density of MUT virus will increase (often from negative to positive) following the initiation of drug treatment. The simple exponential model, therefore, provides a conservative minimal estimate for the prevalence of MUT virus at the initiation of treatment.
Estimation of fitness of MUT populations following drug withdrawal.
The fitness of the MUT population was estimated from the analysis of the replacement of the MUT population with the WT virus in the absence of drugs. This approach assumes replication is continuous in time, where s, the selection coefficient, is the fitness difference between the WT and MUT populations. The selection coefficient can be estimated from a group of data points at various times by linear regression analysis of ln[p(t)/q(t)], with time as an independent variable (29), where p is the frequency of the more fit WT variant and q is the frequency of the less fit MUT variant. When the lack of detectable WT virus during the follow-up period prevented an estimate of s by the above method, the upper limit of s was estimated by 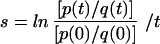 , where p(0)/q(0), the proportion of WT, was estimated to be no less than the mutation rate, 3 × 10−5; p(t)/q(t) was estimated as the sensitivity of detection of the differential hybridization assay, 0.01 (10); and t (in days) is the time of follow-up.
, where p(0)/q(0), the proportion of WT, was estimated to be no less than the mutation rate, 3 × 10−5; p(t)/q(t) was estimated as the sensitivity of detection of the differential hybridization assay, 0.01 (10); and t (in days) is the time of follow-up.
Sequence analysis of the protease-coding region of patient plasma RNA.
RNA was extracted from plasma virus and PCR amplified as described above. One microliter of this reaction was diluted to 10 μl and amplified for another 40 cycles with the same primers. Unincorporated primers were removed with a Centricon-100 (Ambicon, Inc., Beverly, Mass.), and PCR products were sequenced directly on a DNA sequencer (Applied Biosystems, Foster City, Calif.), with unlabeled GNR probe as a primer. All sequences were proofread manually and aligned with that of HIV-1SF2 as a consensus sequence. Alternatively, a nested PCR strategy was employed, followed by cloning of the PCR product and sequencing. cDNA synthesis was performed in a 10-μl reaction mixture containing 0.2 μM primer p3-2 (5′-ACCCTGCAGGATGTGGTATTCCTAA-3′, bp 2376), 50 mM Tris-HCl, 75 mM KCl, 200 μM (each) deoxynucleoside triphosphate, 3 mM MgCl2, and 200 U of Moloney murine leukemia virus RT at 37°C for 30 min. Following cDNA synthesis, 0.2 μM primer p5-2 (5′-AGTGTTTCAATTGTGGCAAA-3′, bp 1514 to 1533) was added and PCR was performed in a mixture containing 10 mM Tris-HCl, 50 mM KCl, 200 μM (each) deoxynucleoside triphosphate, 2.5 mM MgCl2, 2.5 U of Taq polymerase, and 1 μg of DNA in a total volume of 100 μl. PCR was performed for a total of 30 cycles in a Perkin-Elmer 9600 Thermocycler under the following conditions: melting at 94°C for 60 s, annealing at 55°C for 60 s, and extension at 72°C for 105 s. Nested PCR employed conditions and primers previously described (15). The amplified products were cloned into a TA cloning vector (Invitrogen Corp., San Diego, Calif.). Plasmid DNA was purified and sequenced by the dideoxy chain termination method.
Phenotypic sensitivity analysis of subject PBMCs.
Peripheral blood mononuclear cell (PBMC) viral supernatants were raised from selected stored specimens. Five million patient PBMCs were cocultured with an equal number of uninfected phytohemagglutinin-stimulated PBMCs in the presence of interleukin 2. Culture supernatants were titered by p24 antigen by standard techniques. For sensitivity testing, 5 × 106 uninfected phytohemagglutinin-stimulated PBMCs were incubated with 5,000 50% tissue culture infective doses of virus. The cells were washed and plated in duplicate onto a 96-well plate containing fivefold dilutions of ritonavir (0.008 to 5 μM) in addition to a drug-free control. Medium containing appropriate concentrations of the drug was changed on day 4, and the p24 antigen in each well was measured on day 7. As each titration was done in duplicate, p24 antigen levels were averaged, and percent inhibition was determined by comparison to the drug-free control.
RESULTS
Identification of resistance mutations associated with rebounds in plasma HIV RNA.
The rapid and dramatic antiviral effect of ritonavir on plasma HIV RNA levels has been well documented (8, 14, 26). We studied a subset of these patients (mean CD4+-cell concentration, 185/mm3) (14, 26) in whom ritonavir monotherapy produced a mean decrease in plasma HIV RNA levels of 1.7 log units within 2 weeks. A rebound in RNA levels within 1 year of initiation of therapy was observed in 8 of 10 subjects, while 2 of 10 maintained viral suppression. For four subjects who had lost viral suppression, ritonavir dosage was escalated to 1,200 mg daily with little impact on plasma RNA levels.
We analyzed plasma HIV RNA from 10 subjects on ritonavir monotherapy for changes at loci identified as responsible for decreased susceptibility to ritonavir (25, 28). The presence of genotypic changes from the WT consensus sequence at various times for the eight subjects who experienced viral load rebounds are presented in Fig. 1. The bDNA assay has been demonstrated to be able to discern threefold changes in plasma RNA levels as significant (48). For the purposes of this analysis, a threefold (0.5 log10) increase in plasma RNA levels above the nadir on two consecutive determinations was employed to define a viral load rebound. The mean increase in plasma RNA levels over the nadir at the time of viral load rebound was 1.3 (range, 0.66 to 2.53) log units. At the time of the viral load rebound, relatively few mutations were detected, primarily at codons 63 and 82. The majority of subjects, however, had changes at codon 63 at baseline. With continued viral replication in the presence of drug after the viral load rebound, additional genotypic changes are observed.
FIG. 1.

Time of development of various mutations in the protease gene during ritonavir monotherapy. BSLN, baseline; RBND, viral load rebound; +56, viral load rebound plus 56 days; >140, viral load rebound plus ≥140 days. The number of subjects evaluated at each time point is indicated above each bar.
Alternatively, we analyzed longitudinal samples so that genotypic changes could be temporally associated with changes in plasma HIV RNA. The total HIV RNA copies per milliliter and determinations of various MUT populations by differential hybridization and sequencing for selected subjects are presented in Fig. 2. By differential hybridization, a change at codon 82 (seven of eight, V82A; one of eight, V82F) was temporally associated with the initial loss of viral suppression.
FIG. 2.




Plasma virus RNA levels and amounts of RNA containing various genotypes in longitudinal samples from subjects on ritonavir monotherapy. Plasma RNA levels were determined by bDNA assay, and amounts of I54V, L63P/A, A71V, V77I, V82A/F, I84V, and L90M genotypes in plasma RNA, determined by differential hybridization as described in Materials and Methods, are labeled as such. Genotypes as determined by differential hybridization are presented below each plot; an asterisk represents the detection of a change from the consensus WT. Changes from the HIV-1SF2 consensus sequence determined by sequencing are enclosed in boxes embedded in the plots. Closed symbols represent values below the limit of quantitation for the bDNA assay and differential hybridization. IC90, 90% inhibitory concentration; d4T, stavudine.
Two patterns of acquisition of the second most common mutation, I54V, representing the I54V-V82A double MUT, were observed. The first was exemplified by subjects 301 and 306, in whom the I54V mutation was acquired well after the appearance of a V82A mutation and viral load rebound and was not associated with a further rise in HIV RNA levels. Both of these subjects also received the lowest dose of ritonavir. In three other subjects receiving higher doses of ritonavir, the I54V mutation (I54V-V82A double MUT) appeared temporally with the V82A mutation; however, the double-MUT population appeared at lower levels and the rate of expansion was slower. In two of the three subjects (304 and 409), complete conversion of the replicating viral population to the double MUT was associated with further increases in viral load, suggesting that the I54V mutation results in a further decrease in susceptibility to ritonavir. The slower expansion of the double-MUT population, however, may indicate reduced fitness.
An L90M mutation was observed in two subjects (301 and 304) only well after the viral load rebound and in the presence of a large number of other mutations. An I84V change was observed in only one subject (310), following the development of the V82A mutation, and was associated with a further increase in viral load.
Three subjects were completely L63P/A at baseline: 306, 313, and 409. Viral RNA was a mixture of L63 WT and L63P MUT at baseline in three subjects, 304, 310, and 401 (Fig. 1), but became completely L63P MUT concomitant with the loss of viral suppression. In four subjects who were mixtures or L63P/A MUT at baseline, initiation of ritonavir therapy coincided with changes to L63 WT, only to return to L63P MUT at the time of the viral load rebound. This is most apparent in the large fluctuations of L63P MUT in subject 310.
As shown in Fig. 2, the phenotypic sensitivity of virus isolated from PBMCs from four subjects (301, 304, 306, and 313) established a progressive decrease in susceptibility to ritonavir with duration of therapy. Sequence analysis of subjects 301 and 304 demonstrated the stepwise accumulation of additional mutations, including K20R, A71V, and L90M. An M46I mutation appeared in subject 304 at day 57 but reverted to M46 WT, while an M36I change was present at baseline and throughout therapy. Similarly, an M46I mutation appeared at the time of rebound in subject 313 by differential hybridization but had reverted to M46 WT 14 days later (data not shown).
In the two subjects who experienced long-term viral suppression below the limit of detection of the bDNA assay, 406 and 408 (Fig. 1), codon 82 remained WT. Furthermore, certain changes from the WT HIV-1SF2 consensus sequence present at baseline, some of which have been associated with resistance to protease inhibitors (7, 28), were lost with therapy in these subjects. Most notable were changes at codons 63, 71, and 77. While this apparent reversion may be affected by the small amounts of RNA introduced into the RT-PCR reactions, the reversion of both subjects 406 and 408, as well as the maintenance of WT sequence in subject 203 (28), argues against this possibility. It is possible, therefore, that this WT virus represents viral replication in a site that is privileged with regard to exposure to ritonavir, such as latently infected cells, brain, or testes.
Kinetic analysis and estimation of fitness from estimates of baseline plasma RNA levels of MUT populations.
Four subjects in the lowest-dosage group, 600 mg/day, experienced rebounds in plasma RNA within 42 days of ritonavir monotherapy. At time points when the V82A MUT was detectable above the WT background, it was possible to quantify the rate of expansion of the MUT population. Due to the frequency of time points during the exponential viral load rebound, a fit to the data at these time points on a log scale allowed the determination of the slope of the line as defined by linear regression (minimally, 4 time points) in three of these subjects. From this, an estimate of the doubling time of the V82A MUT virus was determined (Table 2). It is possible that the longer doubling time estimate for subject 312 is a result of longer intervals between time points at the time of the viral load rebound than in the other subjects.
TABLE 2.
Estimation of doubling times of V82A MUT populations
| Subject | Baseline WT (copies/ml) | Doubling time (days) | 95% Confidence limits
|
r2 | |
|---|---|---|---|---|---|
| Lower | Upper | ||||
| 301 | 386,800 | 2.36 | 1.53 | 5.17 | 0.969 |
| 306 | 435,100 | 2.81 | 1.45 | 4.33 | 0.913 |
| 312 | 123,000 | 4.81 | 3.83 | 6.54 | 0.980 |
As the steady-state level of the MUT population provides a rough estimate of the selective disadvantage to the viability of the virus, we wished to estimate pretreatment levels of the MUT population. We used a variation of the model of Perelson et al. (36) to estimate the baseline prevalence of the V82A MUT (see Materials and Methods). A maximum estimate of s was calculated by using the hybridization data at the early time points for each subject as the limit of detection of the assay to estimate an upper limit to the amount of V82A MUT, above which the MUT would be detected in the hybridization assay. A conservative minimum estimate of s was calculated from the linear regression analysis described above. Both the CD4+ model and the linear extrapolation are presented graphically for these subjects in Fig. 2. The estimated s values are presented in Table 3. The CD4+ model estimated fitness for the V82A MUT population at 99% that of the WT population for subjects 306 and 312, with minimum estimates on the order of 94 and 90%, respectively. Subject 301 had estimates of fitness that were much less, to the point that the linear extrapolation produced a value that was less than the mutation rate of HIV (24). Plasma virus from this subject also had no genotypic changes from the consensus WT at the initiation of therapy, as opposed to that from subject 306, which was completely L63P at baseline.
TABLE 3.
Estimation of relative fitness from expansion of the V82A population
| Subject | Baseline RNA (equivalents/ml) |
s value
|
||
|---|---|---|---|---|
| Hybridization (maximum) | CD4+ model | Linear extrapolation (minimum) | ||
| 301 | 386,800 | 0.0013 | 0.148 | 4.22a |
| 306 | 435,100 | 0.0002 | 0.010 | 0.059 |
| 312 | 123,000 | 0.0005 | 0.010 | 0.103 |
Represents a prevalence less than the rate of mutation.
Impact of the presence of RT T215Y/F ZDV resistance mutation on development of the V82A ritonavir resistance mutation.
Seven of the 10 subjects’ baseline samples demonstrated baseline ZDV genotypic resistance mutations, including the RT T215Y/F mutation. In order to assess the impacts of ritonavir treatment and the development of the V82A protease resistance mutation on the RT T215Y/F locus, selected longitudinal samples from these subjects were analyzed for the amount of T215Y/F MUT. The amount of virus containing the RT T215Y/F mutation for each of the seven subjects, along with the amount of virus containing the V82A mutation, is presented in Fig. 3. The rapid change from V82 WT to V82A MUT in the protease gene in these subjects had no impact on the T215Y/F MUT of the RT. The appearance of T215 WT sequences was observed in four subjects (see below). Only subject 408, a long-term responder, experienced a complete reversion to T215 WT (day 328 of ritonavir therapy).
FIG. 3.

Impact of development of the V82A protease resistance mutation on the RT T215Y/F ZDV resistance mutation in the indicated subjects.
Fitness of MUT virus following drug withdrawal.
The relative fitness of a MUT population in the absence of drugs can be calculated by standard formulas to model the effect of replacement of the less fit (MUT) genotype by the more fit (WT) genotype. By knowing the change in the proportion of the MUT and WT populations over time, it was possible to calculate the selective disadvantage (s) of the RT T215Y/F MUT in four subjects in which T215 WT sequences became detectable (Table 4). Only time points at which WT virus was detectable were used for the regression analysis in order to get a more accurate estimate of the rate of change.
TABLE 4.
Relative fitness of HIV populations containing the protease V82A or RT T215Y/F mutation following drug withdrawal
| Subject | Mutationa | Day | s | 95% Confidence limit
|
r2 | Additional genotypes | |
|---|---|---|---|---|---|---|---|
| Lower | Upper | ||||||
| 408 | RT T215Y/F | 395 | 0.030 | 0.012 | 0.048 | 0.998 | M41 WT, K70 WT |
| 409 | RT T215Y/F | 311 | 0.039 | −0.006 | 0.083 | 0.992 | M41 WT, K70 WT |
| 312 | RT T215Y/F | 231 | 0.018 | −0.057 | 0.078 | 0.797 | M41L MUTc, K70 WT |
| 304 | RT T215Y/F | 140 | 0.021 | −0.101 | 0.120 | 0.549 | M41L MUT, K70 WT |
| 310 | RT T215Y/F | 313 | ≤0.019b | M41L MUT, K70 WT | |||
| 306 | RT T215Y/F | 178 | ≤0.033b | M41 WT, K70 WT | |||
| 301 | RT T215Y/F | 184 | ≤0.032b | ||||
| 401 | PRd V82A | 198 | <0.029b | I54V MUT, L63P MUT | |||
| 409 | PR V82A | 42 | <0.138b | I54V MUT, L63P MUT | |||
Probe hybridization interrogates a specific mutation; however, this viral population most likely represents virus that is multiply MUT following extended therapy.
See Materials and Methods for estimation of upper limit of s value.
Boldface indicates change from consensus WT.
PR, protease.
In those subjects in which WT sequences were not detectable during the time of follow-up, an upper bound for s was calculated. To approximate the greatest rate of increase of the WT population, the proportion of the WT population at the end of the study period was taken to be the limit of sensitivity of the differential hybridization assay, 1%. Similarly, the proportion of the WT 1 day into drug withdrawal was conservatively estimated to be the mutation rate of the virus. Limited follow-up data on the stability of the RT T215Y/F from three additional subjects and the V82A MUT from two subjects following withdrawal from ZDV and ritonavir therapy, respectively, was available. Estimates of the upper limit of s were on the order of 0.03, except in subject 409, who had the shortest follow-up. Clearly, with longer follow-up, estimates of the upper limit of s decrease.
Factors affecting the durability of the virologic response.
A multifactor one-way analysis of variance of the time to return of the HIV plasma RNA levels towards baseline with respect to the drug dosage, maximal decrease in RNA levels from baseline, baseline RNA levels, nadir RNA levels, and T215Y/F in this small cohort of 10 subjects found only the baseline plasma HIV RNA level (r = −0.85; P = 0.002), the HIV RNA level at the nadir (r = −0.88; P = 0.0007), and the drug dosage (r = 0.96; P < 0.00001) significantly correlated with the time to return to baseline RNA levels (Spearman rank order correlation). Data from 16 additional ritonavir-treated subjects with respect to baseline viral RNA levels and drug dosage were included to increase the power, and the statistical analysis was repeated. Baseline RNA levels remained weakly, but significantly, correlated (r2 = 0.45; P = 0.0001), as did HIV RNA levels at the nadir (r2 = 0.38; P = 0.0008), while drug dosage lost significance (r2 = 0.16; P = 0.04) (Pearson product-moment correlation), although area under the curve (AUC) and trough levels of drug concentration (Cmin) were not available for analysis. The impacts of baseline and nadir RNA levels on the durability of the virologic response are presented in Fig. 4. Inspection of the plot of the HIV RNA levels at the nadir reveals that no subject in this study had a virologic response longer than 60 days unless plasma HIV RNA levels fell below 2,000 copies/ml. Interestingly, both the baseline viral RNA levels (r2 = 0.55; P < 0.00001) and the extent of decrease of RNA from baseline (r2 = 0.40; P = 0.0006) were correlated with the level of plasma RNA at the nadir, while drug dose was not significant. Again, AUC and Cmin were not available for analysis.
FIG. 4.

Impact of baseline and nadir plasma RNA levels on duration of virologic response.
DISCUSSION
Recent clinical studies demonstrate dramatic decreases in plasma HIV RNA levels and dramatic increases in CD4+ cells with ritonavir monotherapy (8, 26). After extended ritonavir monotherapy, however, plasma virus RNA levels began to rise in many subjects, indicating a loss of susceptibility to the drug. Molla et al. (28) have previously reported on an ordered accumulation of mutations during ritonavir monotherapy based on their frequency of appearance. In order to identify genotypic changes which were associated temporally with rebounds in the plasma HIV RNA levels in vivo, differential hybridization of the RT-PCR product derived from plasma virus RNA (10, 16) was coupled with plasma RNA levels in longitudinal samples from 10 patients. Using this approach, we have been able to define three types of mutations: primary, secondary, and compensatory viability.
Primary resistance mutations were temporally and quantitatively associated with the initial loss of suppression of viral replication. For ritonavir monotherapy, the appearance of the V82A/F mutation was identified as the primary mutation in all eight subjects who experienced viral load rebounds (Fig. 1 and 2) and was not observed in those subjects with a prolonged virologic response. Secondary mutations were associated with subsequent additional losses in viral suppression. In two subjects who had higher doses of ritonavir (304 and 409), a secondary rebound in circulating plasma viral RNA levels was associated with the acquisition of an I54V mutation and conversion of the replicating viral population to I54V-V82A double MUT. The I84V mutation, which was the first change observed with in vitro selection (25), was observed in only one subject, 310, as a secondary resistance mutation.
Two patterns of acquisition of the I54V-V82A double mutation were observed wherein the appearance of I54V was greatly delayed at lower drug doses and was not associated with changes in plasma HIV RNA levels. However, at higher drug doses, the appearance of the I54V secondary mutation was much more temporally related to the appearance of the V82A primary mutation and the return of plasma HIV RNA levels to baseline values. This suggests that at higher drug doses, the V82A MUT alone may require additional genotypic changes, i.e., I54V or I84V, to overcome the selective pressure of the drug.
The coincident expansion of the L63P and V82A populations (Fig. 1 and 2) suggests that L63P may play a role in resistance to ritonavir in vivo. Baseline viral populations from many of these subjects are completely L63P/A or represent mixtures, yet potent and durable virologic response is observed with therapy (e.g., subjects 310, 401, 408, and 409) indicating that this mutation is not a primary resistance mutation. In vitro studies suggest L63P may compensate for changes around the active site, such as at codons 82 and 84 (25). Individual changes at codons 46, 63, and 71 in a HIV-1NL4-3 backbone produce phenotypic sensitivities to ritonavir identical to that of WT virus (25). It is possible that the observed changes at codon 63 may restore viability to a replication-challenged V82A/F MUT. Indeed, a lack of viability of virus possessing changes at V82 or V82 and I84 has been reported (25, 34, 38). Changes at codon 10 (38) or 71 (34) have been proposed to compensate for a reduced viability of virus from site-directed mutagenesis studies in vitro. Furthermore, with purified enzyme preparations, the V82A protease exhibited a reduced enzymatic activity (23, 31, 34), while a secondary A71T change was able to restore some of the enzymatic activity (23, 34). The L63P MUT appeared to improve the in vitro growth kinetics of the V82F-I84V double MUT (25). It is, therefore, possible that a change from the consensus leucine at codon 63 may represent a compensatory viability mutation.
Continued viral replication in the presence of drug resulted in the accumulation of additional genotypic changes. Phenotypic sensitivity of PBMC isolates also demonstrate a progressive loss of susceptibility with continued ritonavir therapy. Two subjects eventually developed the L90M mutation, a primary saquinavir resistance mutation (17, 18), indicating the evolution of cross-resistance to other protease inhibitors. The L90M mutation, however, was not detected until long after the initial viral load rebound and several additional changes had occurred. It is, therefore, possible that a switch in therapy shortly after the initial viral load rebound may prevent the potential for cross-resistance in these subjects.
Several groups have estimated virus turnover in vivo by measuring the decline of plasma virus levels with the initiation of a potent antiretroviral therapy (14, 36, 49). We wished to investigate the in vivo kinetics of expansion of the V82A MUT population. Linear regression analysis of the amount of V82A MUT virus in the plasma during the first-order logarithmic expansion resulted in estimates of 2.4 to 4.8 days (Table 2) for the doubling time of the V82A MUT population. The estimated doubling times most likely represent overestimates and could be improved with more frequent samplings. These doubling times are very similar to those calculated for the Y181C nevirapine resistance mutation (13). Consequently, assuming an average doubling time of 3 days and no delay due to pharmacologic delays, etc., 19 generations, or 57 days, would be required for a resistant mutation with a predrug prevalence of 3 × 10−5, the mutation rate of HIV (24), to dominate the replicating viral population. Almost identical estimates are obtained with the CD4 model.
Coffin has proposed that a single-point mutation occurs between 104 and 105 times/day in an HIV-infected individual (5). Furthermore, the steady-state level of the MUT should be a function of the forward and backward mutation rates and the selective disadvantage of the mutation to the viability of the virus, of which the latter has the greater impact. We therefore wished to estimate the level of the V82A MUT at the time of initiation of therapy in order to estimate the selective disadvantage of V82A MUT virus in vivo. To this end, we used a mathematical model similar to that of Perelson et al. based on CD4+ T cells as potential target cells (36). The limit of detection of the differential hybridization assay was used to define an upper limit of the amount of V82A MUT virus at the initiation of therapy and, therefore, a maximum fitness. A linear extrapolation during the exponential expansion of the V82A MUT population back to baseline was employed to define a lower limit of the pretreatment levels of resistant virus. This method does not take into account such considerations as a delay in antiviral effects due to the time required for drug absorption, distribution, and penetration into the target cells nor the delay resulting from the mechanism of the protease inhibitor, in which only subsequent rounds of infection are prevented. The estimated s values of the V82A MUTs derived from estimates of the prevalence of the MUT population at the initiation of drug treatment (Table 3) are consistent with the interpretation that these MUTs were present before drug therapy. Under mutation-selection balance, the expected steady-state frequency of a MUT in a host prior to drug therapy will be roughly μ/s, where μ (∼3 × 10−5 [24]) is the mutation rate and s is the selective disadvantage of the resistance mutation in the absence of drugs (5, 29). The estimates for the initial frequency of V82A mutation (Table 2) in the two subjects (306 and 312) fall within the range that one would expect from mutation-selection theory (i.e., s ∼ 0.01). The somewhat lower predrug frequency of the V82A mutation in subject 301 (whose virus was WT at all amino acids in the protease prior to drug therapy) may reflect the lower fitness of the V82A MUT in the absence of compensatory mutations.
An alternative approach to determining the relative fitness of a resistant viral population in vivo is to determine the rate of reversion to WT following drug withdrawal. As part of the protocol, subjects were removed from previous antiretroviral therapies 2 weeks prior to initiation of ritonavir therapy. The removal from ZDV therapy allowed an estimation of the selective disadvantage of the RT T215Y/F mutation. No significant change in the proportion of RT T215Y/F ZDV resistance mutations was observed in those subjects who experienced a rebound in plasma virus RNA levels and rapid development of the V82A mutation in the protease (Fig. 3), indicating the independence of the target genes. The lack of reversion of the RT T215Y/F genotype with ritonavir monotherapy is very similar to the extended periods required for reversion of this phenotype following drug withdrawal (1, 3, 43). So, while dramatic turnover of sequences in the protease gene is observed with ritonavir therapy, the sequences of the RT appear to be quite stable in the absence of any selective pressure on this gene. This is to be expected from a model of quasispecies of two loci under independent selective pressures. Similar results have been reported by Brown and Cleland (4) regarding the independent evolution of the env and pol genes during ZDV therapy. This suggests that therapy with a protease inhibitor will most likely not be successful in reversing RT inhibitor resistance. Furthermore, the independence of these loci suggests that effective combination therapies directed towards different genetic targets will require multiple, independent genetic changes in a single genome in order to completely escape viral suppression.
Linear regression analysis of the changes in the proportions of the T215Y/F MUT and WT populations following withdrawal from ZDV therapy resulted in estimates of s as ∼0.02 to 0.04 (Table 4). In three subjects, in whom RT 215 WT sequences could not be detected throughout the course of study, only estimates of the upper limit of s were possible. One disadvantage of this approach is that additional mutations in the replicating viral population not detected by the probe used to interrogate the amino acid in question may increase the fitness of the population. Interestingly, s values from those subjects who harbored virus that was doubly MUT with M41L and T215Y/F appeared to have a slightly greater fitness, suggesting that the M41L mutation may impart additional fitness as well as additional resistance. The advantage of this approach, however, is the in vivo nature of the results.
Two subjects with limited follow-up were withdrawn from ritonavir. Plasma virus from these subjects (401 and 409) was found to remain completely V82A MUT at 198 and 42 days, respectively, following withdrawal from drug. An estimate of the upper limit for the selective disadvantage for the V82A mutation relative to WT at codon 82 in the subject with the greatest follow-up, 401, was approximately 0.03. The larger upper limit estimate for the selective disadvantage for the V82A mutation in subject 409 is a reflection of the shorter time following drug withdrawal. The estimates of the fitness of the expanding V82A MUT population, based on the CD4+-T-cell model, are in general agreement with these estimates. As described above, with continued ritonavir therapy following the viral load rebound, additional mutations accumulate. Indeed, our hybridization data reveals the presence of a triple (I54V-L63P-V82A) MUT population in both of these subjects at the time of drug cessation (Fig. 2). It is therefore possible that the accumulation of additional mutations served to decrease the pleiotropic cost of the V82A mutation in the absence of drug.
The lack of a significant fitness loss in the final population of both ZDV- and ritonavir-resistant MUTs points to a potentially serious future problem with respect to antiviral resistance. It suggests that in patients in whom multiply mutated drug-resistant variants have appeared, the resistant strains will not quickly recede following the withdrawal of therapy. The amount of time for a resistant strain to decrease to a given level will depend critically on the selection coefficient s. For a selective disadvantage of 0.02, the time required for the density of the resistant virus to drop to <0.01% of its original level in the complete absence of drug will be on the order of 461 days.
Finally, the duration of virologic response to ritonavir monotherapy, as monitored by plasma HIV RNA levels, was correlated with plasma RNA levels at both baseline and the nadir (Fig. 4) and weakly correlated with drug dosage. It is likely that evaluation of AUC or Cmin may result in a better correlation with duration of therapy. Danner et al. have previously reported a clear relation between increasing dosage and the duration of the response (8). Molla et al. (28) have reported both the AUC and trough levels were more significantly associated with the rate of selection of mutations in the protease during ritonavir therapy than was drug dose, most likely due to intrasubject variation in the pharmacokinetics of ritonavir. Interestingly, Havlir et al. have reported an improved durability of virologic response in some subjects with a higher dosage of nevirapine despite the detection of virus with reduced phenotypic sensitivity to nevirapine (12). Similarly, if drug trough levels can be maintained well above the 99% inhibitory concentration of the V82A/F MUT virus, extended suppression of viral replication may be possible.
While the correlation of plasma HIV RNA levels at the nadir with the time to increase of plasma RNA toward baseline levels was significant (r2 = 0.38; P = 0.0008), Fig. 4 illustrates the significance of this correlation. In this study, no subjects in whom plasma virus RNA levels were not suppressed below 2,000 copies/ml experienced a virologic response greater than 60 days. While there were subjects whose plasma virus RNA levels were suppressed below this level that had short durations of viral suppression, this may have been confounded by other factors, such as drug dose and baseline levels of viral RNA. It is interesting to note that this time frame is in good agreement with our estimates of the time required for a preexisting resistance mutation at a prevalence of 3 × 10−4 to 1 × 10−6 to dominate the replicating viral population (see Results), given a doubling time on the order of 3 days.
In conclusion, we have extended the observations of Molla et al. (28) by identifying mutations that were associated with initial rebounds in plasma HIV RNA levels (primary mutations) and mutations that were associated with subsequent additional rebounds in plasma HIV RNA levels (secondary mutations). During ritonavir monotherapy, the V82A/F mutation was identified as a primary mutation while I54V and I84V were identified as secondary mutations. With continued ritonavir therapy, additional genotypic changes were observed and associated with further losses in phenotypic susceptibility. An L90M saquinavir resistance mutation was occasionally observed, which may prevent subsequent therapy with alternative protease inhibitors.
The doubling time of the expanding V82A MUT population ranged from 2.4 to 4.8 days. As a consequence, HIV RNA levels frequently returned to baseline in a very short period of time, often weeks. The fitness of the V82A MUT, as well as the RT T215Y/F ZDV-resistant MUT, was estimated to be ∼2 to 4% less than that of WT following drug withdrawal, suggesting reversion to WT may not occur rapidly. The duration of the virologic response was correlated with plasma RNA levels at the nadir. No subject with plasma RNA levels >2,000 copies/ml at the nadir experienced a virologic benefit greater than 60 days, consistent with doubling time estimates for complete conversion of the replicating viral population. Complete suppression of viral replication and a long-term virologic response were associated with low baseline viral loads and higher drug levels. These results argue for the early initiation of effective therapy when plasma viremia is low to thoroughly suppress viral replication and prevent the development of drug-resistant variants.
REFERENCES
- 1.Albert J, Wahlberg J, Lundeberg J, Cox S, Sandström E, Wahren B, Uhlén M. Persistence of azidothymidine-resistant human immunodeficiency virus type 1 RNA genotypes in posttreatment sera. J Virol. 1992;66:5627–5630. doi: 10.1128/jvi.66.9.5627-5630.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boucher C A B, Cammack N, Schipper P, Schuurman R, Rouse P, Wainberg M A, Cameron J M. High-level resistance to (−) enantiomeric 2′-deoxy-3′-thiacytidine in vitro is due to one amino acid substitution in the catalytic site of human immunodeficiency virus type 1 reverse transcriptase. Antimicrob Agents Chemother. 1993;37:2231–2234. doi: 10.1128/aac.37.10.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boucher C A B, van Leeuwen R, Kellam P, Schipper P, Tijnagel J, Lange J M A, Larder B A. Effects of discontinuation of zidovudine treatment on zidovudine sensitivity of human immunodeficiency virus type 1 isolates. Antimicrob Agents Chemother. 1993;37:1525–1530. doi: 10.1128/aac.37.7.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown A L B, Cleland A. Independent evolution of the env and pol genes of HIV-1 during zidovudine therapy. AIDS. 1996;10:1067–1073. [PubMed] [Google Scholar]
- 5.Coffin J M. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 6.Condra J H, Holder D J, Schleif W A, Blahy O M, Danovich R M, Gabryelski L J, Graham D J, Laird D, Quintero J C, Rhodes A, Robbins H L, Roth E, Shivaprakash M, Yang T, Chodakewitz J A, Deutsch P J, Leavitt R Y, Massari F E, Mellors J W, Squires K E, Steigbigel R J, Teppler H, Emini E A. Genetic correlates of in vivo viral resistance to indinavir, a human immunodeficiency virus type 1 protease inhibitor. J Virol. 1996;70:8270–8276. doi: 10.1128/jvi.70.12.8270-8276.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Condra J H, Schleif W A, Blahy O M, Gabryelski L J, Graham D J, Quintero J C, Rhodes A, Robbins H L, Roth E, Shivaprakash M, Titus D, Yang T, Teppler H, Squires K E, Deutsch P J, Emini E A. In vivo emergence of HIV-1 variants resistant to multiple protease inhibitors. Nature. 1995;374:569–571. doi: 10.1038/374569a0. [DOI] [PubMed] [Google Scholar]
- 8.Danner S A, Carr A, Leonard J M, Lehman L M, Gudiol F, Gonzales J, Raventos A, Rubio R, Bouza E, Pintado V, Aguado A G, Delomas J G, Delgado R, Borleffs J C C, Hsu A, Valdes J M, Boucher C A B, Cooper D A, Gimeno C, Clotet B, Tor J, Ferrer E, Martinez P L, Moreno S, Zancada G, Alcami J, Noriega A R, Pulido F, Glassman H N. A short-term study of the safety, pharmacokinetics, and efficacy of ritonavir, an inhibitor of HIV-1 protease. N Engl J Med. 1995;333:1528–1533. doi: 10.1056/NEJM199512073332303. [DOI] [PubMed] [Google Scholar]
- 9.deJong M D, Veenstra J, Stilianakis N I, Schuurman R, Lange J M A, deBoer R J, Boucher C A B. Host-parasite dynamics and outgrowth of virus containing a single K70R amino acid change in reverse transcriptase are responsible for the loss of human immunodeficiency virus type 1 RNA load suppression by zidovudine. Proc Natl Acad Sci USA. 1996;93:5501–5506. doi: 10.1073/pnas.93.11.5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eastman P S, Boyer E, Mole L, Kolberg J, Urdea M, Holodniy M. Nonisotopic hybridization assay for determination of relative amounts of genotypic human immunodeficiency virus type 1 zidovudine resistance. J Clin Microbiol. 1995;33:2777–2780. doi: 10.1128/jcm.33.10.2777-2780.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao Q, Gu Z X, Parniak M A, Li X G, Wainberg M A. In vitro selection of variants of human immunodeficiency virus type 1 resistant to 3′-azido-3′-deoxythymidine and 2′,3′-dideoxyinosine. J Virol. 1992;66:12–19. doi: 10.1128/jvi.66.1.12-19.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Havlir D, McLaughlin M M, Richman D D. A pilot study to evaluate the development of resistance to nevirapine in asymptomatic human immunodeficiency virus-infected patients with CD4 cell counts of > 500/mm3: AIDS Clinical Trials Group protocol 208. J Infect Dis. 1995;172:1379–1383. doi: 10.1093/infdis/172.5.1379. [DOI] [PubMed] [Google Scholar]
- 13.Havlir D V, Eastman S, Gamst A, Richman D D. Nevirapine-resistant human immunodeficiency virus: kinetics of replication and estimated prevalence in untreated patients. J Virol. 1996;70:7894–7899. doi: 10.1128/jvi.70.11.7894-7899.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho D D, Neumann A U, Perelson A S, Chen W, Leonard J M, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 15.Ho D D, Toyoshima T, Mo H, Kempf D J, Norbeck D, Chen C M, Wideburg N E, Burt S K, Erickson J W, Singh M K. Characterization of human immunodeficiency virus type 1 variants with increased resistance to a C2-symmetric protease inhibitor. J Virol. 1994;68:2016–2020. doi: 10.1128/jvi.68.3.2016-2020.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holodniy M, Mole L, Margolis D, Moss J, Dong H, Boyer E, Urdea M, Kolberg J, Eastman S. Determination of human immunodeficiency virus RNA in plasma and cellular viral DNA genotypic zidovudine resistance and viral load during zidovudine-didanosine combination therapy. J Virol. 1995;69:3510–3516. doi: 10.1128/jvi.69.6.3510-3516.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacobsen H, Hanggi M, Ott M, Duncan I B, Owen S, Andreoni M, Vella S, Mous J. In vivo resistance to a human immunodeficiency virus type 1 proteinase inhibitor: mutations, kinetics, and frequencies. J Infect Dis. 1996;173:1379–1387. doi: 10.1093/infdis/173.6.1379. [DOI] [PubMed] [Google Scholar]
- 18.Jacobsen H, Yasargil K, Winslow D L, Craig J C, Krohn A, Duncan I B, Mous J. Characterization of human immunodeficiency virus type 1 mutants with decreased sensitivity to proteinase inhibitor Ro 31-8959. Virology. 1995;206:527–534. doi: 10.1016/s0042-6822(95)80069-7. [DOI] [PubMed] [Google Scholar]
- 19.Kern D, Collins M, Fultz T, Detmer J, Hamren S, Peterkin J J, Sheridan P, Urdea M, White R, Yeghiazarian T, Todd J. An enhanced-sensitivity branched-DNA assay for quantification of human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol. 1996;34:3196–3202. doi: 10.1128/jcm.34.12.3196-3202.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kohl N E, Emini E A, Schleif W A, Davis L J, Heimbach J C, Dixon R A, Scolnick E M, Sigal I S. Active human immunodeficiency virus protease is required for viral infectivity. Proc Natl Acad Sci USA. 1988;85:4686–4690. doi: 10.1073/pnas.85.13.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larder B A, Kellam P, Kemp S D. Zidovudine resistance predicted by direct detection of mutations in DNA from HIV-infected lymphocytes. AIDS. 1991;5:137–144. doi: 10.1097/00002030-199102000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Larder B A, Kemp S D. Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT) Science. 1989;246:1155–1158. doi: 10.1126/science.2479983. [DOI] [PubMed] [Google Scholar]
- 23.Loeb D D, Swanstrom R, Everitt L, Manchester M, Stamper S E, Hutchison C A D. Complete mutagenesis of the HIV-1 protease. Nature. 1989;340:397–400. doi: 10.1038/340397a0. [DOI] [PubMed] [Google Scholar]
- 24.Mansky L M, Temin H M. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol. 1995;69:5087–5094. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Markowitz M, Mo H, Kempf D J, Norbeck D W, Bhat T N, Erickson J W, Ho D D. Selection and analysis of human immunodeficiency virus type 1 variants with increased resistance to ABT-538, a novel protease inhibitor. J Virol. 1995;69:701–706. doi: 10.1128/jvi.69.2.701-706.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markowitz M, Saag M, Powderly W G, Hurley A M, Hsu A, Valdes J M, Henry D, Sattler F, Lamarca A, Leonard J M, Ho D D. A preliminary study of ritonavir, an inhibitor of HIV-1 protease, to treat HIV-1 infection. N Engl J Med. 1995;333:1534–1539. doi: 10.1056/NEJM199512073332204. [DOI] [PubMed] [Google Scholar]
- 27.McLean A R, Nowak M A. Competition between zidovudine-sensitive and zidovudine-resistant strains of HIV. AIDS. 1992;6:71–79. doi: 10.1097/00002030-199201000-00009. [DOI] [PubMed] [Google Scholar]
- 28.Molla A, Korneyeva M, Gao Q, Vasavanonda S, Schipper P, Mo H-M, Markowitz M, Chernyavskiy T, Niu P, Lyons N, Hsu A, Granneman G, Ho D, Boucher C, Leonard J, Norbeck D, Kempf D. Ordered accumulation of mutations in HIV protease confers resistance to ritonavir. Nat Med. 1996;2:760–766. doi: 10.1038/nm0796-760. [DOI] [PubMed] [Google Scholar]
- 29.Nagylaki T. Introduction to theoretical population genetics. New York, N.Y: Springer-Verlag; 1992. [Google Scholar]
- 30.Nowak M A, Bonhoeffer S, Shaw G M, May R M. Anti-viral drug treatment: dynamics of resistance in free virus and infected cell populations. J Theor Biol. 1997;184:203–217. doi: 10.1006/jtbi.1996.0307. [DOI] [PubMed] [Google Scholar]
- 31.Otto M J, Garber S, Winslow D L, Reid C D, Aldrich P, Jadhav P K, Patterson C E, Hodge C N, Cheng Y S. In vitro isolation and identification of human immunodeficiency virus (HIV) variants with reduced sensitivity to C-2 symmetrical inhibitors of HIV type 1 protease. Proc Natl Acad Sci USA. 1993;90:7543–7547. doi: 10.1073/pnas.90.16.7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pachl C, Todd J A, Kern D G, Sheridan P J, Fong S J, Stempien M, Hoo B, Besemer D, Yeghiazarian T, Irvine B, Kolberg J, Kokka R, Neuwald P, Urdea M S. Rapid and precise quantification of HIV-1 RNA in plasma using a branched DNA signal amplification assay. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;8:446–454. doi: 10.1097/00042560-199504120-00003. [DOI] [PubMed] [Google Scholar]
- 33.Partaledis J A, Yamaguchi K, Tisdale M, Blair E E, Falcione C, Maschera B, Myers R E, Pazhanisamy S, Futer O, Cullinan A B, Stuver C M, Byrn R A, Livingston D J. In vitro selection and characterization of human immunodeficiency virus type 1 (HIV-1) isolates with reduced sensitivity to hydroxyethylamino sulfonamide inhibitors of HIV-1 aspartyl protease. J Virol. 1995;69:5228–5235. doi: 10.1128/jvi.69.9.5228-5235.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patick A K, Rose R, Greytok J, Bechtold C M, Hermsmeier M A, Chen P T, Barrish J C, Zahler R, Colonno R J, Lin P-F. Characterization of a human immunodeficiency virus type 1 variant with reduced sensitivity to an aminodiol protease inhibitor. J Virol. 1995;69:2148–2152. doi: 10.1128/jvi.69.4.2148-2152.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng C, Ho B K, Chang T W, Chang N T. Role of human immunodeficiency virus type 1-specific protease in core protein maturation and viral infectivity. J Virol. 1989;63:2550–2556. doi: 10.1128/jvi.63.6.2550-2556.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perelson A S, Neumann A U, Markowitz M, Leonard J M, Ho D D. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 37.Richman D D, Havlir D, Corbeil J, Looney D, Ignacio C, Spector S A, Sullivan J, Cheeseman S, Barringer K, Pauletti D, Shih C-K, Myers M, Griffin J. Nevirapine resistance mutations of human immunodeficiency virus type 1 selected during therapy. J Virol. 1994;68:1660–1666. doi: 10.1128/jvi.68.3.1660-1666.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rose R E, Gong Y F, Greytok J A, Bechtold C M, Terry B J, Robinson B S, Alam M, Colonno R J, Lin P F. Human immunodeficiency virus type 1 viral background plays a major role in development of resistance to protease inhibitors. Proc Natl Acad Sci USA. 1996;93:1648–1653. doi: 10.1073/pnas.93.4.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saag M S, Emini E A, Laskin O L, Douglas J, Lapidus W I, Schleif W A, Whitley R J, Hildebrand C, Byrnes V W, Kappes J C, Anderson K W, Massari F E, Shaw G M. A short-term clinical evaluation of L-697,661, a non-nucleoside inhibitor of HIV-1 reverse transcriptase. L-697,661 Working Group. N Engl J Med. 1993;329:1065–1072. doi: 10.1056/NEJM199310073291502. [DOI] [PubMed] [Google Scholar]
- 40.Sanchez-Pescador R, Power M D, Barr P J, Steimer K S, Stempien M M, Brown-Shimer S L, Gee W W, Renard A, Randolph A, Levy J A, Dina D, Luciw P A. Nucleotide sequence and expression of an AIDS-associated retrovirus (ARV-2) Science. 1985;227:484–492. doi: 10.1126/science.2578227. [DOI] [PubMed] [Google Scholar]
- 41.Schmit J-C, Ruiz L, Clotet B, Raventos A, Tor J, Leonard J, Desmyter J, de Clercq E, Vandamme A-M. Resistance-related mutations in the HIV-1 protease gene of patients treated for 1 year with the protease inhibitor ritonavir (ABT-538) AIDS. 1996;10:995–999. doi: 10.1097/00002030-199610090-00010. [DOI] [PubMed] [Google Scholar]
- 42.Schuurman R, Nijhuis M, Vanleeuwen R, Schipper P, Dejong D, Collis P, Danner S A, Mulder J, Loveday C, Christopherson C, Kwok S, Sninsky J, Boucher C A B. Rapid changes in human immunodeficiency virus type 1 RNA load and appearance of drug-resistant virus populations in persons treated with lamivudine (3TC) J Infect Dis. 1995;171:1411–1419. doi: 10.1093/infdis/171.6.1411. [DOI] [PubMed] [Google Scholar]
- 43.Smith M S, Koerber K L, Pagano J S. Long-term persistence of zidovudine resistance mutations in plasma isolates of human immunodeficiency virus type 1 of dideoxyinosine-treated patients removed from zidovudine therapy. J Infect Dis. 1994;169:184–188. doi: 10.1093/infdis/169.1.184. [DOI] [PubMed] [Google Scholar]
- 44.St. Clair M H, Martin J L, Tudor-Williams G, Bach M C, Vavro C L, King D M, Kellam P, Kemp S D, Larder B A. Resistance to ddI and sensitivity to AZT induced by a mutation in HIV-1 reverse transcriptase. Science. 1991;253:1557–1559. doi: 10.1126/science.1716788. [DOI] [PubMed] [Google Scholar]
- 45.Stilianakis N I, Boucher C A B, De Jong M D, Van Leeuwen R, Schuurman R, De Boer R J. Clinical data sets of human immunodeficiency virus type 1 reverse transcriptase-resistant mutants explained by a mathematical model. J Virol. 1997;71:161–168. doi: 10.1128/jvi.71.1.161-168.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tisdale M, Kemp S D, Parry N R, Larder B A. Rapid in vitro selection of human immunodeficiency virus type 1 resistant to 3′-thiacytidine inhibitors due to a mutation in the YMDD region of reverse transcriptase. Proc Natl Acad Sci USA. 1993;90:5653–5656. doi: 10.1073/pnas.90.12.5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tisdale M, Myers R E, Maschera B, Parry N R, Oliver N M, Blair E D. Cross-resistance analysis of human immunodeficiency virus type 1 variants individually selected for resistance to five different protease inhibitors. Antimicrob Agents Chemother. 1995;39:1704–1710. doi: 10.1128/aac.39.8.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Todd, J., C. Pachl, R. White, T. Yeghiazarian, P. Johnson, B. Taylor, M. Holodniy, D. Kern, S. Hamren, D. Chernoff, and M. Urdea. 1995. Performance characteristics for the quantitation of plasma HIV-1 RNA using branched DNA signal amplification technology. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 10(Suppl):S35–S44. [PubMed]
- 49.Wei X, Ghosh S K, Taylor M E, Johnson V A, Emini E A, Deutsch P, Lifson J D, Bonhoeffer S, Nowak M A, Hahn B H, Saag M S, Shaw G M. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]