

Abstract
Nonsense-mediated RNA decay (NMD) is a highly conserved RNA turnover pathway that selectively degrades RNAs harbouring truncating mutations that prematurely terminate translation, including nonsense, frameshift and some splice-site mutations. Recent studies show that NMD shapes the mutational landscape of tumours by selecting for mutations that tend to downregulate the expression of tumour suppressor genes but not oncogenes. This suggests that NMD can benefit tumours, a notion further supported by the finding that mRNAs encoding immunogenic neoantigen peptides are typically targeted for decay by NMD. Together, this raises the possibility that NMD-inhibitory therapy could be of therapeutic benefit against many tumour types, including those with a high load of neoantigen-generating mutations. Complicating this scenario is the evidence that NMD can also be detrimental for many tumour types, and consequently tumours often have perturbed NMD. NMD may suppress tumour generation and progression by degrading subsets of specific normal mRNAs, including those encoding stress-response proteins, signalling factors and other proteins beneficial for tumours, as well as pro-tumour non-coding RNAs. Together, these findings suggest that NMD-modulatory therapy has the potential to provide widespread therapeutic benefit against diverse tumour types. However, whether NMD should be stimulated or repressed requires careful analysis of the tumour to be treated.
While the impact of transcriptional dysregulation in cancer is widely appreciated, the role of post-transcriptional dysregulation in cancer is much less well understood. mRNAs undergo many post-transcriptional processes, including capping, polyadenylation, splicing, nuclear export, editing and translation, all of which are tightly regulated. Perturbation or disruption of any of these steps can lead to disease, including cancer1–8.
Another post-transcriptional step that, when dysregulated, can influence cancer is RNA turnover. While it is often assumed that steady-state mRNA levels are rarely determined by RNA turnover but instead mainly by the rate of RNA synthesis (transcription), there is little empirical evidence for this. Indeed, studies have shown that regulated RNA turnover influences the expression dynamics of large numbers of transcripts9–13. Regulated mRNA turnover confers regulatory properties that complement transcriptional regulation, such as providing rapid shut-off of gene expression14, and thus it is not surprising that RNA turnover is a major regulator of gene expression.
The critical role of mRNA turnover in defining the level of gene expression predicts that perturbations in mRNA turnover will have a profound impact on normal biological processes, and thus cause disease. Indeed, a growing number of studies have implicated dysregulated RNA decay as influencing many diseases, including cancer15–17. In this Review, we focus on how cancer is specifically influenced by nonsense-mediated RNA decay (NMD), a highly conserved and selective RNA turnover pathway. One of the roles of NMD is to promote the decay of specific mRNAs15,18, just as transcription factors control the synthesis of specific RNAs. Among these NMD target mRNAs are many that encode proteins critical for processes intimately associated with malignancy, including stemness and differentiation, proliferation, the integrated stress response, the unfolded protein response and autophagy15,19,20.
In addition to normal mRNAs, aberrant RNAs harbouring mutations are often degraded by NMD21,22. In particular, NMD recognizes premature termination codons (PTCs), which can be created by several different types of mutations, including nonsense mutations and most frameshift mutations. PTCs are also frequently created by mutations that alter RNA splicing, leading, for example, to intron retention, a common mechanism to inactivate tumour suppressor genes23. Thus, a large spectrum of aberrant RNAs are degraded by NMD.
Tumour cells typically express many RNAs bearing PTC-generating mutations, and thus it has long been hypothesized that the NMD pathway influences tumour growth and tumour progression24–26. For example, NMD has been proposed to promote cancer by virtue of its capacity to degrade mRNAs encoding mutant (but still functional) tumour suppressor proteins24,26–31. More recent genome-wide studies — which we discuss in detail herein — confirm and extend these findings. For example, these studies show that NMD has a profound impact on the mutagenic landscape of tumours, leading to selection for mutations that downregulate tumour suppressor genes, while leaving oncogenic genes relatively intact32–34.
Another class of PTC-bearing RNAs expressed in many tumours includes those encoding so-called neoantigen peptides35. Since the PTCs in these RNAs are created by somatic mutations that typically arise long after the immune tolerance phase of fetal development36, the encoded neoantigen peptides are often highly immunogenic and thus can trigger a potent antitumour immune response. However, this antitumour effect is opposed by NMD, which recognizes the truncating mutations and thus degrades the neoantigen-encoding mRNA. This raises the possibility that inhibition of the NMD pathway will increase neoantigen expression and antitumour immune responses35,37, which we discuss herein.
Recent studies suggest that NMD not only promotes malignancy but can also inhibit malignancy. Loss-of-function mutations in NMD factor genes have been found to be enriched in some tumours, and restoration of NMD in these tumours inhibits malignancy38–44. In this Review, we present this evidence and also consider what might be responsible for the opposing effects of NMD in tumour progression.
The NMD pathway
Originally discovered as a curious phenomenon — the rapid decay of Saccharomyces cerevisiae mRNAs harbouring PTCs22 — NMD has since been firmly established as a highly conserved RNA turnover pathway in organisms spanning the phylogenetic scale18,45,46 (FIG. 1a).
Fig. 1 |. The nonsense-mediated RNA decay pathway.
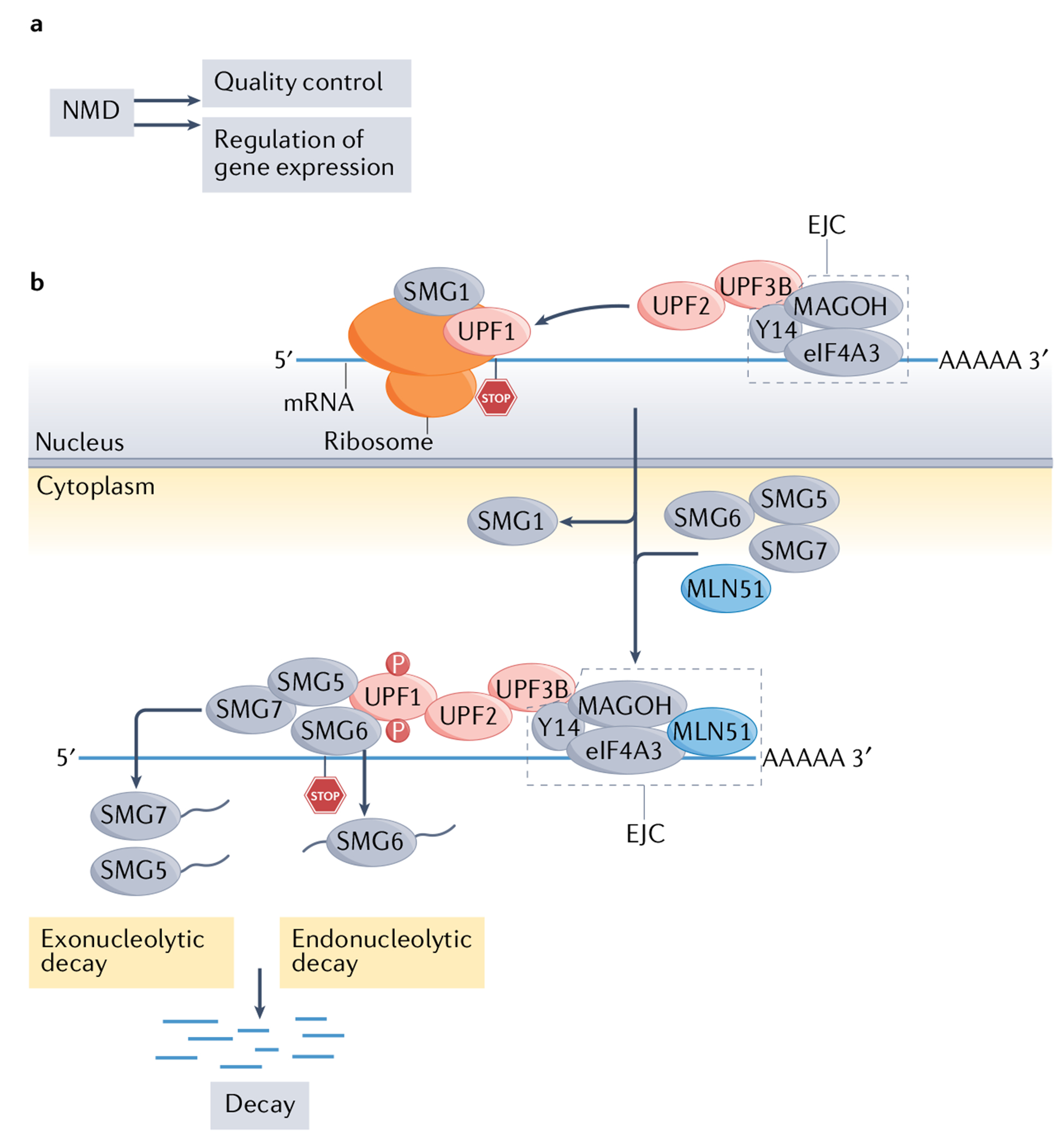
a | The two broad functions of the nonsense-mediated RNA decay (NMD) pathway. b | Simplified view of the molecular mechanism of NMD in mammals. Up-frameshift 1 (UPF1) is an RNA helicase recruited to all mRNAs that remains stably bound only to NMD target mRNAs by an unknown mechanism205; thus, high UPF1 occupancy is regarded as a reliable feature of NMD target mRNAs18. UPF1 also binds to UPF2, which triggers the former to undergo a conformational change that activates its RNA-helicase activity206. UPF2 also binds to UPF3B, which, in turn, interacts with the exon-junction complex (EJC)207, a set of proteins (consisting of eukaryotic translation initiation factor 4A-III (eIF4A3), Y14 (also known as RBM8A) and MAGOH45) that bind near exon–exon junctions in RNAs208 after they are spliced in the nucleus. EJC-bound RNAs are then transported to the cytoplasm, where accessory proteins, including the EJC protein MLN51 (also known as CASC3) are recruited. The formation of an EJC-core NMD factor complex triggers RNA decay, but other mechanisms can also elicit NMD (see FIG. 2). During this chain of events, the protein kinase SMG1 phosphorylates UPF1, which is thought to commit an mRNA to degradation, since phosphorylated UPF1 binds to the RNA endonuclease SMG6, which is known to cleave NMD target mRNAs18. Phosphorylated UPF1 also recruits the SMG5–SMG7 heterodimer104, which interacts with decapping and deadenylating enzymes to further foster RNA decay. NMD is regulated by many factors, including eIF2α phosphorylation during cellular stress, microRNAs and specific regulatory proteins (not shown)18,165,166.
Many proteins have been identified that function in NMD. The first of such NMD factors to be discovered were the proteins up-frameshift 1 (Upf1), Upf2 and Upf3 in S. cerevisiae47,48. Subsequent studies identified orthologues of these proteins that function in NMD in Caenorhabditis elegans49. The UPF proteins were later found to have conserved roles in NMD in all eukaryotes, including fungi, plants and mammals50. Among more complex eukaryotes, several additional proteins were found to function in NMD. The first of these more ‘specialized’ NMD factors to be discovered were those encoded by suppressor with morphological effects on genitalia 1 (smg-1), smg-5, smg-6 and smg-7, identified in a genetic screen in C. elegans49. To date, more than 20 proteins have been shown to have roles in mammalian NMD, some of which are illustrated in FIG. 1b. The molecular choreography of these factors has been dissected in detail and has been well covered by several excellent reviews18,45,46.
The ability of NMD to selectively degrade PTC-bearing mRNAs has been widely hypothesized to indicate that NMD is a quality control mechanism that protects cells from truncated, potentially harmful proteins that would otherwise be produced at high levels51 (FIG. 1a). Indeed, there is evidence that some disease phenotypes are blunted by NMD52,53. However, it has become clear that NMD functions not only as an RNA surveillance mechanism but also as a gene regulatory mechanism that degrades subsets of normal (that is, non-mutant) mRNAs (FIG. 1a). The ability of NMD to regulate normal gene expression was demonstrated first at the genome-wide level in S. cerevisiae54 and later in mammalian cells55. Loss or disruption of NMD leads to dysregulation of between ~5% and 20% of the genes in different organisms, but what proportion of these genes transcribe direct NMD target RNAs remains unclear54–56.
The discovery of widespread normal (non-mutated) NMD target mRNAs led to two key questions. First, how are these normal mRNAs recognized as NMD targets? Second, what are the physiological consequences of the ability of NMD to degrade subsets of normal mRNAs? For the former question, it has been found — in mammalian cells — that NMD is triggered by a stop codon terminating in the main open reading frame within specific contexts, such as upstream of an exon–exon junction or a long 3′ untranslated region (UTR) (FIG. 2b). Although the second question has not yet been fully delineated, it is clear that loss of NMD compromises or completely perturbs several biological processes, implying that NMD has important roles in governing these processes18,57,58. For example, global knockout or depletion of any of a number of NMD factors causes developmental defects, including embryonic lethality, in organisms spanning the phylogenetic scale15,19,59–66. Many studies have also demonstrated that NMD functions in the brain, including during brain development, and in neural differentiation and neural synaptic functions15,67–69. Other physiological roles of NMD are summarized in recent reviews15,18,58,70,71, and are not covered here.
Fig. 2 |. Signals that trigger nonsense-mediated RNA decay and contexts that permit nonsense-mediated RNA decay escape.
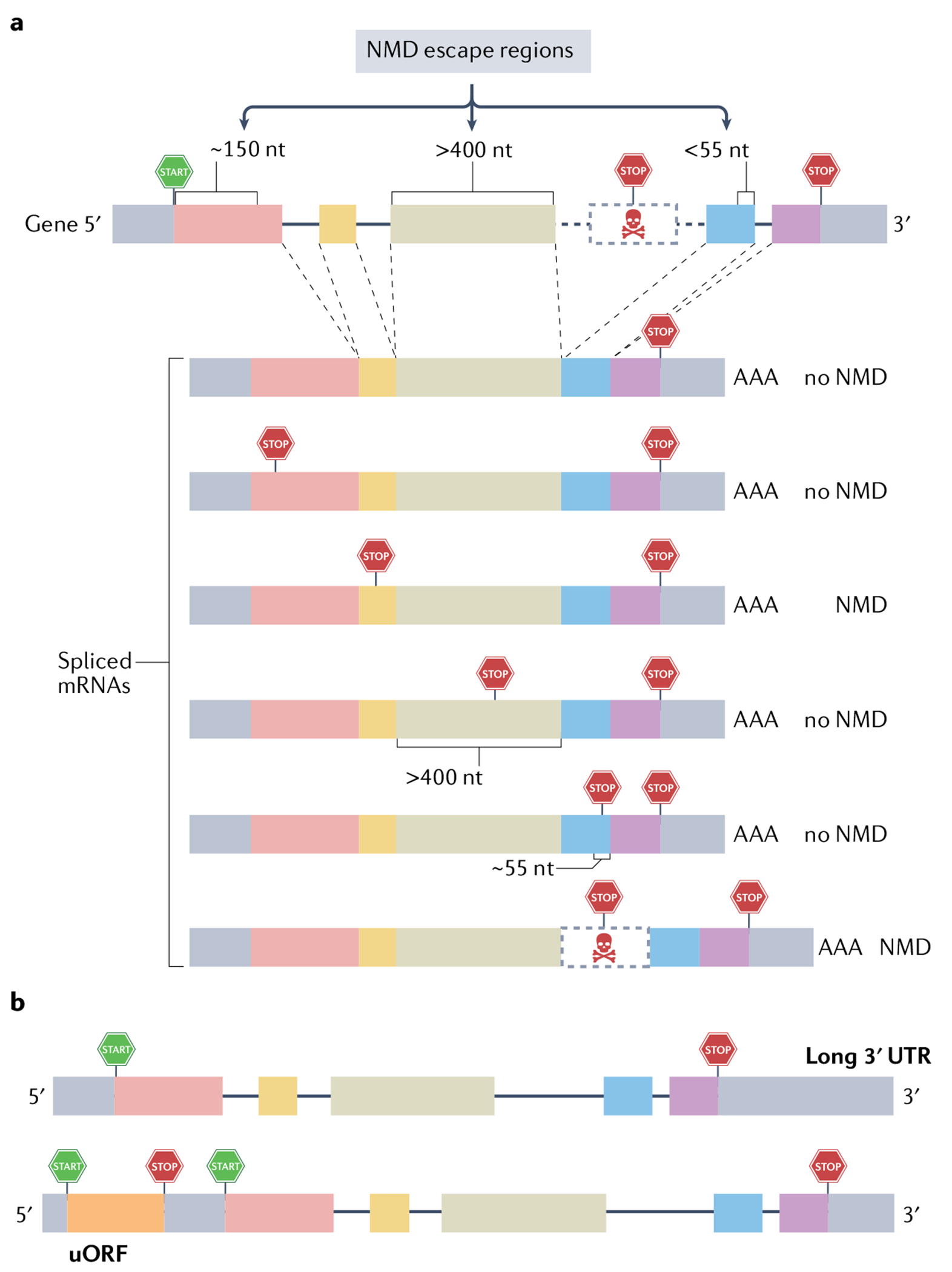
a | In-frame stop codons often, but not always, elicit nonsense-mediated RNA decay (NMD) when upstream of an exon–exon junction. Top: A schematic of a generic gene, showing the position of the translation initiation (‘START’) and termination (‘STOP’) codons defining the main open reading frame (ORF). Bottom: Spliced mRNAs with premature termination codons (PTCs) at different positions, and their fate with respect to whether they are degraded by NMD. Mutations that generate PTCs at most positions upstream of an exon–exon junction trigger NMD. However, this is not universal. For example, PTCs (1) within ~150 nucleotides (nt) of the translation initiation codon, (2) within a large middle exon (more than 400 nt) or (3) located less than 55 nt upstream of the last exon–exon junction typically do not trigger NMD (labelled as ‘NMD escape regions’). The final spliced mRNA shown is an alternatively spliced mRNA harbouring a PTC-bearing alternative (‘poison’) exon, which triggers NMD because the PTC is upstream of an exon–exon junction. Many other alternative splicing events (for example, use of an alternative splice acceptor or donor, not shown) will elicit NMD if this leads to a frameshift and consequent generation of a PTC upstream of an exon–exon junction. Of note, some genes have their normal stop codon in a position that elicits NMD (that is, more than 55 nt upstream of an exon-exon junction); thus, their encoded RNAs are reduced in level by NMD, presumably for the purposes of regulation. b | Long 3′ untranslated regions (UTRs) can also induce NMD, but no particular 3′ UTR length consistently elicits NMD, so this NMD-inducing feature must be tested empirically. Likewise, short upstream ORFs (uORFs) only sometimes trigger NMD, and therefore must also be tested empirically.
While the mechanism by which NMD functions in most biological events is poorly understood, specific NMD target mRNAs have been shown — through rescue experiments — to be critical for the role of NMD in some biological processes, including early embryonic development, neural differentiation and the endoplasmic reticulum stress pathway72–74. This provides strong evidence that the ability of NMD to regulate normal gene expression (as opposed to its ‘RNA surveillance role’ degrading aberrant RNAs) is critical for biological events. An added level of complexity is the evidence that NMD is not a simple linear pathway. Rather, it appears to consist of several branches, each of which depends on different factors and may promote the decay of different sets of transcripts75–78. These NMD branches may also have different functions. For example, loss of the UPF3B-dependent branch of the NMD pathway has been shown to cause intellectual disability and neural defects in humans79,80 and mice68,69.
NMD supports and promotes malignancy
NMD shapes the mutational landscape
‘NMD rules’ defined in tumours.
A central tenet of the NMD pathway is that while mutations that generate a PTC can trigger NMD, many PTC-generating mutations do not elicit NMD (FIG. 2a). Distinguishing between mutations that do or do not elicit NMD is critical for understanding the role of NMD in cancer. While past molecular studies in cell lines defined some rules dictating whether a PTC-generating mutation triggers NMD81–86 (FIG. 2a), it has not been clear whether these rules apply in vivo, and whether there are additional NMD rules. Lindeboom et al.32 investigated these issues by evaluating — genome-wide — the mutational landscape and associated expression pattern of genes in almost 10,000 human tumours. Tumours were ideal for their investigation, as (1) tumours commonly acquire mutations and (2) tumours provide a relatively neutral environment for defining NMD rules given that most of the mutations generated in tumour cells are passenger mutations that do not drive clonal selection87. Lindeboom et al. scored a PTC as being targeted by NMD if the mRNA transcribed from the gene is present at a lower level in tumours bearing the mutation than in tumours of the same type that lack the mutation. Their analysis demonstrated that mutations that generate PTCs in most middle exon positions are associated with decreased mRNA levels, indicative of NMD. In contrast, PTCs in the last exon are associated with normal mRNA levels, in agreement with the well-established ‘last exon rule’ of NMD escape (see FIG. 2a). PTCs in the penultimate exon were also found to be associated with normal mRNA levels if they were closer than approximately 55 nucleotides to the last exon, thereby verifying the well-established ‘55-nucleotide boundary’ rule of NMD escape (FIG. 2a). Indeed, Lindeboom et al. found that almost half of the variation in NMD efficiency across PTCs observed in human tumours could be accounted for by the last exon and 55-nucleotide boundary rules32. They also verified at the genome-wide level that PTCs only a short distance from the start codon often fail to trigger NMD (FIG. 2a), likely due to translation reinitiation, on the basis of earlier studies88. They also obtained evidence that PTCs in long exons (more than 400 nucleotides) tend not to trigger NMD (FIG. 2a). Other NMD-triggering rules were also documented, but 29% of the variation in expression could not be explained, implying that additional NMD rules remain to be identified. Indeed, it has become clear that non-mutant mRNAs can be degraded by NMD by signals in addition to the last exon and 55-nucleotide boundary rules, such as a long 3′ UTR89 (FIG. 2b).
Follow-up work in another cohort of tumour samples showed that half of PTCs (51%) elicit strong NMD, about one quarter (27%) elicit weak NMD and about one quarter (22%) do not trigger NMD33. However, these percentages are highly variable across genes, with a surprisingly large proportion of genes (36%) encoding mRNAs that largely escape NMD when PTCs are introduced into most positions (more than 75%) in the coding sequence. Together, these genomic data allowed the generation of a resource — called ‘NMDetective’ — that predicts whether NMD is triggered by a given PTC-generating mutation33. This resource, along with the recent identification of specific cis elements in RNAs that allow NMD escape89,90, has great potential to be used for precision medicine opportunities in cancer treatment, as well as for diagnosis and treatment of other human diseases impacted by genes harbouring PTC-inducing mutations.
Tumour suppressor genes are targeted by NMD in tumours.
Tumorigenesis is a Darwinian process in which positive selection, negative selection and genetic drift determine the frequency of genetically heterogeneous clones within a tumour mass91. This predicts that PTC-generating mutations in genes that elicit an NMD response favourable for the tumour will be selected for. In support, early studies recognized that tumour suppressor genes are a likely major target of this type of selection mechanism. In particular, tumour suppressor genes in tumour cells were found to accumulate a higher ratio of nonsense and insertion and deletion (indel) frameshift mutations (both of which generate PTCs) relative to missense mutations than do most other genes24–26,92,93. To address the frequency at which this occurs, Lindeboom et al.32 analysed genome-wide expression data from thousands of tumours, evaluating PTC-inducing mutations relative to synonymous mutations for each gene. This analysis verified that tumour suppressor genes acquire PTCs in their coding regions more frequently than most genes in tumours. Importantly, their analysis also showed that tumour suppressor genes in tumours are enriched in PTC-inducing mutations that trigger NMD (‘NMD-elicit’ mutations) as opposed to those that do not elicit NMD32 (FIG. 3). Together, these results support a model in which tumour progression involves selecting for mutations that elicit the decay of mRNAs encoding tumour suppressor proteins.
Fig. 3 |. The dual role of nonsense-mediated RNA decay in cancer.
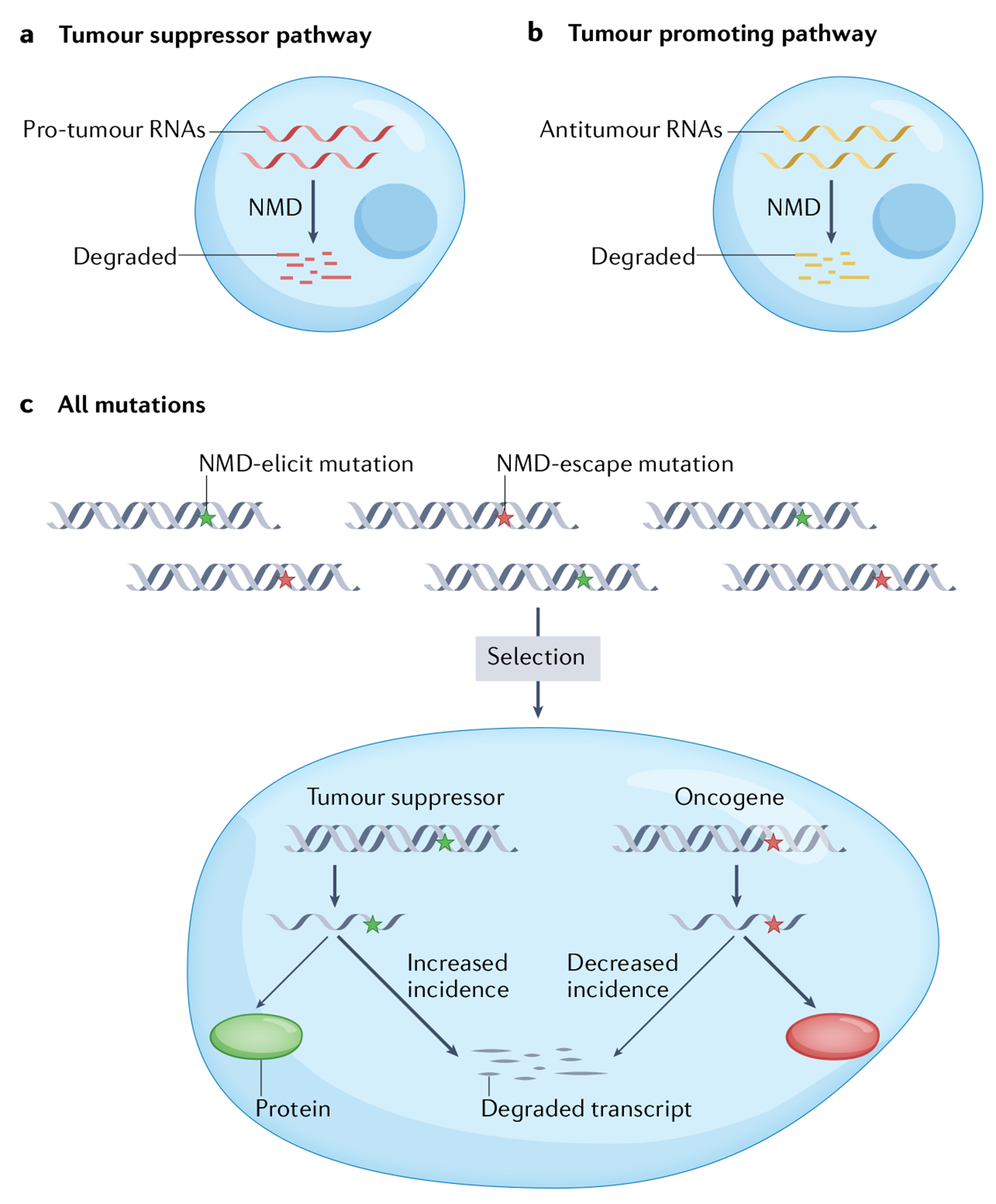
a | Many lines of empirical evidence suggest that nonsense-mediated RNA decay (NMD) is unfavourable to tumours (see the main text). While the mechanism by which NMD inhibits tumours is largely unknown, there is increasing evidence that NMD acts in this capacity by degrading pro-tumour RNAs, including mRNAs encoding proteins involved in signalling pathways and stress responses, as well as non-coding RNAs, including long non-coding RNAs (see FIG. 5). b | NMD can also promote cancer. NMD is thought to favour malignancy through its ability to downregulate the expression of mRNAs encoding both normal and mutant tumour suppressor proteins, as well as other proteins that arrest tumour cell proliferation and/or progression, or lead to programmed cell death. NMD is also known to degrade mutant mRNAs encoding immunogenic neoantigens that would otherwise drive immune-mediated rejection of tumours. c | Randomly generated premature termination codon (PTC) mutations either trigger NMD (‘NMD-elicit’ mutations) or do not trigger NMD (‘NMD-escape’ mutations), depending on their position in an mRNA (see FIG. 2). Advantageous PTC mutations have been shown to be selected for during tumour evolution. Thus, NMD-sensitive PTC mutations tend to be selected for in tumour suppressor genes to decrease their expression. In contrast, NMD-insensitive PTC mutations are enriched in oncogenes to preserve their expression.
In another genome-wide study, Hu et al.34 also found tumours frequently downregulate tumour suppressor genes through NMD. Intriguingly, they discovered that different tumour types tend to differ with regard to which tumour suppressor genes most frequently harbour NMD-elicit mutations. Other gene classes were also found to be targeted by NMD-elicit mutations. In hypermutated tumours, NMD-elicit mutations were found to be enriched in genes encoding proteins involved in DNA repair, chromatin remodelling and/or modifications, and RNA metabolism. This raises the possibility that downregulation of genes involved in these processes is advantageous for hypermutated tumours. In the case of stomach adenocarcinoma with hypermutations, NMD-elicit mutations were commonly enriched in three specific genes involved in RNA metabolism34 — eukaryotic translation initiation factor 5B (EIF5B), La ribonucleoprotein 4B (LARP4B) and PTEN94–96. Among these, PTEN is among one of the most frequently inactivated tumour suppressor genes across all human tumours97. In this regard, it is notable that a recent study showed that tumours driven by PTEN PTC mutations are good candidates to be treated with PTC readthrough-inducing compounds98.
The finding that NMD commonly downregulates mutant tumour suppressor genes suggests that NMD is an important factor promoting tumour progression. In support of this, a recent study33 obtained statistical evidence that NMD more often contributes to disease than protects from disease, including cancer.
Evidence that oncogenes escape NMD.
Given that oncogenes are advantageous to tumours, one would expect tumours to select against mutations that reduce oncogene expression. Consistent with this hypothesis, Lindeboom et al.32 showed that oncogenes in tumours tend to avoid PTC-creating mutations that elicit NMD (FIG. 3). Interestingly, Kishor et al.89 identified a specific example of a mutationally activated oncogene that escapes NMD. Their study focused on the BCL2 pro-survival gene, which is juxtaposed next to the immunoglobulin heavy chain enhancer as a result of translocation in a specific subset of B cell lymphomas. While the translocated immunoglobulin enhancer triggers transcriptional activation of BCL2 in B cells, this translocation also introduces multiple immunoglobulin exons downstream of the BCL2 coding region, which could potentially compromise this transcriptional induction by triggering exon-junction complex-dependent NMD. However, NMD is prevented by specific sequences in the immunoglobulin 3′ UTR (a high density of heterogeneous nuclear ribonucleoprotein L-binding sites), which Kishor et al. showed are essential for high expression of the BCL2 oncogene in B cell lymphomas.
Implications and complexities.
The ‘two-hit’ model of cancer progression posits that biallelic suppression or inactivation of tumour suppressor genes is needed to confer a fitness advantage to cancer cells99. This is traditionally thought to occur via mutations99 that abolish the function of the gene product or transcriptionally inactivate the gene (for example, via DNA methylation100). The weight of evidence now indicates that by destabilizing tumour suppressor mRNAs, NMD is potentially another means to achieve the two-hit mechanism. However, a caveat is that while NMD can reduce the level of some mRNAs harbouring a PTC by more than 30-fold101–103, NMD downregulates most PTC-bearing mRNAs by only threefold to tenfold104. Thus, it is critical to test empirically — through rescue experiments — whether tumour suppressor downregulation mediated by NMD is sufficient to have a functional effect105. To our knowledge, there is currently no direct evidence that the ability of NMD to suppress tumour suppressor gene expression has an impact on tumour formation or progression. A second caveat is that PTC-generating mutations in some tumour suppressor genes, such as TP53 and BRCA1, can generate dominant-negative proteins106–108. By destabilizing such mRNAs, NMD would attenuate the dominant-negative effect, potentially hindering rather than promoting tumours with such mutations (see the section entitled ‘NMD as a tumour suppressor pathway’).
NMD impairs tumour immunogenicity
Neoantigens have potential to elicit tumour rejection.
The field of cancer immunology has historically focused on the ability of single-nucleotide variants (SNVs), including missense mutations, to drive tumour immunity. The concept has been that SNVs lead to the generation of novel amino acids that, in turn, generate novel antigens. However, frameshift indels have the potential to create far greater antigenic diversity than SNVs because the frameshift usually elicits changes in multiple amino acids (FIG. 4). Indeed, studies have indicated that frameshift mutations are far more important for generating tumour immunogenicity than SNVs109,110.
Fig. 4 |. Nonsense-mediated RNA decay degrades mRNAs encoding neoantigens.
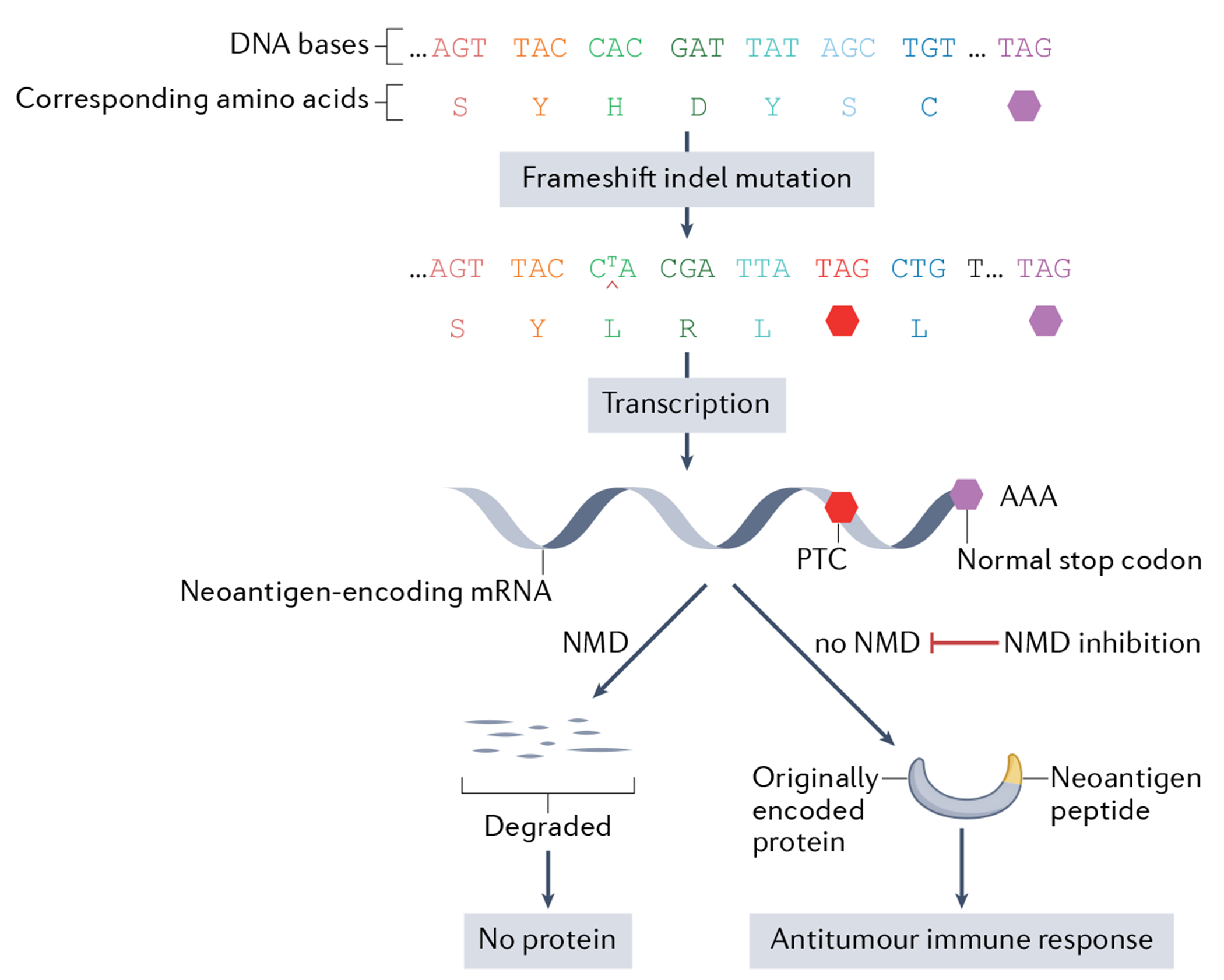
Frameshift insertion and deletion (indel) mutations typically generate novel amino acid sequences at the carboxy terminus of proteins called ‘neoantigens’ (for simplicity, the example in the figure shows a frameshift mutation that generates only three novel carboxy-terminal amino acids). Because neoantigens are regarded as foreign by the immune system, they elicit immune responses, including cytotoxic T cell-mediated death of tumour cells that express these foreign peptides. Mitigating this antitumour action is the fact that frameshift indel mutations typically create a premature termination codon (PTC), which usually leads to the neoantigen-encoding mRNA being rapidly degraded by nonsense-mediated RNA decay (NMD) (see FIG. 2 for positions of PTCs that either elicit or not elicit NMD). Evidence from experimental models indicates that one can reverse the decay of neoantigen-encoding mRNAs by inhibiting NMD. This raises the possibility of using NMD inhibition therapy in patients with cancer to increase the expression of neoantigens on their tumour cells as a means to elicit an effective antitumour immune response.
A frameshift mutation typically results in a truncated protein with a novel amino acid sequence at the carboxy terminus (FIG. 4). This type of neoantigen is commonly generated by tumour cells. In particular, tumours with microsatellite instability (MSI) more frequently generate frameshift mutations because they have defective mismatch repair111. As a result, the tumour-specific antigens which are generated have the potential to be recognized as foreign when presented by major histocompatibility complex (MHC) proteins on the surface of tumour cells112.
As a testament to their importance, Turajlic et al.109 found that frameshift indels generated approximately three times more predicted neoantigens than non-synonymous SNVs. While frameshift indels are relatively infrequent (approximately four per tumour), the few that are generated tend to more likely be immunogenic (that is, generate immunoreactive neoantigens) compared with other types of mutations109. Indeed, neoantigens encoded as a result of frameshift indels are predicted to elicit a ninefold stronger immune response than non-synonymous SNVs109. As evidence that this is relevant to tumour progression, frameshift mutation incidence in MSI tumours was recently shown to be inversely correlated with the predicted immunogenicity of the resulting peptides, suggesting elimination of cell clones with highly immunogenic frameshift peptides113.
Together, these findings raise the possibility that neoantigens produced as a result of frameshift indels are attractive candidates for personalized tumour vaccines. Consistent with this, frameshift indels are associated with a strong response to immune checkpoint inhibitors (ICIs), which confer remarkable antitumour efficacy in subsets of patients with cancer114.
While frameshift indels have great potential to elicit antitumour immunity, the vast majority of neoantigen-generating mutations appear to have no immunogenic effect, on the basis of at least two lines of evidence. First, while hundreds of high-affinity neoantigens are predicted in a typical tumour sample, peptide screens and mass spectrometry studies have detected only a few neoantigens per tumour115,116. Second, tumours responsive to ICI drugs tend to elicit only a modest number of T cell clones117. Thus, while neoantigen burden is a major determinant of tumour immunogenicity, underscored by recent clinical experience with ICI therapy118, most patients’ tumours express few neoantigens, and hence are hyporesponsive to immunotherapy. As we discuss next, NMD has a major role in reducing neoantigen expression in tumours.
Neoantigens are downregulated by NMD.
Frameshift indels typically generate a downstream PTC (FIG. 4). Thus, mRNAs with frameshift indels are often degraded by NMD, thereby reducing the expression of neoantigens (FIG. 4). The corollary of this is that ‘NMD-escape mutations’ — frameshift mutations that generate a PTC in a context where they fail to be recognized by the NMD pathway — allow high expression of a neoantigen. Therefore, whether a frameshift-induced PTC triggers NMD or, instead, evades NMD is critical for determining whether a neoantigen is produced at sufficient level to elicit an immune response. Litchfield et al.37 assessed this issue in a melanoma dataset from The Cancer Genome Atlas (TCGA). They found that frameshift indel mutations predicted to escape NMD (for example, penultimate and last exon mutations) were significantly depleted among all types of frameshift indels in these tumours. Instead, frameshift indel mutations were more likely to occur at positions expected to trigger NMD (for example, in middle exon positions). This implies that NMD-escape mutations are negatively selected in melanomas, leading to the downregulation of most frameshift indel-containing mRNAs encoding neoantigens.
A challenge for oncologists is to determine which patients with cancer should receive ICI therapy. Tumours that express high levels of neoantigens would be expected to be most responsive to ICI therapy. This implies that ICI therapy would be most effective against tumours with neoantigens generated from mRNAs harbouring NMD-escape PTC mutations. To address this hypothesis, Litchfield et al.37 examined the frequency of NMD-escape mutations across a pan-cancer dataset derived from several studies. They found that NMD-escape mutation count was significantly associated with clinical benefit from immunotherapy across both ICI and adoptive cell therapy modalities. Even patients with tumours that had a low tumour mutation burden (TMB) had an NMD-escape mutation count that was significantly associated with clinical benefit from ICI treatment.
To assess whether tumour responsiveness to immunotherapy is linked to neoantigen-induced immunity, Litchfield et al.37 examined data from two antitumour personalized vaccine studies and one ICI study in which functional assays were performed to assess immune reactivity to the patients’ neoantigens. They found that 15 of 19 frameshift indel mutations encode neoantigen peptides that elicit T cell reactivity in vitro. The 15 reactive peptides were significantly longer than the four non-reactive peptides, consistent with the known rules of antigen presentation119. Using a pan-cancer dataset, Lindeboom et al.33 found that tumours with a high frequency of NMD-escape frameshift mutations display significantly higher immune reactivity than tumours harbouring NMD-inducing frameshift mutations. This raised the possibility that this subset of tumours with higher immunoreactivity may also respond better to immunotherapy. Indeed, patients who responded had tumours with significantly higher numbers of NMD-escape frameshift mutations in comparison with patients who did not respond. Furthermore, consideration of the number of NMD-escape frameshift mutations along with TMB was a better predictor of immunotherapy responsiveness than TMB alone33.
If NMD promotes tumour progression by reducing immunogenicity, tumours with perturbed NMD should be subject to increased immune attack. In support of this, Lindeboom et al.33 found that deleterious mutations in the NMD factor gene UPF1 were associated with higher immune filtration in a cohort of patients with endometrial carcinoma. As further evidence, a study examining 17 cancer types obtained bioinformatics evidence that elevated NMD is inversely correlated with stronger immune cytolytic activity120.
Collectively, the results from these studies have several implications. First, the data strongly suggest that NMD suppresses tumour immunosurveillance. Second, the data indicate that an assay of NMD-escape mutation status may be of prognostic value in determining whether a patient should undergo immunotherapy for cancer. Third, the finding that tumours with a high frequency of NMD-escape mutations have a high probability of responding to ICI treatment raises the prospect that patients who fall below the TMB threshold and thus were previously regarded as low priority for ICI therapy might be upgraded to higher priority if they have frequent NMD-escape frameshift mutations.
NMD suppression increases tumour immunogenicity and inhibits tumorigenesis.
The preceding findings predict that NMD inhibition will drive elevated neoantigen expression and consequent immune rejection of tumours. In support of this prediction, Pastor et al.35 found that knockdown of NMD factors inhibited tumour growth and increased T cell infiltration in two metastatic subcutaneous mouse tumour models. However, it was not determined whether increased neoantigen expression was responsible for the increased T cell infiltration. It was also not established whether the increased T cell infiltration was responsible for the decreased tumour growth. Thus, it will be critical for future studies to address whether NMD inhibition is capable of increasing tumour-specific neoantigen expression and consequent immune attack.
A step in this direction was recently taken by Becker et al.121, who found that inhibition of NMD (in response to the clinically licensed drug 5-azacytidine, which, among other functions, acts through MYC to inhibit NMD122) stabilizes frameshift mutation-bearing transcripts and increases the presentation of neoantigen epitopes by class I MHC molecules on MSI tumours. As part of their work, they identified a highly recurrent frameshift indel mutation in MSI colorectal cancer (CRC); immunization with the corresponding neoantigen induced a strong cytolytic T cell response in mice. This study raises the possibility that NMD inhibition therapy will be useful clinically to treat CRC, a notion further supported by two other findings: (1) a chemical NMD inhibitor that antagonizes the exon-junction complex factor eIF4A3 inhibits HCT-116 CRC growth in mice123 and (2) the NMD inhibitor amlexanox124 reduces the growth of MSI CRC cells in vivo but not microsatellite-stable CRC cells, which have fewer PTC mutations125.
NMD magnitude is elevated in some tumour types.
The notion that NMD promotes tumours predicts that NMD magnitude will be elevated in tumours. In support of this, analysis of TCGA pan-cancer datasets revealed that core NMD factor genes (UPF1, UPF2, UPF3B, SMG1, SMG5, SMG6 and SMG7) tend to be amplified in diverse tumour types120. In further support, primary MSI CRC tumours tend to more highly express several NMD factors compared with normal adjacent mucosa125–127, and CRC tumours that overexpress UPF1 tend to have a poor prognosis127. Conversely, UPF3A, which has been reported to act, in part, as an NMD repressor128, is downregulated in clear cell renal cell carcinoma129, and is frequently mutationally inactivated in MSI CRC tumours130. Together, these data suggest that some tumour types have elevated NMD, which is also supported by bioinformatics evidence120. However, it is important to note that, at present, there is no direct evidence that tumours have increased NMD magnitude relative to adjacent normal tissue.
NMD is a tumour suppressor pathway
Tumours with impaired NMD
In the preceding section, we presented the case that NMD promotes tumours. In this section, we present the case for the opposite–that NMD can act to limit tumour progression (FIG. 3). As discussed in detail below, evidence suggests that this is achieved through the ability of NMD to destabilize mRNAs encoding ‘pro-tumour’ proteins. To circumvent this action, it would be advantageous for tumours to downregulate NMD. Indeed, many studies have reported evidence that tumours have impaired NMD. One of the first studies to make this claim was by Wang et al.38, who found that the human PC3 prostate cancer cell line had repressed NMD magnitude (as measured using an NMD reporter and endogenous NMD substrates) when grown as a three-dimensional tumour in vivo compared with when grown as a monolayer in vitro. This interesting finding suggested that the in vivo tumour microenvironment could inhibit NMD. A possible mechanism for this is through hypoxia, as in vivo tumours are often hypoxic, and it has been demonstrated that hypoxia inhibits NMD131. Additional support for this mechanism is the finding by Wang et al.38 that PC3 cells grown as tumours in vivo tend to harbour eIF2α in a phosphorylated state, which is a hallmark of cellular stress responses, including in response to hypoxia.
Subsequent studies obtained additional evidence that tumours have impaired NMD. The core NMD factor UPF1 (FIG. 1b) has been shown to be decreased in level in many different tumour types, including lung adenocarcinoma, gastric cancer, hepatocellular carcinoma (HCC), pancreatic adenosquamous carcinoma (ASC), ovarian cancer, glioma and inflammatory myofibroblastic tumours39–44,132–134. All of these tumour types were reported to have reduced UPF1 mRNA and/or UPF1 levels relative to adjacent normal tissue. Furthermore, two specific regions of the UPF1 gene have been reported to be frequently mutated in ASC and inflammatory myofibroblastic tumours, but not adjacent normal tissue, suggesting that UPF1 downregulation in some tumour types results from somatic mutations41,43. While subsequent work has led to the suggestion that UPF1 mutations in ASC tumours are not ‘cancer drivers’135, this question needs further investigation, including in-depth analysis of tumours rigorously determined to be ASC (as this is a rare tumour that is often misdiagnosed). In addition to UPF1, mutations in other NMD factor genes have been linked with malignancy in a tissue specific context. For example, truncating mutations in the NMD gene SMG7 have been shown to be significantly associated with gastric and colorectal tumours136. Conversely, UPF3A was recently reported to be more highly expressed in CRC metastases than in primary CRC tumours137. Given that UPF3A can act as an NMD repressor128, this raises possibility that NMD suppression may foster metastatic tumour cell movement or the growth of metastatic lesions. In further support of this, a recent analysis of 710 breast tumours revealed that those with high UPF3A expression had a statistically shorter period of metastasis-free survival than did those with low UPF3A expression138.
The evidence that many tumour types have misregulated NMD factor levels and/or NMD factor gene mutations suggests that this property was selected for because NMD suppresses some forms of cancer. Functional experiments support this hypothesis38–40,42,44,132,133. For example, forced expression of the NMD factor UPF1 was found to reduce the malignant phenotype of a lung adenocarcinoma cell line42, gastric cancer cell lines39, an ovarian cancer cell line132, glioma cell lines133, an HCC cell line44 and HCC tumours in vivo40. Manipulation of UPF1 levels was found to alter many different tumour-associated features, including proliferation, apoptosis, invasion, migration and formation of metastases39,40,42,44.
How might NMD suppress malignancy?
The above studies make the case that NMD is decreased in tumours because this RNA turnover pathway has the potential to suppress tumour generation and progression. By what mechanism might this be achieved? Given the increasing evidence that NMD influences normal biological processes through its ability to promote the decay of specific mRNAs72–74, it stands to reason that NMD would also inhibit tumours by the same mechanism. Thus, the prevailing hypothesis is that NMD protects cells from malignant conversion and progression by degrading pro-tumour RNAs (FIG. 3).
In support of this, there is abundant evidence that NMD degrades mRNAs encoding pro-tumour proteins. Genome-wide studies have identified scores of known or suspected NMD target mRNAs encoding proteins with the potential to promote cancer93. For example, a multipronged approach was used to identify 749 high-confidence NMD target mRNAs (expressed in a bone osteosarcoma cell line) that gene ontology analysis showed encode proteins relevant to cancer, including those involved in cell proliferation, apoptosis, and cell migration and invasion38. Other studies have identified high-confidence and putative NMD target mRNAs encoding proteins enriched in cancer-related processes in other tumour cell lines55,139–144. Scores of NMD target mRNAs encoding proteins involved in cancer-associated processes have also been identified in non-tumour cells145,146.
One potential mechanism by which NMD could inhibit tumour progression is by degrading mRNAs that function in signalling pathways known to influence cancer development (FIG. 5). For example, the transforming growth factor-β (TGFβ) signalling pathway component SMAD7, whose mRNA is targeted by NMD to promote the stem-like state73,146, may function downstream of NMD to suppress HCC tumorigenesis40. Since SMAD7 is a negative regulator of TGFβ signalling, NMD is thought to suppress tumorigenesis by promoting TGFβ signalling in this scenario. In contrast, other evidence suggests that NMD confers an antitumour effect by instead inhibiting TGFβ signalling. In particular, the NMD factor UPF1 has been shown — through gain-of-function and loss-of-function studies — to inhibit the epithelial-to-mesenchymal transition (EMT) through inhibition of TGFβ signalling in lung adenocarcinoma cell lines42 and established HCC tumours134. Exactly how EMT is inhibited is unclear, but NMD is known to target mRNAs encoding several EMT regulators, including the hallmark EMT protein TWIST1 (REF.147) (mutations in which portent more aggressive disease in renal cancer148), the tumour suppressor ROBO1 (which is encoded by an mRNA degraded by UPF1 as a result of frameshifting in colon cancer149) and the proto-oncoprotein serine/arginine-rich splicing factor 1 (SRSF1) (whose levels are indirectly regulated by NMD through alternative splicing150).
Fig. 5 |. Putative mechanisms by which nonsense-mediated RNA decay suppresses malignancy.
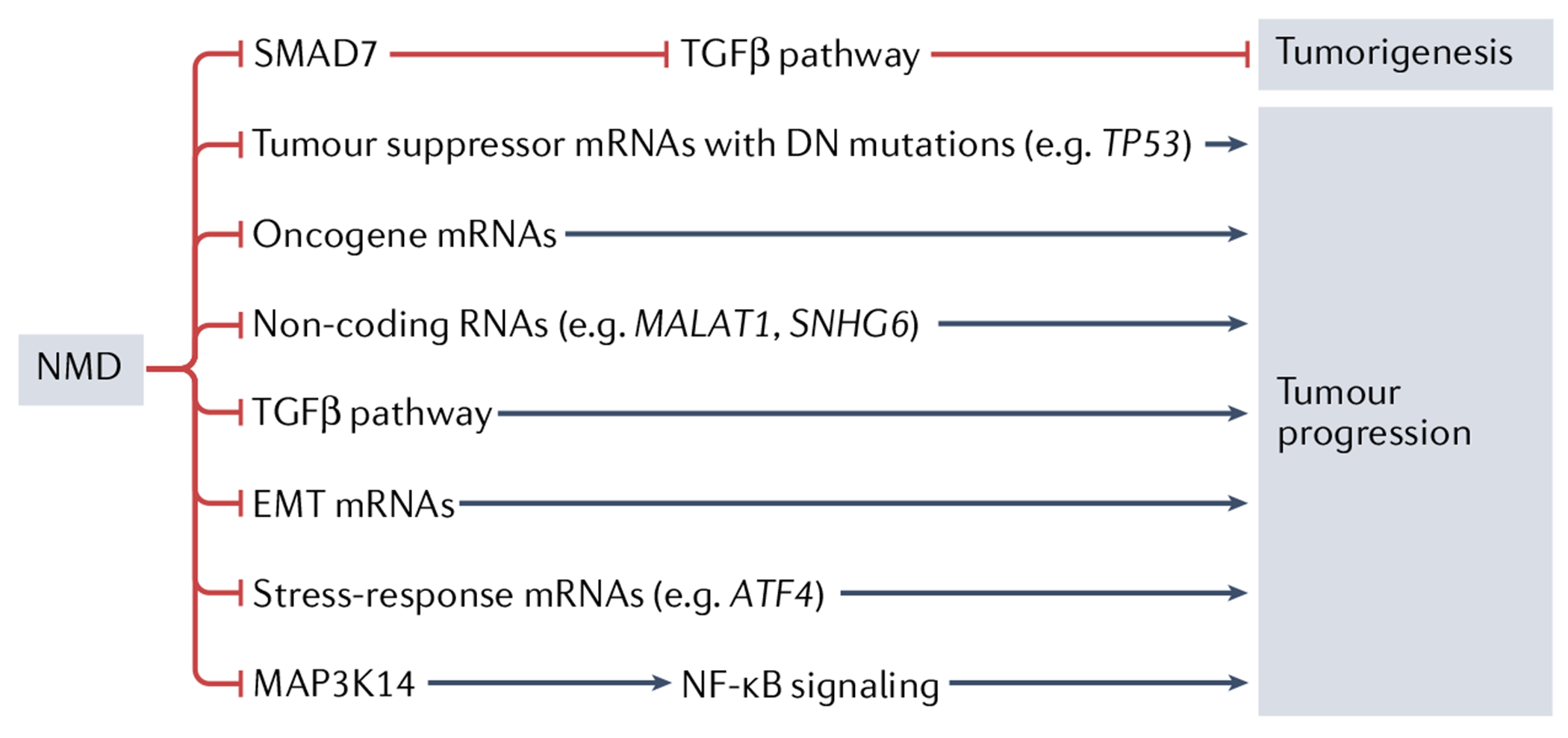
One potential mechanism by which nonsense-mediated RNA decay (NMD) may suppress tumour growth and/or progression is through the known ability of NMD to destabilize mRNAs encoding mutant tumour suppressor proteins harbouring dominant-negative (DN) activity. NMD also destabilizes normal mRNAs encoding some tumour-promoting proteins, raising the possibility that this is another mechanism by which NMD suppresses malignancy. Evidence suggests that NMD may also suppress tumours by degrading pro-tumour non-coding RNAs, including the long non-coding RNA MALAT1 and small nucleolar RNA host gene 6 (SNHG6). NMD also destabilizes many mRNAs encoding stress-response proteins (for example, activating transcription factor 4 (ATF4) mRNA), raising the possibility that this reduces the ability of tumours to cope with the cellular stress known to be elicited by the tumour microenvironment. Evidence suggests that NMD can inhibit malignancy by either promoting or suppressing transforming growth factor-β (TGFβ) signalling, depending on the tumour type. NMD promotes TGFβ signalling by destabilizing the mRNA encoding the negative TGFβ signalling regulator SMAD7. It is not known how NMD inhibits TGFβ signalling, but one conseguence of this action is reduced epithelial-to-mesenchymal transition (EMT). EMT is potentially further reduced by NMD as a result of its known ability to destabilize several mRNAs encoding EMT-promoting factors. Nuclear factor-κB (NF-κB) signalling has been suggested to have a role in inflammatory myofibroblastic tumours through the action of the NF-κB activator MAP3K14 (also known as NIK), which is encoded by an NMD target mRNA41.
Another mechanism by which NMD has been suggested to inhibit tumours is by degrading tumour-promoting non-coding RNAs151–153. While non-coding RNAs do not encode long proteins, many have short open reading frames, and thus some are targeted for decay by NMD through the stop codon at the terminus of these short open reading frames154–157. One long non-coding RNA (lncRNA) that is a candidate to be degraded by NMD to inhibit tumour formation is MALAT1. This lncRNA is upregulated in many different cancers, including non-small-cell lung carcinoma, breast cancer, HCC and gastric cancer158–161. As evidence for causality in cancer, overexpression of MALAT1 was found to reverse UPF1-dependent inhibition of gastric cancer progression39. However, how exactly UPF1 regulates MALAT1 is unclear, as, at present, there is only indirect evidence that it is an NMD target39, and earlier evidence suggests it is degraded by another RNA decay pathway — Staufen-mediated mRNA decay162,163. Another non-coding RNA whose expression may be silenced by UPF1 to suppress tumour formation is the HCC-promoting small nucleolar RNA host gene 6 (SNHG6)164.
Another level of regulation stems from the fact that NMD is suppressed under many conditions, including in response to eIF2α phosphorylation, microRNAs and NMD itself38,141,142,165–167. Of direct relevance to cancer, it has been shown that NMD is inhibited by overexpression of the oncogene MYC, and evidence was obtained that many ‘MYC targets’ are actually NMD targets indirectly regulated by MYC168. This raises the intriguing possibility that activation of MYC in tumours drives their progression, in part, through downregulation of NMD.
Finally, many NMD target RNAs encode proteins involved in cellular stress responses20,38,74,93,131, leading to the interesting possibility that inhibited NMD promotes tumorigenesis by driving chronic stabilization of mRNAs encoding specific stress-adaptation proteins (BOX 1). It is well established that many disease states, including cancer, are associated with long-term activation of stress-response pathways169,170.
Box 1 |. The influence of tumour heterogeneity and cellular stress on nonsense-mediated RNA decay.
Tumours are heterogeneous. Genetically divergent cell populations develop as tumours grow and accumulate additional genomic lesions and/or epigenetic alterations. Layered upon this, different regions of a tumour are subjected to different microenvironmental stresses. This, in turn, influences the transcriptional programmes induced in each cell. Even systemic stresses, such as chemotherapy, may differentially influence cells within a tumour mass, dependent on their microenvironment and mutational status209.
The impact of tumour heterogeneity on nonsense-mediated RNA decay (NMD) is unknown, but it is reasonable to hypothesize that it will lead to alterations in NMD magnitude (with high and low NMD magnitude leading to NMD target mRNAs being downregulated and upregulated, respectively) in different cells (including both tumour cells and stromal cells) within the same tumour. In support of this, many factors have been demonstrated to regulate NMD magnitude165,166. One of the most robust regulators of NMD is cellular stress20,93. Indeed, a likely source of variation in tumours is cellular stress, which most tumours must adapt to for them to continue to develop and thrive210-213. NMD is not only regulated by cellular stress, but, in turn, NMD is known to shape cellular stress responses both in vitro and in vivo17,20,74,93,214. Indeed, many mRNAs encoding stress-response genes are confirmed NMD targets38,74,131. Thus, crosstalk between NMD and cellular stress is likely to dictate stress responses in tumours, ultimately shaping their heterogeneity and consequent malignant potential. Given that cellular stress can occur transiently215-219, NMD magnitude is also likely to change dynamically. Thus, it is critical that NMD magnitude reporters are developed and applied to tumours (see the main text) to determine shifts in NMD strength in different tumour cell populations within a tumour at different points of their evolution.
Much of the evidence that NMD is a tumour suppressor pathway revolves around expression analysis and functional studies conducted on UPF1, as described above. While there is considerable evidence that UPF1 is downregulated or mutated in many tumour types39–44,132–134, in most cases it was not determined whether this was accompanied by reduced NMD magnitude. Because UPF1 mRNA itself is an NMD target141,142, reduced levels of UPF1 could potentially reflect enhanced NMD, not reduced NMD. While this possibility is mitigated by the studies described above showing functional effects of forced UPF1 expression39,40,42,44,132–134, it remains to be determined in most studies whether decreased UPF1 levels translate to reduced NMD efficiency. UPF1 is rate limiting for NMD in some biological contexts131 but not others141. Thus, while NMD magnitude has been shown to be suppressed in some tumour types5,38,40, this has not been assessed for most tumour types with downregulated or mutated NMD factors.
Interpretation of UPF1 gain-of-function and loss-of-function studies is further complicated by the fact that UPF1 functions not only in NMD but also in other pathways, including other RNA turnover pathways171–173. This leads to the interesting possibility that perturbation of UPF1 promotes tumours by mechanisms other than (or in addition to) NMD. In support of this concept, haploinsufficiency of another NMD factor gene, Smg1, has been shown to predispose mice to tumour formation through a mechanism independent of NMD174.
NMD therapy
In the preceding sections, we summarized the evidence that NMD has critical roles in malignancy. This raises the possibility that modulating the activity of NMD will be of therapeutic benefit in treating some forms of cancer. However, for such therapeutic potential to be realized, it will be critical to determine the role of NMD at different stages of tumour development. Once specific NMD target RNAs that are functionally important in a given tumour cell stage and cell type are defined, this will allow the emergence of approaches to determine whether NMD-stimulatory versus NMD-inhibitory therapy is called for in a given patient with cancer (FIG. 6). For example, transcriptome analysis will reveal whether dysregulated pro-tumour or antitumour NMD target RNAs dominate in a given tumour. Further information will be gleaned from tumour DNA sequencing, which is already an area of focus in precision medicine175. In particular, DNA sequencing of the tumour will reveal the profile of PTC-generating mutations, including whether they are likely to trigger NMD. Together, genome-wide DNA and RNA information for a given tumour, coupled with knowledge of the underlying mechanisms by which NMD influences tumorigenesis, will likely generate predictions as to whether to provide NMD-inhibitory therapy, NMD-stimulatory therapy or neither for a given patient.
Fig. 6 |. Nonsense-mediated RNA decay modulatory therapy.

Shown is a step-by-step approach which may be used in the future to determine whether a given tumour is a candidate for nonsense-mediated RNA decay (NMD)-stimulatory therapy or NMD-inhibitory therapy. Genomic and transcriptomic sequencing analysis will provide genome-wide information on mutations and transcript Levels, which will define both normal and mutant mRNAs downregulated by NMD in a given tumour. With use of an algorithm, the data from a given tumour will allow the tumour to be defined as either having an ‘NMD pro-tumour’ phenotype or an ‘NMD antitumour’ phenotype, on the basis of what is known — in the future — about the role of NMD target mRNAs in the particular type of tumour being analysed (as determined by standard pathology analysis). Another factor that will be considered is the overall NMD magnitude in the tumour, which will be determined from transcriptome analysis. Low NMD magnitude (inferred by upregulated NMD target mRNAs) would suggest treatment with NMD-stimulatory therapy. High NMD magnitude (inferred by downregulated NMD target mRNAs) would suggest treatment with NMD-inhibitory therapy.
Another important diagnostic assay will be a means to measure NMD magnitude. High NMD magnitude reduces the levels of NMD target RNAs, while low NMD magnitude has the opposite effect, both of which are likely to have profound effects on tumour cells. One approach to examine NMD magnitude is to use ‘reporter vectors’ that generate a visible and quantitative signal (such as from GFP) from a gene that either contains or lacks a PTC. Several NMD reporters have been generated and used to measure NMD magnitude in cell lines in vitro176–178, but they have not yet been applied to tracking NMD magnitude in tumours in vivo (for example, tumours injected into mice). Another approach to determine NMD magnitude is to analyse shifts in the expression of normal NMD target mRNAs. This approach can be used to measure NMD magnitude in human tumour biopsy samples, and it avoids the inherent bias of examining only a single reporter mRNA. This method has already been used to demonstrate NMD variability between lung tumours from different individuals179. To use this approach to examine NMD heterogeneity within tumours, single-cell RNA-sequencing analysis will be required. Single-cell RNA-sequencing analysis also has the potential to determine shifts in NMD magnitude during tumour progression, including differences that might arise as primary tumours evolve to form metastatic lesions.
Towards the goal of suppressing NMD in patients with cancer, many therapeutic agents that inhibit NMD have been identified, including pateamine A180, wortmannin181, rapamycin182, amlexanox183, NMDI-1 (REF.184), 5-azacytidine122, AKT inhibitors185, homoharringtonine (also known as omacetaxine mepesuccinate)186 and other small molecules187,188. Some of these are clinically licensed drugs already used to treat cancer. For example, 5-azacytidine is used to treat myelodysplastic syndrome189. Derivatives of rapamycin that have mTOR inhibitor activity (for example, everolimus and sirolimus) are widely used to treat cancer182,190. Homoharringtonine is a protein synthesis inhibitor and cell cycle inhibitor that is used to treat leukaemia and other forms of cancer191. Small molecules modelled on the NMD inhibitor wortmannin181,190,192 are currently in late-stage clinical trials for cancer. Thus, it will be critical in the future to determine whether some of the antitumour effects of these chemotherapeutic drugs stem from their ability to inhibit NMD.
Since loss of NMD is known to perturb various biological processes15,18,20,46, it will be important to also identify drugs that inhibit specific NMD branches75–78, rather than the entire NMD pathway. In this regard, it has been reported that knockdown of the NMD factor SMG8 causes less deleterious effects than knockdown of other NMD factors193.
To treat tumours that have been selected to have weak NMD, NMD-stimulatory therapy will be required. Only one NMD activator has been identified to date — tranilast194 — and thus an important area of future research will be to identify other NMD activators.
Single drug approaches have a limited record of success and durability in treating cancer195. Thus, we expect that NMD-modulatory agents will be used mainly as adjuvants with other approaches. In this Review, we have discussed the likely benefits of using NMD inhibitors in combination with ICI therapies to elicit strong immune responses against tumours35,121,123. NMD-modulatory therapy might also exhibit efficacy when used in combination with chemotherapeutic agents; indeed, there is evidence that NMD inhibition and/or NMD stimulation has synergistic antitumour activity in vitro when paired with use of doxorubicin or the targeted therapy sorafenib39,134,196. A further benefit of NMD modulation is that it may ‘homogenize’ NMD magnitude across the tumour cells in a given tumour, thereby making it more susceptible to co-administered chemotherapeutic agents. Of note, NMD has been reported to promote chemoresistance127, perhaps by degrading the mRNA encoding the multidrug resistance protein ATP-binding cassette subfamily C member 2 (ABCC2)134, or by promoting stemness73, an intrinsically chemoresistant state. This provides a further justification for NMD-inhibitory therapy in combination with chemotherapy.
Perspectives and conclusions
There are many outstanding questions that remain to be resolved. For example, which specific steps of tumorigenesis are influenced by NMD? Does NMD promote some stages of tumour development and hinder other stages? The ability of NMD to destabilize mRNAs encoding proapoptotic proteins and other tumour suppressor proteins24,26,27,72,196–199 suggests NMD may be critical for early stages of tumorigenesis when mutagenic activation of oncogenes initially makes tumour cells susceptible to apoptosis200,201. In contrast, the ability of NMD to suppress neoantigen expression might become relevant only after a tumour has sufficiently expanded to have the potential to induce a significant immune response.
To address these issues, it will be important to understand the molecular mechanism of action of NMD in different settings and cellular contexts. One fundamental question will be whether NMD affects tumours through its ability to destabilize mutant PTC-bearing mRNAs or normal mRNAs. While there is considerable evidence that NMD degrades mutant PTC-bearing mRNAs encoding tumour suppressors and neoantigens24,26,27,35, there has been little evidence that NMD actually promotes tumours by this mechanism. Efforts should also be directed towards examining the possibility that NMD also promotes cancer through downregulating specific normal RNAs.
For example, NMD is known to degrade normal RNAs encoding proapoptotic regulators, including growth arrest-specific 5 (GAS5), BAK1, BCL3, GADD45A and GADD45B (REFS72,196,197), but it remains to be seen whether NMD-directed degradation of any of these proapoptotic RNAs provides a significant boost to tumour formation or tumour survival. Conversely, it has been hypothesized that there is selection to suppress NMD as a means to stabilize mRNAs encoding proteins involved in adaption to cellular stress, but this remains to be directly tested (BOX 1).
Another major class of NMD targets is alternatively spliced mRNAs. Given that most genes undergo alternative RNA splicing, the ability of NMD to degrade specific subsets of alternatively spliced mRNAs has the potential to have a large impact on malignancy. A prominent example of this is myeloid malignancies harbouring a recurrent mutation in SRSF2, which encodes an RNA splicing regulatory factor. This mutant SRSF2 not only has aberrant RNA splicing activity but it activates NMD to degrade specific mRNAs, including an alternatively spliced isoform of the mRNA encoding the tumour suppressor enhancer of zeste homologue 2 (EZH2), leading to disrupted haematopoiesis and likely consequent formation of myeloid malignancies202,203. The ability of NMD to destabilize alternatively spliced mRNAs has also been shown to influence EMT150. As another example, it was recently shown that two alternatively spliced forms of p53 — p53β and p53γ — both of which are largely insensitive to repression by the p53 negative regulator MDM2, are encoded by mRNAs destabilized by NMD204. Evidence that this regulation by NMD is physiologically relevant comes from the finding that inhibition of NMD in mouse non-small-cell lung cancer and glioblastoma cell lines increased the expression of these p53 isoforms concordant with increased tumour cell apoptosis, increased tumour radiosensitivity and other known p53-dependent tumour-associated events204.
We conclude that NMD is an evolutionarily conserved pathway with a dichotomous role in cancer. Consistent with the well-recognized role of NMD as an RNA surveillance pathway that ameliorates disease51, evidence suggests that NMD acts to prevent cancer incidence and progression through the ability of NMD to destabilize RNAs that either directly promote tumours (for example, some lncRNAs) or encode pro-tumour proteins (for example, dominant-negative tumour suppressor proteins). In contrast, NMD appears to promote tumour progression by destabilizing RNAs encoding proteins that suppress malignancy, including tumour suppressor proteins and immunogenic proteins harbouring neoantigens generated as a result of PTC mutations recognized by NMD. As we learn more about how NMD influences different tumour types and the different steps of tumorigenesis and tumour progression, it will hopefully become possible to rationally predict the best therapeutic approach to successfully treat a given tumour with NMD-modulatory agents.
Acknowledgements
This study was funded by NIH R01 HD093846 (M.F.W.), as well as NIH R01 CA247562 and NIH R01 CA244182 (D.G.S.).
Glossary
- RNA turnover
Degradation of RNAs; typically achieved through the action of specific nucleases.
- Premature termination codons
(PTCs). Stop codons created by a mutation that prematurely terminates translation. PTC-bearing genes encode truncated proteins.
- Nonsense mutations
Substitutions of a single base pair that leads to the appearance of a stop codon where previously there was a codon specifying an amino acid.
- Frameshift mutations
insertions and deletions downstream of the initiator codon that are not a multiple of 3, thereby shifting the reading frame. This results in a sequence of amino acids different from that in the original protein. Typically, a premature termination codon is also generated, which results in decay of the mRNA by nonsense-mediated RNA decay.
- Neoantigen
Non-self amino acid residue generated as a result of somatic mutations, including frameshift mutations. Such sequences are recognized as foreign when they are generated after the immune tolerance phase of fetal development.
- Untranslated region
(UTR). The region of an mRNA upstream and downstream of the coding region. The 5′ UTR is upstream of the initiator codon and the 3′ UTR is downstream of the stop codon.
- Passenger mutations
Mutations that have no discernible effect on cell fitness but are associated with clonal expansion simply because they occur in the same genome that harbours driver mutations.
- Missense mutations
Genetic alterations in which a single base pair substitution alters the genetic code in a way that produces an amino acid that is different from the usual amino acid at that position.
- Synonymous mutations
Changes in the DNA sequence that alter the codon but do not change the encoded amino acid (due to redundancy of the genetic code).
- PTC readthrough-inducing compounds
Small molecules that enable the ribosomal machinery to read a stop codon as a codon encoding an amino acid, often resulting in the expression of a full-length functional protein.
- Exon-junction complex
A protein complex deposited near most exon–exon junctions following RNA splicing. it stimulates nonsense-mediated RNA decay and also regulates several other post-transcriptional events.
- Dominant-negative proteins
Mutant proteins that antagonize the function of the wild type protein.
- Microsatellite instability
(MSI). Deficient DNA mismatch repair in tumours, which as a consequence makes them prone to hypermutation.
- Immune checkpoint inhibitors
(ICIs). Drugs that block checkpoint proteins (negative regulatory proteins) in immune cells. ICI therapy derepresses immune function and thereby enhances adaptive immune responses (that is, T cell and B cell responses).
- Epithelial-to-mesenchymal transition
(EMT). A critical developmental process co-opted by tumours in which epithelial cells acquire mesenchymal traits, including their migratory and invasive properties.
- Staufen-mediated mRNA decay
An mRNA decay pathway in competition with nonsense-mediated RNA decay that depends on the RNA-binding protein Staufen 1 or Staufen 2 and the RNA helicase up-frameshift 1 (UPF1).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Galloway A & Cowling VH mRNA cap regulation in mammalian cell function and fate. Biochim. Biophys. Acta Gene Regul. Mech 1862, 270–279 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pisera A, Campo A & Campo S Structure and functions of the translation initiation factor eIF4E and its role in cancer development and treatment. J. Genet. Genomics 45, 13–24 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Stavraka C & Blagden S The La-related proteins, a family with connections to cancer. Biomolecules 5, 2701–2722 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghigna C. et al. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol. Cell 20, 881–890 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Karni R. et al. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol 14, 185–193 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dvinge H, Kim E, Abdel-Wahab O & Bradley RK RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 16, 413–430 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kohler A & Hurt E Gene regulation by nucleoporins and links to cancer. Mol. Cell 38, 6–15 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Hautbergue GM RNA nuclear export: from neurological disorders to cancer. Adv. Exp. Med. Biol 1007, 89–109 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Cheadle C. et al. Control of gene expression during T cell activation: alternate regulation of mRNA transcription and mRNA stability. BMC Genomics 6, 75 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alonso CR A complex ‘mRNA degradation code’ controls gene expression during animal development. Trends Genet. 28, 78–88 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Neff AT, Lee JY, Wilusz J, Tian B & Wilusz CJ Global analysis reveals multiple pathways for unique regulation of mRNA decay in induced pluripotent stem cells. Genome Res. 22, 1457–1467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munchel SE, Shultzaberger RK, Takizawa N & Weis K Dynamic profiling of mRNA turnover reveals gene-specific and system-wide regulation of mRNA decay. Mol. Biol. Cell 22, 2787–2795 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hao S & Baltimore D The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol 10, 281–288 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schoenberg DR & Maquat LE Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet 13, 246–259 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaffrey SR & Wilkinson M F Nonsense-mediated RNA decay in the brain: emerging modulator of neural development and disease. Nat. Rev. Neurosci 19, 715–728 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolin SL & Maquat LE Cellular RNA surveillance in health and disease. Science 366, 822–827 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nogueira G, Fernandes R, Garcia-Moreno JF & Romao L Nonsense-mediated RNA decay and its bipolar function in cancer. Mol. Cancer 20, 72 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurosaki T, Popp MW & Maquat LE Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol 20, 406–420 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hwang J & Maquat LE Nonsense-mediated mRNA decay (NMD) in animal embryogenesis: to die or not to die, that is the question. Curr. Opin. Genet. Dev 21, 422–430 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goetz AE & Wilkinson M Stress and the nonsense-mediated RNA decay pathway. Cell Mol. Life Sci 74, 3509–3531 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang JC & Kan YW beta 0 thalassemia, a nonsense mutation in man. Proc. Natl Acad. Sci. USA 76, 2886–2889 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Losson R & Lacroute F Interference of nonsense mutations with eukaryotic messenger RNA stability. Proc. Natl Acad. Sci. USA 76, 5134–5137 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jung H. et al. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat. Genet 47, 1242–1248 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Perrin-Vidoz L, Sinilnikova OM, Stoppa-Lyonnet D, Lenoir GM & Mazoyer S The nonsense-mediated mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing premature termination codons. Hum. Mol. Genet 11, 2805–2814 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Ware MD et al. Does nonsense-mediated mRNA decay explain the ovarian cancer cluster region of the BRCA2 gene? Oncogene 25, 323–328 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Karam R. et al. The NMD mRNA surveillance pathway downregulates aberrant E-cadherin transcripts in gastric cancer cells and in CDH1 mutation carriers. Oncogene 27, 4255–4260 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Anczukow O. et al. Does the nonsense-mediated mRNA decay mechanism prevent the synthesis of truncated BRCA1, CHK2, and p53 proteins? Hum. Mutat 29, 65–73 (2008). [DOI] [PubMed] [Google Scholar]; This study is the first to show different roles for PTCs in tumour suppressor genes. PTCs could elicit NMD or destabilize the final protein product, leading in either case to lower overall protein levels.
- 28.Zientek-Targosz H. et al. Transformation of MCF-10A cells by random mutagenesis with frameshift mutagen ICR191: a model for identifying candidate breast-tumor suppressors. Mol. Cancer 7, 51 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ionov Y, Nowak N, Perucho M, Markowitz S & Cowell JK Manipulation of nonsense mediated decay identifies gene mutations in colon cancer cells with microsatellite instability. Oncogene 23, 639–645 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Ivanov I, Lo KC, Hawthorn L, Cowell JK & Ionov Y Identifying candidate colon cancer tumor suppressor genes using inhibition of nonsense-mediated mRNA decay in colon cancer cells. Oncogene 26, 2873–2884 (2007). [DOI] [PubMed] [Google Scholar]; This study uses a novel screen — involving mutagenesis followed by NMD inhibition — to identify known and novel candidate tumour suppressor genes.
- 31.Rossi MR et al. Identification of inactivating mutations in the JAK1, SYNJ2, and CLPTM1 genes in prostate cancer cells using inhibition of nonsense-mediated decay and microarray analysis. Cancer Genet. Cytogenet 161,97–103 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Lindeboom RG, Supek F & Lehner B The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet 48, 1112–1118 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study analyses the mutational landscape and gene expression patterns of ~10,000 human tumours, allowing ‘NMD rules’ to be defined that dictate whether a PTC mutation elicits NMD. The authors also use these datasets to identify classes of genes undergoing positive and negative selection for ‘NMD-elicit’ PTC mutations in tumours.
- 33.Lindeboom RGH, Vermeulen M, Lehner B & Supek F The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet 51, 1645–1651 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study examines the impact of NMD on cancer and genetic diseases. It also generates NMDetective, a resource that predicts whether NMD is triggered by a given PTC-generating mutation.
- 34.Hu Z, Yau C & Ahmed AA A pan-cancer genome-wide analysis reveals tumour dependencies by induction of nonsense-mediated decay. Nat. Commun 8, 15943 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This genome-wide study classifies tumours with respect to classes of genes enriched in ‘NMD-elicit’ mutations. This revealed that tumour types differ with respect to the profile of tumour suppressor genes that tended to be downregulated by NMD.
- 35.Pastor F, Kolonias D, Giangrande PH & Gilboa E Induction of tumour immunity by targeted inhibition of nonsense-mediated mRNA decay. Nature 465, 227–230 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports the discovery that inhibiting NMD suppresses tumour cell growth and increases T cell infiltration in various subcutaneous and metastatic mouse tumour models.
- 36.Nossal GJ Cellular mechanisms of immunologic tolerance. Annu. Rev. Immunol 1, 33–62 (1983). [DOI] [PubMed] [Google Scholar]
- 37.Litchfield K. et al. Escape from nonsense-mediated decay associates with anti-tumor immunogenicity. Nat. Commun 11, 3800 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows — at the genome-wide level — that frameshift mutated transcripts (encoding neoantigens) that are insensitive to NMD tend to elicit antitumour immune responses.
- 38.Wang D et al. Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol. Cell Biol 31, 3670–3680 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that inhibited NMD promotes tumorigenesis. The authors also provide evidence that the tumour microenvironment inhibits NMD in vivo.
- 39.Li L. et al. The human RNA surveillance factor UPF1 modulates gastric cancer progression by targeting long non-coding RNA MALAT1. Cell Physiol. Biochem 42, 2194–2206 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Chang L. et al. The human RNA surveillance factor UPF1 regulates tumorigenesis by targeting Smad7 in hepatocellular carcinoma. J. Exp. Clin. Canc. Res 10.1186/s13046-016-0286-2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu J. et al. The nonsense-mediated RNA decay pathway is disrupted in inflammatory myofibroblastic tumors. J. Clin. Invest 126, 3058–3062 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao L. et al. Human nonsense-mediated RNA decay regulates EMT by targeting the TGF-ss signaling pathway in lung adenocarcinoma. Cancer Lett. 403, 246–259 (2017). [DOI] [PubMed] [Google Scholar]; This study reports evidence that NMD inhibits EMT through regulation of TGFβ signalling in lung adenocarcinomas, thereby providing a potential mechanism by which NMD suppresses malignancy.
- 43.Liu C. et al. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat. Med 20, 596–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou Y. et al. UPF1 inhibits the hepatocellular carcinoma progression by targeting long non-coding RNA UCA1. Sci. Rep 9, 6652 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang YF, Imam JS & Wilkinson MF The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem 76, 51–74 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Supek F, Lehner B & Lindeboom RGH To NMD or not to NMD: nonsense-mediated mRNA decay in cancer and other genetic diseases. Trends Genet. 10.1016/j.tig.2020.11.002 (2020). [DOI] [PubMed] [Google Scholar]
- 47.Leeds P, Peltz SW, Jacobson A & Culbertson MR The product of the yeast UPF1 gene is required for rapid turnover of mRNAs containing a premature translational termination codon. Genes Dev. 5, 2303–2314 (1991). [DOI] [PubMed] [Google Scholar]
- 48.Gonzalez CI, Bhattacharya A, Wang W & Peltz SW Nonsense-mediated mRNA decay in Saccharomyces cerevisiae. Gene 274, 15–25 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Pulak R & Anderson P mRNA surveillance by the Caenorhabditis elegans smg genes. Genes Dev. 7, 1885–1897 (1993). [DOI] [PubMed] [Google Scholar]
- 50.Lloyd JPB The evolution and diversity of the nonsense-mediated mRNA decay pathway. F1000Res 7, 1299 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holbrook JA, Neu-Yilik G, Hentze MW & Kulozik AE Nonsense-mediated decay approaches the clinic. Nat. Genet 36, 801–808 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Inoue K. et al. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet 36, 361–369 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Thein SL et al. Molecular basis for dominantly inherited inclusion body beta-thalassemia. Proc. Natl Acad. Sci. USA 87, 3924–3928 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lelivelt MJ & Culbertson MR Yeast Upf proteins required for RNA surveillance affect global expression of the yeast transcriptome. Mol. Cell Biol 19, 6710–6719 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F & Dietz HC Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet 36, 1073–1078 (2004). [DOI] [PubMed] [Google Scholar]
- 56.He F. et al. Genome-wide analysis of mRNAs regulated by the nonsense-mediated and 5’ to 3’ mRNA decay pathways in yeast. Mol. Cell 12, 1439–1452 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Karousis ED & Muhlemann O Nonsense-mediated mRNA decay begins where translation ends. Cold Spring Harb. Perspect. Biol 10.1101/cshperspect.a032862 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nasif S, Contu L & Muhlemann O Beyond quality control: the role of nonsense-mediated mRNA decay (NMD) in regulating gene expression. Semin. Cell Dev. Biol 75, 78–87 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Vicente-Crespo M & Palacios IM Nonsense-mediated mRNA decay and development: shoot the messenger to survive? Biochem. Soc. Trans 38, 1500–1505 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Medghalchi SM et al. Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum. Mol. Genet 10, 99–105 (2001). [DOI] [PubMed] [Google Scholar]
- 61.Li T. et al. Smg6/Est1 licenses embryonic stem cell differentiation via nonsense-mediated mRNA decay. EMBO J. 34, 1630–1647 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mcllwain DR et al. Smg1 is required for embryogenesis and regulates diverse genes via alternative splicing coupled to nonsense-mediated mRNA decay. Proc. Natl Acad. Sci. USA 107, 12186–12191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Longman D, Plasterk RH, Johnstone IL & Caceres JF Mechanistic insights and identification of two novel factors in the C. elegans NMD pathway. Genes Dev. 21, 1075–1085 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alonso CR & Akam M A Hox gene mutation that triggers nonsense-mediated RNA decay and affects alternative splicing during Drosophila development. Nucleic Acids Res. 31, 3873–3880 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anastasaki C, Longman D, Capper A, Patton EE & Caceres JF Dhx34 and Nbas function in the NMD pathway and are required for embryonic development in zebrafish. Nucleic Acids Res. 39, 3686–3694 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Metzstein MM & Krasnow MA Functions of the nonsense-mediated mRNA decay pathway in Drosophila development. PLoS Genet. 2, e180 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Colak D, Ji SJ, Porse BT & Jaffrey SR Regulation of axon guidance by compartmentalized nonsense-mediated mRNA decay. Cell 153, 1252–1265 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang L. et al. A Upf3b-mutant mouse model with behavioral and neurogenesis defects. Mol. Psychiatry 23, 1773–1786 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tan K. et al. The role of the NMD factor UPF3B in olfactory sensory neurons. eLife 10.7554/eLife.57525 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Han X. et al. Nonsense-mediated mRNA decay: a ‘nonsense’ pathway makes sense in stem cell biology. Nucleic Acids Res. 46, 1038–1051 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lejeune F. Nonsense-mediated mRNA decay at the crossroads of many cellular pathways. BMB Rep. 50, 175–185 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nelson JO, Moore KA, Chapin A, Hollien J & Metzstein MM Degradation of Gadd45 mRNA by nonsense-mediated decay is essential for viability. eLife 10.7554/eLife.12876 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lou CH et al. Posttranscriptional control of the stem cell and neurogenic programs by the nonsense-mediated RNA decay pathway. Cell Rep. 6, 748–764 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Karam R. et al. The unfolded protein response is shaped by the NMD pathway. EMBO Rep. 16, 599–609 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chan WK et al. An alternative branch of the nonsense-mediated decay pathway. EMBO J. 26, 1820–1830 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gehring NH et al. Exon-junction complex components specify distinct routes of nonsense-mediated mRNA decay with differential cofactor requirements. Mol. Cell 20, 65–75 (2005). [DOI] [PubMed] [Google Scholar]
- 77.Mabin JW et al. The exon junction complex undergoes a compositional switch that alters mRNP structure and nonsense-mediated mRNA decay activity. Cell Rep. 25, 2431–2446 e2437 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yi Z, Sanjeev M & Singh G The branched nature of the nonsense-mediated mRNA decay pathway. Trends Genet. 37, 143–159 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tarpey PS et al. Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat. Genet 39, 1127–1133 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nguyen LS, Wilkinson MF & Gecz J Nonsense-mediated mRNA decay: inter-individual variability and human disease. Neurosci. Biobehav. Rev 46, 175–186 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nagy E & Maquat LE A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem. Sci 23, 198–199 (1998). [DOI] [PubMed] [Google Scholar]
- 82.Cirulli ET et al. A whole-genome analysis of premature termination codons. Genomics 98, 337–342 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Montgomery SB, Lappalainen T, Gutierrez-Arcelus M & Dermitzakis ET Rare and common regulatory variation in population-scale sequenced human genomes. PLoS Genet. 7, e1002144 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.MacArthur DG et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 335, 823–828 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lappalainen T. et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rivas MA et al. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science 348, 666–669 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Martincorena I. et al. Universal patterns of selection in cancer and somatic tissues. Cell 173, 1823 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pereira FJ et al. Resistance of mRNAs with AUG-proximal nonsense mutations to nonsense-mediated decay reflects variables of mRNA structure and translational activity. Nucleic Acids Res. 43, 6528–6544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kishor A, Ge Z & Hogg JR hnRNP L-dependent protection of normal mRNAs from NMD subverts quality control in B cell lymphoma. EMBO J. 10.15252/embj.201899128 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identifies specific cis elements in RNAs that allow NMD escape. The authors show that this escape mechanism permits survival of B cell lymphomas through stabilization of the mRNA encoding the oncoprotein BCL-2.
- 90.Ge Z, Quek BL, Beemon KL & Hogg JR Polypyrimidine tract binding protein 1 protects mRNAs from recognition by the nonsense-mediated mRNA decay pathway. eLife 10.7554/eLife.11155 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Greaves M & Maley CC Clonal evolution in cancer. Nature 481, 306–313 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mort M, Ivanov D, Cooper DN & Chuzhanova NA A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat 29, 1037–1047 (2008). [DOI] [PubMed] [Google Scholar]
- 93.Gardner LB Nonsense-mediated RNA decay regulation by cellular stress: implications for tumorigenesis. Mol. Cancer Res 8, 295–308 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pestova TV et al. The joining of ribosomal subunits in eukaryotes requires eIF5B. Nature 403, 332–335 (2000). [DOI] [PubMed] [Google Scholar]
- 95.Schaffler K. et al. A stimulatory role for the La-related protein 4B in translation. RNA 16, 1488–1499 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yang R. et al. La-related protein 4 binds poly(A), interacts with the poly(A)-binding protein MLLE domain via a variant PAM2w motif, and can promote mRNA stability. Mol. Cell Biol 31, 542–556 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Leslie NR & Downes CP PTEN function: how normal cells control it and tumour cells lose it. Biochem. J 382, 1–11 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Luna S. et al. A global analysis of the reconstitution of PTEN function by translational readthrough of PTEN pathogenic premature termination codons. Hum. Mutat 10.1002/humu.24186 (2021). [DOI] [PubMed] [Google Scholar]
- 99.Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc. Natl Acad. Sci. USA 68, 820–823 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jain PK Epigenetics: the role of methylation in the mechanism of action of tumor suppressor genes. Ann. N. Y. Acad. Sci 983, 71–83 (2003). [DOI] [PubMed] [Google Scholar]
- 101.Gudikote JP & Wilkinson MF T-cell receptor sequences that elicit strong down-regulation of premature termination codon-bearing transcripts. EMBO J. 21, 125–134 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Buhler M, Paillusson A & Muhlemann O Efficient downregulation of immunoglobulin mu mRNA with premature translation-termination codons requires the 5’-half of the VDJ exon. Nucleic Acids Res. 32, 3304–3315 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gudikote JP, Imam JS, Garcia RF & Wilkinson MF RNA splicing promotes translation and RNA surveillance. Nat. Struct. Mol. Biol 12, 801–809 (2005). [DOI] [PubMed] [Google Scholar]
- 104.Kurosaki T. et al. A post-translational regulatory switch on UPF1 controls targeted mRNA degradation. Genes Dev. 28, 1900–1916 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Park S, Supek F & Lehner B Higher order genetic interactions switch cancer genes from two-hit to one-hit drivers. Nat. Commun 12, 7051 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.de Vries A. et al. Targeted point mutations of p53 lead to dominant-negative inhibition of wild-type p53 function. Proc. Natl Acad. Sci. USA 99, 2948–2953 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Boettcher S. et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 365, 599–604 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sylvain V, Lafarge S & Bignon YJ Dominant-negative activity of a Brca1 truncation mutant: effects on proliferation, tumorigenicity in vivo, and chemosensitivity in a mouse ovarian cancer cell line. Int. J. Oncol 20, 845–853 (2002). [PubMed] [Google Scholar]
- 109.Turajlic S. et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 18, 1009–1021 (2017). [DOI] [PubMed] [Google Scholar]; Using a genome-wide screening approach, this study evaluates the relationship between neoantigens and immune responses in cancer. Among the findings is the observation that frameshift-inducing deletions generated more predicted neoantigens than non-synonymous SNVs.
- 110.Hacohen N, Fritsch EF, Carter TA, Lander ES & Wu CJ Getting personal with neoantigen-based therapeutic cancer vaccines. Cancer Immunol. Res 1, 11–15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kloor M & von Knebel Doeberitz M The immune biology of microsatellite-unstable cancer. Trends Cancer 2, 121–133 (2016). [DOI] [PubMed] [Google Scholar]
- 112.Marrack P & Kappler J The T cell receptor. Science 238, 1073–1079 (1987). [DOI] [PubMed] [Google Scholar]
- 113.Ballhausen A. et al. The shared frameshift mutation landscape of microsatellite-unstable cancers suggests immunoediting during tumor evolution. Nat. Commun 11, 4740 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Waldman AD, Fritz JM & Lenardo MJ A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat. Rev. Immunol 20, 651–668 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Robbins PF et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med 19, 747–752 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bassani-Sternberg M. et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun 7, 13404 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Roh W. et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med 10.1126/scitranslmed.aah3560 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cristescu R. et al. Tumor mutational burden predicts the efficacy of pembrolizumab monotherapy: a pan-tumor retrospective analysis of participants with advanced solid tumors. J. Immunother. Cancer 10.1136/jitc-2021-003091 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Livingstone AM & Fathman CG The structure of T-cell epitopes. Annu. Rev. Immunol 5, 477–501 (1987). [DOI] [PubMed] [Google Scholar]
- 120.Zhao B & Pritchard JR Evolution of the nonsense-mediated decay pathway is associated with decreased cytolytic immune infiltration. PLoS Comput. Biol 15, e1007467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Becker JP et al. NMD inhibition by 5-azacytidine augments presentation of immunogenic frameshift-derived neoepitopes. iScience 24, 102389 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that inhibiting NMD stabilizes frameshifted neoantigen-encoding mRNAs and increases the presentation of some of the encoded neoantigens by class I human leukocyte antigen (HLA) molecules. The authors show that one of the recurrent frameshift mutations in an MSI CRC tumour encodes a highly immunogenic neoantigen that elicits strong cytotoxic T cell responses.
- 122.Bhuvanagiri M. et al. 5-azacytidine inhibits nonsense-mediated decay in a MYC-dependent fashion. EMBO Mol. Med 6, 1593–1609 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mizojiri R. et al. Discovery of novel 5-(piperazine-1-carbonyl)pyridin-2(1H)-one derivatives as orally eIF4A3-selective inhibitors. ACS Med. Chem. Lett. 8, 1077–1082 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gonzalez-Hilarion S. et al. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J. Rare Dis 7, 58 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bokhari A. et al. Targeting nonsense-mediated mRNA decay in colorectal cancers with microsatellite instability. Oncogenesis 7, 70 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.El-Bchiri J. et al. Nonsense-mediated mRNA decay impacts MSI-driven carcinogenesis and anti-tumor immunity in colorectal cancers. PLoS ONE 3, e2583 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhu C et al. UPF1 promotes chemoresistance to oxaliplatin through regulation of TOP2A activity and maintenance of stemness in colorectal cancer. Cell Death Dis. 12, 519 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shum EY et al. The antagonistic gene paralogs Upf3a and Upf3b govern nonsense-mediated RNA decay. Cell 165, 382–395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gotoh M. et al. Comprehensive exploration of novel chimeric transcripts in clear cell renal cell carcinomas using whole transcriptome analysis. Genes Chromosomes Cancer 53, 1018–1032 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Michalak M. et al. (Phospho)proteomic profiling of microsatellite unstable CRC cells reveals alterations in nuclear signaling and cholesterol metabolism caused by frameshift mutation of NMD regulator UPF3A. Int. J. Mol. Sci 10.3390/ijms21155234 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gardner LB Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol. Cell Biol 28, 3729–3741 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pei CL, Fei KL, Yuan XY & Gong XJ LncRNA DANCR aggravates the progression of ovarian cancer by downregulating UPF1. Eur. Rev. Med. Pharmacol. Sci 23, 10657–10663 (2019). [DOI] [PubMed] [Google Scholar]
- 133.Lv ZH, Wang ZY & Li ZY LncRNA PVT1 aggravates the progression of glioma via downregulating UPF1. Eur. Rev. Med. Pharmacol. Sci 23, 8956–8963 (2019). [DOI] [PubMed] [Google Scholar]
- 134.Zhang H, You Y & Zhu Z The human RNA surveillance factor Up-frameshift 1 inhibits hepatic cancer progression by targeting MRP2/ABCC2. Biomed. Pharmacother 92, 365–372 (2017). [DOI] [PubMed] [Google Scholar]
- 135.Polaski JT et al. The origins and consequences of UPF1 variants in pancreatic adenosquamous carcinoma. eLife 7554/eLife.62209 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jo YS, Song SY, Kim MS, Yoo NJ & Lee SH Frameshift mutations of SMG7 essential for nonsense-mediated mRNA decay in gastric and colorectal cancers. Pathol. Oncol. Res 23, 221–222 (2017). [DOI] [PubMed] [Google Scholar]
- 137.Bao X, Huang Y, Xu W & Xiong G Functions and clinical significance of UPF3a expression in human colorectal cancer. Cancer Manag. Res 12, 4271–4281 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sirkisoon SR et al. Interaction between STAT3 and GLI1/tGLI1 oncogenic transcription factors promotes the aggressiveness of triple-negative breast cancers and HER2-enriched breast cancer. Oncogene 37, 2502–2514 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tani H. et al. Identification of hundreds of novel UPF1 target transcripts by direct determination of whole transcriptome stability. RNA Biol. 9, 1370–1379 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schmidt SA et al. Identification of SMG6 cleavage sites and a preferred RNA cleavage motif by global analysis of endogenous NMD targets in human cells. Nucleic Acids Res. 43, 309–323 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Huang L. et al. RNA homeostasis governed by cell type-specific and branched feedback loops acting on NMD. Mol. Cell 43, 950–961 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yepiskoposyan H, Aeschimann F, Nilsson D, Okoniewski M & Muhlemann O Autoregulation of the nonsense-mediated mRNA decay pathway in human cells. RNA 17, 2108–2118 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cho H. et al. SMG5-PNRC2 is functionally dominant compared with SMG5-SMG7 in mammalian nonsense-mediated mRNA decay. Nucleic Acids Res. 41, 1319–1328 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Karousis ED, Gypas F, Zavolan M & Muhlemann O Nanopore sequencing reveals endogenous NMD-targeted isoforms in human cells. Genome Biol. 22, 223 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lykke-Andersen S. et al. Human nonsense-mediated RNA decay initiates widely by endonucleolysis and targets snoRNA host genes. Genes Dev. 28, 2498–2517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lou CH et al. Nonsense-mediated RNA decay influences human embryonic stem cell fate. Stem Cell Rep. 6, 844–857 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.You B. et al. Androgen receptor promotes renal cell carcinoma (RCC) vasculogenic mimicry (VM) via altering TWIST1 nonsense-mediated decay through lncRNA-TANAR. Oncogene 40, 1674–1689 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rasti A. et al. Cytoplasmic expression of Twist1, an EMT-related transcription factor, is associated with higher grades renal cell carcinomas and worse progression-free survival in clear cell renal cell carcinoma. Clin. Exp. Med 18, 177–190 (2018). [DOI] [PubMed] [Google Scholar]
- 149.Rossello-Tortella M. et al. Epigenetic loss of the transfer RNA-modifying enzyme TYW2 induces ribosome frameshifts in colon cancer. Proc. Natl Acad. Sci. USA 117, 20785–20793 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Valacca C. et al. Sam68 regulates EMT through alternative splicing-activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. J. Cell Biol 191, 87–99 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.He J & Ma X Interaction between lncRNA and UPF1 in tumors. Front. Genet 12, 624905 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chen BL, Wang HM, Lin XS & Zeng YM UPF1: a potential biomarker in human cancers. Front. Biosci 26, 76–84 (2021). [DOI] [PubMed] [Google Scholar]
- 153.Andjus S, Morillon A & Wery M From yeast to mammals, the nonsense-mediated mRNA decay as a master regulator of long non-coding RNAs functional trajectory. Noncoding RNA 10.3390/ncrna7030044 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Aspden JL et al. Extensive translation of small open reading frames revealed by Poly-Ribo-Seq. eLife 3, e03528 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ruiz-Orera J, Messeguer X, Subirana JA & Alba MM Long non-coding RNAs as a source of new peptides. eLife 3, e03523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Anderson DM et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 160, 595–606 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Guerra-Almeida D & Nunes-da-Fonseca R Small open reading frames: how important are they for molecular evolution? Front. Genet 11, 574737 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gutschner T. et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 73, 1180–1189 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Meseure D. et al. Prognostic value of a newly identified MALAT1 alternatively spliced transcript in breast cancer. Br. J. Cancer 114, 1395–1404 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Malakar P. et al. Long noncoding RNA MALAT1 promotes hepatocellular carcinoma development by SRSF1 upregulation and mTOR activation. Cancer Res. 77, 1155–1167 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lee NK et al. MALAT1 promoted invasiveness of gastric adenocarcinoma. BMC Cancer 17, 46 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gong C, Kim YK, Woeller CF, Tang Y & Maquat LE SMD and NMD are competitive pathways that contribute to myogenesis: effects on PAX3 and myogenin mRNAs. Genes Dev. 23, 54–66 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Park E & Maquat LE Staufen-mediated mRNA decay. Wiley Interdiscip. Rev. RNA 4, 423–435 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chang L. et al. Upregulation of SNHG6 regulates ZEB1 expression by competitively binding miR-101-3p and interacting with UPF1 in hepatocellular carcinoma. Cancer Lett. 383, 183–194 (2016). [DOI] [PubMed] [Google Scholar]
- 165.Karam R, Wengrod J, Gardner LB & Wilkinson MF Regulation of nonsense-mediated mRNA decay: implications for physiology and disease. Biochim. Biophys. Acta 1829, 624–633 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Huang L & Wilkinson MF Regulation of nonsense-mediated mRNA decay. Wiley Interdiscip. Rev. RNA 3, 807–828 (2012). [DOI] [PubMed] [Google Scholar]
- 167.Bruno IG et al. Identification of a microRNA that activates gene expression by repressing nonsense-mediated RNA decay. Mol. Cell 42, 500–510 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wang D, Wengrod J & Gardner LB Overexpression of the c-myc oncogene inhibits nonsense-mediated RNA decay in B lymphocytes. J. Biol. Chem 286, 40038–40043 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hetz C & Saxena S ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol 13, 477–491 (2017). [DOI] [PubMed] [Google Scholar]
- 170.Zhao L & Ackerman SL Endoplasmic reticulum stress in health and disease. Curr. Opin. Cell Biol 18, 444–452 (2006). [DOI] [PubMed] [Google Scholar]
- 171.Chawla R. et al. Human UPF1 interacts with TPP1 and telomerase and sustains telomere leading-strand replication. EMBO J. 30, 4047–4058 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ngo GHP, Grimstead JW & Baird DM UPF1 promotes the formation of R loops to stimulate DNA double-strand break repair. Nat. Commun 12, 3849 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lavysh D & Neu-Yilik G UPF1-mediated RNA decay — Danse Macabre in a cloud. Biomolecules 10.3390/biom10070999 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Roberts TL et al. Smg1 haploinsufficiency predisposes to tumor formation and inflammation. Proc. Natl Acad. Sci. USA 110, E285–E294 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gupta D. et al. Case series of outcomes in advanced cancer patients with single pathway alterations receiving N-of-one therapies. NPJ Precis. Oncol 6, 18 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Alexandrov A, Shu MD & Steitz JA Fluorescence amplification method for forward genetic discovery of factors in human mRNA degradation. Mol. Cell 65, 191–201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Boelz S, Neu-Yilik G, Gehring NH, Hentze MW & Kulozik AE A chemiluminescence-based reporter system to monitor nonsense-mediated mRNA decay. Biochem. Biophys. Res. Commun 349, 186–191 (2006). [DOI] [PubMed] [Google Scholar]
- 178.Paillusson A, Hirschi N, Vallan C, Azzalin CM & Muhlemann O A GFP-based reporter system to monitor nonsense-mediated mRNA decay. Nucleic Acids Res. 33, e54 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wang M. et al. Assessing the activity of nonsense-mediated mRNA decay in lung cancer. BMC Med. Genomics 10, 55 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dang Y. et al. Inhibition of nonsense-mediated mRNA decay by the natural product pateamine A through eukaryotic initiation factor 4AIII. J. Biol. Chem 284, 23613–23621 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yamashita A, Ohnishi T, Kashima I, Taya Y & Ohno S Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev. 15, 2215–2228 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Martinez-Nunez RT et al. Modulation of nonsense mediated decay by rapamycin. Nucleic Acids Res. 45, 3448–3459 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bordonaro M & Lazarova D Amlexanox and UPF1 modulate Wnt signaling and apoptosis in HCT-116 colorectal cancer cells. J. Cancer 10, 287–292 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Durand S. et al. Inhibition of nonsense-mediated mRNA decay (NMD) by a new chemical molecule reveals the dynamic of NMD factors in P-bodies. J. Cell Biol 178, 1145–1160 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Palma M. et al. A role for AKT1 in nonsense-mediated mRNA decay. Nucleic Acids Res. 49, 11022–11037 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhao J. et al. Molecular profiling of individual FDA-approved clinical drugs identifies modulators of nonsense-mediated mRNA decay. Mol. Ther. Nucleic Acids 27, 304–318 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Martin L. et al. Identification and characterization of small molecules that inhibit nonsense-mediated RNA decay and suppress nonsense p53 mutations. Cancer Res. 74, 3104–3113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Iwatani-Yoshihara M. et al. Discovery and characterization of a eukaryotic initiation factor 4A-3-selective inhibitor that suppresses nonsense-mediated mRNA decay. ACS Chem. Biol 12, 1760–1768 (2017). [DOI] [PubMed] [Google Scholar]
- 189.Kungwankiattichai S. et al. Maintenance with hypomethylating agents after allogeneic stem cell transplantation in acute myeloid leukemia and myelodysplastic syndrome: a systematic review and meta-analysis. Front. Med 9, 801632 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Pal M, Ishigaki Y, Nagy E & Maquat LE Evidence that phosphorylation of human Upfl protein varies with intracellular location and is mediated by a wortmannin-sensitive and rapamycin-sensitive PI 3-kinase-related kinase signaling pathway. RNA 7, 5–15 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Yakhni M. et al. Homoharringtonine, an approved anti-leukemia drug, suppresses triple negative breast cancer growth through a rapid reduction of anti-apoptotic protein abundance. Am. J. Cancer Res 9, 1043–1060 (2019). [PMC free article] [PubMed] [Google Scholar]
- 192.Pedersen S, Celis JE, Nielsen J, Christiansen J & Nielsen FC Distinct repression of translation by wortmannin and rapamycin. Eur. J. Biochem 247, 449–456 (1997). [DOI] [PubMed] [Google Scholar]
- 193.Usuki F. et al. Inhibition of SMG-8, a subunit of SMG-1 kinase, ameliorates nonsense-mediated mRNA decay-exacerbated mutant phenotypes without cytotoxicity. Proc. Natl Acad. Sci. USA 110, 15037–15042 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Xu W. et al. Reactivation of nonsense-mediated mRNA decay protects against C9orf72 dipeptide-repeat neurotoxicity. Brain 142, 1349–1364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.CMP Health Care Media. Cancer Management:A Multi-disciplinary Approach, Medical, Surgical & Radiation Oncology. 13th edn (CMP Health Care Media, 2011). [Google Scholar]
- 196.Popp MW & Maquat LE Attenuation of nonsense-mediated mRNA decay facilitates the response to chemotherapeutics. Nat. Commun 6, 6632 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study is among the first to provide proof of principle that NMD-modulatory therapy exhibits efficacy in promoting tumour cell death when used in combination with other chemotherapeutic agents.
- 197.Tani H, Torimura M & Akimitsu N The RNA degradation pathway regulates the function of GAS5 a non-coding RNA in mammalian cells. PLoS ONE 8, e55684 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Cowen LE & Tang Y Identification of nonsense-mediated mRNA decay pathway as a critical regulator of p53 isoform beta. Sci. Rep 7, 17535 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Reddy JC et al. WT1-mediated transcriptional activation is inhibited by dominant negative mutant proteins. J. Biol. Chem 270, 10878–10884 (1995). [DOI] [PubMed] [Google Scholar]
- 200.McMahon SB MYC and the control of apoptosis. Cold Spring Harb. Perspect. Med 4, a014407 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Jinesh GG, Sambandam V, Vijayaraghavan S, Balaji K & Mukherjee S Molecular genetics and cellular events of K-Ras-driven tumorigenesis. Oncogene 37, 839–846 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kim E. et al. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 27, 617–630 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Rahman MA, Lin KT, Bradley RK, Abdel-Wahab O & Krainer AR Recurrent SRSF2 mutations in MDS affect both splicing and NMD. Genes Dev. 34, 413–427 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Gudikote JP et al. Inhibition of nonsense-mediated decay rescues p53beta/gamma isoform expression and activates the p53 pathway in MDM2-overexpressing and select p53-mutant cancers. J. Biol. Chem 10.1016/j.jbc.2021.101163 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides evidence that inhibiting NMD triggers tumour cell apoptosis and enhanced tumour radiosensitivity by stabilizing two alternatively spliced forms of p53 that are largely insensitive to repression by the p53 negative regulator MDM2.
- 205.Lee SR, Pratt GA, Martinez FJ, Yeo GW & Lykke-Andersen J Target discrimination in nonsense-mediated mRNA decay requires Upf1 ATPase activity. Mol. Cell 59, 413–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kurosaki T & Maquat LE Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci 129, 461–467 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Buchwald G. et al. Insights into the recruitment of the NMD machinery from the crystal structure of a core EJC-UPF3b complex. Proc. Natl Acad. Sci. USA 107, 10050–10055 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Bono F, Ebert J, Lorentzen E & Conti E The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell 126, 713–725 (2006). [DOI] [PubMed] [Google Scholar]
- 209.Ni Y. et al. The role of tumor-stroma interactions in drug resistance within tumor microenvironment. Front. Cell Dev. Biol 9, 637675 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Al Tameemi W, Dale TP, Al-Jumaily RMK & Forsyth NR Hypoxia-modified cancer cell metabolism. Front. Cell Dev. Biol 7, 4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Daskalaki I, Gkikas I & Tavernarakis N Hypoxia and selective autophagy in cancer development and therapy. Front. Cell Dev. Biol 6, 104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Senft D & Ronai ZA Adaptive stress responses during tumor metastasis and dormancy. Trends Cancer 2, 429–442 (2016). [DOI] [PubMed] [Google Scholar]
- 213.Saavedra-Garcia P. et al. Systems level profiling of chemotherapy-induced stress resolution in cancer cells reveals druggable trade-offs. Proc. Natl Acad. Sci. USA 10.1073/pnas.2018229118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Popp MW & Maquat LE Nonsense-mediated mRNA decay and cancer. Curr. Opin. Genet Dev 48, 44–50 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Ruhle A. et al. Hypoxia dynamics on FMISO-PET in combination with PD-1/PD-L1 expression has an impact on the clinical outcome of patients with head-and-neck squamous cell carcinoma undergoing chemoradiation. Theranostics 10, 9395–9406 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Saxena K & Jolly MK Acute vs. chronic vs. cyclic hypoxia: their differential dynamics, molecular mechanisms, and effects on tumor progression. Biomolecules 10.3390/biom9080339 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Oakes SA Endoplasmic reticulum stress signaling in cancer cells. Am. J. Pathol 190, 934–946 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Friedl P & Alexander S Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147, 992–1009 (2011). [DOI] [PubMed] [Google Scholar]
- 219.Fendt SM, Frezza C & Erez A Targeting metabolic plasticity and flexibility dynamics for cancer therapy. Cancer Discov. 10, 1797–1807 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]